

Abstract
Tissue barriers maintain homeostasis, protect underlying tissues, are remodeled during organogenesis and injury and limit aberrant proliferation and dissemination. In this context, endothelial and epithelial intercellular junctions are the primary targets of various cues. This cellular adaptation requires plasticity and dynamics of adhesion molecules and the associated cytoskeleton, as well as the adhesive-linked signaling platforms. It is therefore not surprising that the guidance molecules from the Semaphorin family arise as novel modifiers of epithelia and endothelia in development and diseases. This review will focus on the actions of Semaphorins, and their cognate receptors, Plexins and Neuropilins, on epithelial and endothelial barrier properties.
Keywords: barrier, cadherin, permeability, semaphorin, tight junction
Historically characterized for their roles in the developing central nervous system (CNS), Semaphorins have emerged as multifaceted guidance proteins that control biological responses in the epithelia and endothelia under physiological and pathological circumstances. In this review, we discuss their influence and mode of action in the regulation of tissue barriers, especially at the level of intercellular junctions.
Vertebrate Semaphorins and their Receptors
Semaphorins were identified more than 20 years ago as proteins, which provide a wide repertoire of attractive and repulsive signals that orchestrate axon outgrowth.1,2 A large number of these guidance molecules combines with multiple receptors and co-receptors, increasing thereby both specificity and complexity of the Semaphorin mode of action (Fig. 1). Therefore, the elucidation of signaling cascades activated by these molecules is the subject of intense research.

Figure 1. Schematic representation of vertebrate Semaphorins and their receptors, Plexins and Neuropilins. Each semaphorin contains one sema and one PSI domains. Class III Semaphorins are secreted and possess a C-terminus basic-rich domain. They signal through Plexin (Plx) A and Neuropilins (NRP), except for Sema3E that only required PlxD1. Class IV Semaphorins are transmembrane proteins and operate through PlxD1 and PlxB. Sema5A is also transmembrane and characterized by thrombospondin repeats, and binds to PlxB3. Transmembrane class VI Semaphorins use PlxA, while GPI-anchored Sema7A signals through PlxC1.
Semaphorins
In vertebrates, there are 20 semaphorin genes,2 sub-divided into classes III to VII. Class III semaphorins (Sema3A to G) are secreted, while Semaphorins from subclasses IV to VI exist as transmembrane proteins and Sema7A contains a glycosylphosphatidylinositol (GPI) anchor. Each Semaphorin possesses a 500 amino acid extracellular domain, called semaphorin (sema) composed of a seven-bladed β propeller domain, essential for signaling. This domain is followed by a cysteine-rich domain (PSI), involved in receptor binding ability.3 Conversely, the C-terminal tail features additional sequence motifs that diverge among classes: class III contains both an immunoglobulin-like and a basic residue-rich domains, whereas class V are distinguished by thrombospondin repeats4 (Fig. 1). Semaphorins co-opt two kinds of transmembrane receptors (Plexins and Neuropilins), which mediate downstream signaling.
Plexins
Plexins are the main cell surface receptors for Semaphorin signal transduction. Plexins comprise nine members in mammals and are subdivided into four classes: class A (A1–4), class B (B1–3), PlexinC1 and PlexinD15 (Fig. 1). Unlike Semaphorins, Plexin architecture is conserved throughout the family. The extracellular region is composed by one sema domain and two or three PSI and IPT (immunoglobulin shared by Plexins and transcription factors) repeats. Class III and VI Semaphorins mainly bind to PlexinA. Most of the class III Semaphorins requires a co-receptor from the Neuropilin family (NRP, see paragraph 1.3), while the class VI Semaphorins directly interact with PlexinA through their respective sema domains, which in turn conveys intracellular signaling.5 The Plexin cytoplasmic region contains a Rho and Ras-family-specific GTPase-activating protein (GAP) domain.6,7 Although several models have been proposed, the exact role of the Ras-GAP activity in Semaphorin signaling remains obscure.5,8,9 However, it has recently been characterized that this GAP domain can be activated by dimerization after Semaphorin binding to Plexin ectodomains.10 This process could in turn inhibit Rap GTP-binding proteins and affect downstream pathways. Additionally, Plexins B and D bear PDZ (post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1) and zonula occludens-1 protein (ZO-1))-binding motifs that are most likely involved in protein-protein interactions and downstream signaling.
Neuropilins
The Neuropilin receptors, namely NRP-1 and -2 are only found in vertebrates, where they exist as multiple isoforms, including soluble ones. They operate as receptors or co-receptors for diverse ligands, ranging from class III Semaphorins, heparin-binding proteins and growth factors, such as vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF) and placental growth factor 2 (PlGF).11-13 These single transmembrane proteins include five extracellular domains and a short cytoplasmic tail. Their extracellular region contains two complement-like6 (CUB) domains, also designated a1 and a2 domains, two FV/FVIII coagulation factor-like domains, also known as b1 and b2, and a single meprin/A5-protein/PTPμ (MAM) domain, also called c. The a1/a2 domains are involved in Semaphorin binding, while the b1/b2 part is necessary for VEGF binding11,14. More specifically, the domain a1 is essential for the stabilization of the class III Sema3-PlexinA interaction and subsequently allows Semaphorins signaling through PlexinA14 (Fig. 1); with the exception of Sema3E which acts via PlexinD1 and can signal in the absence of Neuropilins in endothelial cells.15 The cytoplasmic tail does not harbor enzyme activity per se but rather a PDZ-binding motif, whose function requires further investigation.
General mode of action of Semaphorins
Semaphorins have been implicated in a wide variety of functions instrumental in the development of the CNS. These include neuron migration,16 axonal pruning,17,18 synapse formation19 and synaptic transmission.20
Neurons, as with epithelial or endothelial cells, are polarized cells. Indeed, neurons harbor a unique long axon, which is directed by a highly motile structure, named the growth cone, and, multiple shorter dendrites, whose formation is controlled, among others, by Semaphorin 3A (Sema3A). Indeed, this decisive Semaphorin was first characterized for its ability to promote dendrite formation via the inhibition of axon specification.21 The Sema3A co-receptor NRP-1 favors the accumulation of cyclic GMP (cGMP) over AMP and the further inhibition of PKA and GSK3β activities, two crucial signaling pathways for axon development.21 Through this cGMP/AMP balance, Sema3A repels growth cone movement, while stimulating dendrite outgrowth. This action is thought to rely on a protein named Nervy that links PKA to the Sema3A receptor PlexinA. Alternatively, it employs a different set of neuropilin co-receptors.22,23
Another important aspect of Semaphorin action in neuron biogenesis is its impact on the actin cytoskeleton and extracellular matrix adhesion. For instance, class III Semaphorins released into the surrounding environment of a growth cone, triggered Plexin GAP activity upon receptor engagement, which in turn decreases R-Ras activation and integrin attachment.24 This results in the reduction of outgrowth, illustrating the repellent activity of Semaphorins. On the other edge of the growth cone, the absence of Semaphorins allows higher level of R-Ras activity, and therefore the upregulation of integrin binding that facilitates growth.7,25
The leading edge of migrating cells, such as endothelial or epithelial cells, is comparable to the axon growth cone. Any abnormal changes in the expression of Semaphorins might trigger CNS diseases by affecting neuronal structure,26 as demonstrated in the case of regeneration failure where Sema3A inhibits axon growth at sites of injury.27 It also appears that Semaphorins can contribute to tumor progression,28 a point that will be particularly discussed in this review.
