

Abstract
β-Adrenergic receptor blockade reduces total mortality and all-cause hospitalizations in patients with heart failure (HF). Nonetheless, β-blockade does not halt disease progression, suggesting that cAMP-dependent protein kinase (PKA) signaling downstream of β-adrenergic receptor activation may persist through unique post-translational states. In this study, human myocardial tissue was used to examine the state of PKA subunits. As expected, total myosin binding protein-C phosphorylation and Ser23/24 troponin I phosphorylation significantly decreased in HF. Examination of PKA subunits demonstrated no change in type II regulatory (RIIα) or catalytic (Cα) subunit expression, although site specific RIIα (Ser96) and Cα (Thr197) phosphorylation were increased in HF. Further, the expression of type I regulatory subunit (RI) was increased in HF. Isoelectric focusing of RIα demonstrated up to three variants, consistent with reports that Ser77 and Ser83 are in vivo phosphorylation sites. Western blots with site-specific monoclonal antibodies showed increased Ser83 phosphorylation in HF. 8-fluo-cAMP binding by wild type and phosphomimic Ser77 and Ser83 mutant RIα proteins demonstrated reduced Kd for the double mutant as compared to WT RIα. Therefore, failing myocardium displays altered expression and post-translational modification of PKA subunits that may impact downstream signaling.
Keywords: Protein kinase A, Regulatory, Catalytic, Heart, Phosphorylation
Introduction
The use of β-adrenergic receptor (β-AR)1 blocker therapy in heart failure has been demonstrated to improve mortality [1]. These drugs counter the impact of increased circulating levels of catechol-amines that, if left unchecked, eventually lead to reduced density and desensitization of β-ARs during heart failure [2,3]. Consistent with antagonism of the receptor, prior studies have shown strong dephosphorylation of the sarcomeric proteins troponin I (TnI) and myosin binding protein-C (MYBP-C) in samples from explanted hearts [4–7]. These two sarcomeric proteins are key targets of the cAMP-dependent protein kinase (PKA; reviewed in [8,9]) activated subsequent to β-AR stimulation [10,11] and have roles in modulating cardiac muscle contractility (reviewed in [12]). The dephosphorylation of these proteins may be due to desensitization of the β-adrenergic receptors during heart failure or concomitant pharmacological blockade, and despite the role of these sarcomeric proteins in acto-myosin crossbridge cycling and Ca2+ sensitivity of force activation [13,14], the exact mechanism linking their phosphorylation state to the physiological impact of β-adrenergic receptor blockade remains largely unknown.
As a kinase responsible for MYBP-C and TnI phosphorylation, PKA activity plays an important role in cardiac muscle physiology. The PKA holoenzyme is a tetramer composed of a dimer of regulatory subunits and a dimer of catalytic subunits. Two types of regulatory subunits, RI and II, have been described in mammals and two separate genes for each encode α- and β-forms (reviewed in [8]), with α predominant in the heart [15]. The regulatory subunits bind cAMP generated by activated adenylate cyclase, in turn releasing the catalytic subunits and allowing downstream phosphorylation of target proteins. To maintain the holoenzyme in the inactive state, RI and II tether C through inhibitory domains presented either as a pseudosubstrate site in RIα [16] or the phosphorylatable Ser96 in RIIα [17]. For the effector catalytic subunit, three genes encode for Cα/β/γ, with the expression of γ being largely restricted to testis [18]. A newly found member, PrKX, appears atypical through its association with only RIα [19]. Cα is the predominant gene expressed in the heart and although it may encode two isoforms, only one appears to be expressed [20]. A minority of the C subunit expression in the heart is from the β gene, which similarly encodes two isoforms that are present at very low levels upon enrichment [21]. Functionally, the Cα subunit has two well documented phosphorylation sites Thr197 and Ser338 [22,23] that are likely substrates for a two-step phosphorylation-activation mechanism [24]. Of particular interest, phosphorylation of Thr197 significantly increases the km of the enzyme towards substrates without significantly impacting kcat [25].
Currently, there are limited data that examine the PKA subunit expression level and post-translational state in human pathophysiology. Previous reports demonstrated that RIα and IIα were reduced in patients with dilated cardiomyopathy [26,27]. Furthermore, these two regulatory subunits may be mislocalized or misorganized in heart failure [15,26], possibly impacting the selectivity of Cα for its targets following β-AR stimulation. Given the importance of the post-translational status of PKA subunits in defining the enzyme’s function, we have examined their expression level and phosphorylation status in failing vs. donor human myocardium. These data suggest that the post-translational state of Cα and RIIα subunits favor an enzyme with increased km for substrates and a decreased tendency for reassociation into the holoenzyme, respectively. Furthermore, data for RIα validate the existence of two phosphorylation sites, Ser77 and Ser83, with the abundance of the latter being higher in heart failure samples. Further, we demonstrate that an RIα mutant mimicking Ser77/83 phosphorylation shows reduced affinity for 8-fluo-cAMP, opening the possibility that these phosphorylation events may impact type I PKA activity.
Experimental procedures
Tissue collection and sample preparation
Tissue samples were collected from the left ventricle of explanted hearts of patients at the time of cardiac transplantation at the Mayo Clinic, in accordance with IRB #06-005671. Consent was obtained from all patients prior to sample collection. Obtained samples were rapidly frozen in liquid nitrogen upon retrieval and stored at −80 °C until use.
MYBP-C and TnI phosphorylation
To determine the relative overall phosphorylation of TnI and MYBP-C, SDS–PAGE resolved samples were stained with Pro-Q Diamond phosphoprotein stain and then subsequently with Deep Purple total protein stain as previously described [28]. Stained gels were scanned using a Typhoon 9410 and the signals quantified by Image-Quant TL software. Following densitometry, the ratio of the phosphoprotein stain to the total protein stain was reported as the relative extent of phosphorylation. All the available failing and non-failing heart samples were used for analysis, and for statistical comparison, Student’s t-test was used with a P < 0.05 cutoff for significance.
