

Abstract
The purpose of this study was to determine the effects of the histone deacetylase inhibitor, MS-275, on the Fas signaling pathway and susceptibility of osteosarcoma (OS) to Fas ligand (FasL)-induced cell death. OS metastasizes almost exclusively to the lungs. We have shown that Fas expression in OS cells is inversely correlated with their metastatic potential. Fas+ cells are rapidly eliminated when they enter the lungs via interaction with FasL, which is constitutively expressed in the lungs. Fas− OS cells escape this FasL-induced apoptosis and survive in the lung microenvironment. Moreover, upregulation of Fas in established OS lung metastases results in tumor regression. Therefore, agents that upregulate Fas expression or activate the Fas signaling pathway may have therapeutic potential. Treatment of Fas− metastatic OS cell lines with 2 μM MS-275 sensitized cells to FasL-induced cell death in vitro. We found that MS-275 did not alter the expression of Fas on the cell surface; rather it resulted in the downregulation of the anti-apoptotic protein, c-FLIP (cellular FLICE-inhibitory protein), by inhibiting c-FLIP mRNA. Downregulation of c-FLIP correlated with caspase activation and apoptosis induction. Treatment of nu/nu-mice with established OS lung metastases with oral MS-275 resulted in tumor regression, increased apoptosis and a significant inhibition of c-FLIP expression in tumors. Histopathological examination of mice showed no evidence of significant toxicity. Overall, these results suggest that the mechanism by which MS-275 sensitizes OS cells and lung metastases to FasL-induced cell death may be by a direct reduction in the expression of c-FLIP.
Keywords: c-FLIP, Fas, FasL, histone deacetylase inhibitors, MS-275, Entinostat, osteosarcoma
INTRODUCTION
Osteosarcoma (OS) is the most common primary malignant tumor of the bone in pediatric patients. Although survival of patients with nonmetastatic disease has improved dramatically, patients who present with metastasis, primarily to the lung, have a poor prognoses with overall survival rates of less than 20% [1, 2]. Therefore, the need for novel therapeutic strategies for metastatic OS is ongoing.
Our laboratory has demonstrated that the Fas/Fas ligand (FasL) pathway plays an important role in the metastatic potential of OS. We demonstrated in OS in vivo models that while the OS primary tumor in the bone contained a population of both Fas+ and Fas− cells, pulmonary metastases were Fas−, suggesting that Fas expression was inversely correlated with metastatic potential [3]. In addition, pulmonary metastases from patients were found to be Fas−. The lung is one of the few organs to constitutively express FasL [3–6]. We have demonstrated that Fas+ OS cells are cleared in the lung by activation of Fas signaling and apoptosis, while Fas− cells have the ability to evade this and survive to form metastatic lesions [3–6]. In particular, we showed a correlation between Fas expression and the clearance of OS cells from the lung [3]. Fas+ OS cells were cleared within 24 hours while Fas− cells remained. Upregulation of Fas expression in Fas− OS lung metastases resulted in tumor regression indicating that this may have therapeutic potential [4, 7–10].
The Fas/FasL signaling pathway has been implicated in the pathogenesis of several tumor types and malignancies. The Fas receptor is known to induce apoptosis by binding to FasL. Receptor-ligand interaction induces the recruitment of Fas-associated death domain (FADD) and procaspase-8 to form the death-inducing signaling complex (DISC). Interaction of procaspase-8 at the DISC leads to its autocatalytic cleavage and activation, which result in caspase cleavage either via the mitochondrial pathway or by direct activation of the effector caspases. Inhibition of Fas-mediated apoptosis is regulated by FLICE-inhibitory protein (FLIP), the structural homologue of procaspase-8 [11]. Cellular FLIP (c-FLIP) competes with procaspase-8 for recruitment to FADD at the DISC [7]. c-FLIP has been found to be overexpressed in numerous cancer cell lines and primary cells and tissues from patients [12–18]. Since overexpression of c-FLIP is associated with increased resistance to death receptor pathways, several investigators have found that downregulation of c-FLIP results in the sensitization of tumor cell lines to Fas-mediated apoptosis.
Histone deacetylase (HDAC) inhibitors are promising anticancer agents with therapeutic potential against numerous solid and hematological malignancies. Several HDAC inhibitors, including MS-275, are in clinical development for various cancer types. HDAC inhibitors have been identified to induce cell cycle arrest and apoptosis in vitro and in vivo. Specifically, many HDAC inhibitors have been shown to sensitize cells to Fas-mediated apoptosis. However, the specific mechanism of how this may occur has been shown to vary by tumor type and drug. For example, upregulation of Fas or FasL expression has been demonstrated in neuroblastoma, promyelocytic leukemia and uveal melanoma following treatment with the HDAC inhibitors CBHA, apicidin and depsipeptide, respectively [19–22]. Induction of cytotoxicity of acute leukemia cells by the HDAC inhibitor, PCI-24781, has been shown to be dependent on caspase-8 and FADD [23]. Additionally, downregulation of c-FLIP expression by the HDAC inhibitor depsipeptide has been observed in both chronic lymphocytic leukemia (CLL) cells and OS cells [22, 24]. MS-275 has also been shown to inhibit c-FLIP expression CLL cells and was followed by the induction of caspase-dependent apoptosis [25]. However, few studies have examined the use of HDAC inhibitors to treat pediatric solid tumors, including OS [19, 26, 27].
Because of the finding that HDAC inhibitors can activate the Fas pathway in some solid tumors and our data have shown that upregulation of Fas expression in OS cells is therapeutically beneficial, we sought to determine whether the HDAC inhibitor, MS-275, upregulates Fas expression on the surface of OS cells. Our results showed that at subtoxic doses, MS-275 was able to sensitize metastastic OS cell lines to FasL-induced cell death in vitro and induced the regression of established lung metastases in vivo. We identified the mechanism by which this sensitization occurs involves MS-275-induced downregulation of c-FLIP mRNA and protein expression.
