

Abstract
Background
Cotton Verticillium wilt is a serious soil-borne vascular disease that causes great economic loss each year. However, due to the lack of resistant varieties of upland cotton, the molecular mechanisms of resistance to this disease, especially to the pathogen Verticillium dahliae, remain unclear.
Results
We used the RNA-seq method to research the molecular mechanisms of cotton defence responses to different races of Verticillium dahliae by comparing infected sea-island cotton and upland cotton. A total of 77,212 unigenes were obtained, and the unigenes were subjected to BLAST searching and annotated using the GO and KO databases. Six sets of digital gene expression data were mapped to the reference transcriptome. The gene expression profiles of cotton infected with Verticillium dahliae were compared to those of uninfected cotton; 44 differentially expressed genes were identified. Regarding genes involved in the phenylalanine metabolism pathway, the hydroxycinnamoyl transferase gene (HCT) was upregulated in upland cotton whereas PAL, 4CL, CAD, CCoAOMT, and COMT were upregulated in sea-island cotton. Almost no differentially expressed genes in this pathway were identified in sea-island cotton and upland cotton when they were infected with V. dahliae V991 and V. dahliae D07038, respectively.
Conclusions
Our comprehensive gene expression data at the transcription level will help elucidate the molecular mechanisms of the cotton defence response to V. dahliae. By identifying the genes involved in the defence response of each type of cotton to V. dahliae, our data not only provide novel molecular information for researchers, but also help accelerate research on genes involved in defences in cotton.
Keywords: Cotton, Verticillium dahliae, RNA sequencing, Transcriptome analysis, Mechanism of resistance
Background
Cotton is the nature fibre crop with the greatest economic importance. Cotton Verticillium wilt (CVW) is a serious soil-borne vascular disease caused by Verticillium dahliae[1]. The disease can cause cotton yellowing, wilt, defoliating, and finally death [2]. CVW was first reported in upland cotton in the United States, and spread to China on imported American cotton in 1935[3-5]. At present, more than 200 million hectares of cotton are subject to CVW, and the economic loss is tremendous every year, especially defoliated CVW [6]. Currently, no fungicides are available to cure commercial upland cotton (Gossypium hirsutum L., the main cultivated cultivar) once they become infected, although the sea-land cotton species (Gossypium barbadense L., not the main cultivated cultivar) is immune to V. dahliae. Because of the difficulty obtaining a new highly resistant upland cotton breed by cross-breeding of upland cotton and sea-island cotton, no completely suitable high-resistance upland cotton variety exists. The breeding of disease-resistant cultivars remains the primary control method for cotton. At present, the primary method of breeding for disease-resistant cotton is crossbreeding of resistant cotton, which has been used to create a series of varieties with resistance to CVW [7,8].
In recent years, researchers have investigated the molecular mechanisms of resistance to V. dahliae with the aim of ultimately using genetic engineering to breed cultivars resistant to CVW. Resistance to CVW is controlled by a single dominant gene in sea-island cotton, whereas that in upland cotton may be controlled by either a single dominant gene or two dominant genes [5,8-13]. When cotton is infected by the CVW pathogen, the defence system is activated, generating a series of cascade reactions. Three aspects of this system, tissue resistance, physiological and biochemical resistance, and micro-organism resistance, are involved in the mechanism of resistance [14,15]. Several V. dahliae resistance-related genes have been isolated and studied, including the following four groups: cinnamyl alcohol dehydrogenase (CAD), farnesyl diphosphate synthase, isopentenyl diphosphate isomerase, and 3-hydroxy-3-methylglutaryl coenzyme A reductase, the key regulatory enzymes in the synthesis of phytoalexin, which can kill pathogens through increased local concentrations in the plant [16,17] (Group 1); lipid transfer proteins, chitinase, β-1,3 glucanase, nonexpressor of pathogenesis-related genes 1, thionin, gastrodia antifungal protein, and lectin-like protein, which may play important roles in the response to pathogen infections in cotton plants [18,19] (Group 2); phenylalanine ammonia lyase (PAL), cinnamate-4-hydroxylase, peroxisome, polygalacturonase-inhibiting proteins, and laccase, which are defence proteins that can slow down the spread of pathogens [20,21] (Group 3); and resistance gene analogues and defence gene analogues that have been cloned and further transformed by genetic engineering for functional research, such as lipid transfer protein, nonrace-specific disease resistance 1, MAP kinase kinase 2, and others [9,22] (Group 4).
The phenylpropanoid pathway plays a key role in cotton resistance to V. dahliae[8,23-25]. Even so, the molecular mechanisms of V. dahliae resistance are unclear. In recent years, novel, high-throughput, deep-sequencing transcriptome analysis, termed RNA-seq, has made it possible to efficiently generate large-scale expressed sequence tag (EST) libraries and improved the speed of gene discovery [26]. Although RNA-seq technologies have been used for researching gene expression profiles in the resistance response of sea-island cotton to V. dahliae, the phenylpropanoid metabolism pathway and genes associated with lignin metabolism has been found to play a central role [25]. Whether a different response mechanism to the wilt fungus V. dahliae exists in cotton of different races and whether different response mechanisms exist in different cotton species (G. hirsutum L. and G. barbadense L.) are questions for further research. Therefore, in this study, four samples, including ZhongZhiMian KV-1 (G. hirsutum L.) and XinHai 15 (G. barbadense L.) infected with V. dahliae strain V991 (highly toxic) and D07038 (intermediately toxic), respectively, and two uninfected samples include ZhongZhiMian KV-1 (G. hirsutum L.) and XinHai 15 (G. barbadense L.) were sequenced in order to addressed the above issues using RNA-seq technologies.
Results
Illumina sequencing and sequence assembly
A total of 56,836,100 reads (accumulated length, 4,652,696,700 bp; SRA accession number SRX128210) were generated through Illumina sequencing and assembled into 167,545 contigs. Then the contigs were further assembled into 77,212 unigenes, with a mean length of 666 bp. The size distribution indicated that 44,838 (58%) unigenes were 100–500 bp, and 42% unigenes were greater than 500 bp (Figure 1). To evaluate the quality of the data set, the ratio of the gap length to the length of the assembled unigenes was analysed. All of the unigenes showed gap lengths of ≤5%.
Figure 1.

Unigene size distribution. Bracket show the ratio of total unigenes.
Functional annotation and classification
All of the unigenes were compared to the sequences in public databases, including the NCBI nonredundant protein (nr) database, the Clusters of Orthologous Groups (COG) database, the Swiss-Prot protein database, and the KEGG database, using BLASTX with a cutoff e-value of 10-5. A total of 66,208 unigenes (78% of all unigenes) returned a significant BLAST result.
The assembled unigenes were compared against the COG database to phylogenetically analyse widespread domain families. The results revealed 16,789 unigenes with significant homology and assigned them to the appropriate COG clusters. These COG classifications were grouped into 25 functional categories (Figure 2). Among these COG categories, the cluster 'general function’ (5274; 31.4%) represented the largest group, followed by 'transcription’ (2869; 17.1%); 'replication, recombination, and repair’ (2561; 15.3%); 'signal transduction mechanisms’ (2268;13.5%); 'posttranslational modification, protein turnover, chaperones’ (2192; 13.1%); 'translation, ribosomal structure, and biogenesis’ (1752; 10.4%); and 'carbohydrate transport and metabolism’ (1736; 10.3%).