Western blotting
To determine the relative expression levels of total or phosphorylated proteins, multiplex Western blotting was performed with simultaneous monitoring of sarcomeric α-actin as the internal standard. Muscle samples were homogenized in SDS–PAGE sample buffer with protease and phosphatase inhibitors (Roche) and resolved by Bis–Tris SDS–PAGE [29]. When samples were to be dephosphorylated by alkaline phosphatase (50 units/1 h/37 °C), the tissue was homogenized directly into the supplied alkaline phosphatase buffer (Sigma) and the phosphatase inhibitors were omitted. Prepared homogenates were transferred to low fluorescence PVDF membrane or Hybond-P membrane (GE Lifesciences), blocked and incubated simultaneously with a mouse monoclonal antibody against sarcomeric α-actin (Sigma) along with varying combinations of: a polyclonal rabbit antibody against Ser23/24 phosphorylated TnI (Cell Signaling), a monoclonal rabbit antibody against Cα (Epitomics), a monoclonal rabbit antibody against Thr197 phosphorylated Cα (Epitomics), monoclonal mouse antibodies against PKA regulatory subunits type I and IIα (BD Biosciences) or a monoclonal rabbit antibody against Ser96 phosphorylated type IIα regulatory subunit of PKA (Epitomics). To measure the extent of RIα phosphorylation, two custom rabbit monoclonal antibodies raised against Ser77 or Ser83 phosphorylated RIα were used (Epitomics). Following overnight primary antibody incubation and washing, blots were incubated with the appropriate Cy3-labeled, Cy5-labeled, or HRP-labeled secondary antibody (Jackson Immunoresearch/GE Lifesciences), washed, and either developed with ECL reagent or allowed to dry and scanned on a Typhoon 9410 imager at the appropriate channels for Cy3 and Cy5 signal quantitation. The scanned images were analyzed using ImageQuant TL software. For all samples, the quantified signal for the protein of interest was divided by the actin signal to internally control for relative expression. The normalized data were then index to the control group that was set to 1. The signals for phosphorylated species were normalized to actin rather than the total expression of the given protein (e.g. pThr197 Cα normalized to total Cα) because the magnitudes of expression are relevant to the binding equilibria within the cell. To normalize values across different blots, one of the human heart samples was chosen as a standard sample and loaded on all Western blots. All the available failing and non-failing heart samples were used for analysis, and for statistical comparison, Student’s t-test was used with a P < 0.05 cutoff for significance.
Two-dimensional SDS–PAGE
To further examine the post-translational status of the subunits of RIα, 2D SDS–PAGE of homogenates or enriched samples was performed. The procedure was as previously described and used 7 cm 4–7 linear gradient strips [28]. Strips were rehydrated overnight and focused in the first dimension the next morning, followed by second dimension SDS–PAGE and Western blotting. For Western blotting of 2D SDS–PAGE, minimal cross-reaction horseradish peroxidase coupled anti-mouse or anti-rabbit IgG antibodies (Jackson Immunoresearch) were used. Mass spectrometry from silver stained 2D SDS–PAGE was done by the Mayo Clinic Proteomics core (See Supplementary Fig. S1).
cAMP affinity chromatography
To enrich type I PKA regulatory subunits, a modified protocol was used based on a previously published method [30]. Tissue (~20 mg) was homogenized for 3 min on ice in 500 μL of (in mM) 150 NaCl, 20 HEPES, pH 7.4, 2 tris(2-carboxyethyl)phosphine (TCEP), 10 MgCl2, 20 sucrose, 0.1 EDTA, 0.1 NADH, protease and phosphatase inhibitor cocktails (Roche). Following homogenization, the lysate was centrifuged (10,000g/5 min), the supernatant collected and 8-(2-aminoethylamino)-cAMP (8-AEA-cAMP) agarose (equivalent to 0.5 μmol cAMP ligand) was added. The slurry was allowed to rotate at room temperature for 1 h, the non-bound fraction cleared, and the beads were washed three times for 5 min at room temperature with 500 μL of the homogenization buffer. Following washing, bound protein was eluted with 150 μL of 2D SDS–PAGE rehydration buffer [28] by incubating at room temperature for 10 min with occasional vortexing.
Steady state 8-fluo-cAMP binding
cDNA for human RIα was reverse-transcribed and amplified from heart total RNA (Life Technologies, CA) using a forward (5′-acatatggagtctggcagtaccgccg-3′) and reverse primer (5′-ttcagacagacagtgacacaaaactgt-3′) by previously published methods [31]. To mimic phosphorylation at Ser77 and Ser83, single and double mutants to Asp were generated by the Quikchange method (Agilent). The wild type (WT) and phosphomimic RIα proteins (Ser77Asp, Ser83Asp, Ser77/83Asp) were expressed in Rosetta2 (DE3) Escherichia coli (EMD Millipore) grown in LB medium at 37 °C until O.D.600–0.7, and induced overnight at room temperature with the addition of 0.2 mM IPTG. Bacteria were harvested by centrifugation, resuspended in 20 mM KPO4, pH 7.4 buffer and lysed by bead beating using 0.1 mm beads. The supernatant was collected following centrifugation and fractionated on a HiLoad 26/10 Sepharose Q column developed by a gradient from 0 to 0.4 M arginine hydrochloride in 20 mM KPO4, pH 7.4. Fractions containing RIα protein were identified by SDS–PAGE, pooled, and the protein captured by 8-AEA-cAMP affinity chromatography. Following washing with 0.4 M arginine hydrochloride, 20 mM KPO4, pH 7.4 buffer, bound RIα was eluted using 7 M urea, 20 mM KPO4, pH 7.4, 5 mM TCEP. The eluted RIα was concentrated by 10,000 MWCO centrifugal filtration and dialyzed into 25 mM KCl, 10 mM PIPES, pH 7. Steady state binding of 8-fluo-cAMP to purified WT or mutant RIα was measured by fluorescence anisotropy on a Shimadzu RF-5301PC with appropriate polarization filters and excitation/emission wavelengths of 485 and 515 nm. Anisotropy measurements were made as described [32]. A cuvette containing 0.5 μM RIα with 2 nM 8-fluo-cAMP in 150 mM KCl, 20 mM PIPES, pH 7 was serially diluted with 2 nM 8-fluo-cAMP in 150 mM KCl, 20 mM PIPES, pH 7 to measure the anisotropy vs. RIα concentration. Binding curves were fit individually to determine the Kd for each titration, as previously described [28,33].