MATERIALS AND METHODS
Cell Lines and Reagents
The human LM7 OS lung metastatic cell line was created in our laboratory by the repeated intravenous recycling of its parent cell line, SAOS-2, through the lungs of nude mice [3, 28]. The human OS cell line CCH-OS-D was provided by Dr. Dennis Hughes (The University of Texas MD Anderson Cancer Center). Both LM7 and CCH-OS-D cells have been confirmed to have a high metastatic potential and low expression of Fas as compared to SAOS-2 cells, and were therefore utilized in this study. All cells were mouse antibody protection (MAP) tested and were found to be mycoplasma-negative. Cell lines were validated by short tandem repeat (STR) DNA fingerprinting using the AmpF STR Identifier kit according to the manufacturer’s instructions (Applied Biosystems cat 4322288). The STR profiles were compared with known ATCC fingerprints (ATCC.org), to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 and to the MD Anderson fingerprint database. The STR profiles matched either known DNA fingerprints or were unique. The authenticity of the cells was determined by the Characterized Cell Line core at UT MD Anderson Cancer Center. Cell lines were maintained in complete Dulbecco’s modified Eagle’s medium (Whittaker Bioproducts Inc. Walkersville, MD) containing 10% heat-inactivated bovine serum (Intergen, Purchase, NJ) at 37°C in 5% CO2. Recombinant soluble superFasL (sFasL) was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY). MS-275 was a kind gift from Syndax Pharmaceuticals, Inc. (Waltham, MA). The dose of 2 μM MS-275 was found to be the half maximal inhibitory concentration (IC50) in dose response experiments. Additionally, dose response experiments with sFasL was also performed (data not shown). Based on these dose response studies, 2 μM MS-275 and 10 ng/ml sFasL were utilized for subsequent in vitro experiments.
Clonogenic and MTT Assays
OS cells were seeded into six-well culture plates and allowed to attach overnight. Cells were then treated with 10 ng/ml sFasL for 24 hours, 2 μM MS-275 for 48 hours or a combination of both. Following treatment, the medium was removed and replaced with fresh medium. After 10–12 days of incubation, cells were washed with phosphate buffered saline (PBS) and fixed with formalin. Cells were then stained with 0.4% crystal violet for 30 minutes to visualize the colonies. The colonies were counted using the Leica direct laser metal sintering (DLMS) light microscope by averaging eight fields per well. One colony was defined as 50 cells. 3-(4,5-dimethylthiazol-2yl)2,5-diphenyltetrazolium bromide (MTT) assay was used to assess the viability of cells after pretreatment with the caspase inhibitor Z-VAD-fmk (Enzo Life Sciences Inc., Farmingdale, NY) followed by treatment with MS-275 and sFasL. Briefly, 3000 cells were plated on 96-well plates and pre-treated with 20 μM Z-VAD-fmk for 2 hours followed by treatment with 10 ng/ml sFasL for 24 hours, MS-275 for 48 hours or a combination of both MS-275 and sFasL Untreated cells and cells treated with a single agent served as controls. After treatment, MTT was added to the cells at a concentration of 0.08 mg/ml for 4 hours followed by lysis with 0.1 ml of dimethyl sulfoxide (DMSO). Using a microtiter plate reader, the absorbance was measured at 570 nm to calculate the cytotoxicity. These experiments were performed in triplicate and repeated three times.
Western Blot Analysis and Antibodies
Total cell fractions were lysed using radioimmunoprecipitation buffer. For determination of acetyl-histone H3 (AcH3) and histone H3 expression, nuclear fractionation was performed by lysing cells with nuclear extraction buffer containing 50 mM 4-(2 hydroxyethyl)-1-piperazineethanesulfonic acid] - potassium salt (HEPES-K) pH 8.0, 140 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0, 0.40% Igpel CA-630, 0.20% Triton X-100, 10 μg/ml Aprotinin, 10 μg/ml Leupeptin, 2 ng/ml Pepstatin and 0.5 mM phenylmethylsulfonyl fluoride (PMSF). Nuclei were pelleted via centrifugation at 3000 x g for 3 minutes. Pellets were resuspended in sodium dodecyl sulfate (SDS) lysis buffer and sonicated for 15 minutes using the Misonex Sonicator 3000 at a power of 1. Lysates were then centrifuged for 10 minutes at 15,000 x g and the supernatant was collected as the nuclear extract. Equal amounts of protein were separated on 10% SDS- polyacrylamide gel electrophoresis (PAGE) gels and transferred onto a nitrocellulose membrane. Immunoblot analysis was performed using antibodies specific for AcH3 (Millipore Corp., Billerica, Massachusetts), histone H3 (Abcam, Cambridge, UK), caspase-8 (C15) and c-FLIP (NF6) (Enzo Life Sciences Inc., Farmingdale, NY). Loading was confirmed by probing membranes with an anti-β-actin antibody (Sigma Aldrich, Inc.). Western blot analysis for all experiments was repeated three times. Densitometric analysis on immunoblots was performed using the ImageJ software program (version 1.42q; National Institutes of Health).
Caspase-Activity Assays
Colorimetric caspase-8 and caspase-3 activity assays were performed using Ile-Glu-Thr-Asp p-nitroaniline (IETD-pNA) and Asp-Glu-Val-Asp p-nitroaniline (DEVD-pNA) substrates, respectively, according to manufacturer’s instructions (BioVision Inc., Palo Alto, CA). The assays were performed in triplicate.