Effects of Semaphorins on Epithelial Junctions
Epithelia are located at the boundaries of tissues where they form regulated sealing barriers controlling solute diffusion and immune cell passage. To assume their roles, epithelial cells establish specific junctions that maintain barrier integrity. For instance, in the case of the intestine, epithelial cells protect underlying tissues from luminal content, while permitting the absorption and secretion of fluids and ions. In this context, epithelial cells are polarized; i.e cell-cell contacts are organized in complexes distributed specifically along the apical-basal axis. Both tight and adherens junctions, as well as desmosomes are found within epithelial intercellular junctions (Fig. 2).
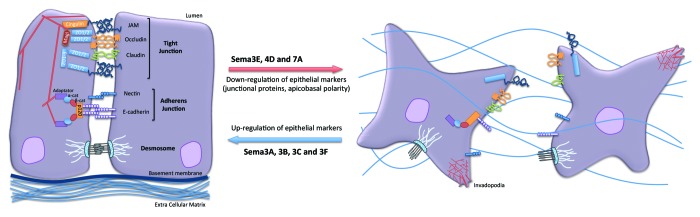
Figure 2. Impact of Semaphorins on the organization of epithelial cell-cell junctions. Specific and distinct adhesion proteins structure the epithelial cell junctions: tight junctions (JAM, occludin and claudins) and adherens junctions (nectin and E-cadherin) are linked to actin cytoskeleton and multiple intracellular adaptators. Upon exposure to Sema3A, 3B, 3C and 3F, cell adhesion can be strengthened, while Sema3E, 4D and 7A provoked dramatic cell-cell junction remodeling, which may ultimately favor tumorigenesis.
Epithelial adherens junctions
Structure and function of E-cadherin
The key protein of adherens junctions (AJ) that is found accumulated at epithelial cell-cell contacts is the transmembrane glycoprotein E-cadherin from the classical cadherin family. Structurally, classical cadherins consist of three different domains: the extracellular domain (ECD), the transmembrane (TM) and the intracellular domain (ICD).29 The ECD is formed by the repetition of five cadherin repeats called EC 1 to 5, from the N-terminal to the C-terminal end. Each EC consists of 110 amino acids organized in β-sheets.30,31 The EC1 domain contains the HAV sequence and is suspected to bear the adhesive specificity and thus to promote homophilic trans-association with adjacent cells,32 while the entire ECD likely engages in heterophilic interaction.33 The ICD is highly conserved among vertebrate cadherins, in terms of sequence, length and cytoplasmic interacting partners. This cytosolic part modulates strength, dynamics and signaling abilities of cadherins at the cell-cell junctions.
E-cadherin cytoplasmic partners
The E-cadherin ICD is connected to the actin cytoskeleton through its association with β-catenin, which in turn binds to α-catenin. Finally, α-catenin interacts with actin and several actin-binding adaptors, such as formin, vinculin, α-actinin, afadin and ZO-1,34,35 that can modulate actin organization, dynamics and polymerization. Cell-cell contacts can also be strengthened through E-cadherin cis-interaction involving the juxtamembrane region where the p120 catenin serves as a linker.36 Importantly, epithelial cell-cell contacts still remain plastic, as E-cadherin can undergo endocytosis, recycling, lateral movements and shedding.37 Tight junctions (TJ) presented in the paragraph below, delimited the apical pole of epithelial cells and accumulated above AJs. At the opposite, basolateral proteins, such as desmosomes, are found below AJs (Fig. 2). Thus, E-cadherin is not uniformly distributed over the cell surface but rather clustered in specific membrane domains within AJs, which serve as signaling platforms.36 Indeed, AJs can also signal through proteins such as Rho GTPases, tyrosine kinase receptors and other lipid modifications.38,39 These interactions contribute to the organization of membrane trafficking and promote polarized growth in regions that can be immediately adjacent or distant from AJs. In this scenario, AJs modulate TJ formation and epithelial polarization and therefore discriminate apical and basolateral subcellular areas.
Overall, one should keep in mind that AJs are not a rigid structure but rather a complex that can integrate and adapt to external changes and morphogenetic movements, including delamination, cell division and epithelial-to-mesenchymal transition (EMT).
Epithelial tight junctions
The epithelium is fastened apically by TJs, which almost completely obstruct the paracellular exchange pathway. TJs therefore contribute to the regulation of the ion and fluid passage, while restricting the diffusion of large molecules. In addition to their role as a barrier, TJs can regulate numerous cellular processes such as polarity, proliferation, differentiation and migration. First identified by electronic microscopy in epithelial cells,40 TJs form typical structures of close apposition between membranes of two adjacent cells. The freeze-fracture method had allowed the observation of focal hemifusion sites associated with intracellular fibrils. This highlights the interplay between transmembrane proteins, cytosolic partners and the cytoskeleton.
Structure and functions of transmembrane proteins
TJs are enriched with many transmembrane proteins that associate to each other and link to scaffolding proteins and the actin cytoskeleton. Three protein families are found in TJs: claudins, occludin and junctional adhesion molecules (JAMs).
Claudins are calcium independent cell-cell adhesion proteins, comprising at least 24 members, which regulate paracellular permeability. They are composed of two extracellular loops that mediate homo- and hetero-typic intercellular junctions, as well as ion and fluid passage selectivity. They are instrumental in the maintenance of barrier integrity, as demonstrated by severe barrier defects in knockout mice lacking individual claudin family members.41 Interestingly, claudins exhibit organ and tissue specific expression patterns, thus forming a large repertoire of TJs with different strength, size and ion specificity.
Occludin was the first protein identified as a TJ component.42 Occludin is a tetraspan membrane protein bearing a MARVEL domain (MAL and related proteins for vesicle trafficking and membrane link).43 Its role is not fully understood, but in vitro studies agree that this junctional protein operates at the epithelial barrier and functioned as a signaling protein.44-46
Finally JAMs, the third group of proteins found at TJs, consist of single transmembrane proteins that belong to the immunoglobulin superfamily.47 They are composed of two extracellular immunoglobulin-like domains, a single transmembrane region and a C-terminal part. Among them, JAM-A, -B, -C and -448 and the Coxsackie and adenovirus receptor (CAR), are found at epithelial junctions. JAMs can be engaged either in either homo- or hetero-philic adhesions within the TJs. However, they cannot form TJs by themselves when expressed in fibroblasts.49 Although their exact function is still uncertain, JAMs, similarly to claudins and occludin, can bind to cytoplasmic scaffolding partners.
Scaffolding proteins
Among all cytosolic proteins involved at the cytoplasmic surface of TJs, the major components are the zonula occludens (ZO), designated as ZO-1, ZO-2 and ZO-3.50-52 These proteins share sequence similarity and molecular organization, as they all contain three PDZ domains and one Src-homology 3 (SH3) domain followed by one guanylate kinase-like (GUK) domain involved in protein-protein interactions. Through the N-terminal PDZ domains, ZOs interact directly with JAMs, occludin and claudins, while they are connected to the cytoskeleton via their C-terminal tail.53 ZO-1 was found to be crucial for epithelial barrier function.54 This protein also serves as scaffold for regulatory proteins such as kinases, GTP exchange factors (GEF) and many transcription factors.55 Many other PDZ-containing proteins have since been discovered to be cytoplasmic components of the TJs, including membrane-associated guanylate kinases (MAGI-1, -2, -3), Multi-PDZ domain protein 1 (MUPP1), cingulin and polarity proteins PAR3/6, and will not be discussed in details here.56
These TJ components, much like the cytoplasmic partners for AJs, are pivotal for the epithelial biology. They exhibit the ability to link many transmembrane proteins to the cytoskeleton, promote signals toward other cellular compartments and adapt the cellular responses to external cues.