Results
Patient population
Twenty total samples were examined in the study, with known patient characteristics summarized in Table 1. Ten samples of heart failure myocardium were obtained from human hearts at the time of transplant. Samples were obtained from 9 males and 1 female (median age 59, range 37–75). Of the 10 transplant patients, 6 had ischemic cardiomyopathy and 4 nonischemic cardiomyopathy, and the average ejection fraction in the group was 18 ± 2%. Although patients were on a variety of medications, all were on β-blocker therapy. Comparator myocardium samples were from donors deceased of non-cardiac causes (10 males, median age 53, range 21–65) and were kindly provided by Dr. Cris dos Remedios, University of Sydney, Australia. Due to the means by which the samples were procured, previous medication history of the donors is unknown.
Table 1.
Patient characteristics.
| Patient | Age | Sex | Ejection fraction (%) | Cause of death/medications |
|---|---|---|---|---|
| Control 1 | 52 | M | Cerebral hemorrhage | |
| Control 2 | 21 | M | Unknown | |
| Control 3 | 55 | M | Aortic aneurysm | |
| Control 4 | 65 | M | Aortic aneurysm | |
| Control 5 | 61 | M | Unknown | |
| Control 6 | 56 | M | Cerebral hemorrhage | |
| Control 7 | 47 | M | Unknown | |
| Control 8 | 54 | M | Crush injury | |
| Control 9 | 41 | M | Unknown | |
| Control 10 | 44 | M | Unknown | |
| HF1 | 58 | F | 10 | Amiodarone, bumetanide, furosemide, losartan, metolazone, metoprolol, milrinone, potassium, simvastatin, spirinolactone |
| HF2 | 60 | M | 10 | Aspirin, carvedilol, furosemide, simvastatin |
| HF3 | 41 | M | 20 | Aspirin, carvedilol, furosemide, lisinopril, potassium |
| HF4 | 72 | M | 10 | Digoxin, ezetimibe, metropolol, milrinone, potassium, pravastatin, spirinolactone |
| HF5 | 55 | M | 15 | Aspirin, carvedilol, digoxin, furosemide, lisinopril, potassium, spirinolactone |
| HF6 | 75 | M | 20 | Amiodarone, carvedilol, digoxin, furosemide, metolazone, potassium, spirinolactone |
| HF7 | 68 | M | 35 | Aspirin, furosemide, lisinopril, metoprolol, simvastatin |
| HF8 | 39 | M | 20 | Amiodarone, aspirin, carvedilol, furosemide, lisinopril, potassium, spirinolactone |
| HF9 | 37 | M | 15 | Amiodarone, aspirin, carvedilol, digoxin, losartan, furosemide, milrinone, spirinolactone, simvastatin |
| HF10 | 74 | M | 25 | Aspirin, carvedilol, digoxin, furosemide, isosorbide dinitrate, lisinopril, niacin |
Phosphorylation of MYBP-C and TnI
The extent of total phosphorylation of MYBP-C and TnI were measured in donor (n = 10) vs. heart failure (n = 10) samples by successive ProQ Diamond phosphoprotein and Deep Purple total protein staining (Fig. 1A and B). There was a statistically significant decrease in the extent of phosphorylation of both MYBP-C (1 ± 0.09 vs. 0.65 ± 0.02) and TnI (1 ± 0.11 vs. 0.27 ± 0.05), as previously reported by others [4–7]. Furthermore, the extent of Ser23/24 phosphorylation of TnI was also significantly decreased in the heart failure samples vs. donors (1 ± 0.12 vs. 0.59 ± 0.05; Fig. 1C, n = 10 per group) whereas the relative amount of TnI vs. actin was unchanged (Fig. 1D). In the heart failure group, no trends were found in the data when analyzing TnI and MyBP-C phosphorylation vs. age or ejection fraction (data not shown).
Fig. 1.

TnI and MYBP-C phosphorylation in heart failure. Myocardial homogenates from heart failure and donor patients were resolved by SDS–PAGE and stained successively with ProQ Diamond phosphoprotein (top panels in A and B) and Deep Purple total protein stain (bottom panels in A and B). There were statistically significant decreases in total phosphorylation of both MYBP-C (A) and TnI (B) in the heart failure samples. Further analyses of site specific Ser23/24 phosphorylation of TnI by Western blotting demonstrated a concurrent 41% decrease in the heart failure samples (C) without a change in total TnI expression (D). *Denotes P < 0.05 vs. donor.
One-dimensional analysis of the PKA catalytic and type II regulatory subunits
The expression level of Cα and its phosphorylation at Thr197 were examined by Western blotting. Although there was a significant increase in Thr197 phosphorylation of the subunit during heart failure (1 ± 0.06 vs. 1.67 ± 0.15; Fig. 2A, n = 10 per group), no significant change in total Cα expression between donor and heart failure samples was observed (1 ± 0.06 vs. 1.14 ± 0.14; Fig. 2B, n = 10 per group). When compared to age or ejection fraction, no trends were observed for Cα expression or phosphorylation (data not shown). For RIIα, the total expression in the myocardium from explanted hearts was not significantly different than donors, although there was a significant increase in Ser96 phosphorylation of the subunit (1 ± 0.22 vs. 1.72 ± 0.28; Fig. 3A, n = 10 per group). Further, no relationship was found between patient characteristics such as age or EF and RIIα expression or phosphorylation (data not shown).
Fig. 2.