Quantitative Real-Time PCR
Total RNA was extracted using Trizol reagent (Life Technologies Inc., Gaithersburg, MD) and subjected to cDNA synthesis using a reverse transcription system (Promega Corp., Madison, WI). The PCR reaction mixture contained 100 ng of reverse-transcribed total RNA, 50 nM forward and reverse primers, and 12.5 μl of SYBR® green buffer (Bio-Rad Laboratories Inc., Hercules, California) in a final volume of 25 μl. Quantitative real-time PCR was carried out in triplicate using the Bio-Rad 105 Real-time PCR detection system. The primer sequences used were 5′ACCG AGACTACGACA GCTTT GTG3′ and 5′CAATGTGAAGATCCAGGAG TGGG3′ for c-FLIP and 5′ATCATGG TTTGAGACCTTCAACA3′ and 5′CATCTCTTGCTCGAAG TCCA3′ for β-actin as a control. The cycling conditions were 3 minutes at 95°C followed by 45 cycles at 57°C for 30 seconds, 95°C for 30 seconds and 1 minute at 60°C. All experiments were repeated three times.
c-FLIP Short Hairpin RNA-Expressing Clones
LM7 OS cells were infected with retroviruses expressing shRNA-cFLIP, shRNA-scrambled or vector pGFP-V-RS (Origene Technologies, Inc., Rockville, MD). To select transfected clones, cells were grown in media containing puromycin. Knockdown was confirmed by western blot analysis using an antibody against c-FLIP (NF6; Enzo Life Sciences Inc., Farmingdale, NY) and by quantitative real time-PCR using primers against c-FLIP (5′ACCGAGACTACGACAGCTTTGTG3′ and 5′CAATGTGAAGATCCAGG AGTG GG3′. The shRNA sequence for c-FLIP knockdown was TGCACAGTTCACCG AGAAGCTGACTTCTT (ID: GI355835) (Origene Technologies, Inc.).
Animal Studies
Female nu/nu mice were purchased from the National Cancer Institute (Bethesda, MD). All animals used for in vivo experiments were housed in standard cages, at five mice per cage and provided with food and water ad libitum. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at The University of Texas MD Anderson Cancer Center. Mice were injected with 2 × 106 LM7 cells via the tail vein. Formation of microscopic and macroscopic lung metastases was verified within 5 - 6 weeks by observation and hematoxylin and eosin (H&E) staining of resected lung tissue. The mice were randomly divided into 2 groups (10 mice/group) and received 20 mg/kg MS-275 or DMSO (as a control) in 0.2 ml by oral gavage every other day for 15 days. Survival studies were conducted using 15 mice per group using the same method described above to establish lung metastases. MS-275- and DMSO-treated mice were observed to assess their survival. The dose of MS-275 used in our in vivo studies was comparable to the dose used in other tumor mouse models [26].
Immunohistochemistry
Lung tissue sections were deparaffinized in xylene, rehydrated, and examined using immunohistochemistry. Sections were incubated with 3% H2O2 for 12 minutes to block exogenous peroxidase and then incubated with PBS containing 10% normal horse serum. Antibodies against AcH3 (Millipore Corp., Billerica, Massachusetts) and FLIP (Abbiotec, San Diego, CA) were applied and left overnight at 4°C. Secondary antibodies labeled with horseradish peroxidase were then applied for 2 hours at room temperature. Slides were then developed with 3,3′-diaminobenzidine (DAB) as a substrate and counterstained with hematoxylin. Negative controls were prepared via omission of the primary antibodies. Paraffinized sections of murine liver and heart tissue were subjected to H&E staining and then pathological analysis to identify any drug-induced toxic effects.
Apoptosis was measured using a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. Lung tissue sections were deparaffinized as described above, incubated with 20 mg/mL proteinase K (Sigma Aldrich, Inc.) for 10 minutes, 3% H2O2 for 12 minutes, and terminal deoxynucleotidyl transferase buffer for 2 minutes at room temperature. Tissue sections were then incubated with terminal transferase (Boehringer-Mannheim Corp., Mannheim, Germany) and biotin-160 (Roche, Indianapolis, IN) in a humidity chamber at 37°C for 1 hour. Following incubation, sections were incubated with 2% bovine serum albumin (BSA) for 10 minutes followed by horseradish peroxidase-conjugated streptavidin at 37°C for 1 hour. The tissue sections were washed twice with double-distilled water, stained with DAB, and counterstained with hematoxylin, as described above.
Statistical Analysis
Statistical comparisons of groups were performed using student t-test and Kaplan-Meier curves were generated using the GraphPad Prism 5 software program. A p-value of less than 0.05 was deemed as statistically significant.
RESULTS
MS-275 Sensitizes OS Cells to FasL-Induced Cell Death
The effects of MS-275 on clonogenic growth were examined in the metastatic OS cell lines, LM7 and CCH-OS-D. Both cell lines had relatively low levels of Fas expression, and as expected, were minimally sensitive to the effects of sFasL (Fig. 1). While MS-275 alone partially inhibited cell survival, the combination of MS-275 and sFasL resulted in a greater inhibition of clonogenic survival (Fig. 1). This suggested that pretreatment of OS cells with MS-275 sensitized Fas− cells to sFasL. To determine the ability of MS-275 and sFasL to induce apoptosis, we examined the caspase activity in cells after treatment. We observed lower levels of full-length caspase-8 following treatment with both MS-275 and sFasL than after treatment with either agent alone (Fig. 2A). In fact, no decrease in full-length caspase-8 was observed in cells incubated with sFasL alone. Because caspase cleavage products do not always equate to caspase activity, we performed caspase activity assays in both cell lines. Once again we saw no significant change in caspase activity in cells treated with sFasL alone. However, pretreatment of cells with MS-275 significantly increased sFasL-induced caspase-8 and caspase-3 activity (Figs. 2B and 2C). Pretreatment with the pan-caspase inhibitor Z-VAD-fmk inhibited cell death following treatment with MS-275 alone and with sFasL, further confirming that MS-275 sensitizes Fas− OS cells to sFasL in a caspase-dependent manner (Fig. 2D).
Figure 1. MS-275 sensitizes OS cells to FasL-induced cell death.