Semaphorins and the epithelial barrier function
At present, knowledge concerning Semaphorins in epithelial barrier function, and in particular their direct action on junctional components, is quite limited. This review will instead present selected cases that support for a role of individual Semaphorin in the reinforcement of epithelial barrier components, or, at the opposite end, favor barrier dismantlement, especially in the course of cancer progression (Fig. 2).
Pro-barrier Semaphorins
During lung organogenesis, layers of progenitors lining the developing airways maintain their epithelial characteristics, while proliferating and differentiating into multiple cell types.57 Many genes that establish and maintain cell polarity and/or cell junctions are regulated.58,59 In this context, it has been shown that class 3 Semaphorins, namely Sema3A, Sema3C and Sema3F, display a specific spatiotemporal distribution in lung epithelial cells. Organ culture analyses have shown that Sema3C and Sema3F positively regulate branching and promote epithelial proliferation, whereas Sema3A leads to a reduced number of terminal pulmonary buds.60 Altogether these data suggest that a repertoire of Semaphorins may orchestrate lung epithelial morphogenesis, a process that might culminate in the regulation of the epithelial barrier architecture.
As Sema3B is concerned, this gene was characterized to be inactivated in lung cancer61,62 and metastatic variants63; while its expression in lung and breast cancers induces growth inhibition and apoptosis activation.64 In a more recent study, the Grainyhead-like (Grhls) transcription factors were proposed to function upstream of Semaphorins in lung development.65 These factors regulate cell-cell adhesion molecule expression, for instance, Grhl3 controls claudin1 and occludin transcription, while Grhl2 positively affects E-cadherin and claudin4 levels. Grhl2 was also described to modulate Sema3B and Sema3C expression, both directly and indirectly, plus that of their corresponding receptor NRP-2. Indeed, Grhl2 downregulation correlated with Sema3B, Sema3C and NRP-2 decrease that in turn could impact gene expression of apical junction proteins. Although the signaling mechanisms involved have not been fully characterized, Sema3B and Sema3C might emerge as important positive mediators of lung epithelial barrier integrity.
As mentioned above, the lung epithelium is also modulated by Sema3F, which is expressed in lung epithelial cells together with NRP-1 and -2. Interestingly, Sema3F and Sema3B map into 3p21.3, a region frequently downregulated in small cell lung cancer66-68 and breast carcinoma.69 In their study, Brambilla E. and colleagues70 describe Sema3F distribution in normal lung and several lung epithelial cancer cell lines. In normal epithelial cells, Sema3F localizes predominantly at the apical face. In the case of low-grade tumor cells, with little to no locomotion, Sema3F accumulated at the interface of adjacent cells, in a region evocative of AJs. In contrast, in high-grade tumor cells, Sema3F expression was considerably reduced and remained mainly in the cytoplasm and in motile regions, such as lamellipodia and protrusions. Nasarre P. and colleagues71 have further focused on the functional role of Sema3F during the course of breast cancer progression. Using poorly to highly motile cancer cells, a different expression pattern of Sema3F and its receptors was first highlighted. Low levels of Sema3F, NRP-1 and NRP-2 were found in motile cells, whereas less motile cells release more Sema3F and NRP-1 was the unique receptor being expressed. Interestingly Sema3F is thought to have a repulsive activity on highly motile cells, operating in an NRP-2-dependent manner. In less motile cancer cells, addition of Sema3F induced an NRP-1-dependent delocalization of membrane E-cadherin and β-catenin. No impact on N-cadherin expression was however detected. It has thus been hypothesized that Sema3F could dampen tumor progression. Although not completely elucidated, the mechanism underlying the switch from high to low Sema3F expression in cancer cells, and its associated redistribution, might involve VEGF, a potential Sema3F binding competitor for Neuropilins. Indeed, loss of Sema3F membrane expression correlated with high levels of VEGF.72 In this scenario, Sema3F reduction and cytoplasmic sequestration could facilitate VEGF receptor activation and associated migratory responses.
Similarly to its putative role in the lung epithelium barrier morphogenesis, Sema3A could affect as well differentiation processes in the corneal epithelium. Sema3A, together with its receptors PlexinA1 and NRP-1 are expressed in the cornea.73 The works by Nishida group74,75 demonstrate that Sema3A released from surrounding corneal fibroblasts triggers an upregulation in the membrane expression of E- and N-cadherins in corneal epithelial cells. In a wound-healing model, NRP-1 and Sema3A levels were increased, in association with epithelial thickening. However, Sema3A was not colocalized within junctional proteins such as ZO-1, occludins, E-cadherin and β-catenin.74
Beside their impact on adhesion molecules, Semaphorins also regulate actin at motile structures, comparable to neuron growth cones.76 Altogether these observations suggest that Semaphorins might emerged as modulators of cell-cell adhesion and migration during development and maturation of specialized epithelia, while their aberrant expression and/or function is involved in the course of cancer progression.
Anti-barrier Semaphorins
In opposition to the roles of class III Semaphorins in epithelial junction reinforcement are other Semaphorins that participate in epithelial junction dissociation (Fig. 2).12
Normal and tumor epithelial liver cells express PlexinB1, which mediates cell scattering upon Sema4D stimulation.77 This mechanism triggers cell-cell dissociation and subsequent cell migration, in a manner similar to the effect of Scatter Factor (SF-1), the ligand for the receptor encoded by the Met proto-oncogene.78 Indeed, while Sema4D acts primarily through PlexinB1, the signal transduction leading to cell dissociation and invasive growth rather implied the tyrosine kinase activity of Met. Moreover, this mechanism was not restricted to the liver but was also unveiled in pancreatic, lung and mammary epithelial tumor cell lines.28 This original signaling complex, which is formed between tyrosine kinase receptors and Plexins is modulated by Semaphorins, and impacts on epithelial cell junctions and therefore aggressiveness of tumor cells.
In a comparable manner, Sema7A was described to be instrumental for EMT in mammary epithelial cells.79 Indeed, Sema7A was suggested to be required for TGF-β-induced EMT through E-cadherin downregulation. Furthermore, the Sema7A promoter contains tandem Ets-binding sites, which can be repressed by Ets-2-repressor factor (ERF), suggesting a point of convergence with TGF-β signaling, which is also inhibited by ERF. Additionally, Sema7A might play a role in cell motility through β1-integrin interaction80 and PlexinC1 signaling.81 Thus, Sema7A appears to be an important modifier of epithelial cell adhesion.
Finally, the role of Sema3E, also known as Semaphorin H, was recently highlighted in epithelial barrier integrity. This signaling molecule, together with its receptor PlexinD1, are dramatically upregulated in high-grade tumor cells, when compared with normal and low-grade tumors,82,83 especially in ovarian84 and colon epithelial cancer cells.85 It has been suggested that Sema3E expression drives cancer and metastasis progression through several crucial steps such as EMT, invasion into the extracellular matrix, extravasation and metastatic colonies formation. For instance, Sema3E and PlexinD1 signaling have been demonstrated to drive Snail1 nuclear relocalization, in correlation with E-cadherin downregulation and vimentin upregulation, crucial events in cancer EMT.84 In addition, Sema3E can activate the ErbB2 tyrosine kinase receptor via its phosphorylation, which in turn triggers MAPK, PLC-γ and PI3K pathways that collectively induce massive cytoskeletal rearrangement and increased cell motility.85 Notably, Sema3E-expressing tumor cells also provoke extracellular matrix degradation, in areas that colocalize with actin-rich structures characterized as invadopodia.85 Likewise, such cells possess filopodia and lamellipodia, a round-shaped morphology and increased migration capacity. Overall, these data converge on the idea that Sema3E can affect epithelial barrier integrity by disrupting among others, E-cadherin.
