

Abstract
Mercury (Hg) is a persistent environmental bioaccumulative metal, with developmental exposure to methylmercury (MeHg) resulting in long-term health effects. We examined the impact of early-life exposure to MeHg and knockdown of skn-1 on dopaminergic (DAergic) neurodegeneration in the nematode Caenorhabditis elegans (C. elegans). SKN-1, a the major stress-activated cytoprotective transcription factors, promotes the transcription of enzymes that scavenge free radicals, synthesizes glutathione (GSH) and catalyzes reactions that increase xenobiotic excretion. Deletions or mutations in this gene suppress stress resistance. Thus, we hypothesized that the extent of MeHg’s toxicity is dependent on intact skn-1 response; therefore skn-1 knockout (KO) worms would show heightened sensitivity to MeHg-induced toxicity compared to wildtype worms. In this study we identified the impact of early-life MeHg exposure on Hg content, stress reactivity and DAergic neurodegeneration in wildtype, and skn-1KO C. elegans. Hg content, measured by Inductively Coupled Plasma Mass Spectrometry (ICP-MS), showed no strain-dependent differences. Reactive oxygen species (ROS) generation was dramatically increased in skn-1KO compared to wildtype worms. Structural integrity of DAergic neurons was microscopically assessed by visualization of fluorescently-labeled neurons, and revealed loss of neurons in skn-1KO and MeHg exposed worms compared to wildtype controls. Dopamine levels detected by High-performance liquid chromatography (HPLC), were decreased in response to MeHg exposure and decreased in skn-1KO worms, and functional behavioral assays showed similar findings. Combined, these studies suggest that knockdown of skn-1 in the nematode increases DAergic sensitivity to MeHg exposure following a period of latency.
Keywords: Methylmercury, skn-1, early-life exposure, NRF2, DAergic neurodegeneration
INTRODUCTION
Methylmercury (MeHg) neurotoxicity has been studied for decades, but the precise molecular mechanism underlying MeHg-induced damage remains elusive. Nuclear factor erythroid 2-related factor (Nrf2) is an important mammalian stress/antioxidant response mediator. Previous studies in our laboratory have shown the upregulation of genes under the control of Nrf2 following MeHg exposure [1]. In C. elegans these stress responses are mediated by skn-1. Inhibition of skn-1 leads to increased sensitivity to stress and deletions or loss-of-function mutations of skn-1 suppress oxidative stress resistance [2–5]. SKN-1 is expressed in the ASI neurons and the gut, as well as in dopaminergic (DAergic) neurons [2, 3]. Due to its involvement in the stress response and its expression pattern, SKN-1 is posited to be an important mediator of MeHg’s toxicity and of dopaminergic (DAergic) loss in neurodegenerative disease [5, 6].
Oxidative stress and lipid peroxidation represent key mechanisms in mediating neuronal death both in neurodegenerative diseases and upon exposure to MeHg [1, 7–11]. Mitochondrial damage is implicated in the loss of DAergic neurons in Parkinson’s disease (PD), corroborated by findings that mitochondrial poisons, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone and 6-hydroxydopamine induce a Parkinsonian-like syndrome [12–15]. Post-mortem PD brains show increased levels of lipid peroxidation, protein oxidation, 3-nitrotyrosine formation, DNA oxidation and breaks, and a decrease in the activities of reactive oxygen species (ROS) scavenging enzymes and glutathione (GSH) peroxidase [16–19]. Combined, there is ample evidence to suggest that MeHg and PD share common molecular mechanisms of DAergic degeneration, including mitochondrial dysfunction, oxidative damage, GSH depletion and -synuclein aggregation [10, 12, 20–24].
Developmental exposure to MeHg has long-term, latent health effects [25–31]. Early-life exposure may remain silent for decades and become unmasked late in life by aging and genetic predisposition [32]. In this study we focused on the role of skn-1 in mediating the late-life occurrence of DAergic neurodegeneration following early-life MeHg exposure, positing heightened DAergic neurodegeneration in skn-1KO compared to wildtype worms. Our studies examined DAergic function, structural integrity and dopamine (DA) levels following knockdown of skn-1 and exposure to MeHg.
MATERIALS AND METHODS
1.1 C. elegans maintenance
C. elegans strains were handled and maintained at 20°C as previously described [33]. Worms were grown on plates containing nematode growth medium (NGM) or 8P and seeded with either Escherichia coli strain OP50 or NA22, respectively, as previously described [33]. The following strains were used: N2 Bristol (wildtype) strain (as a control for all experiments), VC1772 (skn-1(ok2315) IV/nT1[qIs51](IV;V)) strain (referred to as skn-1KO in all experiments), LG326 (skn-1(zu169) IV; geIs7) and cat-2 mutants (TH deficient). Because skn-1 is required for optimal worm development, and exposure to MeHg was carried out at the L1 stage (early lifespan), worms with a homozygous deletion could not be used. The homozygous deletion is lethal, while the VC1772 strain has a heterozygous deletion with a balancer to yield surviving larva. All strains were obtained from the Caenorhabditis Genetics Center, Minneapolis, MN.
1.2 MeHgCl exposure
To obtain a synchronous population prior to exposure, worms were treated with an alkaline bleach solution. Methylmercuric chloride (CH3HgCl; Sigma-Aldrich) exposures (0–50μM) were performed for 30 minutes (min) in synchronized L1 worms to determine appropriate dosing. Five thousand (lifespan, lethality, and behavior), 10,000 (DCF assay), 20,000 (RNA), 50,000 (ICP-MS) or 150,000 (dopamine) nematodes were treated with 0 or 20μM MeHgCl. After exposure, worms were washed three times with M9 buffer (KH2PO4; Na2HPO4; and NaCl) and either plated on seeded NGM plates or collected for immediate analysis. A sample size of six (n = 6) represents the total number of independent worm preparations; each independent experiment was carried out with 5,000–150,000 worms (see above).
1.3 Lethality
Following exposure and washing, 5,000 worms were plated on seeded 60 mm NGM plates and allowed to grow for 24 hours (hrs). Worms were then counted and scored using a grid system. Nematodes on 4 of the 64 grids were counted and the number of worms per grid was averaged and multiplied by 64. Surviving worms were expressed as percent control.