Figure 2.

COG Function Classification of all unigene. A total of 16,789 unigenes showing significant homology to the COGs database have a COG classification among the 25 cateories.
Gene Ontology (GO) assignments were used to classify the functions of the predicted cotton genes. Based on sequence homology, 23,291 sequences were categorised into 44 functional groups (Figure 3). In each of the three main categories (biological process, cellular component, and molecular function) of the GO classification, the major subcategories were as follows: six subcategories for biological process ('biological regulation’ , 'cellular process’ , 'localization’ , 'metabolic process’ , 'regulation of biological process’ , and 'response to stimulus’); three sub-categories for cellular components ('cell’ , 'cell part’ and 'organelle’); and two sub-categories for molecular function ('binding’ and 'catalytic activity’). Only a few genes were clustered in terms of 'biological adhesion’ , 'cell killing’ , 'locomotion’ , 'pigmentation’ , 'rhythmic process’ , 'viral reproduction’ , 'cell junction’ , 'extracellular region’ , 'virion’ , and 'translation regulator activity’.
Figure 3.
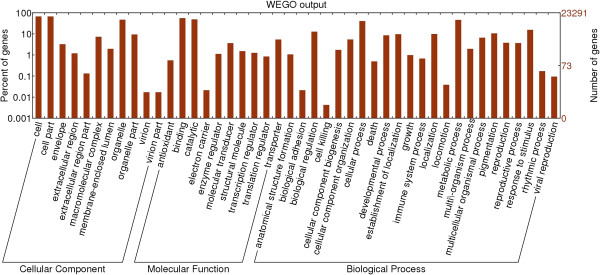
Gene ontology classification of the cotton transcriptome.
Functional classification and pathway assignment were performed using the KEGG database [27]. In total, 28,395 unigenes were assigned to 126 KEGG pathways (see Table 1). The major pathways were 'metabolic pathways’ , 'biosynthesis of secondary metabolites’ , 'plant hormone signal transduction’ , and 'plant-pathogen interaction’; the gene numbers and percentages assigned to these pathways were 5912 (20.82%), 2959 (10.42%), 2243 (7.9%), and 2238 (7.88%), respectively.
Table 1.
Pathway annotation to KEGG
| ID | Pathway | No. of unigenes (28395) |
|---|---|---|
| 1 |
Metabolic pathways |
5912 (20.82%) |
| 2 |
Biosynthesis of secondary metabolites |
2959 (10.42%) |
| 3 |
Plant hormone signal transduction |
2243 (7.9%) |
| 4 |
Plant-pathogen interaction |
2238 (7.88%) |
| 5 |
RNA transport |
1102 (3.88%) |
| 6 |
Spliceosome |
915 (3.22%) |
| 7 |
Ribosome biogenesis in eukaryotes |
907 (3.19%) |
| 8 |
RNA degradation |
818 (2.88%) |
| 9 |
Protein processing in endoplasmic reticulum |
791 (2.79%) |
| 10 |
Purine metabolism |
732 (2.58%) |
| 11 |
Endocytosis |
715 (2.52%) |
| 12 |
Glycerophospholipid metabolism |
666 (2.35%) |
| 13 |
mRNA surveillance pathway |
654 (2.3%) |
| 14 |
Starch and sucrose metabolism |
596 (2.1%) |
| 15 |
Ribosome |
574 (2.02%) |
| 16 |
Ubiquitin mediated proteolysis |
512 (1.8%) |
| 17 |
Phenylpropanoid biosynthesis |
467 (1.64%) |
| 18 |
Ether lipid metabolism |
455 (1.6%) |
| 19 |
Pyrimidine metabolism |
387 (1.36%) |
| 20 |
Circadian rhythm - plant |
384 (1.35%) |
| 21 |
Amino sugar and nucleotide sugar metabolism |
359 (1.26%) |
| 22 |
Phagosome |
357 (1.26%) |
| 23 |
Peroxisome |
355 (1.25%) |
| 24 |
Stilbenoid, diarylheptanoid and gingerol biosynthesis |
346 (1.22%) |
| 25 |
Flavonoid biosynthesis |
342 (1.2%) |
| 26 |
Oxidative phosphorylation |
341 (1.2%) |
| 27 |
Glycolysis/Gluconeogenesis |
320 (1.13%) |
| 28 |
Limonene and pinene degradation |
301 (1.06%) |
| 29 | Phosphatidylinositol signaling system | 290 (1.02%) |
Note: Data only showed the annotation unigenes percent of each pathway more than 1%.
DGE library sequencing
Six cotton digital gene expression (DGE) profiling libraries were sequenced (1 ~ 6, corresponding to the SRA accession numbers SRX128211, SRX128213, SRX128212, SRX128214, SRX128215, and SRX128216, respectively), which generated approximately 11 to 12 million high-quality reads for each library (Table 2). The percentage of clean reads among the raw reads in each library was >99% (Table 3). Among the clean reads, the number of sequences that could be mapped to unigenes ranged from 9.4 to 10.2 million, and the percentage of clean reads was above 79% in the six libraries. As Table 2 shows, the vast majority of these mapped reads were uniquely matched to unigenes (> 55%), and the percentage of multi-position matched reads was 24%.
Table 2.
Statistics of DGE sequencing
| Total reads | 12230124(100%) | 11875055(100%) | 11764630(100%) | 12511454(100%) | 12167879(100%) | 12420918(100%) |
|---|---|---|---|---|---|---|
| Total mapped reads |
9770169(79.89 %) |
9493222(79.94 %) |
9411269(80 %) |
9969428(79.68 %) |
9867697(81.1 %) |
10150382(81.72 %) |
| Perfect match |
6659597(54.45 %) |
6469378(54.48 %) |
6478583(55.07 %) |
6778488(54.18 %) |
6721125(55.24 %) |
6954849(55.99 %) |
| <=2 bp mismatch |
3110572(25.43 %) |
3023844(25.46 %) |
2932686(24.93 %) |
3190940(25.5 %) |
3146572(25.86 %) |
3195533(25.73 %) |
| Unique match |
6853979(56.04 %) |
6623521(55.78 %) |
6692373(56.89 %) |
7004728(55.99 %) |
7165038(58.88 %) |
7483020(60.25 %) |
| Multi-position match |
2916190(23.84 %) |
2869701(24.17 %) |
2718896(23.11 %) |
2964700(23.7 %) |
2702659(22.21 %) |
2667362(21.47 %) |
| Total unmapped reads | 2459955(20.11 %) | 2381833(20.06 %) | 2353361(20 %) | 2542026(20.32 %) | 2300182(18.9 %) | 2270536(18.28 %) |
Note: 1: KV1/D; 2: XH/D; 3: ZKV1/V; 4:XH/V; 5:KV1/U; and 6: XH/U.
Table 3.