To conclude, Semaphorin expression is modulated during physiological and pathological epithelial morphogenesis. Although molecular mechanisms are still under investigation, their signaling activity impacts on cell-cell junction organization. These molecules could thus represent promising candidates for novel therapeutic targets in cancers.86
Effects of Semaphorins on Endothelial Junctions
The vascular endothelium forms an interface between circulating blood and irrigated tissues. Endothelial cells are paramount to vascular biology and control the distribution of molecules and cells, as well as waste release throughout the body. The intercellular endothelial junctions primarily contain TJs and AJs, and restrict bidirectional passage between the blood compartment and underlying tissues. Unlike epithelial junctions (see paragraphs 2.1 and 2.2), endothelial junctions exhibit a more flexible organization, where TJs and AJs are found intertwined along the cell-cell contact zone (Fig. 3).
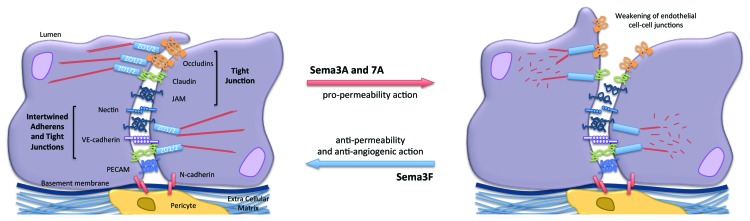
Figure 3. Impact of Semaphorins on the organization of endothelial cell-cell junctions. Endothelial cell-cell junctions are organized around intertwined adherens (nectin and VE-cadherin) and tight junctions (JAM, occludin and claudins). Other non-junctional adhesive proteins, such as PECAM, an endothelial marker, and N-cadherin, which anchors pericytes, might collectively contribute to the endothelial barrier properties. Semaphorins exert either pro- or anti-barrier actions on the endothelial junctions, which in turn alter vascular properties including permeability and angiogenesis.
Endothelial tight junctions
Collectively TJs formed approximately 20% of the total cellular junctions in endothelial cells,87 and allow strong electrical resistance and low permeability.88 Endothelial TJs are organized around adhesion proteins from the same families than those found in epithelial cells, although the isoforms expressed and the associated partners diverge. For instance, the claudin family comprises more than 20 members, however only a few are found in endothelial cells; such as only claudin-3, -5 and -12 that are expressed in the brain endothelium.89 Of note, claudin5 knockout mice have normal TJs with an increased permeability in brain vasculature territories.90
As in epithelial junctions, other transmembrane proteins compose TJs, namely occludin and JAMs. Occludin is particularly enriched in the brain microvasculature,44 in corroboration with stronger tissue barrier function. However, loss of occludin alone in mice is not sufficient in mouse model to impact on vascular permeability.91 Endothelial JAMs, namely JAM-A, -B, -C, ESAM (endothelial cell specific adhesion molecule), CD146 (melanoma cell adhesion molecule MCAM or cell surface glycoprotein MUC18) and PECAM (platelet endothelial cell adhesion molecule or CD31) are also recruited to cell-cell contacts. Their level of expression, localization and interaction with extracellular ligands might vary in response to permeability factors, involved in either angiogenesis and/or immune responses. For instance, histamine and VEGF promote JAM-C relocalization into cell-cell contacts, where it could in turn modulate the stability of the vascular cell-to-cell adhesion molecule, Vascular Endothelial (VE)-cadherin.92
Endothelial adherens junctions
AJs contribute to the initiation and stabilization of cell-cell adhesion, as well as regulation of the actin cytoskeleton and intracellular signaling. They are mainly composed by VE-cadherin, which belongs to the calcium-dependent adhesion molecule family (see paragraph 2.1; Fig. 3). Similarly to classical cadherins, VE-cadherin binds to p120-catenin and β-catenin, which bridges the cadherin-catenin complex to the actin cytoskeleton via α-catenin. VE-cadherin is ubiquitously and specifically expressed in the vascular endothelium. Importantly, VE-cadherin deletion in mice induces embryonic death because of vascular system defects; in other words, VE-cadherin is essential for the formation of stable, mature vessels.93 Moreover, VE-cadherin blocking antibodies provoke an increase of microvascular permeability,94 while its stability at the junctions oppose VEGF-mediated permeability.95 In addition, VE-cadherin modulates signaling pathways that affect the overall TJ architecture and organization.96,97 Hence VE-cadherin is the target of many molecular pathways involved in vascular leakage.98
Vascular permeability is finely tuned in quiescent vessels and adapts to many challenges in the perivascular microenvironment including angiogenesis, shear stress, flow and inflammation.99 The intercellular junctions, and especially VE-cadherin, actively contribute to these modifications in the vascular barrier. Indeed, VE-cadherin integrity can be altered by multiple means, including phosphorylation, destabilization of catenin interaction, internalization, shedding and acto-myosin contractility, all of which ultimately converge on increased endothelial permeability.95,100
Semaphorins and the endothelial barrier
Semaphorin family members have emerged as important molecular modifiers of both neural fate and endothelial biology, largely by operating a fine tuned combination of attractive and repulsive signals.101 Indeed, cancer cells release pro-angiogenic Semaphorins, such as Sema4D, that are able to promote endothelial migration and vascular invasion within the tumor mass.28,102-104 By contrast, class III Semaphorin expression is generally reduced in cancers; they normally function as general inhibitors of angiogenesis.72,105-109
Semaphorin 3A
Beside its axon guidance properties, Sema3A controls vessel formation by inhibiting integrin function. It is also suspected to act as an anti-angiogenic factor.110 However, while most anti-angiogenic proteins oppose permeability, Sema3A, paradoxically, increases vascular permeability (Fig. 4). The signaling mechanisms mediating this effect involve the destabilization of VE-cadherin through tyrosine and serine phosphorylations.111,112 From a molecular standpoint, Sema3A permeability action depends on NRP-1 and PlexinA1 receptors, independently of VEGF. However, divergent downstream molecular pathways have been reported depending on the endothelial context, i.e. macro- vs. micro-vascular cells.111,112 In a first study, Sema3A was found to induce permeability via the PI3Kγ-δ/Akt pathway, independently of Src via NRP-1.111 Another study from our lab established that tumor-released Sema3A provoked serine 665 phosphorylation of VE-cadherin and endothelial junction disorganization, thereby leading to increased endothelial permeability.112 In microvascular brain endothelial cells, Sema3A operates via Src phosphorylation and activation, recruitment of Set, a potent inhibitor of the serine/threonine Protein Phosphatase 2A (PP2A), and inhibition of PP2A phosphatase activity.112 The Sema3A pathway is then subsequently involved in the dissociation of a constitutive PP2A/VE-cadherin interaction that might operate to secure VE-cadherin junctions in quiescent endothelial cells.112 To conclude, Sema3A exerts pro-permeability action through targeting VE-cadherin at the endothelial junctions as a means to modulate local disruption of the endothelial barrier.