PKA Catalytic subunit expression in heart failure. The total expression level and phosphorylation of Cα at Thr197 were examined by multiplex Western blotting. The extent of Thr197 phosphorylation was significantly higher by 67% in the heart failure samples (A). However, this was not reflected in the total Cα expression levels, as myocardium from both donor and failed hearts demonstrated similar levels (B). *Denotes P < 0.05 vs. donor.
Fig. 3.

Expression of type II PKA Regulatory subunits. The expression level of Ser96 phosphorylated (A) and total RIIα (B) in donor vs. heart failure myocardium. Expression of Ser96 phosphorylated RIIα significantly increased by 72% without an accompanying significant change in total RIIα expression. *Denotes P < 0.05 vs. donor.
Expression and post-translational status of the type I PKA regulatory subunit
In the heart failure myocardium, there was a significant increase in the expression of RI (1 ± 0.07 vs. 1.45 ± 0.09; Fig. 4A, n = 10 per group). Myocardium predominantly expresses RIα, and examination of its post-translational status required novel tools. A limited number of prior publications have found two sites of phosphorylation of RIα at Ser77 and Ser83 [34,35], and this was consistent with 2D Western blots wherein up to three isoelectric variants were observed in total homogenates from two donor and two failing myocardium (Fig. 4B). A similar pattern of RIα isoelectric variants could be observed by enriching cAMP-binding proteins from a heart failure patient sample using 8-AEA-cAMP chromatography (Fig. 4C, left) followed by 2D SDS–PAGE (Fig. 4C, right). Mass spectrometry analyses confirmed the identity of the labeled spots as RIα and supported phosphorylation at Ser77 and 83 (Supplementary Fig. S1). To examine the extent of phosphorylation at Ser77 and Ser83, two rabbit monoclonal antibodies, pSer77 and pSer83, were generated on contract (Epitomics) and validated. ELISA experiments using singly and doubly phosphorylated peptides showed that the antibodies demonstrated the expected selectivity for phosphorylated Ser77 or 83 (Fig. 5A). In rat myocardial homogenates prepared with alkaline phosphatase treatment, both pSer77 and pSer83 antibodies showed a reduction in signal when compared to untreated homogenates (Fig. 5B). The antibodies were also used in 2D SDS–PAGE Western blotting with the 8-AEA-cAMP chromatography enriched protein sample (Fig. 5C). As expected, the pSer77 and pSer83 monoclonal antibodies identified a subset of the RIα isoelectric variants, whereas the antibody against total RI identified all three (Fig. 5C, right). Using the pSer77 or pSer83 antibodies, the relative abundance of phosphorylation at these sites in the two sample sets was measured by Western blotting. The extent of Ser77 phosphorylation was not different between donor and heart failure groups but trended towards increase (1 ± 0.21 vs. 1.51 ± 0.28; P = 0.08, Fig. 6A, n = 10 per group). However, there was a statistically significant increase in the extent of Ser83 phosphorylation (1 ± 0.28 vs. 2.10 ± 0.45; Fig. 6B, n = 10 per group). The increase in Ser83 phosphorylation was evident when absolute expression levels were compared, but was not maintained if it was normalized to total RI expression. Therefore, the proportion of Ser83 phosphorylated RI did not change, but absolute levels were increased. For all expression and phosphorylation parameters measured, there were no correlations between patient age or ejection fraction (data not shown).
Fig. 4.

Expression of type I PKA Regulatory subunits. In failing myocardium, the relative expression of RIα significantly increased by 45% over donor samples (A, *P < 0.05 vs. donor). To scout for possible post-translational modifications of RIα, 2D Western blotting was done on two different donor and two different heart failure samples and showed up to three isoelectric variants of RIα (B). The presence of three isoelectric variants was additionally confirmed by capture of cAMP-binding proteins from a homogenate prepared from the HF1 sample by 8-AEA-cAMP-agarose beads (C, left). The fraction of the homogenate not bound to beads, the subsequent washes and the eluted fraction from the beads were analyzed by 1D SDS–PAGE and silver staining. Subsequently, the eluted fraction was analyzed by 2D SDS–PAGE and silver staining (C, right) which recapitulated the observed RIα pattern (denoted by arrowheads) as well capturing RIIα (denoted by *).
Fig. 5.
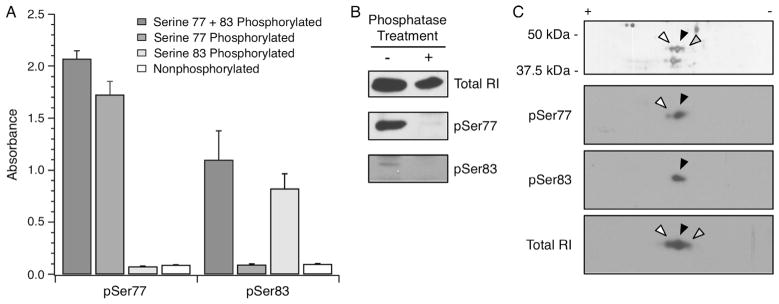
RIα phosphospecific antibody validation. Two rabbit monoclonal antibodies specific for Ser77 or Ser83 phosphorylated RIα were validated. (A) The sequence CAGCTRTDS77REDEIS83PPPNP was synthesized into four peptides to yield non-phosphorylated, Ser77 or Ser83 singly phosphorylated, and Ser77/83 doubly phosphorylated peptides. The culture supernatant from the pSer77 and pSer83 hybridomas were used in ELISA (65) and exhibited high specificity for the intended phosphorylated peptides. (B) Rat myocardial homogenates were left untreated or treated with alkaline phosphatase, followed by blotting with total RI, pSer77 or pSer83 antibodies. A net loss of signal for the Ser77 and Ser83 phosphorylated RIα was observed following alkaline phosphatase treatment. (C) A human myocardium homogenate enriched by 8-AEA-cAMP affinity chromatography for RIα was resolved by 2D SDS–PAGE. The white, black and grey arrowheads represent successively basic spots verified to be RIα by mass spectrometry (data not shown). 2D Western blotting of the same sample showed that the pSer77 and pSer83 antibodies identified the relatively acidic isoelectric variants, whereas the antibody against total RI identified all three isoelectric variants. The spots are aligned with arrowheads in accordance with the accompanying silver stained gel.