1.4 Lifespan
For lifespan determination, 40 nematodes from each dose group were picked to a fresh NGM plate 24 hrs following exposure. The worms were counted each day and scored as live or dead (worms were censored if dead bodies could not be located). Live C. elegans were picked to new plates each day during the egg-laying period of their lifecycle; once egg-laying ceased they were picked every other day until no live C. elegans remained.
1.5 Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) for Mercury content
The protocol used for ICP-MS was carried out as previously described [33]. Briefly, 50,000 worms were treated with MeHgCl, then washed three times with M9. Samples were then air-dried until a dehydrated pellet was obtained. Next, the dried sample was transferred to pre-weighed Teflon jars and reweighed. Concentrated acids HNO3, HCl and H2SO4 were added, and the samples were placed overnight in heat block at 100°C under the fume hood. The following day, digested samples were reweighed and placed in pre-weighed metal-free 15mL centrifuge tubes. They were analyzed by ICP-MS with a Hg limit-of-detection in the parts per trillion range.
1.6 DCF assay for oxidative stress
Synchronized L1s were treated with MeHg as described earlier, and washed in M9 buffer. The formation of reactive oxygen species (ROS) was evaluated with 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate at 1 mM for one hr in the dark. The worms were then washed in M9, 4 additional times. The supernatants were transferred to a 96-well plate and their fluorescence levels (excitation: 485 nm; emission: 535 nm) detected with a FLEXstation III (Molecular Devices) pre-heated to 37°C. Fluorescence in each well was measured at time 0 and 1 hr after the addition of MeHg. Values from all readings were adjusted for background signal and reported as percent fluorescence relative to their respective control strain.
1.7 mRNA isolation
Following MeHg exposure, 20,000 synchronized L1 stage worms were washed 3 times with M9 and frozen at −80°C until RNA purification. Worm pellets were suspended in Trizol (200μL/100μL pellet); the protein and other impurities were separated from nucleic acids with chloroform. The pellets were washed with 70% EtOH; DNA was digested with RNAse free DNAse kit (Ambion). RNA concentrations were determined with a ND-1000 spectrophotometer (NanoDrop®; OD 260nm).
1.8 cDNA synthesis
Reverse transcription reactions were performed in 20 μL following the manufacturer’s protocol (Applied Biosystems). A 10μL reaction mixture containing 10X RT buffer, 25X dNTP mix (100mM), 10X random primers, Multiscribe reverse transcriptase and sterile nuclease-free water was added to 10μL of RNA (75ng). The contents were mixed gently and incubated at 25°C for 10 min, 37°C for 120 min followed by inactivation by heating at 85°C for 5 min. Synthesized cDNA was stored at −20°C.
1.9 Semi-quantitative determination of transcript levels by Real-time PCR
Real-time PCR was conducted in a Gene Amp 7300 sequence detection system (Applied Biosystems) in a 96-well plate (MicroAmp™ Fast). The relative quantification was determined with the 2−ΔΔCt method [34]. gpd-1 (mammalian:GAPDH) was used as an internal control. Oligonucleotide sequences for skn-1: 5′AGTGTCGGCGTTCCAGATTTC3′ and 5′GTCGACGAATCTTGCGAATCA3′; gst-4: 5′TGCTCAATGTGCCTTACGAG3′ and 5′AGTTTTTCCAGCGAGTCCAA3′; gpd-1 (housekeeping): 5′CAATGCTTCCTGCACCACTA3′ and 5′CTCCAGAGCTTTCCTGATGG3′. The specificity of each primer pair was confirmed by the identification of a single PCR product of predicted size on 1.5% (w/v) agarose gels. Oligonucleotide sequences were designed based on published literature.
1.10 Dopamine measured via High-Performance Liquid Chromotagraphy (HPLC)
One hundred fifty thousand synchronized L1 worms were collected and washed three times in M9. Next, they were pelleted and the supernatant removed. Tubes were immediately frozen in liquid nitrogen and stored at −80°C. For each tube, the worm pellet was re-suspended in lysis buffer containing EDTA to scavenge free metal ions and then sonicated to disrupt cell membranes. A portion of the lysate was used for protein levels and analyzed via bicinchoninic acid (BCA) assay. Isoproterenol, dihydroxybenzylamine (internal standard), Tris buffer and Al2O3 was added to the remaining lysate, which was applied to the alumina to bind DA. After 30 min, the buffer was removed and the Al2O3 was washed three times with water. Next, the alumina was eluted with 0.1 acetic acid and the collected samples were injected into the biogenic amine HPLC chromatograph. To correct for inter-sample variations in the extraction efficiency, the ratio of DA to isoproternol was estimated, and the total DA content calculated relative to protein levels.
1.11 Basal slowing behavioral analysis
The behavioral analysis was adapted from a previously published protocol [35]. Well-fed L4 worms from the same exposure group were placed on two sets of 60×15mm plates: one with ~20μL bacteria spread in a ring with an inner diameter of ~1cm and an outer diameter of ~3.5cm and one without bacteria (both incubated at 37°C overnight and cooled to room temperature prior to assay). Bacterial mechanosensation induces the dopamine-mediated slowing of locomotion in the presence of food (bacteria) and can be measured by counting the number of body bends per 20-second interval. Locomotor rates were compared between well-fed worms placed on plates of food versus those placed on plates in the absence of food; this ratio is referred to as the change (Δ) in body bends/20 seconds. A lower value represents diminished slowing on food, indicating deficits in DAergic function. The cat-2 strain is tyrosine hydroxylase (TH) deficient and therefore defective in bacterial mechanosensation, representing a positive control [35].
1.12 Microscopy
To examine skn-1 protein expression, 10–15 worms in M9 were mounted on 4% agarose pads and anaesthetized with 0.2% tricaine/0.02% tetramisole in M9. skn-1::GFP (LG326) worms were selected by fluorescence microscopy 24–48 hrs following exposure. Fluorescence was determined with an epifluorescence microscope (Nikon Eclipse 80i, Nikon Corporation, Tokyo, Japan) equipped with a Lambda LS Xenon lamp (Sutter Instrument Company) and Nikon Plan Fluor 20× dry and Nikon Plan Apo 60 × 1.3 oil objectives.