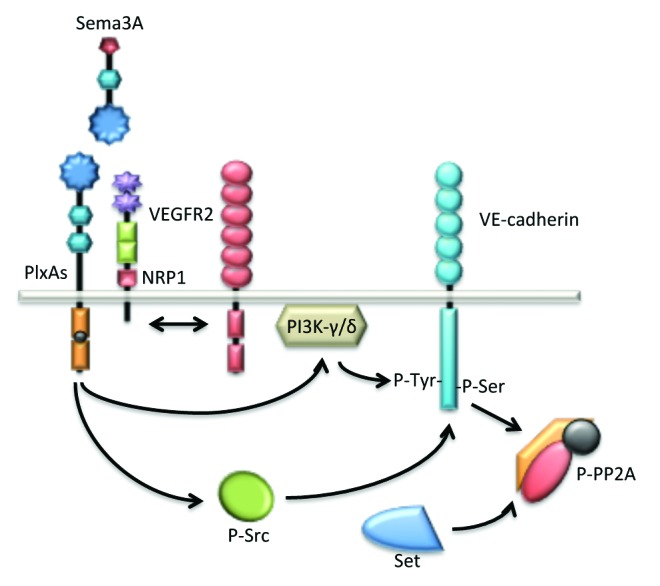
Figure 4. Sema3A enhances endothelial permeability through VE-cadherin destabilization. Sema3A directly augments endothelial permeability by the mean of PlxA1 and NRP1 receptors. Further downstream signaling relies on the activation of the kinase activities bear by PI3K-γ/δ and Src. These pathways culminate at the phosphorylation and destabilization of VE-cadherin. Interestingly, in quiescent endothelial cells, VE-cadherin is associated with the phosphatase PP2A, a mechanism possibly involved in locking the adherens junctions. In this model, Src-directed PP2A phosphorylation and inactivation contribute in turn to VE-cadherin destabilization upon Sema3A challenge.
Semaphorin 3F
Sema3F is a well-characterized repulsive molecule in endothelial cells,72,113,114 and could be a potential candidate for anti-angiogenic therapies. However, it has been recently proposed that hypoxia opposes Sema3F anti-angiogenic action and signaling activity in endothelial cells by downregulating the levels of NRP-2 115. This ultimately favors VEGF-paracrine function and therefore angiogenesis. Nonetheless, in poorly vascularized tumors, such as schwannomas, Sema3F overexpression could enhance pericyte coverage and decrease vascular permeability.115,116 Therefore, these findings suggest that depending on the tumor context, Sema3F could act as a vascular normalization agent.
Semaphorin 7A
Sema7A is reported to take part in immune responses and inflammation.117,118 Interestingly, Sema7A increases transmigration of polymorphonuclear neutrophils across endothelial layers.119 This effect relies on the endothelial expression of Sema7A, rather than a direct action on leukocytes. Moreover, Evan’s blue experiments demonstrated that hypoxia-induced vascular leakage was attenuated in Sema7A knockout mice.119 Likewise, mice lacking Sema7A exhibited decreased blood-brain barrier permeability upon West Nile virus infection and were thus protected.120 These results were also recapitulated in human cell models for West Nile virus infection using a Sema7A blocking antibody, thus pointing toward a role of Sema7A at the crossroads between endothelial biology and immunology.
Class III Semaphorins in particular were recently highlighted for their anti-angiogenic properties, as well as their remarkable ability to impact on endothelial barrier properties (Fig. 3). Although the signaling mechanisms are not fully elucidated, this interplay between Semaphorins and intercellular junctions opens a new field of investigation in endothelial biology.
Epithelial and endothelial junctions function as keystones in the organization and integrity of tissue barriers. They accomplish this by modulating the biology of the epithelia and endothelia through a finely tuned combination of multiple inside-out and outside-in signals. In pathological conditions, such as cancer progression, misexpression of junctional proteins and/or their dysfunction are frequently observed. In this context, Semaphorins could guide the dismantlement of cell-cell contacts, or, at the opposite end, reinforce the intercellular junctions. Although research efforts are still required to better understand the combinatory actions of Semaphorins, Plexins and Neuropilins and their associated molecular signaling in epithelial and endothelial barriers, targeting them will offer new opportunities for future cancer therapies.
Acknowledgments
The authors thank members of JG laboratory, in particular Dr Julie Dwyer, for comments on the manuscript, and Fondation ARC pour la Recherche sur le Cancer, Fondation pour la Recherche Medicale, Ligue Nationale contre le Cancer Comite de Paris, Centre National pour la Recherche Scientifique, Marie Curie Actions within the 7th framework program for research and Universite Paris Descartes for financial support.
Disclosure of Potential Conflicts of Interest
The authors have declared that no competing interests exist.
Footnotes
Previously published online: www.landesbioscience.com/journals/tissuebarriers/article/23272
References
- 1.Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993;75:217–27. doi: 10.1016/0092-8674(93)80064-L. [DOI] [PubMed] [Google Scholar]
- 2.Semaphorin Nomenclature Committee Unified nomenclature for the semaphorins/collapsins. Cell. 1999;97:551–2. doi: 10.1016/S0092-8674(00)80766-7. [DOI] [PubMed] [Google Scholar]
- 3.Janssen BJ, Robinson RA, Pérez-Brangulí F, Bell CH, Mitchell KJ, Siebold C, et al. Structural basis of semaphorin-plexin signalling. Nature. 2010;467:1118–22. doi: 10.1038/nature09468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capparuccia L, Tamagnone L. Semaphorin signaling in cancer cells and in cells of the tumor microenvironment--two sides of a coin. J Cell Sci. 2009;122:1723–36. doi: 10.1242/jcs.030197. [DOI] [PubMed] [Google Scholar]
- 5.Nogi T, Yasui N, Mihara E, Matsunaga Y, Noda M, Yamashita N, et al. Structural basis for semaphorin signalling through the plexin receptor. Nature. 2010;467:1123–7. doi: 10.1038/nature09473. [DOI] [PubMed] [Google Scholar]
- 6.Hu H, Marton TF, Goodman CS. Plexin B mediates axon guidance in Drosophila by simultaneously inhibiting active Rac and enhancing RhoA signaling. Neuron. 2001;32:39–51. doi: 10.1016/S0896-6273(01)00453-6. [DOI] [PubMed] [Google Scholar]
- 7.Oinuma I, Ishikawa Y, Katoh H, Negishi M. The Semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science. 2004;305:862–5. doi: 10.1126/science.1097545. [DOI] [PubMed] [Google Scholar]
- 8.He H, Yang T, Terman JR, Zhang X. Crystal structure of the plexin A3 intracellular region reveals an autoinhibited conformation through active site sequestration. Proc Natl Acad Sci U S A. 2009;106:15610–5. doi: 10.1073/pnas.0906923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell CH, Aricescu AR, Jones EY, Siebold C. A dual binding mode for RhoGTPases in plexin signalling. PLoS Biol. 2011;9:e1001134. doi: 10.1371/journal.pbio.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, He H, Srivastava N, Vikarunnessa S, Chen YB, Jiang J, et al. Plexins are GTPase-activating proteins for Rap and are activated by induced dimerization. Sci Signal. 2012;5:ra6. doi: 10.1126/scisignal.2002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell. 1997;90:739–51. doi: 10.1016/S0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- 12.Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/S0092-8674(00)80063-X. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, et al. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell. 1999;99:59–69. doi: 10.1016/S0092-8674(00)80062-8. [DOI] [PubMed] [Google Scholar]
- 14.Janssen BJ, Malinauskas T, Weir GA, Cader MZ, Siebold C, Jones EY. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat Struct Mol Biol. 2012;19:1293–9. doi: 10.1038/nsmb.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, et al. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science. 2005;307:265–8. doi: 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Sima J, Jin M, Wang KY, Xue XJ, Zheng W, et al. Semaphorin-3A guides radial migration of cortical neurons during development. Nat Neurosci. 2008;11:36–44. doi: 10.1038/nn2018. [DOI] [PubMed] [Google Scholar]