Different components of the raw reads in each sample
| Summary | Clean reads only | Only adaptor | Containing N | Low quality |
|---|---|---|---|---|
| 1 |
12230124(99.39%) |
54281(0.44%) |
788(0.01%) |
19459(0.16%) |
| 2 |
11875055(99.45%) |
45599(0.38%) |
0 |
19899(0.17%) |
| 3 |
11764630(99.38%) |
53254,0.45%) |
745(0.01%) |
19561(0.17%) |
| 4 |
12511454(99.42%) |
51023(0.41%) |
0 |
21851(0.17%) |
| 5 |
12167879(99.42%) |
49895(0.41%) |
749(0.01%) |
20211(0.17%) |
| 6 | 12420918(99.48%) | 49511,0.40%) | 1094(0.01%) | 14766(0.12%) |
Note: 1: KV1/D; 2: XH/D; 3: ZKV1/V; 4:XH/V; 5:KV1/U; and 6: XH/U.
Gene expression variation among the different samples
First, to evaluate the DGE data, we analysed the distribution of unigene coverage in each sample, which is the number of clean reads that aligned to the reference unigenes. As shown in Figure 4, most unigene coverage was >50% (1, 69% of all unigenes; 2, 70% of all unigenes; 3, 69% of all unigenes; 4, 78% of all unigenes; 5, 67% of all unigenes; and 6, 66% of all unigenes). Second, the number of clean reads was calculated and the gene expression level was calculated using the reads per kb per million reads method for each unigene, and then the differentially expressed genes (DEGs) were identified in different samples.
Figure 4.

Distribution of reference unigenes’ coverage in each sample. 1: KV1/D; 2: XH/D; 3: ZKV1/V; 4:XH/V; 5:KV1/U; and 6: XH/U.
Variation in gene expression was identified based on comparisons of the above. The following significant DEGs were identified: (a) between samples 1 (KV1/D) and 5 (KV1/U), 4013 and 2373 genes were up- and downregulated, respectively; (b) between samples 3 (KV1/V) and 5 (KV1/U), 3393 and 2507 genes were up- and downregulated, respectively; (c) between samples 2 (XH/D) and 6 (XH/U), 7871 and 2988 genes were up- and downregulated, respectively; and (d) between samples 4 (XH/V) and 6 (XH/U), 7998 and 3494 genes were up- and downregulated, respectively (Figure 5).
Figure 5.

Numbers of differnet expressed unigenes in each comparison. The numbers on column showed quantity of up-regulated (blue) and down-regulated (red) unigenes. 1: KV1/D; 2: XH/D; 3: ZKV1/V; 4:XH/V; 5:KV1/U; and 6: XH/U.
Genes related to resistance to CVW resistance
Based on the above comparisons, the DEGs were functionally analysed using MapMan software. The results showed that a total of 1975 genes occurred simultaneously in the four comparisons above (a–d), including 1535 upregulated genes and 440 downregulated genes. These genes were principally associated with cell wall, lipids, and secondary metabolism and so on (Additional file 1). To identify cotton genes most associated with resistance to CVW, an absolute value of the fold change times > =5 was used as a threshold, resulting in 44 DEGs being screened out (including 41 upregulated and 3 downregulated genes, Table 4).
Validation of RNA-Seq-based gene expression
To validate the assembled sequences and the expression profiles obtained by RNA-Seq, real-time RT-PCR was performed on 20 of the above DEGs. Gene expression levels in V991 and the controls were compared using qRT-PCR. Of the 20 genes, all but gene 12 (id: unigene 64452) were detected. For the 19 detected genes, the upregulation trend obtained in real-time RT-PCR expression was in agreement with the RNA-Seq data, except for genes 17 and 20 (unigene 51427 and unigene 12430). However, for most of the genes, the upregulated range in real-time RT-PCR was lower than in the DGE data (Figure 6).
Figure 6.

Expression pattern of the selected genes in G.hirsutum. (A) Gene expression data for DGE analysis. The fold changes of the genes were shown on the y-axis. (B) The qRT-PCR analysis of gene expression data.V991 mean ZhongZhiMian KV-1 infected by V991, blank show ZhongZhiMian KV-1 uninfected. Error bars represent SE for three independent experiments.
Discussion
Cotton is an economically important crop, and cotton CVW causes significant economic losses. Although some cotton cultivars resistant to V. dahliae have been generated using conventional breeding techniques, the molecular mechanisms of disease resistance in cotton are still unclear. Xu et al. [25] found that lignin metabolism is a key pathway in the resistance of cotton, but the authors mainly analysed the RNA-seq results by analysing the resistance mechanism of the resistant Gossypium barbadense in contrast to the susceptible Gossypium hirsutum. Are other key genes involved in the resistance of cotton? In this study, we used the commercial upland cotton (Gossypium hirsutum) variety ZhongZhiMian KV-1 and the sea-island cotton Xinhai 15, both highly resistant to V. dahliae, to study the resistance relationships between different cotton species and different V. dahliae strains.
Using transcriptome sequence analysis, 56,836,100 reads were obtained, corresponding to about 4.7 Gb of raw sequence data. Then the raw sequences were further assembled using the short-reads assembly program Trinity and used to predict unigenes. In spite of the genome sequence of a diploid cotton Gossypium raimondi had been published, whose progenitor is the putative contributor of the D-subgenome to cotton species Gossypium barbadense and Gossypium hirsutum[28,29], due to the lack of A-genome sequence information, the D-genome sequence were not used in our bioinformation analysis in this study, and the quality of these assembled unigenes was confirmed by qRT-PCR. The results showed that signals for 19 out of 20 tested genes were detected, demonstrating the high quality of the assembled results, and also indicating that relatively short reads from Illumina sequencing can be effectively assembled and used for novel gene discovery. The 66208 predicted unigenes were subjected to BLAST annotation; 78% of the unigenes returned a significant BLAST result. About 20% of the predicted unigenes could not be assigned annotation information, which may be caused by the limited information about the genomes or transcriptomes of cotton and its related species. Some unigenes were too short to allow statistically meaningful matches. From the KEGG annotation results, 2238 unigenes (7.88% of all KEGG annotation unigenes) were found to be involved in the group 'plant-pathogen interaction’, which may be associated with the defence response of cotton to V. dahliae. This group was the fourth largest group in our results, which suggests that it is feasible through the immense capacity of Illumina sequencing to discover genes related to cotton resistance to V. dahliae in metabolic pathways.
To understand the regulatory pathways and molecular mechanisms of resistance to V. dahliae, we created six DGE libraries to analyse gene expression patterns following inoculation with V. dahliae. About 80% of the reads were mapped to our transcriptome reference database. The high proportion of mapped reads supports the validity of our analysis. The quality of the DGE libraries was also confirmed by qRT-PCR analysis. Because the cotton disease resistance-related genes may begin to be expressed after inoculation with V. dahlia, cotton seedlings of different species inoculated with different races of V. dahliae were compared to controls. Different genes may be involved in disease resistance, and the common genes in the four comparison groups reduced the number of DEGs that may be related to cotton disease resistance. There were no replicated data using the Audic-Claverie method in DGE data analysing, but the results are credible because we applied a series of correction method, such as: a false discovery rate of <0.001 and an absolute value of the log 2 ratio >1 as the threshold were used for judging the significance of the gene expression differences, and combined with the quality results of the DGE libraries been further confirmed by qRT-PCR.