To examine morphological changes in neurons as a result of MeHg exposure, worms expressing Pdat-1::mCherry in DAergic neurons (N2 and VC1772) were visualized by confocal microscopy. Worms were treated with 0 or 20μM MeHg and visualized 96 hrs following exposure. Approximately 20 worms were mounted on 4% agarose pads in M9 and anaesthetized with 0.2% tricane and 0.02% tetramisole. Images were captured through Plan-Apochromat 20x objective on a LSM510 confocal microscope (Carl Zeiss MicroImaging, Inc) scanning every 200 nm for XZ sections. Images were processed with the Zeiss LSM Image Browser.
1.13 Statistical analysis
Statistics were carried out with GraphPad Prism. Briefly, we used a sigmoidal dose-response model with a top constraint at 100% to draw the curves and determine the LD50. The Kaplan-Meier method was used to estimate survival curves. Curves were compared with the log rank test or Wald test of coefficients from a Cox proportional hazards regression. Statistical analysis of significance was carried out by one-way analysis of variance (ANOVA) for change in DA-mediated locomotor activity. Two-way ANOVA for dopamine, ROS, skn-1, gst-4 and MeHg content. When p values in the ANOVA were <0.05, the source of the statistical difference was determined by post-hoc Bonferroni or student’s t-test. Error bars in all the figures represent ±SEM.
RESULTS
2.1 MeHg-induced lethality (Figure 1)
Figure 1.
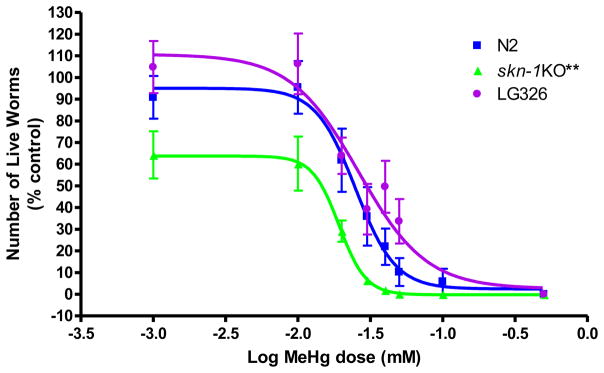
Dose-response curve for MeHg-induced lethality in N2 wildtype and skn-1KO worms was used to determine appropriate doses for the remainder of the experiments Worms were exposed to MeHg in early-life (L1 stage) for 30 minutes and counted 24 hours following exposure. skn-1KO worms (LD50= 19 μM, n=7) were more sensitive to MeHg than N2 wildtype worms (LD50 for N2 = 25 μM; n=8). LG326, in which skn-1 is linked to GFP, (skn-1::GFP) had an LD50 = 26 μM; n=10.
To determine appropriate MeHg concentrations for dosing in our studies, N2 and skn-1KO C. elegans were treated with MeHg (0–50μM) for 30 min and counted 24 hrs later. Assessment of lethality following 30 min exposure revealed a significant leftward shift for skn-1KOs compared to N2 wildtype worms (**p<0.004) and a similar significant leftward shift for skn-1KOs compared to LG326 worms (**p<0.001). LD50 for N2, skn-1 KO, and LG326 were 25, 19 and 26μM, respectively. These data suggest that skn-1KO worms are more sensitive to MeHg than N2 wildtypes. It is noteworthy that doses applied in the present study are of physiological relevance to mammalian systems. For example, perinatal MeHg treatment in rats resulted in observable effects with Hg concentrations at 0.1 μgHg/g brain [36], and continuous pre- and postnatal exposure in rats was associated with brain Hg concentrations of 0.5 μg/g at birth and 0.04 μg/g at weaning [37]. The threshold for observable clinical effects in humans is ~0.3μg/g [36].
2.2 Mercury content (Figure 2)
Figure 2.
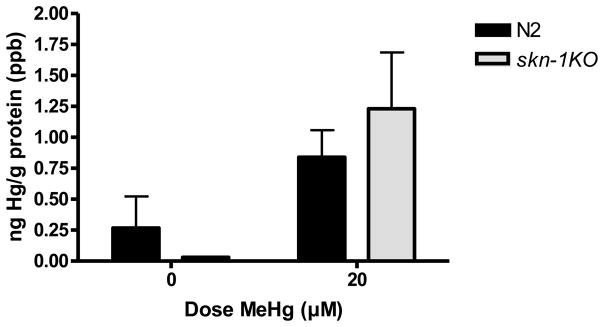
Hg content in wildtype and skn-1KO worms. There was a significant increase in Hg content in MeHg exposed worms, as measured by ICP-MS, yielding a significant dose effect (*p=0.0003). Hg content was indistinguishable between N2 wildtype and skn-1KO strains. Results are presented as mean ± SEM of 4–5 independent experiments.
To determine if there were differences in MeHg retention mediated by skn-1KO, we assessed Hg content immediately following exposure. ICP-MS quantification of Hg content revealed a significant dose effect (**p=0.0003), but no strain effect (p=0.958, n.s.), or a dose x strain interaction (p=0.438, n.s.). There were no strain differences in Hg content.
2.3 Lifespan (Figure 3)
Figure 3.

Lifespan is affected in skn-1KO but not wildtype worms after early life exposure to MeHg. N2 wildtype worms do not show a decrease in lifespan following MeHg exposure. skn-1KOs exhibit reduced lifespan overall (compared to wildtype worms) (*p<0.05) with a further reduction in lifespan following 20 μM MeHg exposure (^p=0.006). Plotted values represent averages of three independent experiments, and the curves represent the best sigmoidal fit using log-rank statistics.
To determine long-term sensitivity to MeHg and to discern if its effect is exacerbated by KO of skn-1, we performed lifespan measurements. Log-rank test revealed that MeHg had a significant effect on lifespan in skn-1KO (*p=0.006) worms, which was not seen in wildtype worms (p=0.61). Knockout of skn-1 alone had an effect on lifespan, which was exacerbated by exposure to MeHg (fig. 3).