- 17.Renaud J, Kerjan G, Sumita I, Zagar Y, Georget V, Kim D, et al. Plexin-A2 and its ligand, Sema6A, control nucleus-centrosome coupling in migrating granule cells. Nat Neurosci. 2008;11:440–9. doi: 10.1038/nn2064. [DOI] [PubMed] [Google Scholar]
- 18.Bagri A, Cheng HJ, Yaron A, Pleasure SJ, Tessier-Lavigne M. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell. 2003;113:285–99. doi: 10.1016/S0092-8674(03)00267-8. [DOI] [PubMed] [Google Scholar]
- 19.Leslie JR, Imai F, Fukuhara K, Takegahara N, Rizvi TA, Friedel RH, et al. Ectopic myelinating oligodendrocytes in the dorsal spinal cord as a consequence of altered semaphorin 6D signaling inhibit synapse formation. Development. 2011;138:4085–95. doi: 10.1242/dev.066076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahay A, Kim CH, Sepkuty JP, Cho E, Huganir RL, Ginty DD, et al. Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci. 2005;25:3613–20. doi: 10.1523/JNEUROSCI.5255-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shelly M, Cancedda L, Lim BK, Popescu AT, Cheng PL, Gao H, et al. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron. 2011;71:433–46. doi: 10.1016/j.neuron.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terman JR, Kolodkin AL. Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science. 2004;303:1204–7. doi: 10.1126/science.1092121. [DOI] [PubMed] [Google Scholar]
- 23.Wolman MA, Liu Y, Tawarayama H, Shoji W, Halloran MC. Repulsion and attraction of axons by semaphorin3D are mediated by different neuropilins in vivo. J Neurosci. 2004;24:8428–35. doi: 10.1523/JNEUROSCI.2349-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oinuma I, Katoh H, Negishi M. Molecular dissection of the semaphorin 4D receptor plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. 2004;The Journal of neuroscience24:11473–80. doi: 10.1523/JNEUROSCI.3257-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keely PJ, Rusyn EV, Cox AD, Parise LV. R-Ras signals through specific integrin alpha cytoplasmic domains to promote migration and invasion of breast epithelial cells. J Cell Biol. 1999;145:1077–88. doi: 10.1083/jcb.145.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mann F, Chauvet S, Rougon G. Semaphorins in development and adult brain: Implication for neurological diseases. Prog Neurobiol. 2007;82:57–79. doi: 10.1016/j.pneurobio.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, et al. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med. 2006;12:1380–9. doi: 10.1038/nm1505. [DOI] [PubMed] [Google Scholar]
- 28.Serini G, Maione F, Giraudo E, Bussolino F. Semaphorins and tumor angiogenesis. Angiogenesis. 2009;12:187–93. doi: 10.1007/s10456-009-9138-4. [DOI] [PubMed] [Google Scholar]
- 29.Ivanov DB, Philippova MP, Tkachuk VA. Structure and functions of classical cadherins. Biochemistry (Mosc) 2001;66:1174–86. doi: 10.1023/A:1012445316415. [DOI] [PubMed] [Google Scholar]
- 30.Harrison OJ, Corps EM, Berge T, Kilshaw PJ. The mechanism of cell adhesion by classical cadherins: the role of domain 1. J Cell Sci. 2005;118:711–21. doi: 10.1242/jcs.01665. [DOI] [PubMed] [Google Scholar]
- 31.Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grübel G, et al. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–37. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Sivasankar S, Nelson WJ, Chu S. Resolving cadherin interactions and binding cooperativity at the single-molecule level. Proc Natl Acad Sci U S A. 2009;106:109–14. doi: 10.1073/pnas.0811350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimoyama Y, Tsujimoto G, Kitajima M, Natori M. Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. Biochem J. 2000;349:159–67. doi: 10.1042/0264-6021:3490159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bershadsky A. Magic touch: how does cell-cell adhesion trigger actin assembly? Trends Cell Biol. 2004;14:589–93. doi: 10.1016/j.tcb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol. 2010;12:533–42. doi: 10.1038/ncb2055. [DOI] [PubMed] [Google Scholar]
- 36.Anastasiadis PZ, Reynolds AB. The p120 catenin family: complex roles in adhesion, signaling and cancer. J Cell Sci. 2000;113:1319–34. doi: 10.1242/jcs.113.8.1319. [DOI] [PubMed] [Google Scholar]
- 37.Yap AS, Crampton MS, Hardin J. Making and breaking contacts: the cellular biology of cadherin regulation. Curr Opin Cell Biol. 2007;19:508–14. doi: 10.1016/j.ceb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J. Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism. J Cell Sci. 2008;121:358–68. doi: 10.1242/jcs.017145. [DOI] [PubMed] [Google Scholar]
- 39.Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, et al. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95:892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 40.Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. 1963;17:375–412. doi: 10.1083/jcb.17.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furuse M, Moriwaki K. The role of claudin-based tight junctions in morphogenesis. Ann N Y Acad Sci. 2009;1165:58–61. doi: 10.1111/j.1749-6632.2009.04441.x. [DOI] [PubMed] [Google Scholar]
- 42.Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sánchez-Pulido L, Martín-Belmonte F, Valencia A, Alonso MA. MARVEL: a conserved domain involved in membrane apposition events. Trends Biochem Sci. 2002;27:599–601. doi: 10.1016/S0968-0004(02)02229-6. [DOI] [PubMed] [Google Scholar]
- 44.Hirase T, Staddon JM, Saitou M, Ando-Akatsuka Y, Itoh M, Furuse M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110:1603–13. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- 45.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–42. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beeman N, Webb PG, Baumgartner HK. Occludin is required for apoptosis when claudin-claudin interactions are disrupted. Cell Death Dis. 2012;3:e273. doi: 10.1038/cddis.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–27. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ebnet K, Suzuki A, Ohno S, Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci. 2004;117:19–29. doi: 10.1242/jcs.00930. [DOI] [PubMed] [Google Scholar]
- 49.Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol. 2001;154:491–7. doi: 10.1083/jcb.200103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103:755–66. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gumbiner BM. Membrane glycoprotein choreography. Curr Biol. 1991;1:271–3. doi: 10.1016/0960-9822(91)90081-7. [DOI] [PubMed] [Google Scholar]
- 52.Balda MS, Anderson JM. Two classes of tight junctions are revealed by ZO-1 isoforms. Am J Physiol. 1993;264:C918–24. doi: 10.1152/ajpcell.1993.264.4.C918. [DOI] [PubMed] [Google Scholar]
- 53.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–28. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 54.Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126:741–54. doi: 10.1016/j.cell.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 55.Matter K, Balda MS. Epithelial tight junctions, gene expression and nucleo-junctional interplay. J Cell Sci. 2007;120:1505–11. doi: 10.1242/jcs.005975. [DOI] [PubMed] [Google Scholar]