The inoculated V. dahliae cotton samples compared with uninoculated samples exhibited much more upregulated genes and downregulated genes, this possibly indicates that V. dahliae activates cotton resistance genes during the infection process. Furthermore, the number of upregulated and downregulated genes of G. barbadense was much greater than that in G. hirsutum (both inoculated with V. dahliae races V991 and D07038) in the four compared groups, suggesting that the resistance genes to V.dahliae of the sea-island cotton G. barbadense is more than that of the upland cotton G. hirsutum.
The genes that were upregulated or downregulated more than five fold in the four compared groups are listed in Table 4. It is feasible that the functional genes play key roles in the resistance of cotton to V. dahliae. Of these genes, the gene unigene5503_kv-1 was described as a cellulose synthase-like D gene; cellulose synthase-like genes mediate the synthesis of cell wall (1,3;1,4)-beta-D-glucans and/or are required for cellulose deposition [30-33]. Vega-Sánchez et al. found a cellulose synthase-like protein related to defence responses of rice plants [33]. In our study, the cellulose synthase-like D protein was differentially expressed upon infection of cotton seedlings with V. dahliae. The genes unigene68082_kv-1 and unigene67909_kv-1 were annotated as encoding xyloglucan endotraglucosylase/hydrolase and xyloglucan 6-xylosyltransferase, respectively. As reported previously, xyloglucan 6-xylosyltransferase can catalyse the formation of UDP-D-xylose-D-glucose from UDP-D-xylose in xyloglucan, and xyloglucan endotransglycosylase/hydrolase can degrade xyloglucan [34-37]. Xyloglucans play an important role in cross-linking adjacent cellulose microfibrils to form a cellulose-xyloglucan network that constitutes the major load-bearing structure of the primary cell wall. Xu et al. found that lignin has a key role in the cotton defence response, suggesting that genes related to cotton cell wall components are involved in the defence response to V. dahlia[25]. The gene unigene52341_kv-1 is similar to the terpene synthase 3 gene from Populus trichocarpa. Danner et al. found that terpene synthase 3 can activate both mono- and sesquiterpene synthase in Populus trichocarpa[38], and in cotton, sesquiterpene phytoalexins are elicited in response to bacterial or fungal infection [39,40]. In conclusion, the four genes mentioned above are probably related to cotton resistance. Three of the genes are related to cell-wall compounds and one gene is related to sesquiterpene metabolism, suggesting that the cell wall and sesquiterpene are involved in the resistance of cotton to CVW. These genes are targets of further research in our next study.
Table 4.
Different expression genes screening out
| BinCode | BinName | ID | Description | 1v5 | 2v6 | 4v6 | 3v5 |
|---|---|---|---|---|---|---|---|
| 10.2.1 |
cell wall.cellulose synthesis.cellulose synthase |
unigene5503_kv-1 |
cellulose synthase-like D5 |
13.21 |
14.26 |
13.38 |
13.14 |
| 10.7 |
cell wall.modification |
unigene68082_kv-1 |
xyloglucosyl transferase |
13.07 |
14.76 |
14.18 |
11.22 |
| 16.1.5 |
secondary metabolism.isoprenoids.terpenoids |
unigene52341_kv-1 |
TPS14 (TERPENE SYNTHASE 14) |
11.83 |
13.03 |
14.84 |
13.19 |
| 17.5.1 |
hormone metabolism.ethylene.synthesis-degradation |
unigene5647_kv-1 |
oxidoreductase |
13.82 |
12.63 |
12.65 |
13.61 |
| 20.2.3 |
stress.abiotic.drought/salt |
unigene23926_kv-1 |
early-responsive to dehydration protein-related |
6.41 |
6.27 |
6.15 |
6.44 |
| 20.2.3 |
stress.abiotic.drought/salt |
unigene23927_kv-1 |
early-responsive to dehydration protein-related |
5.25 |
5.98 |
5.96 |
5.39 |
| 26.2 |
misc.UDP glucosyl and glucoronyl transferases |
unigene67909_kv-1 |
XT1 (XYLOSYLTRANSFERASE 1) |
13.73 |
14.14 |
14.56 |
14.13 |
| 26.1 |
misc.cytochrome P450 |
unigene25666_kv-1 |
fatty acid (omega-1)-hydroxylase/ oxygen binding |
5.03 |
9.4 |
8.89 |
5.15 |
| 26.28 |
misc.GDSL-motif lipase |
unigene5707_kv-1 |
LTL1 (LI-TOLERANT LIPASE 1) |
11.21 |
13.81 |
12.54 |
11.52 |
| 27.3.3 |
RNA.regulation of transcription.AP2/EREBP, APETALA2/Ethylene-responsive element binding protein family |
unigene11425_kv-1 |
ABR1 (ABA REPRESSOR1) |
5.06 |
8.78 |
8.51 |
6.77 |
| 27.3.3 |
RNA.regulation of transcription.AP2/EREBP, APETALA2/Ethylene-responsive element binding protein family |
unigene13946_kv-1 |
ap2/EREBP transcription factor |
13.34 |
13.56 |
14.29 |
13.77 |
| 27.3.6 |
RNA.regulation of transcription.bHLH,Basic Helix-Loop-Helix family |
unigene64452_kv-1 |
basic helix-loop-helix (bHLH) family protein |
6.23 |
15.4 |
13.06 |
5.08 |
| 27.3.7 |
RNA.regulation of transcription.C2C2(Zn) CO-like, Constans-like zinc finger family |
unigene56521_kv-1 |
JAZ10 (JASMONATE-ZIM-DOMAIN PROTEIN 10) |
6.13 |
7.61 |
7.35 |
5.54 |
| 27.3.26 |
RNA.regulation of transcription.MYB-related transcription factor family |
unigene54286_kv-1 |
ATRL5 (ARABIDOPSIS RAD-LIKE 5) |
14.31 |
12.55 |
12.98 |
13.43 |
| 28.1.3 |
DNA.synthesis/chromatin structure.histone |
unigene16272_kv-1 |
|
12.43 |
14.56 |
14.31 |
12.18 |
| 29.4 |
protein.postranslational modification |
unigene9429_kv-1 |
MAPKKK17 |
5.27 |
6.13 |
5.81 |
5.18 |
| 29.5.11.4.3.2 |
protein.degradation.ubiquitin.E3.SCF.FBOX |
unigene4485_kv-1 |
|
14.03 |
14.2 |
12.62 |
12.41 |
| 30.3 |
signalling.calcium |
unigene51427_kv-1 |
calmodulin-binding family protein |
6.02 |
15.52 |
12.88 |
5.35 |
| 33.99 |
development.unspecified |
unigene25035_kv-1 |
NAC domain-containing protein 68 |
5.54 |
14.28 |
13.09 |
5.46 |
| 34.3 |
transport.amino acids |
unigene60928_kv-1 |
amino acid/polyamine transporter II |
7.1 |
7.05 |
5.87 |
7.51 |
| 34.18 | transport.unspecified anions | unigene12430_kv-1 | BOR4 (REQUIRES HIGH BORON 4) | 10.18 | 5.7 | 5.05 | 11.99 |
Note: column 5–8 showed the fold change times in four compared groups. 1: KV1/D; 2: XH/D; 3: ZKV1/V; 4:XH/V; 5:KV1/U; and 6: XH/U. The 'not assigned.unknown’ genes were not showed.