2.4 Reactive oxygen species (ROS) levels are elevated in skn-1KO and following MeHg exposure (Figure 4)
Figure 4.

Reactive oxygen species (ROS) levels are elevated in skn-1KOs and in N2 wildtype worms following early life MeHg exposure. (A) Immediately following MeHg exposure. Wildtype worms exhibit significantly higher ROS upon MeHg exposure (*p=0.04). Both untreated and treated skn-1KO worms (**p=0.0012;*p=0.018, respectively) show significant increase in ROS production compared to N2 wildtype untreated worms. skn-1KO MeHg treated worms are not significantly different from their respective untreated controls (p=0.5262, n.s.). *denotes significance from 0μM MeHg N2 wildtype (*p<0.05). #denotes significance from N2 wildtype 20 μM MeHg (#p<0.05). (B) One hour after 30 min MeHg exposure. Wildtype worms exhibit significantly higher ROS 1hr following MeHg exposure (*p=0.04). Both untreated and treated skn-1KO worms (***p=0.0001;*p=0.02, respectively) have significant ROS compared to wildtype untreated 1 hr following 30 minute early life exposure. skn-1KO MeHg treated worms were not significantly different from their respective untreated controls (p=0.4517, n.s.). NaNP=sodium nitroprusside (500μM) used as a positive control was significantly different from the 0 μM MeHg control (**p=0.002). *denotes significance from 0 μM MeHg wildtype (*p<0.05). #denotes significance from wildtype 20 μM MeHg (#p<0.05).
We hypothesized that early-life exposure to MeHg would increase toxicity reflecting increased ROS production. The DCF assay showed a significant increase in ROS levels in wildtype strains immediately following (fig. 4A) and 1hr after (fig. 4B) 20μM MeHg exposure. skn-1KO worms had significantly increased ROS levels (compared to wildtype worms) even in the absence of MeHg exposure. A greater increase in ROS levels was noted in skn-1KO MeHg exposed worms compared to skn-1KO unexposed worms, but it did not attain statistically significance (fig. 4). ROS generation immediately upon MeHg exposure was strain- (**p=0.0002), but not dose- (p=0.468, n.s.) or dose x strain-dependent (p=0.562, n.s); at 1hr the ROS increased in all strains (strain effect **p=0.0002; dose p=0.375; dose x strain interaction p=0.504). Sodium nitroprusside (NaNP) was used as a positive control and MeHg in the absence of worms was used as a negative control (not shown). Post-hoc analysis showed that immediately following MeHg exposure wildtype worms exhibited significantly higher ROS levels compared to unexposed wildtype controls (*p=0.04). Both untreated and treated skn-1KO worms (**p=0.0012;*p=0.018, respectively) had significantly increased ROS levels compared to wildtype untreated worms. skn-1KO treated worms were not significantly different from their respective untreated controls (p=0.5262, n.s.). One hour after MeHg exposure, wildtype worms exhibited significantly higher ROS levels compared to unexposed wiltype controls (*p=0.04). Both untreated and treated skn-1KO worms (***p=0.0001;*p=0.02, respectively) had significantly higher ROS levels compared to wildtype untreated, and skn-1KO treated worms were not significantly different from their respective untreated controls (p=0.4517, n.s.). Collectively, this data suggests that skn-1 plays an important role in mitigating oxidative stress even under basal conditions and in the absence of MeHg exposure.
2.5 MeHg induces SKN-1-mediated increase in Phase II detoxification genes (Supplemental Figure 1)
skn-1-regulated genes were assessed to validate knockdown of skn-1 and activation of skn-1 in wildtype MeHg exposed worms. Whole extracts were used to probe for skn-1 and gst-4 transcriptional activity. Using qRT-PCR, we show that wildtype worms significantly upregulate skn-1 (*p<0.05; supplemental fig. 1A) and gst-4 mRNA levels (*p<0.05; supplemental fig. 1B) in response to MeHg, but skn-1KO worms do not. Differences in the average threshold cycle (ΔCt) were determined and normalized to the expression of gpd-1. These data suggest that wildtype worms mounted an skn-1-mediated antioxidant response, whereas skn-1KO worms failed to do so in response to MeHg. Analysis of skn-1 mRNA, revealed a significant strain (**p=0.002), and dose effect (**p=0.002), but not a dose x strain interaction (supplemental fig. 1A). gst-4 mRNA analysis showed a significant dose effect (***p=0.0007), strain effect (***p=0.001) and a dose x strain interaction (**p=0.007) (supplemental fig 1B). Post-hoc analysis showed skn-1 levels were significantly induced in wildtype worms following MeHg exposure (**p=0.003) but not in skn-1KO worms. There was a significant difference in skn-1 mRNA between the strains in 0 μM control worms (**p=0.003). skn-1 mRNA levels in wildtype 20 μM MeHg versus skn-1KO 20 μM MeHg were significantly different (^p=0.02). Post-hoc for gst4 mRNA levels showed that wildtype 0 μM MeHg was significantly different from wildtype 20 μM MeHg (**p=0.001) and from skn-1KO 0μM MeHg (**p=0.002). There was also a significant difference between N2 20μM and skn-1KO 20μM MeHg exposed worms (^p=0.01). This data showed that optimal stress responsiveness was absent in our skn-1KO worms.
2.6 skn-1 is induced 24 hrs after exposure to MeHg (Figure 5)
Figure 5.

SKN-1 induction in the gut following early life exposure to MeHg. LG326 worms (skn-1::GFP) were exposed to MeHg to visualize SKN-1 protein induction. SKN-1 expression in the gut can be seen in the 20 μM MeHg exposure group 24 hrs following exposure.
Intestinal accumulation of skn-1 in C. elegans is apparent following 20 μM MeHg exposure, this accumulation was absent in 0 and 10μM MeHg exposed worms. SKN-1 is present in ASI nuclei under normal conditions, and accumulates in intestinal nuclei in response to oxidative stress (An and Blackwell, 2003). The accumulation of skn-1 in the intestine of worms exposed to 20μM MeHg suggests activation of skn-1 is occurring in response to sublethal doses of MeHg.