Fig. 6.

Expression of Ser77 and Ser83 phosphorylated RIα. (A) Using the rabbit monoclonal antibody pSer77, Western blot analyses revealed no significant change in Ser77 phosphorylation in the heart failure samples vs. donor samples. (B) In contrast, the pSer83 monoclonal antibody demonstrated a significant 110% relative increase in expression in the heart failure samples (1 ± 0.28 vs. 2.10 ± 0.45). *Denotes P < 0.05 vs. donor.
Steady state 8-fluo-cAMP binding by WT and mutant RIα
The WT and Ser77Asp, Ser83Asp and Ser77/83Asp RIα proteins were expressed in E. coli and purified based on adaptations of published methods [36,37]. The purified proteins (Fig. 7A) were subsequently used in fluorescence anisotropy experiments to test their steady state affinity for the fluorescent cAMP analog 8-fluo-cAMP [38]. The Kd of WT RIα for 8-fluo-cAMP was 3.8 ± 0.9 nM (n = 3; Fig. 7B), which was significantly decreased from Ser77/83Asp RIα (5.4 ± 0.5 nM; n = 3; P < 0.05) but not Ser77Asp (2.8 ± 0.4 nM; n = 3) or Ser83Asp RIα (4.3 ± 1.2 nM; n = 3).
Fig. 7.
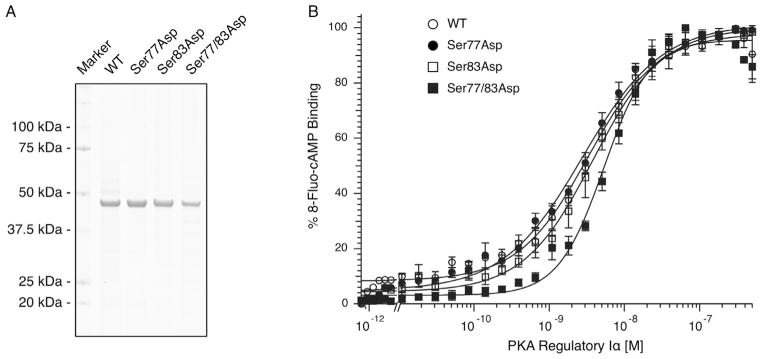
Steady state 8-fluo-cAMP binding by WT and mutant RIα. (A) Wild type and mutant RIα proteins were expressed in E. coli and purified. (B) Using fluorescence anisotropy, 8-fluo-cAMP binding by the various RIα proteins was measured. Average ± S.E.M. data are plotted with an accompanying fit for illustrative purposes. The Kd of Ser77/83Asp RIα was significantly increased compared to WT RIα (P < 0.05).
Discussion
In this study, we examined the expression level and post-translational state of the PKA regulatory and catalytic subunits expressed in failing human myocardium. This pathway is of interest as β-AR blockade has an unequivocal impact on the management of cardiovascular disease [1], but is not sufficient to halt disease progression. Whether this is due to a change in prioritization of intracellular PKA substrates through altered PKA activity, a change in the kinase’s subcellular localization, or a change in expression level and post-translational status remains unclear. This and previous studies using myocardial samples from failing human hearts have consistently observed reduced TnI and MYBP-C phosphorylation (Fig. 1; ([4–7])). For TnI, this was further detailed as an accompanying decrease in Ser23/24 phosphorylation without a change in total TnI expression level (Fig. 1C and D). The observed reduction in phosphorylation of these sarcomeric proteins was expected from prior studies, and is likely explained in part by a combination of reduced β-AR density and β-AR blockade. The latter point is not unequivocal however, since recent hypotheses speculate that β-AR resensitization may be an important mechanism to consider (reviewed in [39]), although clinical data for significant resensitization in the face of β-AR blockade are scant. In fact, the strong dephosphorylation of TnI and MyBP-C observed in this and other studies [5–7], suggests that resensitization, if any, is not sufficient to restore phosphorylation at the level of the contractile filaments. Furthermore, the strong TnI and MyBP-C dephosphorylation observed in this and previous studies suggests that the limitations imposed by our donor group (lack of previous medication history, trend towards younger age (P = 0.09), ex-US population) did not constrain the applicability of our findings. Nonetheless, it is worth noting that it remains difficult to precisely match patient characteristics between the donor and HF cohorts, and our results should be viewed with this caveat in mind.
To better understand the reduced phosphorylation of sarcomeric proteins, relative expression levels of the regulatory and catalytic subunits of PKA were measured (Figs. 2–4). Contrary to a prior report demonstrating reduced expression of both types of regulatory subunit in the soluble fraction of dilated cardiomyopathy tissue samples [27], we observed an increase in total RIα expression but no significant change in RIIα expression in the explanted hearts when compared to donors. At this point, it is unclear if this discrepancy simply represents the fact that the prior study fractionated the tissue prior to analysis whereas this study measured the total content without prior fractionation. At face value, an increase in RIα subunits may intuitively suggest a concomitant increase in Cα expression if the cell is to maintain the stoichiometry of the holoenzyme. Nonetheless, it is unclear if this stoichiometry persists during heart failure. Our results did not demonstrate a statistically significant increase in Cα expression (Fig. 2) despite the change in RIα expression, making it possible that during heart failure PKA subunit dimers may not be expressed in matched quantities. A prior study of Cα overexpression in the heart demonstrated that increased, chronic activation of the catalytic subunit was overtly deleterious [40]. These mice rapidly developed cardiomyopathy and suffered sudden death, which is consistent and expected based on models of β-agonist induced heart failure [41]. Therefore it is possible that in this patient population, the increase in RIα expression is a compensatory mechanism to tilt the equilibrium of type I PKA holoenzyme association–dissociation towards the associated state and to reduce downstream substrate phosphorylation by Cα. By contrast, the increase in Ser96 phosphorylation of RIIα favors the dissociated state of type II PKA [42,43]. Therefore, these data suggest that the development of heart failure was associated with divergent means of regulation of the distinct type I and II holoenzymes.