Other reports have shown that the phenylalanine metabolism pathway is related to the cotton defence response to V. dahliae. Therefore, in this study, we also analysed this pathway and identified some DEGs. Different cotton species and races of CVW were used in our study. The DEGs were nearly the same for sea-island cotton and upland cotton infected with different races of V. dahliae, but very different when comparing the two groups of sea-island cotton and the two groups of upland cotton. As shown in Figure 7, only one gene, hydroxycinnamoyl transferase (HCT), was upregulated and showed a strong change in expression when upland cotton was infected by V. dahliae (V991 or D07038), but five other genes (phenylalanine ammonia-lyase [PAL], 4-coumarate [4CL], cinnamyl alcohol dehydrogenase [CAD], caffeoyl-CoA O-methyltransgerase [CCoAOMT], and caffeoyl O-methyltransgerase [COMT]) were upregulated and obviously changed in sea-island cotton infected by V. dahliae (including V991 and D07038). As the reports, HCT can affect lignification in alfalfa, and lignin plays a key role in the cotton defence response, so the same function may exit in upland cotton [41].The results suggest that HCT is related to the upland cotton defence response to V. dahlia, but has little role in the sea-island cotton defence response, and the other five genes (especially the most upregulated gene, CCoAOMT) may be related to the defence response of sea-island cotton but not that of upland cotton. In other words, different disease-resistance mechanisms may exist in sea-island cotton and upland cotton. This may be the reason that the genes HCT and CCoAOMT were not identified.
Figure 7.
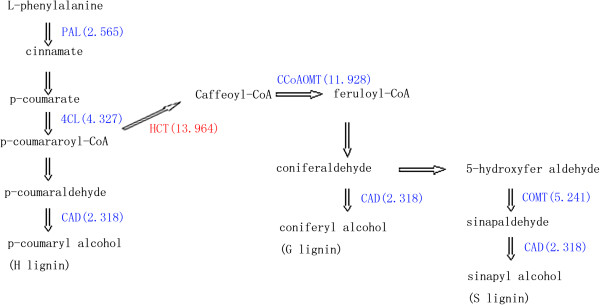
The unigenes involved in the phenylpropanoid pathway. Red show up-regulated in compared group of infected and uninfected in upland cotton G.hirsutum. Blue display up-regulated in compared group of infected and uninfected in sea island cotton G. barbadense. Digital in bracket mean the one biggest fold change times of annotating unigenes. hydroxycinnamoyl transferase (HCT), phenylalanine ammonia-lyase (PAL), 4-coumarate (4CL), cinnamyl alcohol dehydrogenase (CAD), caffeoyl-CoA O-methyltransgerase (CCoAOMT), caffeoyl O-methyltransgerase (COMT).
Conclusions
Although the molecular functions of the cotton defence response to V. dahliae and the associated signal transduction pathways remain largely unknown, the present transcriptome analysis provides valuable information regarding cotton resistance, which may facilitate future investigations of the detailed regulatory mechanisms and pathways. Using Illumina sequencing technology, we obtained about 5 Gb of raw sequence data and predicted 77,212 assembled unigenes, 66,208 of which were able to be annotated. Finally 44 differentially expressed genes were identified. The results suggest that HCT in upland cotton and CCoAOMT, PAL, 4CL, CAD, and COMT in sea-island cotton are involved in cotton defence response to V. dahliae. We believe that our data not only provide novel molecular information, but will also help to accelerate research into gene functions of cotton defence responses.
Methods
Preparation of material
One highly aggressive strain of the defoliating fungus V. dahliae, V991, and one intermediately aggressive strain, D07038, from the Institute of Plant Protection (IPPCAAS) and the Cotton Institute of the Chinese Academy of Agriculture Sciences(CICAAS), respectively, were used for inoculation [2]. The cotton varieties ZhongZhiMian KV-1 (G. hirsutum L., from IPPCAAS) and XinHai 15 (G. barbadense L.,from the Economic Crop Institute of the Xinjiang Academy of Agriculture Sciences,ECIXAAS, and CICAAS) resistant to V. dahliae were grown in sterilized soil (a mix of peat and sawdust) in a natural environment [42]. Each seedling was inoculated with 10 mL V. dahliae spore suspension of 2 × 107 spores per mL by watering injured roots from plants at the two-true-leaf growth stage. Control plants were not inoculated but were otherwise treated and sampled with distilled water in the same way. Plants from six individual seedlings were collected for each treatment at each sampling time point (including 24, 48, and 96 h after inoculation) after washed by 75% alcohol and sterile water.
cDNA preparation for Illumina sequencing
Total RNA was extracted using a Plant RNA EASYspin Plus Kit (aidlab, Peking, China) according to the manufacturer’s instructions. Six RNA samples (for each sample, all time points of each individual treatment were mixed), including four samples of ZhongZhiMian KV-1 (G. hirsutum) and XinHai 15 (G. barbadense) infected with V. dahliae strain V991 (highly toxic) and D07038 (intermediately toxic), respectively. Two plant samples treated with sterile water were further verified for the quantity of RNA using an ultraviolet spectrometer and electrophoresis on a denaturing formaldehyde agarose gel. The samples were as follows: 1, ZhongZhiMian KV-1 infected with D07038 (named: KV1/D); 2, XinHai 15 infected with D07038(named: XH/D); 3, ZhongZhiMian KV-1 infected with V991(named: KV1/V); 4, XinHai 15 infected with V991(named: XH/V); 5, uninfected ZhongZhiMian KV-1(named: KV1/U); and 6, uninfected XinHai 15(named: XH/U). All samples were mixed equally for transcriptome sequencing, but subjected individually to DGE sequencing. Oligo(dT) beads were used to isolate poly(A) + mRNA from total RNA. Fragmentation buffer was added to disrupt the mRNA into short fragments. These short fragments were used as templates with random hexamer primer to synthesise first-strand cDNA. Second-strand cDNA was synthesised by adding buffer, dNTPs, RNase, and DNA polymerase I. The resulting short fragments were purified with a QiaQuick PCR extraction kit and resolved with EB buffer for end reparation and addition of a poly(A) tail. Next, the short fragments were connected with sequencing adapters. Following agarose gel electrophoresis, suitable fragments were selected as templates for PCR. Finally, the library was sequenced using an Illumina HiSeq™ 2000.