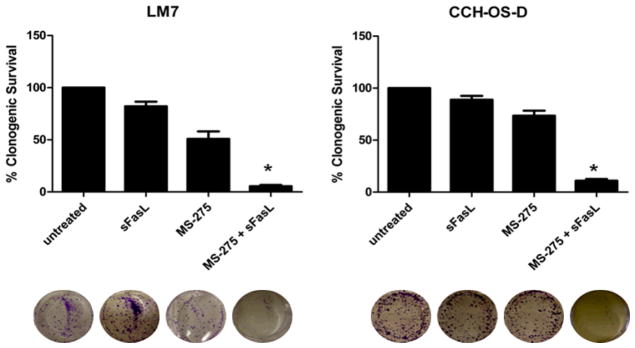
LM7 and CCH-OS-D cells were treated with 2 μM MS-275 for 48 hours with or without 10 ng/ml sFasL for 24 hours. Drug was removed at the end of the treatment period and replaced with fresh medium. Cytotoxicity was quantified 15 days later by counting colonies following crystal violet staining. Colonies were counted as 50 cells per colony. *P < 0.05 compared to control and either agent alone, n=3.
Figure 2. Effect of MS-275 on caspase activity in OS cells.
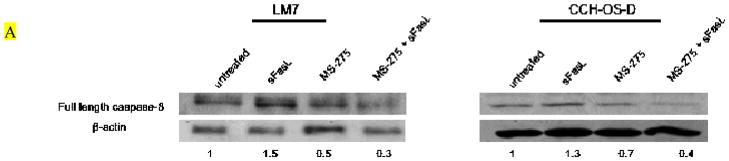

LM7 and CCH-OS-D cells were treated with 2 μM MS-275 for 48 hours with or without 10 ng/ml sFasL for 24 hours. Cell lysates were separated by 10% SDS-PAGE. (A) Immunoblot analysis was done using an antibody against pro-caspase-8. Numbers represent densitometry changes relative to β-actin and compared to the untreated sample. (B) Caspase-8 and (C) caspase-3 activity assays following treatment. *P-values are compared to control and either agent alone. (D) LM7 and CCH-OS-D cells were pretreated with 20 μM Z-VAD-fmk for 2 hours followed by treatment with 2 μM MS-275 for 48 hours with or without sFasL for 24 hours. MTT assay was performed to assess cytotoxicity. *p < 0.02, n=3.
MS-275 Induces Accumulation of Acetylated Histone H3
To confirm that MS-275 functions as a HDAC inhibitor, we examined changes in acetylated histone H3 levels by western blot at the dose that inhibited clonogenic survival. MS-275 induced a time-dependent increase in acetylated histone H3 which peaked at 48 hours, the timepoint at which MS-275 was able to both inhibit clonogenic survival and sensitize cells to sFasL. The total levels of histone H3 remained unaltered (Fig. 3).
Figure 3. MS-275 induces accumulation of acetylated histone H3.
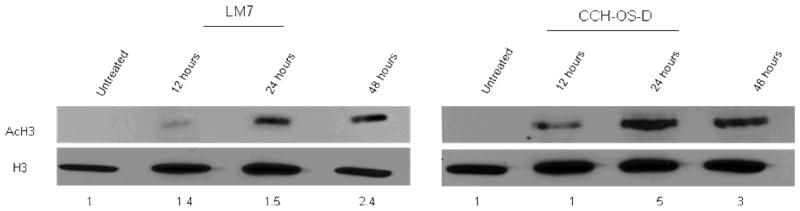
LM7 and CCH-OS-D cells were treated with 2 μM MS-275 for the indicated times. Histones were then extracted by lysing cells with nuclear extraction buffer followed by centrifugation and sonication of lysates. Lysates were then subjected to separation on 10% SDS-PAGE followed by immunoblot analysis with antibodies against acetylated H3 and total histone H3. Numbers represent densitometry changes relative to histone H3 and compared to the untreated sample.
Effect of MS-275 c-FLIP mRNA and Protein Expression
We previously demonstrated that in both LM7 and CCH-OS-D cells, Fas expression was unaltered after treatment with MS-275 [29]. This suggested that the mechanism by which MS-275 sensitizes OS cells to sFasL is not mediated through induction Fas expression on the cell surface but may involve mediators of the Fas/FasL signaling pathway downstream of the Fas receptor itself. Quantitative real time-PCR and western blot analysis showed that cells treated with MS-275 had decreased levels of c-FLIP mRNA and protein (Figs. 4A and 4B). All OS cell lines tested expressed only the c-FLIPL isoform and not the c-FLIPS (data not shown).
Figure 4. MS-275 downregulates c-FLIP expression in OS cells.
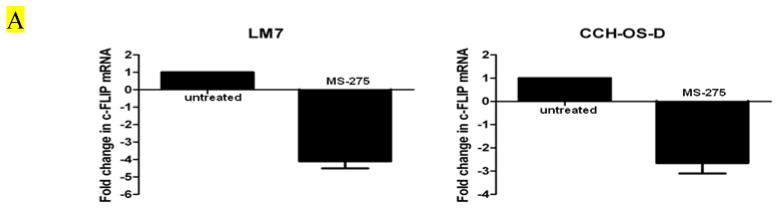
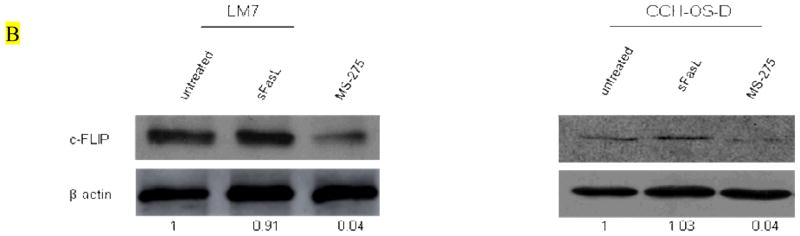
(A) OS cells were treated with 2μM MS-275 for 24 hours. RNA was extracted and analyzed by quantitative real-time PCR using primers specific for c-FLIP. (B) OS cells were treated with 2μM MS-275 for 48 hours. Cells were lysed and analyzed by western blot using an anti-c-FLIP antibody.