In addition to changes in expression level, Cα subunits are post-translationally modified with important consequences for PKA structure–function. Although total Cα expression was not different between groups, there was a statistically significant increase in phosphorylation at Thr197 (Fig. 2). Phosphorylation at Thr197 is known to impact the km of the enzyme for its substrates [25], yet the physiological mechanism regulating in vivo phosphorylation–dephosphorylation of the Thr197 site in the activation loop is not fully described. The catalytic subunit is believed to be constitutively phosphorylated at Thr197 through a post-translational trans-mechanism following Ser338 phosphorylation during translation [24]. In addition, this site may also be regulated through alternate kinases such as phosphoinositide-dependent protein kinase [44,45]. Conversely, the putative dephosphorylation mechanism is less clear. Prior studies have demonstrated that Thr197 phosphorylated Cα is very resistant to in vitro and in vivo dephosphorylation unless Cys199 is oxidized [46,47]. The increased phosphorylation at Thr197 in the heart failure cohort may therefore be a reflection of an altered redox state in these cardiomyocytes [48], more specifically a reductive stress [49,50].
At this point it is not clear how the post-translational state of RIα contributes to the activation of PKA in the examined cohorts. The 2D SDS–PAGE results suggested two potential phosphorylation sites (Fig. 4B), consistent with prior reports of phosphorylation at Ser77 and Ser83 [34,35]. The existence of these phosphorylation sites in vivo was further confirmed by Western blots with Ser77 and Ser83 phosphospecific antibodies (Figs. 5 and 6), and only phosphorylation at Ser83 was observed to change significantly in the heart failure cohort if absolute levels were compared. This phosphorylation site was previously identified through an examination of RIα purified from bovine striated muscles, yet a functional significance to the post-translational modification was not assigned [34]. An analysis of cAMP-interacting proteins from mouse ventricular tissue had also identified Ser77 as a phosphorylation site [35], but again, a functional characterization of the site had not been undertaken. We examined the affinity of WT and Ser77 and/or Ser83Asp RIα mutants to begin the functional characterization of these sites (Fig. 7). As these sites are not directly part of the cAMP binding motifs, it is not surprising that, individually, phosphomimics of Ser77 or Ser83 did not significantly impact the affinity for the cAMP analog. Nonetheless, the double mutant Ser77/83Asp did show decreased affinity for 8-fluo-cAMP. Although the mechanism is currently unclear, we hypothesize that the double mutation at this N-linker region may cause a subtle modulation of the RIα subunit structure to adjust the affinity of the cAMP binding domains for the second messenger.
Identification of the two phosphorylated serine residues in vivo suggests that there may be multiple signaling inputs into the regulation of type I PKA. These sites may be targets of kinases that independently modify PKA signaling through alternative cellular inputs, as neither site is phosphorylated by Cα itself (data not shown). Both sites are distal to the cAMP binding domains that reside towards the COOH-terminus of the primary sequence [51], and are just upstream of the “hinge” region of RIα that houses the pseudosubstrate site for Cα [8]. Accordingly, these sites are part of the N-linker domain that is often disordered in structural determinations, suggesting that it may be a dynamic structure [9,52–55]. Therefore, in addition to the effect on 8-fluo-cAMP binding, phosphorylation at these sites may alter the association–dissociation properties of the type I holoenzyme, may impact the association with the accompanying A-kinase anchoring proteins [56], or may influence the in vivo turnover of RIα [34]. The latter point is interesting, given that Ser83 resides within a PEST sequence [57,58], and therefore phosphorylation at this site may impact RIα stability during heart failure, especially given the subunit’s relative instability in vivo [59,60]. Additional experiments will be required to examine the role of these phosphorylation sites on PKA structure–function, and how they contribute to intracellular signaling in the normal and failing heart.
In sum, these analyses demonstrate that the expression and post-translational status of PKA is altered during heart failure. Despite β-AR blocker therapy, this patient population demonstrated increased RIα expression, an RIIα post-translational status that favored release of the catalytic subunit, and a catalytic subunit that was predisposed to higher affinity for its substrates through increased Thr197 phosphorylation. Although it is currently difficult to quantify the contributions of each finding, it is noteworthy that two of these changes support a net increase in intracellular PKA activity. Nonetheless, the low phosphorylation levels of sarcomeric PKA targets (Fig. 1) suggests that the intracellular effects dictated by the change in RIIα/Cα post-translational state are not ubiquitous. In effect, the subcellular localization of the impacted PKA subunits may prompt their activity towards novel targets or signaling pathways that promote cardiac hypertrophy or cardioprotection [61,62] rather than contributing to contractility through sarcomeric protein phosphorylation. This may explain why prior experiments have not found differences in basal or cAMP-activated PKA activity in homogenates from failing and nonfailing human myocardium [63,64], as these assays no longer preserve the intracellular localization of the various pools of PKA holoenzyme. Conversely, the reduced phosphorylation of sarcomeric targets may reflect their dependence on type I PKA activity, and the observed increase in RIα expression may be acting to sequester the catalytic subunits that are proximal to these substrates. A decrease in the cAMP affinity due to increased Ser77/83 phosphorylation of RIα would additionally contribute to this scenario. In either case, these findings raise the possibility that signaling pathways initiated during heart failure may be able to modulate the activity of PKA through preferential post-translational modification of its subunits, even during pharmacological β-AR blockade. Of particular interest for future studies are the phosphorylation sites at Ser77 and Ser83 of RIα, especially Ser83 which was increased in the heart failure cohort. Experiments focused on identifying the impact of these post-translational modifications will provide much needed insight into the signaling pathways governing their phosphorylation and dephosphorylation, and how they contribute to mechanisms that regulate PKA activity and its role in the etiology of heart disease.