2.7 DA content (Figure 6)
Figure 6.

DA levels are decreased in skn-1KO compared to N2 wildtype worms following early life exposure to MeHg. Total DA levels were measured by HPLC immediately following 30 min early life MeHg exposure (L1 stage). There was a significant difference between the 0 μM MeHg N2 wildtype worms and all other strains/doses (*p<0.05;**p<0.005;***p<0.0001). All doses and strains were significantly different from cat-2s (tyrosine hydroxlase deficient worms; used as a control).
Prenatal MeHg exposure studies in animals have described altered DAergic function [38, 39]. Accordingly, we examined the extent of DAergic neurodegeneration in response to early-life MeHg exposure. Worms’ DA levels were determined immediately after MeHg exposure, when the worms were at the L1 stage. DA levels were correlated to the DA-mediated behavioral data which was collected 72 hrs post exposure (fig. 7) and the structural data collected at 96 hrs post exposure (fig. 8). There was no significant effect of dose (p=0.51); or dose x strain interaction (p=0.11), there was a significant strain effect (**p<0.005). There was a trend towards decreased DA content in MeHg exposed N2 wildtype worms, and no significant difference in skn-1KO worms. These findings corroborate the functional behavioral DA readout, which was measured 72 hrs following exposure (fig. 6). Post-hoc analyses revealed a significant difference between the 0 μM MeHg wildtype worms and all other strains/doses (*p<0.05;**p<0.005; ***p<0.0001). DA content from treated and untreated wildtype and skn-1KO worms were significantly different from cat2 mutants.
Figure 7.

MeHg has an effect on DA function as shown by the Basal Slowing Response. Loss-of-function of DAergic neurons is present later in life (72 hrs) following a single, early-life (L1 stage) exposure to 20μM MeHg. For each strain, locomotion rates in the absence and presence of bacteria (supplemental fig. 2) were calculated, and results are presented as change (Δ) body bends/20 seconds. Higher values indicate functional, while lower values indicate dysfunctional DAergic neurons. cat-2 mutants (TH deficient) unexposed worms were significantly different from all MeHg-treated wildtype worms (*p<0.05). **denotes significance from 0μM MeHg N2 wildtype control (*p<0.05, ***p<0.0005). ## denotes significance from 0μM skn-1KO (#p<0.05). Error bars represent the mean ± SEM of 25–35 individual worms.
Figure 8.
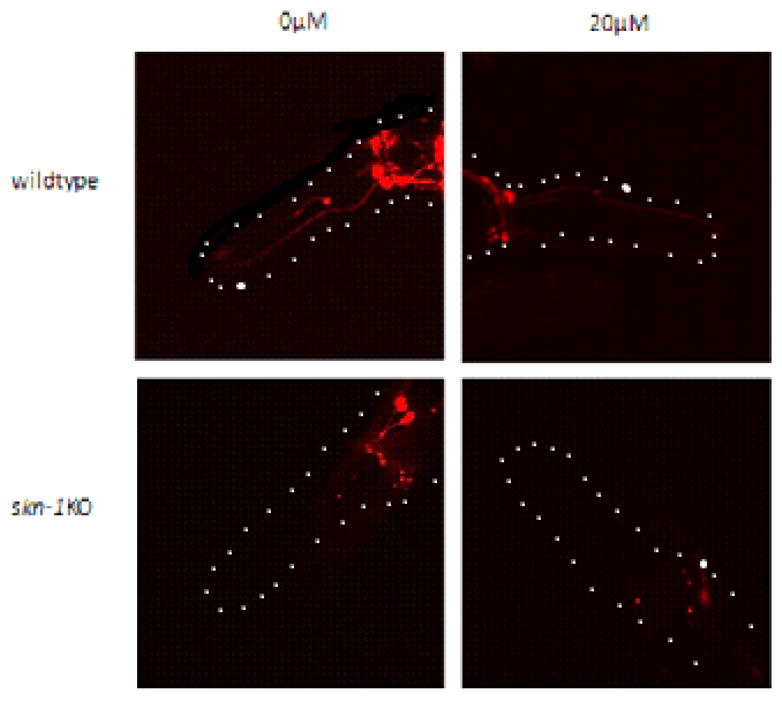
Degeneration of DAergic neurons and loss of fluorescence are visible in skn-1KOs and treated N2 wildtype worms later in life (96 hrs) following a single early-life MeHg exposure. DAergic neurons, labeled with mCherry, in N2 wildtype and skn-1KO worms following exposure to 0 or 20 μM MeHg. CEP: cephalic (from nerve ring to tip of nose), ADE: anterior deirid DAergic neurons.
2.8 Basal slowing behavioral analysis (Figure 7)
We analyzed DA-mediated behavior 72 hrs post exposure (adult stage) to determine if there was a decrease in functional DA. For each strain, locomotion rates in the absence and presence of bacteria (supplemental fig. 2) were calculated, and results are presented as change (Δ) body bends/20 seconds (fig. 8). Higher values indicate functional, while lower values indicate dysfunctional DAergic neurons. When the change in locomotor activity (Δ body bends/20 seconds) was compared by one-way ANOVA, it was statistically significant (***p<0.0001). Post-hoc analysis showed all doses/strains were significantly different from 0 μM MeHg wildtype (*p<0.05, ***p<0.0005) and all doses/strains were significantly different from 0 μM MeHg skn-1KOs (#p<0.05). cat-2 mutants unexposed worms were significantly different from all MeHg-treated wildtype worms (*p<0.05).
2.9 Degeneration and loss of fluorescence in skn-1KO DAergic neurons after low dose MeHg exposure (Figure 8)
We assessed the structural integrity of mCherry-labeled DAergic neurons in wildtype and skn-1KO worms, 96 hrs after MeHg exposure. MeHg exposed wildtype worms showed loss of fluorescence similar to skn-1KO worms. In skn-1KO MeHg exposed worms, the loss of fluorescence was exacerbated compared to skn-1KO unexposed worms and wildtype MeHg exposed worms. This data suggests that skn-1KO worms are susceptible to DAergic neurodegeneration following short, early life exposure to MeHg. CEP: cephalic, ADE: anterior deirid DAergic neurons.