Analysis of transcriptome sequencing data
The raw data from the images were collected using Illumina GA Pipeline 1.6 by removing low-quality reads (reads with unknown sequences 'N’), adaptor sequence fragments, and empty reads. Next, de novo assembly of the transcriptome into unigenes was carried out with Trinity, a short-read assembly program [43]. In brief, Trinity firstly combines reads with certain length of overlap to form longer fragments, which are called contigs. Then the reads are mapped back to contigs; with paired-end reads it is able to detect contigs from the same transcript as well as the distances between these contigs. Finally, Trinity connects the contigs, and get sequences that cannot be extended on either end. Such sequences are defined as Unigenes. Subsequently, a Basic Local Alignment Search Tool (BLAST) BLASTx alignment (e-value < 0.00001) was performed for the unigenes with protein databases, including the non-redundant (nr), Swiss-Prot, Kyoto Encyclopaedia of Genes and Genomes (KEGG), and COG databases, and the best alignments were used to decide the sequence direction of the unigenes. If the results from the different databases conflicted with each other, a priority order of nr, Swiss-Prot, KEGG, and COG was followed to decide the sequence direction of the unigenes. When a unigene was not aligned in any of the above databases, ESTScan was used to predict its coding regions and decide its sequence direction [44]. In the final step, using nr annotation, the Blast2GO program was used to obtain the Gene Ontology (GO) and KEGG annotations for the unigenes [45]. After the GO annotation was obtained for each unigene, WEGO software was used to classify the unigenes by function and to determine the distribution of gene functions in the species at the macro level [46]. All raw transcriptome data have been deposited in SRA (NCBI).
Analysis and mapping of DGE reads
The raw image data were transformed by base calling into sequence data. To map the DGE reads, the sequenced raw data were filtered to remove adaptor sequences, low-quality sequences (reads with unknown sequences 'N’), empty reads. For reads annotation, clean reads sequences were mapped to our transcriptome reference database, allowing no more than two nucleotide mismatch [43,47]. Gene coverage is the percentage of a gene covered by reads. This value is equal to the ratio of the base number in a gene covered by unique mapping reads to the total bases number of that gene. For gene expression analysis, the number of expressed sequence was calculated and normalised to the number of Reads per Kb per Million reads (RPKM).
Identification of differentially expressed genes
To compare the differences in gene expression, the reads frequency in each DGE library was statistically analysed according to the method of Audic and Claverie [48]. We used a false discovery rate of <0.001 and an absolute value of the log 2 ratio >1 as the threshold for judging the significance of the gene expression differences [49]. To determine the genes related to disease resistance, the inoculated V. dahliae cotton samples (samples 1 to 4) were compared to water-inoculated cotton samples (samples 5 and 6). Next, the differentially expressed genes were subjected to GO and KEGG Ontology (KO) enrichment analysis [27]. At the same time, the differentially expressed genes sequences of each group were uploaded to the Mercator web application to assign MapMan bins, and then differentially expressed genes between the four compared groups of related pathways were screened and analysed using MapMan software [50].
Data validation by qPCR
Total RNA was extracted as described for DGE library preparation and sequencing. Total RNA (1 μg) from each sample was reverse-transcribed in a 10 μL reaction using an AMV RNA PCR Kit 3.0 (Takara). The sequences of the primers used are shown in Table 5. The 18S rRNA gene and UBQ7 gene of cotton were used as internal control genes. qRT-PCR was performed using a SYBR Premix Ex Taq™ Kit (Takara) according to the manufacturer’s protocol. The selected genes were verified using a Bio-Rad iQ5 real-time PCR detection system with a cycling temperature of 57°C and with a single peak on the melting curve to ensure a single product. At least three replicates were tested per sample.
Table 5.
primers for qRT-PCR
| Description | id | ID | F | R |
|---|---|---|---|---|
| Celullose synthase-like D protein |
unigene5503_kv-1 |
1 |
CAATGCGTGAGTCCAAGT |
AAAGCCATTAGCACCTGA |
| Xyloglucan endotraglucosylase/hydrolase |
unigene68082_kv-1 |
2 |
GCAACTGAAGATGGCAAAT |
GACGAGTAAGCCGAGCAA |
| Terpene synthase 3 |
unigene52341_kv-1 |
3 |
ATTGGAGTTTGCTAGGGATC |
AGGAAATGGGCTTTGTGA |
| Oxidoreductase |
unigene5647_kv-1 |
4 |
TGAAGTTGGGTGGGAGAA |
CAGGCTGTGGACATTTGG |
| Early-responsive to dehydration protein-related |
unigene23926_kv-1 |
5 |
TTGATGCTGAAACCGCTTAT |
ATTGCCTCTTCCCTGTCCT |
| Early-responsive to dehydration protein-related |
unigene23927_kv-1 |
6 |
TTATGCTACCGTGACACCT |
CAACAGCCAGAGTAATGAGA |
| Xyloglucan 6-xylosyltransferase |
unigene67909_kv-1 |
7 |
CGAAAGGGAAGGTTAGAG |
TAAACTATGGCGGACTGA |
| Cytochrome P450 |
unigene25666_kv-1 |
8 |
TTGGTGGAGATGAAATGTG |
TGGACTAGAATGGGTAAGC |
| GSDL-motif lipase |
unigene5707_kv-1 |
9 |
TGCCATACTTGAGCCCTGAC |
TAGCCGCTGTGCCTGTTG |
| AP2/ERF domain-containing transcription factor |
unigene11425_kv-1 |
10 |
GGGCAGCCGAGATAAGAG |
TTGGGTCAGTAGATGTAGGAAT |
| AP2/ERF domain-containing transcription factor |
unigene13946_kv-1 |
11 |
TCCGTTGCTTCGTATCCT |
AACCCTTCATCTTTGACCAC |
| Symbiotic ammonium transporter |
unigene64452_kv-1 |
12 |
GAAAGCCAATCTAACAATC |
TCAACAAAGCCAGTCGTA |
| Protein TIFY 9 |
unigene56521_kv-1 |
13 |
AGGGCTACATCCGAGAAT |
AGGAGGACTTGCCACTTT |
| MYB transcription factor |
unigene54286_kv-1 |
14 |
TCAGTCGAGGAAGTAAGAA |
GTATAGGGATTTGACCAGA |
| Mitogen-activated protein kinase kinase kinase 17 |
unigene9429_kv-1 |
15 |
AGTGTCGTCGTCAGTTTCC |
GGTATTTCAGGCACATCAG |
| F-box family protein-like |
unigene4485_kv-1 |
16 |
GCTTTAGCTTCGGCTGTC |
CCAAATCGCTTTCACTCA |
| Calmodulin-binding-like protein |
unigene51427_kv-1 |
17 |
TTGGTTGCTCGTGATGGA |
TTGCCCGTATGAGTTGTC |
| NAC domain protein |
unigene25035_kv-1 |
18 |
AGTGATCGGGATGAAGAAA |
CGGCTGGATTATAGTGGTC |
| Amino acid transporter |
unigene60928_kv-1 |
19 |
TGAGTTTGGAGGGCTTAC |
GCATTGAACTGTAGAGGGTA |
| Anion exchanger family protein |
unigene12430_kv-1 |
20 |
TACTTCCTGGTATGTTTCG |
ATTCTCCTCTGTGCGTTG |
| 18 s |
L24145 |
21 |
TCGTAGTTGGACTTAGGGTGGG |
CAAATGCTTTCGCAGTTGTTCG |
| UBQ7 | DQ116441 | 22 | GAAGGCATTCCACCTGACCAAC | CTTGACCTTCTTCTTCTTGTGCTTG |
Competing interests
The authors declare that they have no competing financial interests.
Authors’ contributions
YFC conceived the study. HZJ, QS, XYZ , XHH, YZS,YLY and XMD participated in experiment materials preparation. XYZ, QS, WNW participated in RNA extraction. QS analyzed data and performed qRT-PCR. YFC, QS wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
The distribution of different expression genes in MapMan software function annotation. Note: column 1 showed BinCode ID. Column 2 meant the bin name of corresponding Bincode id or metabolic pathways name.