Supplementary Material
Acknowledgments
We thank Dr. John Shabb for the rabbit polyclonal antibody against Ser83 phosphorylated PKA regulatory I and Dr. Cris dos Remedios for the non-heart failure human myocardium samples. We thank Dr. Frank Brozovich for critical reading of the manuscript. Y.S.H. was supported by T32HL007111. J.A. was supported by R25 GM75148 to the Mayo Graduate School. This work was supported by HL078845, HL078845-S1 and the Marie Ingalls Cardiovascular Research Fund to O.O.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.abb.2013.08.002.
Footnotes
Abbreviations used: β-AR, beta-adrenergic receptor; Cα, PKA catalytic subunit α; MYBP-C, myosin binding protein-C; PKA, cAMP-dependent protein kinase; RIα, type Iα regulatory subunit; RIIα, type IIα regulatory subunit; TCEP, tris(2-carboxy-ethyl)phosphine; TnI, troponin I.
Disclosures
None.
References
- 1.Hjalmarson A, Goldstein S, Fagerberg B, Wedel H, Waagstein F, Kjekshus J, Wikstrand J, El Allaf D, Vítovec J, Aldershvile J, Halinen M, Dietz R, Neuhaus KL, Jánosi A, Thorgeirsson G, Dunselman PH, Gullestad L, Kuch J, Herlitz J, Rickenbacher P, Ball S, Gottlieb S, Deedwania P. JAMA. 2000;283:1295–1302. doi: 10.1001/jama.283.10.1295. [DOI] [PubMed] [Google Scholar]
- 2.Chidsey CA, Braunwald E, Morrow AG. Am J Med. 1965;39:442–451. doi: 10.1016/0002-9343(65)90211-1. [DOI] [PubMed] [Google Scholar]
- 3.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 4.Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PA. Circulation. 1997;96:1495–1500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- 5.Messer AE, Jacques AM, Marston SB. J Mol Cell Cardiol. 2006;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 6.Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, Tsang VT, Marston SB. J Mol Cell Cardiol. 2008;45:209–216. doi: 10.1016/j.yjmcc.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 7.van der Velden J, Narolska NA, Lamberts RR, Boontje NM, Borbély A, Zaremba R, Bronzwaer JGF, Papp Z, Jaquet K, Paulus WJ, Stienen GJM. Cardiovasc Res. 2005;69:876–887. doi: 10.1016/j.cardiores.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Taylor SS, Buechler JA, Yonemoto W. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 9.Taylor SS, Ilouz R, Zhang P, Kornev AP. Nat Rev Mol Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solaro RJ, Moir AJ, Perry SV. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 11.Garvey JL, Kranias EG, Solaro RJ. Biochem J. 1988;249:709–714. doi: 10.1042/bj2490709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solaro RJ. J Biol Chem. 2008;283:26829–26833. doi: 10.1074/jbc.R800037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Circ Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 14.Stelzer JE, Patel JR, Walker JW, Moss RL. Circ Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 15.Krall J, Taskén K, Staheli J, Jahnsen T, Movsesian MA. J Mol Cell Cardiol. 1999;31:971–980. doi: 10.1006/jmcc.1999.0926. [DOI] [PubMed] [Google Scholar]
- 16.Poteet-Smith C, Shabb J, Francis S, Corbin J. J Biol Chem. 1997;272:379. doi: 10.1074/jbc.272.1.379. [DOI] [PubMed] [Google Scholar]
- 17.Carmichael DF, Geahlen RL, Allen SM, Krebs EG. J Biol Chem. 1982;257:10440–10445. [PubMed] [Google Scholar]
- 18.Beebe SJ, Oyen O, Sandberg M, Frøysa A, Hansson V, Jahnsen T. Mol Endocrinol. 1990;4:465–475. doi: 10.1210/mend-4-3-465. [DOI] [PubMed] [Google Scholar]
- 19.Diskar M, Zenn HM, Kaupisch A, Kaufholz M, Brockmeyer S, Sohmen D, Berrera M, Zaccolo M, Boshart M, Herberg FW, Prinz A. J Biol Chem. 2010;285:35910–35918. doi: 10.1074/jbc.M110.155150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desseyn JL, Burton KA, McKnight GS. Proc Natl Acad Sci USA. 2000;97:6433–6438. doi: 10.1073/pnas.97.12.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thullner S, Gesellchen F, Wiemann S, Pyerin W, Kinzel V, Bossemeyer D. Biochem J. 2000;351:123–132. doi: 10.1042/0264-6021:3510123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shoji S, Parmelee DC, Wade RD, Kumar S, Ericsson LH, Walsh KA, Neurath H, Long GL, Demaille JG, Fischer EH, Titani K. Proc Natl Acad Sci USA. 1981;78:848–851. doi: 10.1073/pnas.78.2.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoji S, Titani K, Demaille JG, Fischer EH. J Biol Chem. 1979;254:6211–6214. [PubMed] [Google Scholar]
- 24.Keshwani MM, Klammt C, Klammt C, von Daake S, Ma Y, Kornev AP, Choe S, Insel PA, Taylor SS. Proc Natl Acad Sci. 2012;109:E1221–1229. doi: 10.1073/pnas.1202741109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steichen JM, Iyer GH, Li S, Saldanha SA, Deal MS, Woods VL, Taylor SS. J Biol Chem. 2010;285:3825–3832. doi: 10.1074/jbc.M109.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aye TT, Soni S, van Veen TAB, van der Heyden MAG, Cappadona S, Varro A, de Weger RA, de Jonge N, Vos MA, Heck AJR, Scholten A. J Mol Cell Cardiol. 2012;52:511–518. doi: 10.1016/j.yjmcc.2011.06.003. http://www.sciencedirect.com/science/article/pii/S0022282811002239. [DOI] [PubMed] [Google Scholar]
- 27.Zakhary DR, Moravec CS, Stewart RW, Bond M. Circulation. 1999;99:505–510. doi: 10.1161/01.cir.99.4.505. [DOI] [PubMed] [Google Scholar]