DISCUSSION
MeHg is an environmental contaminant of concern due to its widespread release and contamination of fish [40, 41]. The nervous system is the primary target of MeHg and environmental exposure, particularly in fish-eating populations, can be detrimental. While adults are susceptible to MeHg exposure, the developmental period in humans and rodents is a window of high vulnerability [42, 43] and perinatal exposure to MeHg presents with a less specific pattern of damage than that of adult exposure, shown to by concentration-, duration of exposure- and stage of development-dependent [44]. Latent effects following MeHg exposure have also been described [29]. Therefore, it is important to address the effects of early life exposure to MeHg on later life endpoints like neurodegeneration. Our findings revealed that early-life MeHg exposure led to decreases in DA-mediated behavior seen at 72 hrs which positively correlated to DAergic degeneration at 96 hrs (adult life stage).
MeHg is known to produce oxidative stress and the skn-1 (Nrf2 homolog) signaling pathway is involved in oxidative stress resistance and eradication of oxidants [1, 4, 45–48]. Given that oxidative stress mediates both MeHg toxicity and the development of PD [10, 12, 20, 23, 24], we assessed the role of skn-1 in MeHg-induced oxidative stress response and its contribution to DAergic toxicity. While other neuronal subtypes are also susceptible to MeHg, emphasis was directed to DAergic neurons based on their expression of skn-1 [3] and the documented importance of skn-1 in attenuating MeHg toxicity [1]. We focused on addressing whether MeHg exposures that occur early in the lifespan have deleterious effects later in life.
Assessment of lethality 24 hrs after 30 min exposure to MeHg during the L1 period indicated that skn-1KO worms are more sensitive to MeHg exposure (fig. 1). This heightened sensitivity is likely not due to increased uptake of Hg in skn-1KO worms as there were no strain differences in Hg content immediately after exposure (fig. 2).
Lifespan was significantly affected by MeHg exposure in both the wildtype and skn-1KO strains. Consistent with other published studies, skn-1KOs had a significant decrease in lifespan compared to wildtype worms (fig. 3) [2]; this decrease was further exacerbated by MeHg exposure. Many environmental contaminants have a significant impact on lifespan through generation of ROS [49–53]. The increase in ROS levels following MeHg exposure is consistent with earlier reports [10]. More striking is the increased ROS production measured in the skn-1KO untreated strain compared to the untreated wildtype strain. This increase at baseline has been documented in the human cell-line and murine literature following silencing or knockdown of Nrf2 [54, 55] suggesting that skn-1 is important in regulating ROS levels under basal conditions as well as after presentation of a stimulus.
skn-1 knockdown was confirmed by measuring RNA content (supplemental fig. 1A) for skn-1 and gst-4, a gene under the control of skn-1 (supplemental fig. 1B). skn-1 and gst-4 levels increased following MeHg exposure in wildtype worms, but did not increase in skn-1KO worms, confirming the knockdown (supplemental fig. 1A/B). skn-1::GFP worms assessed at 24 hrs showed gut florescence in the 20μM MeHg exposed worms, indicating activation of skn-1 (fig. 5). SKN-1 is present in ASI nuclei under normal conditions, and accumulates in intestinal nuclei in response to oxidative stress [2]. Under basal conditions, glycogen synthase kinase 3 (gsk3; mammalian: Keap1) inhibits skn-1 nuclear localization [56]. Following oxidative stress, skn-1 is liberated from gsk3 repression and translocates to the nucleus, leading to transcription of Phase II detoxification genes [57]. Inhibition of skn-1 leads to increased sensitivity to stress [2].
Our studies were designed to establish novel and translational information on mechanisms associated with DAergic neurodegeneration, especially after early life exposure to MeHg. We are reporting the presence of irregularities in DAergic neurons 96 hrs following 30 min exposure (at L1 stage) to 20μM MeHg (fig. 8). C. elegans possess eight DAergic neurons; four cephalic (CEP) neurons, two anterior deirid (ADE) neurons, and two postdeirid (PDE) neurons (fig. 8; PDE not shown). We show degeneration of the DAergic neurons 96 hrs following exposure to MeHg (fig. 8); however, skn-1KO worms show degeneration even in the absence of MeHg exposure. These results indicating loss of DAergic neurons in skn-1KO worms are corroborated by an Nrf2-knockout mouse model, which showed 23% greater nigral DAergic neuron loss as measured by positive stain for TH, than Nrf2+/+ mice [6]. It is not surprising that MeHg exposed skn-1KOs show the most robust loss in DAergic neuron integrity given the evidence for the role of Nrf2 in the DAergic system. Nrf2 induction in transgenic mice brains has been shown to be protective of MPTP damage in the nigrostriatal DAergic pathway [4] and Nrf2 has been shown to translocate to the nucleus in the substantia nigra of PD patients, although it has proved to be insufficient for protection [5]. Another fact that cannot be overlooked is that Nrf2 activity decreases with age while PD susceptibility increases with age [58]. The decrease in fluorescence in our model could be explained by decreases in DAT, as the mCherry florephore is under the control of dat-1 promoter. Another group reported decreases in total DAT activity in MeHg exposed murine embryonic stem cells as well as decreases in TH+ cells and downregulation of DA receptors [59] suggesting that the dopamine system is particularly susceptible to MeHg.
The decrease in DA in wildtype worms (fig. 6) suggests that MeHg has significant effects on DAergic neuron function. The absence of effect of MeHg on skn-1KO worm DA levels is interesting, given our hypothesis that skn-1KO worms should be more susceptible to DAergic changes following exposure (fig. 6). The lack of MeHg effect in this strain may be due to the early time point in which the DA levels were measured (i.e. immediately rather than days after exposure), not allowing time for a measureable effect. Additionally, DA metabolites may present a more complete understanding of DA vulnerability [60] but we are unable to measure DA metabolites in our model. It is known that dopamine can oxidize and form ROS and reactive quinones and that those quinones can covalently bind to cysteine sites [61]. The dopamine transporter has several cysteine residues and therefore is a target for covalent modification by reactive quinones and ROS [62]. This type of modification to DA transporters could explain the altered state of the DAergic system seen after MeHg exposure. The decreased basal levels of DA in skn-1KO control worms compared to wildtype controls (fig. 6) have been reported in murine literature showing low basal levels of TH immunoreactivity in Nrf2−/− mice [45].