Selective Knockdown of c-FLIP Increases Sensitivity of OS Cells to FasL-Induced Cell Death
To determine the relationship between MS-275-induced sensitization of OS cells to FasL and the downregulation of c-FLIP expression following treatment, we selectively decreased the expression of c-FLIP in OS cells by small hairpin-mediated knockdown of c-FLIP (data not shown). MTT assay was performed to assess cytotoxicity following treatment of negative control, scrambled control and shFLIP transduced cells with sFasL. Our data demonstrate that knockdown of c-FLIP significantly increased the sensitivity of OS cells to FasL-induced cell death (Fig. 5).
Figure 5. c-FLIP knockdown in OS cells increases the sensitivity to FasL-induced cell death.
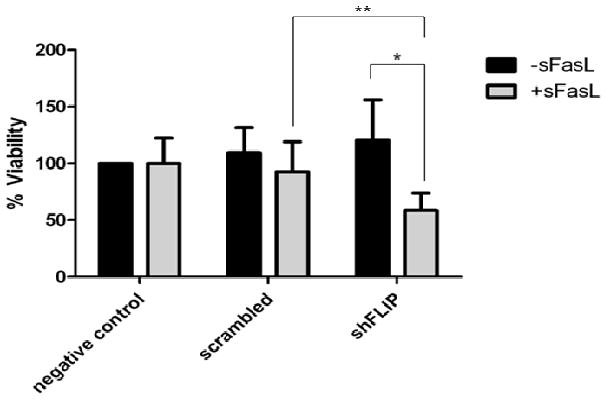
LM7 OS cells were transduced with shRNA to c-FLIP, scrambled and negative control vectors. Cells were then treated with control media or sFasL and MTT assay was performed in triplicate to measure cytotoxicity. *p=0.029, **p=0.049
Effect of Oral Administration of MS-275 on LM7 OS Lung Metastases In Vivo
Since our results indicated that MS-275 sensitized OS cells to FasL-induced cell death in vitro we investigated the effects of orally administering MS-275 in vivo. The lung epithelium constitutively expresses FasL; therefore, MS-275 would be expected to induce tumor cell death and regression of OS lung metastases. To ensure that the drug was effectively distributed to the metastatic tumors in the lung, tumor sections were analyzed by immunohistochemistry for acetylated histone H3. As shown in Fig. 6A, tumor sections obtained from MS-275-treated mice had increased acetylated histone H3 expression. Tumor sections from MS-275–treated mice had significantly higher levels of TUNEL positivity and lower levels of c-FLIP expression than did DMSO-treated mice, which were consistent with our in vitro data (Figs. 6B and 6C).
Figure 6. Effect of oral administration of MS-275 on OS lung metastases.


Mice were injected intravenously with 2×106 LM7 cells. Treatment with 20 mg/ml MS-275 via oral gavage was initiated 42 days after tumor cell injection and continued every other day for 15 days. Mice were then sacrificed, and their lungs were extracted and analyzed using immunohistochemistry. Lung tissue sections were stained with antibodies specific for (A) AcH3, (B) TUNEL, and (C) c-FLIP (magnification, 400x). (D) Murine liver and heart tissue sections were analyzed using H&E staining.
There was no evidence of significant organ toxicity observed following MS-275 treatment. MS-275–treated mice did experience intermittent diarrhea during treatment; however, this also occurred in the DMSO-treated mice. Examination of liver and heart tissue sections showed no evidence of irreversible cellular injury. Heart sections were entirely unremarkable. Liver sections from MS-275–treated mice had slightly larger and more metabolically active nuclei than did those from DMSO-treated mice (Fig. 6D). In addition, liver sections from the MS-275-treated mice showed foci of mild steatosis. These are reversible cellular changes presumed to represent drug effect. Microscopic foci of probable acute inflammation were observed in liver sections from both treated and control mice with comparably low frequency.
As anticipated, treatment of mice with LM7 OS lung metastases with MS-275 also resulted in an inhibition of tumor growth. Oral administration of MS-275 significantly reduced the mean tumor surface area (p = 0.006), lung weight (p < 0.05) and number of micrometastases (p < 0.05) (Figs. 7A and 7B). The mean number of visible metastases did not differ significantly between the two groups, however, which may have resulted from the fact that the metastatic nodules in the control DMSO-treated group were extremely large and may have contained multiple smaller tumor nodules.
Figure 7. MS-275 treatment inhibits OS lung metastases.

Mice with LM7 lung metastastases received MS-275 as described in Fig. 7 (A and B). Mice were sacrificed at the end of therapy, and their lungs were extracted, weighed, and assessed for tumor volume. (C) Mice with LM7 lung metastases received 20 mg/kg MS-275 every other day via oral gavage. Long-term survival was assessed from the first day of treatment until death. The mice were observed and deaths were recorded daily. Mice were sacrificed when they became moribund, and their deaths were recorded (p = 0.002).
Given that oral administration of MS-275 was effective in inducing tumor cell apoptosis and inhibiting the growth of OS lung metastases, we investigated whether administration of the same dose of MS-275 affected the overall survival of mice. Mice treated with 20 mg/kg MS-275 every other day had a significantly increased survival when compared to the DMSO-treated control group (p = 0.026) (Fig. 7C).
DISCUSSION
Our laboratory previously showed that OS lung metastases were Fas negative in both mouse models and patient samples [3, 4, 9]. Our models utilized an intravenous tumor cell implantation method to establish lung metastases since it well understood that bone sarcoma metastasis occurs via the bloodstream rather than the lymphatic system [28]. We also have demonstrated that Fas+ OS cells are rapidly cleared from the lung microenvironment by the FasL+ lung epithelium, leaving only Fas− cells to form metastatic tumors [3, 5]. In contrast, OS lung metastases in FasL-deficient mice were comprised of both Fas+ and Fas− cells [6]. These data provided the basis for our conclusion that Fas expression on OS cells and the FasL+ lung microenvironment play a critical role in the metastatic potential of OS. We also demonstrated that inducing the re-expression of Fas using aerosol therapy resulted in the regression of OS lung metastases [5, 9, 10, 29]. There was no therapeutic effect, however, in FasL-deficient mice once again underscoring the importance of the lung microenvironment. The results presented herein extend our previous findings and indicate that the HDAC inhibitor MS-275 sensitizes OS to FasL-mediated cell death and may have potential as a therapeutic agent for the treatment of OS lung metastases.