Contributor Information
Quan Sun, Email: sunquan3810@163.com.
Huaizhong Jiang, Email: jianghz@cqupt.edu.cn.
Xiaoyan Zhu, Email: zhuxiaoyans@sina.com.
Weina Wang, Email: wangwn1@126.com.
Xiaohong He, Email: hexh@cqupt.edu.cn.
Yuzhen Shi, Email: syzmb@yahoo.com.cn.
Youlu Yuan, Email: youluyuan@hotmail.com.
Xiongming Du, Email: dxm630723@163.com.
Yingfan Cai, Email: yingfancai@gmail.com.
Acknowledgements
We acknowledge the Beijing Genomics institute at Shenzhen for its assistance in original data processing and related bioinformatics analysis and thank the National Medium-term Gene Bank of Cotton in China, Professor Guiliang Jian in IPP, Professor Xueyuan Li in ECIXAAS, Dr.Heqin Zhu in CICAAS for the resource materials supply. This work was supported by the Natural Science Foundation of China (NSFC grant No. 31071461) and Chongqing Natural Science Foundation (cstc2012jjA80037;cstc2011AB1095) and Chongqing Municipal Commission of Education (KJ130502, KJ110506).
References
- Sal'kova EG, Guseva NN. The role of pectolyitc enzymes of the Verticillium dahliae fungus in the development of cotton wilt. Dokl Akad Nauk SSSR. 1965;14:515–522. [PubMed] [Google Scholar]
- Sink KC, Grey WE. A root-injection method to assess Verticillium wilt resistance of peppermint (Mentha × piperita L.) and its use in identifying resistant somaclones of cv. Black Mitcham. Euphytica. 1999;14(3):223–230. doi: 10.1023/A:1003591908308. [DOI] [Google Scholar]
- Du W-S, Du X, Ma Z. Progress of Inheritance and Molecular Biology of Verticillium wilt resistance in Cotton. Cotton Sci. 2002;14(5):55–61. [Google Scholar]
- Bugbee W, Sappenfield W. Effect of Verticillium wilt on cotton yield, fiber properties, and seed quality. Crop Sci. 1970;14(6):649–652. doi: 10.2135/cropsci1970.0011183X001000060011x. [DOI] [Google Scholar]
- Cai Y, Xiaohong H, Mo J, Sun Q, Yang J, Liu J. Molecular research and genetic engineering of resistance to Verticillium wilt in cotton: a review. Afr J Biotechnol. 2009;14(25):7363–7372. [Google Scholar]
- Jing YL, Liu YB, Fan WF, Xiao BC. Advance in the Study on Verticillium Wilt of Cotton and It’s Breeding for Resistance. Acta Agric Boreali-occidentalis Sin. 1999;14(3):106–110. [Google Scholar]
- Jian G, Lu M. Study on the method of breeding cotton for resistance to Verticillium dahliae. Acta Phytopathologica Sin. 2004;14(4):70–74. [Google Scholar]
- Ma C, Jian G, Zheng C. The advances in cotton breeding resistance to Fusarium and Verticillium wilts in China during past fifty years. Agric Sci China. 2002;14(5):508–513. [Google Scholar]
- Qi J, Ma C, Zhao L, Liu S. Study on Heredity of Verticillium wilt resistance of G. barbadense L. Acta Gossypii Sin. 2000;14(4):169–171. [Google Scholar]
- Barrow JR. Heterozygosity in inheritance of Verticillium wilt tolerance in cotton. Phytopathology. 1970;14(2):301–303. doi: 10.1094/Phyto-60-301. [DOI] [Google Scholar]
- Verhalen LM, Brinkerhoff L, Fun K-C, Morrison WC. A quantitative genetic study of Verticillium wilt resistance among selected lines of upland cotton. Crop Sci. 1971;14(3):407–412. doi: 10.2135/cropsci1971.0011183X001100030029x. [DOI] [Google Scholar]
- Jiang F, Zhao J, Zhou L, Guo W, Zhang T. Molecular mapping of Verticillium wilt resistance QTL clustered on chromosomes D7 and D9 in upland cotton. Sci China Ser C, Life Sci / Chin Acad Sci. 2009;14(9):872–884. doi: 10.1007/s11427-009-0110-8. [DOI] [PubMed] [Google Scholar]
- Wang L, Dai X. Progress on Molecular Research of Cotton Verticillium wilt resistances. Mol Plant Breed. 2003;14(1):97–102. [Google Scholar]
- Smit F, Dubery IA. Cell wall reinforcement in cotton hypocotyls in response to a Verticillium dahliae elicitor. Phytochemistry. 1997;14(5):811–815. doi: 10.1016/S0031-9422(96)00595-X. [DOI] [Google Scholar]
- Zhou Y, Du H, Yuan H, Zhang Y, Zhu B. Isolation and purification of antifungal protein from Paenibacillus to Verticillium dahliae. Cotton Sci. 2007;14(2):98–101. [Google Scholar]
- Gaffe J, Bru J-P, Causse M, Vidal A, Stamitti-Bert L, Carde J-P, Gallusci P. LEFPS1, a tomato farnesyl pyrophosphate gene highly expressed during early fruit development. Plant Physiol. 2000;14(4):1351–1362. doi: 10.1104/pp.123.4.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannanov RN. The use of natural bio-agents for the control of cotton phytopathogens. Meded Rijksuniv Gent Fak Landbouwkd Toegep Biol Wet. 2001;14(3a):183–186. [PubMed] [Google Scholar]
- Cheng H, Jian G, Ni W, Yang H, Z-x WANG, W-j SUN, B-l ZHANG, WANG X-f MAC, S-r JIA. Increase of Fusarium-and Verticillium-resistance by transferring chitinase and glucanase gene into cotton. Sci Agric Sin. 2005;14(6):1160–1166. [Google Scholar]
- Zhang Y, Wang X, Cheng C, Gao Q, Liu J, Guo X. Molecular cloning and characterization of GhNPR1, a gene implicated in pathogen responses from cotton (Gossypium hirsutum L.) Biosci Rep. 2008;14:7–14. doi: 10.1042/BSR20070028. [DOI] [PubMed] [Google Scholar]
- Smith CG, Rodgers MW, Zimmerlin A, Ferdinando D, Bolwell GP. Tissue and subcellular immunolocalisation of enzymes of lignin synthesis in differentiating and wounded hypocotyl tissue of French bean (Phaseolus vulgaris L.) Planta. 1994;14(2):155–164. doi: 10.1007/BF01089030. [DOI] [Google Scholar]
- H-k HUANG, Y-y WU, W-x XU, Q-y ZHOU. Cloning, Sequencing and Expression Profile Analysis of A Polygalacturonase-inhibiting Protein (PGIP) Gene in Cotton. J Henan Agric Sci. 2008;14:012. [Google Scholar]
- Wang HM, Lin ZX, Zhang XL, Chen W, Guo XP, Nie YC, Li YH. Mapping and quantitative trait loci analysis of Verticillium wilt resistance genes in cotton. J Integr Plant Biol. 2008;14(2):174–182. doi: 10.1111/j.1744-7909.2007.00612.x. [DOI] [PubMed] [Google Scholar]
- Pomar F, Novo M, Bernal MA, Merino F, Barceló AR. Changes in stem lignins (monomer composition and crosslinking) and peroxidase are related with the maintenance of leaf photosynthetic integrity during Verticillium wilt in Capsicum annuum. New Phytol. 2004;14(1):111–123. doi: 10.1111/j.1469-8137.2004.01092.x. [DOI] [PubMed] [Google Scholar]