The basal slowing behavior, assessed 72 hrs following exposure (fig. 7), confirms the loss of functional DAergic neurons. Functional DAergic neurons are required for food sensing (mechanosensation) [35]. This data is consistent with the images taken at 96 hrs (fig. 8) and the trends in DA levels immediately following exposure (fig. 6).
Neuronal differentiation and establishment of connectivity occurs early in the lifespan and could be disrupted by exposure to MeHg. These connections, or lack thereof, may not be apparent until the brain has completed the developmental period therefore we do not see them until after a period of latency. Acute and chronic MeHg exposures have been shown to produce latent toxicity (reviewed in [29]). Others have postulated that microtubule function may be altered by MeHg preferential binding to sulfhydryl groups [63] and this may result in altered cytoskeletal architecture, which in our studies, is described as degeneration. Another explanation for the latency period is that these effects may not be seen until after a period of normal decline due to aging thereby exposing the MeHg-induced damage to the DAergic system.
Taken together and consistent with the Barker hypothesis [64, 65], these results indicate that acute early-life exposure to MeHg confers DAergic neurodegeneration later in life. Furthermore, knockdown of skn-1 amplifies MeHg’s effect. Future studies should aim to address if Hg is still present at later time periods to determine what role, if any, they are playing in the degenerative process as well as establish a timeline of cell death of the DAergic neurons. It would also be beneficial to examine other neuronal subtypes to determine if there is additional damage.
Supplementary Material
Acknowledgments
This manuscript was supported by the National Institute of Environmental Health Sciences (NIEHS) [R01ES07331, T32ES007028] and the National Institutes of Health Loan Repayment Program. We would like to acknowledge the Caenorhabditis Genetic Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440), for providing the strains used in this manuscript.
Literature Cited
- 1.Ni M, et al. Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicol Sci. 2010;116(2):590–603. doi: 10.1093/toxsci/kfq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003;17(15):1882–93. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanduyn N, et al. SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci. 2010;118(2):613–24. doi: 10.1093/toxsci/kfq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaidery NA, et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid Redox Signal. 2013;18(2):139–57. doi: 10.1089/ars.2011.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey CP, et al. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66(1):75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lastres-Becker I, et al. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum Mol Genet. 2012;21(14):3173–92. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- 7.Gorell JM, et al. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20(2–3):239–47. [PubMed] [Google Scholar]
- 8.Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem. 1995;41(12 Pt 2):1819–28. [PubMed] [Google Scholar]
- 9.Recchia A, et al. Alpha-synuclein and Parkinson’s disease. FASEB J. 2004;18(6):617–26. doi: 10.1096/fj.03-0338rev. [DOI] [PubMed] [Google Scholar]
- 10.Aschner M, et al. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res. 2007;40(3):285–91. doi: 10.1590/s0100-879x2007000300001. [DOI] [PubMed] [Google Scholar]
- 11.Zhang P, et al. In vitro protective effects of pyrroloquinoline quinone on methylmercury-induced neurotoxicity. Environ Toxicol Pharmacol. 2009;27(1):103–10. doi: 10.1016/j.etap.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Chiueh CC, et al. In vivo generation of hydroxyl radicals and MPTP-induced dopaminergic toxicity in the basal ganglia. Ann N Y Acad Sci. 1994;738:25–36. doi: 10.1111/j.1749-6632.1994.tb21786.x. [DOI] [PubMed] [Google Scholar]
- 13.Dabbeni-Sala F, et al. Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity. FASEB J. 2001;15(1):164–170. doi: 10.1096/fj.00-0129com. [DOI] [PubMed] [Google Scholar]
- 14.De Iuliis A, et al. A proteomic approach in the study of an animal model of Parkinson’s disease. Clin Chim Acta. 2005;357(2):202–9. doi: 10.1016/j.cccn.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 15.Sherer TB, et al. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179(1):9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- 16.Riederer P, et al. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52(2):515–20. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 17.Sian J, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348–55. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 18.Sian J, et al. Glutathione-related enzymes in brain in Parkinson’s disease. Ann Neurol. 1994;36(3):356–61. doi: 10.1002/ana.410360306. [DOI] [PubMed] [Google Scholar]
- 19.Sofic E, et al. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett. 1992;142(2):128–30. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- 20.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515–7. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 21.Cantuti-Castelvetri I, et al. Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol Aging. 2005;26(10):1343–55. doi: 10.1016/j.neurobiolaging.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38(5):518–20. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 23.Swerdlow RH, et al. Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol. 1996;40(4):663–71. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- 24.Gorell JM, et al. Occupational metal exposures and the risk of Parkinson’s disease. Neuroepidemiology. 1999;18(6):303–8. doi: 10.1159/000026225. [DOI] [PubMed] [Google Scholar]
- 25.Gao Y, et al. Effects of methylmercury on postnatal neurobehavioral development in mice. Neurotoxicol Teratol. 2008;30(6):462–7. doi: 10.1016/j.ntt.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Lin FM, Malaiyandi M, Sierra CR. Toxicity of methylmercury: effects on different ages of rats. Bull Environ Contam Toxicol. 1975;14(2):140–8. doi: 10.1007/BF01701304. [DOI] [PubMed] [Google Scholar]
- 27.do Maia CS, et al. Inhibitory avoidance acquisition in adult rats exposed to a combination of ethanol and methylmercury during central nervous system development. Behav Brain Res. 2010;211(2):191–7. doi: 10.1016/j.bbr.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 28.Uchino M, et al. Neurologic features of chronic Minamata disease (organic mercury poisoning) certified at autopsy. Intern Med. 1995;34(8):744–7. doi: 10.2169/internalmedicine.34.744. [DOI] [PubMed] [Google Scholar]