At therapeutically achievable doses, MS-275 sensitized OS cells to FasL-induced cell death in vitro as indicated by a reduction in clonogenic growth and an increase in caspase cleavage/activity. Pretreatment of cells with the caspase inhibitor z-VAD-fmk decreased the sensitivity of cells to FasL following MS-275-treatment, suggesting that the mechanism is caspase-dependent, which is an integral component of Fas signaling. Blocking the Fas signaling pathway using FADD-dominant negative transfection of OS cells also inhibited the ability of MS-275 to sensitize cells to FasL-induced cell death [30]. Taken together, these results implicate a role for the Fas signaling pathway in the mechanism of action of MS-275. Upregulating cell surface Fas is one way to sensitize cells to FasL. However, we observed no change in Fas expression, as detected by flow cytometry, when OS cells were treated with MS-275 [30]. Therefore, we evaluated the effect of MS-275 treatment on the downstream mediators of the Fas pathway. Interestingly, we observed a downregulation in c-FLIP expression both in vitro and in vivo, implicating a role for c-FLIP in the mechanism of action. Further, knockdown of c-FLIP expression in OS cells, increased the sensitivity to FasL. This suggests that MS-275-mediated sensitization to FasL may be a consequence of the downregulation of c-FLIP following MS-275 treatment.
The inhibitor of apoptosis, c-FLIP, has been shown to regulate Fas-mediated apoptosis [31]. HDAC inhibitors such as dacinostat, panobinostat, depsipeptide, valproic acid and droxinostat have been shown to decrease c-FLIP expression and increase death-receptor mediated apoptosis in leukemia, breast cancer, pancreatic cancer and hepatoma cell lines [24, 32–35]. Consistent with our findings, the pan-HDAC inhibitor, depsipeptide, also sensitized OS cells to Fas-mediated apoptosis and involved the downregulation of c-FLIP [22]. Depsipeptide is a selective class I/II HDAC inhibitor with more specificity for class I than class II HDACs [36, 38]. MS-275 is also selective for class I HDACs [37, 38]. Similar to depsipeptide, we demonstrated that MS-275 induced the downregulation of c-FLIP. Taken together these data support the hypothesis that the downregulation of c-FLIP may be related to class I HDAC inhibition. We report for the first time that OS cells with high metastatic potential and low Fas expression are sensitized in vitro to FasL-induced cell death by MS-275 with a synonymous downregulation of c-FLIP mRNA and protein. These findings are consistent with those reported above but are unique in that we demonstrated increased cell death and downregulated c-FLIP both in vitro and in vivo. We also report for the first time that oral administration of MS-275 decreases c-FLIP expression in OS lung metastases.
MS-275 has exhibited preclinical activity in several tumor models, including pediatric cancers [26, 30]. However, we observed the novel finding that oral administration of MS-275 in mice with established OS lung metastases resulted in increased tumor histone acetylation, tumor cell apoptosis, and tumor regression. MS-275–treated mice had fewer and smaller lung metastases compared with control DMSO-treated mice. Oral MS-275 also increased the overall survival in mice with established OS lung metastases. Importantly, the dose of MS-275 that produced antitumor activity did not cause significant toxic effects. The current clinical trials with MS-275 utilize an oral formulation. Our data showing the activity of oral MS-275 against established OS lung metastases indicate that this agent may have therapeutic potential for patients with relapsed OS in the lung. Visualizing and tracking the metastatic behavior of cells in vivo will allow us to better understand the response to MS-275 treatment and the effect of the tumor microenvironment on this response [39]. Experiments using in vivo imaging to track tumor response to therapy in real time are currently underway to validate our findings.
CONCLUSION
In summary, our data show that oral MS-275 is effective against OS pulmonary metastases as judged by a reduction in tumor size, number of metastases and an increase in survival. MS-275 has demonstrated therapeutic activity in several preclinical models and clinical trials either as a single agent or in combination with chemotherapy [37]. Our data is unique however, as we show for the first time that in addition to sensitizing OS cells to FasL, a class I-specific HDAC inhibitor affected the expression of c-FLIP both in vitro and in vivo and had a therapeutic effect in metastatic OS. We conclude that this downregulation of c-FLIP was associated with the sensitization of OS cells to FasL-induced cell death. Our findings indicate that MS-275 may have therapeutic potential for patients with OS lung metastases and also suggest c-FLIP as a possible new therapeutic target.
Acknowledgments
This work was supported by National Cancer Institute grant R01 CA42992 (E.S.K.) and National Institutes of Health core grant CA16672. This research is supported in part by the National Institutes of Health through MD Anderson’s Cancer Center Support Grant CA016672. We thank Dr. Peter Ordentlich and Syndax Pharmaceuticals, Inc. for providing the MS-275 compound. We would also like to express our gratitude to the American Legion Auxiliary for the Fellowship Award to K.R-B. This paper includes data that was presented in a dissertation (by KR-B) to The University of Texas MD Anderson Cancer Center and UT Health Graduate School of Biomedical Sciences: Bindal, Krithi R., “The histone deacetylase inhibitor, MS-275, sensitizes metastatic osteosarcoma to FasL-induced cell death: a role for c-FLIP” (2012). UT GSGS Dissertations and Theses. Paper 218.