- 28.Han YS, Ogut O. PLoS One. 2010;5:e9528. doi: 10.1371/journal.pone.0009528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hachmann JP, Amshey JW. Models of protein modification in Tris–glycine and neutral pH Bis–Tris gels during electrophoresis: effect of gel pH. Anal Biochem. 2005;342:237–245. doi: 10.1016/j.ab.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 30.Hanke SE, Bertinetti D, Badel A, Schweinsberg S, Genieser HG, Herberg FW. New Biotechnol. 2010;28:294–301. doi: 10.1016/j.nbt.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Ogut O, Hossain MM, Jin JP. J Biol Chem. 2003;278:3089–3097. doi: 10.1074/jbc.M205853200. [DOI] [PubMed] [Google Scholar]
- 32.Jameson DM, Mocz G. Methods Mol Biol. 2005;305:301–322. doi: 10.1385/1-59259-912-5:301. [DOI] [PubMed] [Google Scholar]
- 33.Pizarro GO, Ogut O. Biochemistry. 2009;48:7533–7538. doi: 10.1021/bi900669m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boeshans K, Resing K, Hunt J, Ahn N, Shabb J. Protein Sci. 1999;8:1515–1522. doi: 10.1110/ps.8.7.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scholten A, van Veen TAB, Vos MA, Heck AJR. J Proteome Res. 2007;6:1705–1717. doi: 10.1021/pr060601a. [DOI] [PubMed] [Google Scholar]
- 36.León DA, Dostmann WR, Taylor SS. Biochemistry. 1991;30:3035–3040. doi: 10.1021/bi00226a008. [DOI] [PubMed] [Google Scholar]
- 37.Gibson RM, Ji-Buechler Y, Taylor SS. J Biol Chem. 1997;272:16343–16350. doi: 10.1074/jbc.272.26.16343. [DOI] [PubMed] [Google Scholar]
- 38.Moll D, Prinz A, Gesellchen F, Drewianka S, Zimmermann B, Herberg FW. J Neural Transm. 2006;113:1015–1032. doi: 10.1007/s00702-006-0515-5. [DOI] [PubMed] [Google Scholar]
- 39.Vasudevan NT, Mohan ML, Goswami SK, Naga Prasad SV. Cell Cycle. 2011;10:3684–3691. doi: 10.4161/cc.10.21.18042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Circ Res. 2001;89:997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 41.Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Circ Res. 2010;107:1094–1101. doi: 10.1161/CIRCRESAHA.110.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rangel-Aldao R, Rosen OM. J Biol Chem. 1976;251:3375–3380. [PubMed] [Google Scholar]
- 43.Rangel-Aldao R, Rosen OM. J Biol Chem. 1977;252:7140–7145. [PubMed] [Google Scholar]
- 44.Cheng X, Ma Y, Moore M, Hemmings BA, Taylor SS. Proc Natl Acad Sci USA. 1998;95:9849–9854. doi: 10.1073/pnas.95.17.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cauthron RD, Carter KB, Liauw S, Steinberg RA. Mol Cell Biol. 1998;18:1416–1423. doi: 10.1128/mcb.18.3.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bechtel PJ, Beavo JA, Krebs EG. J Biol Chem. 1977;252:2691–2697. [PubMed] [Google Scholar]
- 47.Humphries KM, Deal MS, Taylor SS. J Biol Chem. 2005;280:2750–2758. doi: 10.1074/jbc.M410242200. [DOI] [PubMed] [Google Scholar]
- 48.Christians ES, Benjamin IJ. Am J Physiol Heart Circ Physiol. 2012;302:H24–H37. doi: 10.1152/ajpheart.00903.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Min X, Li C, Benjamin IJ, Qian B, Zhang X, Ding Z, Gao X, Yao Y, Ma Y, Cheng Y, Liu L. Hypertension. 2010;55:1412–1417. doi: 10.1161/HYPERTENSIONAHA.109.147066. [DOI] [PubMed] [Google Scholar]
- 51.Titani K, Sasagawa T, Ericsson LH, Kumar S, Smith SB, Krebs EG, Walsh KA. Biochemistry. 1984;23:4193–4199. doi: 10.1021/bi00313a028. [DOI] [PubMed] [Google Scholar]
- 52.Ilouz R, Bubis J, Wu J, Yim YY, Deal MS, Kornev AP, Ma Y, Blumenthal DK, Taylor SS. Proc Natl Acad Sci. 2012;109:12443–12448. doi: 10.1073/pnas.1209538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boettcher AJ, Wu J, Kim C, Yang J, Bruystens J, Cheung N, Pennypacker JK, Blumenthal DA, Kornev AP, Taylor SS. Structure. 2011;19:265–276. doi: 10.1016/j.str.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim C, Xuong NH, Taylor SS. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 55.Kim C, Cheng CY, Saldanha SA, Taylor SS. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 56.Scott JD, Santana LF. Circulation. 2010;121:1264–1271. doi: 10.1161/CIRCULATIONAHA.109.896357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rogers S, Wells R, Rechsteiner M. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 58.Rechsteiner M, Rogers SW. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 59.Steinberg RA, Agard DA. J Biol Chem. 1981;256:11356–11364. [PubMed] [Google Scholar]
- 60.Steinberg RA, Agard DA. J Biol Chem. 1981;256:10731–10734. [PubMed] [Google Scholar]
- 61.Yoo BS, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L, Rockman HA. Am J Physiol Heart Circ Physiol. 2009;297:H1377–86. doi: 10.1152/ajpheart.00504.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. J Clin Invest. 2007;117:2445–2458. doi: 10.1172/JCI31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirchhefer U, Schmitz W, Scholz H, Neumann J. Cardiovasc Res. 1999;42:254–261. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 64.Böhm M, Reiger B, Schwinger RH, Erdmann E. Cardiovasc Res. 1994;28:1713–1719. doi: 10.1093/cvr/28.11.1713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.