- 29.Weiss B, Clarkson TW, Simon W. Silent latency periods in methylmercury poisoning and in neurodegenerative disease. Environ Health Perspect. 2002;110(Suppl 5):851–4. doi: 10.1289/ehp.02110s5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshida M, et al. Emergence of delayed methylmercury toxicity after perinatal exposure in metallothionein-null and wild-type C57BL mice. Environ Health Perspect. 2008;116(6):746–51. doi: 10.1289/ehp.10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida M, et al. Neurobehavioral effects of combined prenatal exposure to low-level mercury vapor and methylmercury. J Toxicol Sci. 2011;36(1):73–80. doi: 10.2131/jts.36.73. [DOI] [PubMed] [Google Scholar]
- 32.Landrigan PJ, et al. Early environmental origins of neurodegenerative disease in later life. Environ Health Perspect. 2005;113(9):1230–3. doi: 10.1289/ehp.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helmcke KJ, et al. Characterization of the effects of methylmercury on Caenorhabditis elegans. Toxicol Appl Pharmacol. 2009;240(2):265–72. doi: 10.1016/j.taap.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26(3):619–31. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 36.Burbacher TM, Rodier PM, Weiss B. Methylmercury developmental neurotoxicity: a comparison of effects in humans and animals. Neurotoxicol Teratol. 1990;12(3):191–202. doi: 10.1016/0892-0362(90)90091-p. [DOI] [PubMed] [Google Scholar]
- 37.Newland MC, Rasmussen EB. Aging unmasks adverse effects of gestational exposure to methylmercury in rats. Neurotoxicol Teratol. 2000;22(6):819–28. doi: 10.1016/s0892-0362(00)00107-0. [DOI] [PubMed] [Google Scholar]
- 38.Dare E, et al. Effects of prenatal exposure to methylmercury on dopamine-mediated locomotor activity and dopamine D2 receptor binding. Naunyn Schmiedebergs Arch Pharmacol. 2003;367(5):500–8. doi: 10.1007/s00210-003-0716-5. [DOI] [PubMed] [Google Scholar]
- 39.Reed MN, Newland MC. Gestational methylmercury exposure selectively increases the sensitivity of operant behavior to cocaine. Behav Neurosci. 2009;123(2):408–17. doi: 10.1037/a0014595. [DOI] [PubMed] [Google Scholar]
- 40.Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol. 2006;36(8):609–62. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- 41.Clarkson TW, Magos L, Myers GJ. The toxicology of mercury--current exposures and clinical manifestations. N Engl J Med. 2003;349(18):1731–7. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- 42.Castoldi AF, et al. Neurodevelopmental toxicity of methylmercury: Laboratory animal data and their contribution to human risk assessment. Regul Toxicol Pharmacol. 2008;51(2):215–29. doi: 10.1016/j.yrtph.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Goulet S, Dore FY, Mirault ME. Neurobehavioral changes in mice chronically exposed to methylmercury during fetal and early postnatal development. Neurotoxicol Teratol. 2003;25(3):335–47. doi: 10.1016/s0892-0362(03)00007-2. [DOI] [PubMed] [Google Scholar]
- 44.Rodier PM. Developing brain as a target of toxicity. Environ Health Perspect. 1995;103(Suppl 6):73–6. doi: 10.1289/ehp.95103s673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rojo AI, et al. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58(5):588–98. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 46.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266(18):11632–9. [PubMed] [Google Scholar]
- 47.Enomoto A, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59(1):169–77. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen T, et al. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem. 2005;280(37):32485–92. doi: 10.1074/jbc.M503074200. [DOI] [PubMed] [Google Scholar]
- 49.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Finley EJ, et al. Free Radic Biol Med. 2013. Manganese neurotoxicity and the role of reactive oxygen species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hagen TM. Oxidative stress, redox imbalance, and the aging process. Antioxid Redox Signal. 2003;5(5):503–6. doi: 10.1089/152308603770310149. [DOI] [PubMed] [Google Scholar]
- 52.Li J, Holbrook NJ. Common mechanisms for declines in oxidative stress tolerance and proliferation with aging. Free Radic Biol Med. 2003;35(3):292–9. doi: 10.1016/s0891-5849(03)00308-3. [DOI] [PubMed] [Google Scholar]
- 53.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R18–36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 54.Kim HL, Seo YR. Molecular and genomic approach for understanding the gene-environment interaction between Nrf2 deficiency and carcinogenic nickel-induced DNA damage. Oncol Rep. 2012;28(6):1959–67. doi: 10.3892/or.2012.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang B, et al. Deficiency in the nuclear factor E2-related factor 2 renders pancreatic beta-cells vulnerable to arsenic-induced cell damage. Toxicol Appl Pharmacol. 2012;264(3):315–23. doi: 10.1016/j.taap.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2001;20(29):3906–17. doi: 10.1038/sj.onc.1204506. [DOI] [PubMed] [Google Scholar]
- 57.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 58.Suh JH, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101(10):3381–6. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zimmer B, et al. Sensitivity of dopaminergic neuron differentiation from stem cells to chronic low-dose methylmercury exposure. Toxicol Sci. 2011;121(2):357–67. doi: 10.1093/toxsci/kfr054. [DOI] [PubMed] [Google Scholar]
- 60.Itier JM, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12(18):2277–91. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- 61.Berman SB, Zigmond MJ, Hastings TG. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem. 1996;67(2):593–600. doi: 10.1046/j.1471-4159.1996.67020593.x. [DOI] [PubMed] [Google Scholar]
- 62.Chen R, et al. Direct evidence that two cysteines in the dopamine transporter form a disulfide bond. Mol Cell Biochem. 2007;298(1–2):41–8. doi: 10.1007/s11010-006-9348-7. [DOI] [PubMed] [Google Scholar]
- 63.Johansson C, et al. Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox Res. 2007;11(3–4):241–60. doi: 10.1007/BF03033570. [DOI] [PubMed] [Google Scholar]
- 64.Barker DJ. The fetal origins of diseases of old age. Eur J Clin Nutr. 1992;46(Suppl 3):S3–9. [PubMed] [Google Scholar]
- 65.Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311(6998):171–4. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.