ABBREVIATIONS
- OS
osteosarcoma
- FasL
Fas ligand
- c-FLIP
cellular FLICE-inhibitory protein
- FADD
Fas-associated death domain
- DISC
death-inducing signaling complex
- HDAC
histone deacetylase
- CLL
chronic lymphocytic leukemia
- STR
short tandem repeat
- CLIMA
cell line integrated molecular authentication database
- sFasL
superFasL
- MTT
3-(4,5-dimethylthiazol-2yl)2,5-diphenyl tetrazolium bromide
- DMSO
dimethyl sulfoxide
- AcH3
acetyl-histone H3
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- AALAS
American Association for Laboratory Animal Science
- H&E
hematoxylin and eosin
- DAB
3-3′-diaminobenzidine
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
- BSA
bovine serum albumin
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest.
References
- 1.Hawkins DS, Arndt CA. Pattern of disease recurrence and prognostic factors in patients with osteosarcoma treated with contemporary chemotherapy. Cancer. 2003;98(11):2447–56. doi: 10.1002/cncr.11799. [DOI] [PubMed] [Google Scholar]
- 2.Eilber F, Giuliano A, Eckardt J, Patterson K, Moseley S, Goodnight J. Adjuvant chemotherapy for osteosarcoma: a randomized prospective trial. J Clin Oncol. 1987;5(1):21–6. doi: 10.1200/JCO.1987.5.1.21. [DOI] [PubMed] [Google Scholar]
- 3.Koshkina NV, Khanna C, Mendoza A, Guan H, DeLauter L, Kleinerman ES. Fas-negative osteosarcoma tumor cells are selected during metastasis to the lungs: the role of the Fas pathway in the metastatic process of osteosarcoma. Mol Cancer Res. 2007;5(10):991–9. doi: 10.1158/1541-7786.MCR-07-0007. [DOI] [PubMed] [Google Scholar]
- 4.Gordon N, Arndt CA, Hawkins DS, Doherty DK, Inwards CY, Munsell MF, Stewart J, Koshkina NV, Kleinerman ES. Fas expression in lung metastasis from osteosarcoma patients. J Pediatr Hematol Oncol. 2005;27(11):611–5. doi: 10.1097/01.mph.0000188112.42576.df. [DOI] [PubMed] [Google Scholar]
- 5.Gordon N, Kleinerman ES. The role of Fas/FasL in the metastatic potential of osteosarcoma and targeting this pathway for the treatment of osteosarcoma lung metastases. Cancer Treat Res. 2009;152:497–508. doi: 10.1007/978-1-4419-0284-9_29. [DOI] [PubMed] [Google Scholar]
- 6.Gordon N, Koshkina NV, Jia SF, Khanna C, Mendoza A, Worth LL, Kleinerman ES. Corruption of the Fas pathway delays the pulmonary clearance of murine osteosarcoma cells, enhances their metastatic potential, and reduces the effect of aerosol gemcitabine. Clin Cancer Res. 2007;13(15 Pt 1):4503–10. doi: 10.1158/1078-0432.CCR-07-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan X, Jia SF, Koshkina N, Kleinerman ES. Intranasal interleukin-12 gene therapy enhanced the activity of ifosfamide against osteosarcoma lung metastases. Cancer. 2006;106(6):1382–8. doi: 10.1002/cncr.21744. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Z, Lafleur EA, Koshkina NV, Worth LL, Lester MS, Kleinerman ES. Interleukin-12 up-regulates Fas expression in human osteosarcoma and Ewing’s sarcoma cells by enhancing its promoter activity. Mol Cancer Res. 2005;3(12):685–91. doi: 10.1158/1541-7786.MCR-05-0092. [DOI] [PubMed] [Google Scholar]
- 9.Jia SF, Worth LL, Densmore CL, Xu B, Duan X, Kleinerman ES. Aerosol gene therapy with PEI: IL-12 eradicates osteosarcoma lung metastases. Clin Cancer Res. 2003;9(9):3462–8. [PubMed] [Google Scholar]
- 10.Jia SF, Worth LL, Densmore CL, Xu B, Zhou Z, Kleinerman ES. Eradication of osteosarcoma lung metastases following intranasal interleukin-12 gene therapy using a nonviral polyethylenimine vector. Cancer Gene Ther. 2002;9(3):260–6. doi: 10.1038/sj.cgt.7700432. [DOI] [PubMed] [Google Scholar]
- 11.Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999;11(2):255–60. doi: 10.1016/s0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 12.Bullani RR, Huard B, Viard-Leveugle I, Byers HR, Irmler M, Saurat JH, Tschopp J, French LE. Selective expression of FLIP in malignant melanocytic skin lesions. J Invest Dermatol. 2001;117(2):360–4. doi: 10.1046/j.0022-202x.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 13.de Hooge AS, Berghuis D, Santos SJ, Mooiman E, Romeo S, Kummer JA, Egeler RM, van Tol MJ, Melief CJ, Hogendoorn PC, Lankester AC. Expression of cellular FLICE inhibitory protein, caspase-8, and protease inhibitor-9 in Ewing sarcoma and implications for susceptibility to cytotoxic pathways. Clin Cancer Res. 2007;13(1):206–14. doi: 10.1158/1078-0432.CCR-06-1457. [DOI] [PubMed] [Google Scholar]
- 14.Elnemr A, Ohta T, Yachie A, Kayahara M, Kitagawa H, Fujimura T, Ninomiya I, Fushida S, Nishimura GI, Shimizu K, Miwa K. Human pancreatic cancer cells disable function of Fas receptors at several levels in Fas signal transduction pathway. Int J Oncol. 2001;18(2):311–6. doi: 10.3892/ijo.18.2.311. [DOI] [PubMed] [Google Scholar]
- 15.Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol. 1998;161(6):2833–40. [PubMed] [Google Scholar]
- 16.Hernandez A, Wang QD, Schwartz SA, Evers BM. Sensitization of human colon cancer cells to TRAIL-mediated apoptosis. J Gastrointest Surg. 2001;5(1):56–65. doi: 10.1016/s1091-255x(01)80014-7. [DOI] [PubMed] [Google Scholar]