- Gayoso C, Pomar F, Novo-Uzal E, Merino F, de Ilárduya ÓM. The Ve-mediated resistance response of the tomato to Verticillium dahliae involves H2O2, peroxidase and lignins and drives PAL gene expression. BMC Plant Biol. 2010;14(1):232. doi: 10.1186/1471-2229-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Zhu L, Tu L, Liu L, Yuan D, Jin L, Long L, Zhang X. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J Exp Bot. 2011;14(15):5607–5621. doi: 10.1093/jxb/err245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge WJ. Next-generation DNA sequencing techniques. New Biotechnol. 2009;14(4):195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;14(suppl 1):D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew H, Paterson JFW, Heidrun G, Hui G, Jerry J, Dianchuan J, Danny L, Kurtis C, Showmaker S, Shengqiang S, Joshua U, Mi-jeong Y, Robert B, Wei C, Adi D-F, Mary V, Duke LG, Jane G, Corrinne G, Kara G, Guanjing H, Tae-ho L, Jingping L, Lifeng L, Tao L, Barry S, Marler J, Justin, Page, Alison. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;14(7429):5. doi: 10.1038/nature11798. [DOI] [PubMed] [Google Scholar]
- Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S. et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;14(10):1098–1103. doi: 10.1038/ng.2371. [DOI] [PubMed] [Google Scholar]
- Burton RA, Wilson SM, Hrmova M, Harvey AJ, Shirley NJ, Medhurst A, Stone BA, Newbigin EJ, Bacic A, Fincher GB. Cellulose synthase-like CslF genes mediate the synthesis of cell wall (1, 3; 1, 4)-ß-D-glucans. Science. 2006;14(5769):1940–1942. doi: 10.1126/science.1122975. [DOI] [PubMed] [Google Scholar]
- Doblin MS, Pettolino FA, Wilson SM, Campbell R, Burton RA, Fincher GB, Newbigin E, Bacic A. A barley cellulose synthase-like CSLH gene mediates (1,3;1,4)-beta-D-glucan synthesis in transgenic Arabidopsis. Proc Natl Acad Sci U S A. 2009;14(14):5996–6001. doi: 10.1073/pnas.0902019106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Xiong G, Li R, Cui J, Tang D, Zhang B, Pauly M, Cheng Z, Zhou Y. Rice cellulose synthase-like D4 is essential for normal cell‒wall biosynthesis and plant growth. Plant J. 2009;14(6):1055–1069. doi: 10.1111/j.1365-313X.2009.04022.x. [DOI] [PubMed] [Google Scholar]
- Vega-Sanchez ME, Verhertbruggen Y, Christensen U, Chen X, Sharma V, Varanasi P, Jobling SA, Talbot M, White RG, Joo M. et al. Loss of Cellulose synthase-like F6 function affects mixed-linkage glucan deposition, cell wall mechanical properties, and defense responses in vegetative tissues of rice. Plant Physiol. 2012;14(1):56–69. doi: 10.1104/pp.112.195495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK, Braam J, Fry SC, Nishitani K. The XTH family of enzymes involved in xyloglucan endotransglucosylation and endohydrolysis: current perspectives and a new unifying nomenclature. Plant Cell Physiol. 2002;14(12):1421–1435. doi: 10.1093/pcp/pcf171. [DOI] [PubMed] [Google Scholar]
- Shao M, Wang X, Ni M, Bibi N, Yuan S, Malik W, Zhang H, Liu Y, Hua S. Regulation of cotton fiber elongation by xyloglucan endotransglycosylase/hydrolase genes. Genet Mol Res. 2011;14:3771–3782. doi: 10.4238/2011.October.27.1. [DOI] [PubMed] [Google Scholar]
- Lee J, Burns TH, Light G, Sun Y, Fokar M, Kasukabe Y, Fujisawa K, Maekawa Y, Allen RD. Xyloglucan endotransglycosylase/hydrolase genes in cotton and their role in fiber elongation. Planta. 2010;14(5):1191–1205. doi: 10.1007/s00425-010-1246-2. [DOI] [PubMed] [Google Scholar]
- Michailidis G, Argiriou A, Darzentas N, Tsaftaris A. Analysis of xyloglucan endotransglycosylase/hydrolase (XTH) genes from allotetraploid (Gossypium hirsutum) cotton and its diploid progenitors expressed during fiber elongation. J Plant Physiol. 2009;14(4):403–416. doi: 10.1016/j.jplph.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Danner H, Boeckler GA, Irmisch S, Yuan JS, Chen F, Gershenzon J, Unsicker SB, Köllner TG. Four terpene synthases produce major compounds of the gypsy moth feeding-induced volatile blend of Populus trichocarpa. Phytochemistry. 2011;14(9):897–908. doi: 10.1016/j.phytochem.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Chen XY, Chen Y, Heinstein P, Davisson VJ. Cloning, expression, and characterization of (+)-delta-cadinene synthase: a catalyst for cotton phytoalexin biosynthesis. Arch Biochem Biophys. 1995;14(2):255–266. doi: 10.1006/abbi.1995.0038. [DOI] [PubMed] [Google Scholar]
- Chen XY, Wang M, Chen Y, Davisson VJ, Heinstein P. Cloning and heterologous expression of a second (+)-delta-cadinene synthase from Gossypium arboreum. J Nat Prod. 1996;14(10):944–951. doi: 10.1021/np960344w. [DOI] [PubMed] [Google Scholar]
- Shadle G, Chen F, Srinivasa Reddy M, Jackson L, Nakashima J, Dixon RA. Down-regulation of hydroxycinnamoyl CoA: shikimate hydroxycinnamoyl transferase in transgenic alfalfa affects lignification, development and forage quality. Phytochemistry. 2007;14(11):1521–1529. doi: 10.1016/j.phytochem.2007.03.022. [DOI] [PubMed] [Google Scholar]
- ZHAO L, S-z WANG, F-j QI, W-w ZHANG, G-l JIAN. Isolation, Characterization and Phylogenetic Analysis of the Resistance Gene Analogues (RGAs) in Upland Cotton (Gossypium hirsutum L.) Zhongzhimian KV-1 [J] Cotton Sci. 2009;14:007. [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;14(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB. 1999;14:138–148. [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;14(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;14(suppl 2):W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;14(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Audic S, Claverie J-M. The significance of digital gene expression profiles. Genome Res. 1997;14(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;14(4):1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. mapman: a user‒driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;14(6):914–939. doi: 10.1111/j.1365-313X.2004.02016.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The distribution of different expression genes in MapMan software function annotation. Note: column 1 showed BinCode ID. Column 2 meant the bin name of corresponding Bincode id or metabolic pathways name.