- 56.Ohno S. Intercellular junctions and cellular polarity: the PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr Opin Cell Biol. 2001;13:641–8. doi: 10.1016/S0955-0674(00)00264-7. [DOI] [PubMed] [Google Scholar]
- 57.Warburton D, El-Hashash A, Carraro G, Tiozzo C, Sala F, Rogers O, et al. Lung organogenesis. Curr Top Dev Biol. 2010;90:73–158. doi: 10.1016/S0070-2153(10)90003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cardoso WV, Lü J. Regulation of early lung morphogenesis: questions, facts and controversies. Development. 2006;133:1611–24. doi: 10.1242/dev.02310. [DOI] [PubMed] [Google Scholar]
- 59.Morrisey EE, Hogan BL. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell. 2010;18:8–23. doi: 10.1016/j.devcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kagoshima M, Ito T. Diverse gene expression and function of semaphorins in developing lung: positive and negative regulatory roles of semaphorins in lung branching morphogenesis. Genes Cells. 2001;6:559–71. doi: 10.1046/j.1365-2443.2001.00441.x. [DOI] [PubMed] [Google Scholar]
- 61.Kuroki T, Trapasso F, Yendamuri S, Matsuyama A, Alder H, Williams NN, et al. Allelic loss on chromosome 3p21.3 and promoter hypermethylation of semaphorin 3B in non-small cell lung cancer. Cancer Res. 2003;63:3352–5. [PubMed] [Google Scholar]
- 62.Ito M, Ito G, Kondo M, Uchiyama M, Fukui T, Mori S, et al. Frequent inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by promoter hypermethylation and allele loss in non-small cell lung cancer. Cancer Lett. 2005;225:131–9. doi: 10.1016/j.canlet.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 63.de Lange R, Dimoudis N, Weidle UH. Identification of genes associated with enhanced metastasis of a large cell lung carcinoma cell line. Anticancer Res. 2003;23(1A):187–94. [PubMed] [Google Scholar]
- 64.Castro-Rivera E, Ran S, Thorpe P, Minna JD. Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast cancer, whereas VEGF165 antagonizes this effect. Proc Natl Acad Sci U S A. 2004;101:11432–7. doi: 10.1073/pnas.0403969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varma S, Cao Y, Tagne JB, Lakshminarayanan M, Li J, Friedman TB, et al. The Transcription Factors Grainyhead-like 2 and NK2-Homeobox 1 Form a Regulatory Loop That Coordinates Lung Epithelial Cell Morphogenesis and Differentiation. J Biol Chem. 2012;287:37282–95. doi: 10.1074/jbc.M112.408401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roche J, Boldog F, Robinson M, Robinson L, Varella-Garcia M, Swanton M, et al. Distinct 3p21.3 deletions in lung cancer and identification of a new human semaphorin. Oncogene. 1996;12:1289–97. [PubMed] [Google Scholar]
- 67.Xiang RH, Hensel CH, Garcia DK, Carlson HC, Kok K, Daly MC, et al. Isolation of the human semaphorin III/F gene (SEMA3F) at chromosome 3p21, a region deleted in lung cancer. Genomics. 1996;32:39–48. doi: 10.1006/geno.1996.0074. [DOI] [PubMed] [Google Scholar]
- 68.Sekido Y, Bader S, Latif F, Chen JY, Duh FM, Wei MH, et al. Human semaphorins A(V) and IV reside in the 3p21.3 small cell lung cancer deletion region and demonstrate distinct expression patterns. Proc Natl Acad Sci U S A. 1996;93:4120–5. doi: 10.1073/pnas.93.9.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Senchenko VN, Liu J, Loginov W, Bazov I, Angeloni D, Seryogin Y, et al. Discovery of frequent homozygous deletions in chromosome 3p21.3 LUCA and AP20 regions in renal, lung and breast carcinomas. Oncogene. 2004;23:5719–28. doi: 10.1038/sj.onc.1207760. [DOI] [PubMed] [Google Scholar]
- 70.Brambilla E, Constantin B, Drabkin H, Roche J. Semaphorin SEMA3F localization in malignant human lung and cell lines: A suggested role in cell adhesion and cell migration. Am J Pathol. 2000;156:939–50. doi: 10.1016/S0002-9440(10)64962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nasarre P, Kusy S, Constantin B, Castellani V, Drabkin HA, Bagnard D, et al. Semaphorin SEMA3F has a repulsing activity on breast cancer cells and inhibits E-cadherin-mediated cell adhesion. Neoplasia. 2005;7:180–9. doi: 10.1593/neo.04481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kessler O, Shraga-Heled N, Lange T, Gutmann-Raviv N, Sabo E, Baruch L, et al. Semaphorin-3F is an inhibitor of tumor angiogenesis. Cancer Res. 2004;64:1008–15. doi: 10.1158/0008-5472.CAN-03-3090. [DOI] [PubMed] [Google Scholar]
- 73.Morishige N, Ko JA, Liu Y, Chikama T, Nishida T. Localization of semaphorin 3A in the rat cornea. Exp Eye Res. 2008;86:669–74. doi: 10.1016/j.exer.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 74.Morishige N, Ko JA, Morita Y, Nishida T. Expression of semaphorin 3A in the rat corneal epithelium during wound healing. Biochem Biophys Res Commun. 2010;395:451–7. doi: 10.1016/j.bbrc.2010.03.124. [DOI] [PubMed] [Google Scholar]
- 75.Ko JA, Akamatsu Y, Yanai R, Nishida T. Effects of semaphorin 3A overexpression in corneal fibroblasts on the expression of adherens-junction proteins in corneal epithelial cells. Biochem Biophys Res Commun. 2010;396:781–6. doi: 10.1016/j.bbrc.2010.04.029. [DOI] [PubMed] [Google Scholar]
- 76.Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–78. doi: 10.1083/jcb.121.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D, et al. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat Cell Biol. 2002;4:720–4. doi: 10.1038/ncb843. [DOI] [PubMed] [Google Scholar]
- 78.Giordano S, Ponzetto C, Di Renzo MF, Cooper CS, Comoglio PM. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature. 1989;339:155–6. doi: 10.1038/339155a0. [DOI] [PubMed] [Google Scholar]
- 79.Allegra M, Zaragkoulias A, Vorgia E, Ioannou M, Litos G, Beug H, et al. Semaphorin-7a reverses the ERF-induced inhibition of EMT in Ras-dependent mouse mammary epithelial cells. Mol Biol Cell. 2012;23:3873–81. doi: 10.1091/mbc.E12-04-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Messina A, Ferraris N, Wray S, Cagnoni G, Donohue DE, Casoni F, et al. Dysregulation of Semaphorin7A/β1-integrin signaling leads to defective GnRH-1 cell migration, abnormal gonadal development and altered fertility. Hum Mol Genet. 2011;20:4759–74. doi: 10.1093/hmg/ddr403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lazova R, Gould Rothberg BE, Rimm D, Scott G. The semaphorin 7A receptor Plexin C1 is lost during melanoma metastasis. Am J Dermatopathol. 2009;31:177–81. doi: 10.1097/DAD.0b013e318196672d. [DOI] [PubMed] [Google Scholar]
- 82.Roodink I, Raats J, van der Zwaag B, Verrijp K, Kusters B, van Bokhoven H, et al. Plexin D1 expression is induced on tumor vasculature and tumor cells: a novel target for diagnosis and therapy? Cancer Res. 2005;65:8317–23. doi: 10.1158/0008-5472.CAN-04-4366. [DOI] [PubMed] [Google Scholar]