- 17.Korkolopoulou P, Goudopoulou A, Voutsinas G, Thomas-Tsagli E, Kapralos P, Patsouris E, Saetta AA. c-FLIP expression in bladder urothelial carcinomas: its role in resistance to Fas-mediated apoptosis and clinicopathologic correlations. Urology. 2004;63(6):1198–204. doi: 10.1016/j.urology.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Nam SY, Jung GA, Hur GC, Chung HY, Kim WH, Seol DW, Lee BL. Upregulation of FLIP(S) by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci. 2003;94(12):1066–73. doi: 10.1111/j.1349-7006.2003.tb01402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glick RD, Swendeman SL, Coffey DC, Rifkind RA, Marks PA, Richon VM, La Quaglia MP. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999;59(17):4392–9. [PubMed] [Google Scholar]
- 20.Klisovic DD, Katz SE, Effron D, Klisovic MI, Wickham J, Parthun MR, Guimond M, Marcucci G. Depsipeptide (FR901228) inhibits proliferation and induces apoptosis in primary and metastatic human uveal melanoma cell lines. Invest Ophthalmol Vis Sci. 2003;44(6):2390–8. doi: 10.1167/iovs.02-1052. [DOI] [PubMed] [Google Scholar]
- 21.Kwon SH, Ahn SH, Kim YK, Bae GU, Yoon JW, Hong S, Lee HY, Lee YW, Lee HW, Han JW. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J Biol Chem. 2002;277(3):2073–80. doi: 10.1074/jbc.M106699200. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe K, Okamoto K, Yonehara S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. 2005;12(1):10–8. doi: 10.1038/sj.cdd.4401507. [DOI] [PubMed] [Google Scholar]
- 23.Rivera-Del Valle N, Gao S, Miller CP, Fulbright J, Gonzales C, Sirisawad M, Steggerda S, Wheler J, Balasubramanian S, Chandra J. PCI-24781, a Novel Hydroxamic Acid HDAC Inhibitor, Exerts Cytotoxicity and Histone Alterations via Caspase-8 and FADD in Leukemia Cells. Int J Cell Biol. 2010;2010:207420. doi: 10.1155/2010/207420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, Shen T, Murphy T, Wickham J, Kanakry C, Lucas DM, Reed JC, Grever MR, Byrd JC. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003;102(2):652–8. doi: 10.1182/blood-2002-12-3794. [DOI] [PubMed] [Google Scholar]
- 25.Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD, Flax EL, Wickham J, Reed JC, Byrd JC, Grever MR. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18(7):1207–14. doi: 10.1038/sj.leu.2403388. [DOI] [PubMed] [Google Scholar]
- 26.Jaboin J, Wild J, Hamidi H, Khanna C, Kim CJ, Robey R, Bates SE, Thiele CJ. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 2002;62(21):6108–15. [PubMed] [Google Scholar]
- 27.Coffey DC, Kutko MC, Glick RD, Butler LM, Heller G, Rifkind RA, Marks PA, Richon VM, La Quaglia MP. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001;61(9):3591–4. [PubMed] [Google Scholar]
- 28.Strauss SJ, Ng T, Mendoza-Naranjo A, Whelen J, Sorensen PH. Understanding micrometastatic disease and Anoikis resistance in ewing family of tumors and osteosarcoma. Oncologist. 2010;15(6):627–35. doi: 10.1634/theoncologist.2010-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koshkina NV, Kleinerman ES. Aerosol gemcitabine inhibits the growth of primary osteosarcoma and osteosarcoma lung metastases. Int J Cancer. 2005;116(3):458–63. doi: 10.1002/ijc.21011. [DOI] [PubMed] [Google Scholar]
- 30.Koshkina NV, Rao-Bindal K, Kleinerman ES. Effect of the histone deacetylase inhibitor SNDX-275 on fas signaling in osteosarcoma cells and the feasibility of its topical application for the treatment of osteosarcoma lung metastases. Cancer. 2011;117 (15):3457–67. doi: 10.1002/cncr.25884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 32.Bijangi-Vishehsaraei K, Saadatzadeh MR, Huang S, Murphy MP, Safa AR. 4-(4-Chloro-2-methylphenoxy)-N-hydroxybutanamide (CMH) targets mRNA of the c-FLIP variants and induces apoptosis in MCF-7 human breast cancer cells. Mol Cell Biochem. 2010;342(1–2):133–42. doi: 10.1007/s11010-010-0477-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo F, Sigua C, Tao J, Bali P, George P, Li Y, Wittmann S, Moscinski L, Atadja P, Bhalla K. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004;64(7):2580–9. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- 34.Kauh J, Fan S, Xia M, Yue P, Yang L, Khuri FR, Sun SY. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One. 2010;5(4):e10376. doi: 10.1371/journal.pone.0010376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuchmann M, Schulze-Bergkamen H, Fleischer B, Schattenberg JM, Siebler J, Weinmann A, Teufel A, Worns M, Fischer T, Strand S, Lohse AW, Galle PR. Histone deacetylase inhibition by valproic acid down-regulates c-FLIP/CASH and sensitizes hepatoma cells towards CD95- and TRAIL receptor-mediated apoptosis and chemotherapy. Oncol Rep. 2006;15(1):227–30. doi: 10.3892/or.15.1.227. [DOI] [PubMed] [Google Scholar]
- 36.Hartlapp I, Pallasch C, Weibert G, Kemkers A, Hummel M, Re D. Depsipeptide induces cell death in Hodgkin lymphoma-derived cell lines. Leuk Res. 2009;33(7):929–36. doi: 10.1016/j.leukres.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 37.Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, Guidotti A. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A. 2006;103(5):1587–92. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19(9):1049–66. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffman R. The multiple uses of flourescent proteins to visualize cancer in vivo. Nature Reviews Cancer. 2005;5(10):796–806. doi: 10.1038/nrc1717. [DOI] [PubMed] [Google Scholar]