- 83.Roodink I, Verrijp K, Raats J, Leenders WP. Plexin D1 is ubiquitously expressed on tumor vessels and tumor cells in solid malignancies. BMC Cancer. 2009;9:297. doi: 10.1186/1471-2407-9-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tseng CH, Murray KD, Jou MF, Hsu SM, Cheng HJ, Huang PH. Sema3E/plexin-D1 mediated epithelial-to-mesenchymal transition in ovarian endometrioid cancer. PLoS One. 2011;6:e19396. doi: 10.1371/journal.pone.0019396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Casazza A, Finisguerra V, Capparuccia L, Camperi A, Swiercz JM, Rizzolio S, et al. Sema3E-Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J Clin Invest. 2010;120:2684–98. doi: 10.1172/JCI42118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rehman M, Tamagnone L. Semaphorins in cancer: Biological mechanisms and therapeutic approaches. Semin Cell Dev Biol. 2012 doi: 10.1016/j.semcdb.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 87.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–99. doi: 10.1016/S1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 88.Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–25. doi: 10.1016/S0166-2236(00)02004-X. [DOI] [PubMed] [Google Scholar]
- 89.Luissint AC, Federici C, Guillonneau F, Chrétien F, Camoin L, Glacial F, et al. Guanine nucleotide-binding protein Gαi2: a new partner of claudin-5 that regulates tight junction integrity in human brain endothelial cells. J Cereb Blood Flow Metab. 2012;32:860–73. doi: 10.1038/jcbfm.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–60. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M, et al. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol. 1998;141:397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Orlova VV, Economopoulou M, Lupu F, Santoso S, Chavakis T. Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J Exp Med. 2006;203:2703–14. doi: 10.1084/jem.20051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–57. doi: 10.1016/S0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 94.May C, Doody JF, Abdullah R, Balderes P, Xu X, Chen CP, et al. Identification of a transiently exposed VE-cadherin epitope that allows for specific targeting of an antibody to the tumor neovasculature. Blood. 2005;105:4337–44. doi: 10.1182/blood-2005-01-0010. [DOI] [PubMed] [Google Scholar]
- 95.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–34. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 96.Walsh TG, Murphy RP, Fitzpatrick P, Rochfort KD, Guinan AF, Murphy A, et al. Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J Cell Physiol. 2011;226:3053–63. doi: 10.1002/jcp.22655. [DOI] [PubMed] [Google Scholar]
- 97.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–34. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- 98.Le Guelte A, Dwyer J, Gavard J. Jumping the barrier: VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biol Cell. 2011;103:593–605. doi: 10.1042/BC20110069. [DOI] [PubMed] [Google Scholar]
- 99.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 100.Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–94. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- 102.Meda C, Molla F, De Pizzol M, Regano D, Maione F, Capano S, et al. Semaphorin 4A exerts a proangiogenic effect by enhancing vascular endothelial growth factor-A expression in macrophages. J Immunol. 2012;188:4081–92. doi: 10.4049/jimmunol.1101435. [DOI] [PubMed] [Google Scholar]
- 103.Basile JR, Gavard J, Gutkind JS. Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem. 2007;282:34888–95. doi: 10.1074/jbc.M705467200. [DOI] [PubMed] [Google Scholar]
- 104.Basile JR, Holmbeck K, Bugge TH, Gutkind JS. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J Biol Chem. 2007;282:6899–905. doi: 10.1074/jbc.M609570200. [DOI] [PubMed] [Google Scholar]
- 105.Sakurai A, Gavard J, Annas-Linhares Y, Basile JR, Amornphimoltham P, Palmby TR, et al. Semaphorin 3E initiates antiangiogenic signaling through plexin D1 by regulating Arf6 and R-Ras. Mol Cell Biol. 2010;30:3086–98. doi: 10.1128/MCB.01652-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chakraborty G, Kumar S, Mishra R, Patil TV, Kundu GC. Semaphorin 3A suppresses tumor growth and metastasis in mice melanoma model. PLoS One. 2012;7:e33633. doi: 10.1371/journal.pone.0033633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maione F, Molla F, Meda C, Latini R, Zentilin L, Giacca M, et al. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J Clin Invest. 2009;119:3356–72. doi: 10.1172/JCI36308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Varshavsky A, Kessler O, Abramovitch S, Kigel B, Zaffryar S, Akiri G, et al. Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res. 2008;68:6922–31. doi: 10.1158/0008-5472.CAN-07-5408. [DOI] [PubMed] [Google Scholar]
- 109.Sabag AD, Bode J, Fink D, Kigel B, Kugler W, Neufeld G. Semaphorin-3D and semaphorin-3E inhibit the development of tumors from glioblastoma cells implanted in the cortex of the brain. PLoS One. 2012;7:e42912. doi: 10.1371/journal.pone.0042912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, et al. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 2003;424:391–7. doi: 10.1038/nature01784. [DOI] [PubMed] [Google Scholar]
- 111.Acevedo LM, Barillas S, Weis SM, Göthert JR, Cheresh DA. Semaphorin 3A suppresses VEGF-mediated angiogenesis yet acts as a vascular permeability factor. Blood. 2008;111:2674–80. doi: 10.1182/blood-2007-08-110205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Le Guelte A, Galan-Moya EM, Dwyer J, Treps L, Kettler G, Hebda JK, et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J Cell Sci. 2012;125:4137–46. doi: 10.1242/jcs.108282. [DOI] [PubMed] [Google Scholar]
- 113.Bielenberg DR, Hida Y, Shimizu A, Kaipainen A, Kreuter M, Kim CC, et al. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest. 2004;114:1260–71. doi: 10.1172/JCI21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guttmann-Raviv N, Shraga-Heled N, Varshavsky A, Guimaraes-Sternberg C, Kessler O, Neufeld G. Semaphorin-3A and semaphorin-3F work together to repel endothelial cells and to inhibit their survival by induction of apoptosis. J Biol Chem. 2007;282:26294–305. doi: 10.1074/jbc.M609711200. [DOI] [PubMed] [Google Scholar]
- 115.Coma S, Shimizu A, Klagsbrun M. Hypoxia induces tumor and endothelial cell migration in a semaphorin 3F- and VEGF-dependent manner via transcriptional repression of their common receptor neuropilin 2. Cell Adh Migr. 2011;5:266–75. doi: 10.4161/cam.5.3.16294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wong HK, Shimizu A, Kirkpatrick ND, Garkavtsev I, Chan AW, di Tomaso E, et al. Merlin/NF2 regulates angiogenesis in schwannomas through a Rac1/semaphorin 3F-dependent mechanism. Neoplasia. 2012;14:84–94. doi: 10.1593/neo.111600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–93. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Suzuki K, Okuno T, Yamamoto M, Pasterkamp RJ, Takegahara N, Takamatsu H, et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature. 2007;446:680–4. doi: 10.1038/nature05652. [DOI] [PubMed] [Google Scholar]
- 119.Morote-Garcia JC, Napiwotzky D, Köhler D, Rosenberger P. Endothelial Semaphorin 7A promotes neutrophil migration during hypoxia. Proc Natl Acad Sci U S A. 2012;109:14146–51. doi: 10.1073/pnas.1202165109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sultana H, Neelakanta G, Foellmer HG, Montgomery RR, Anderson JF, Koski RA, et al. Semaphorin 7A contributes to West Nile virus pathogenesis through TGF-β1/Smad6 signaling. J Immunol. 2012;189:3150–8. doi: 10.4049/jimmunol.1201140. [DOI] [PMC free article] [PubMed] [Google Scholar]