

Abstract
The intestinal epithelium is a dynamic barrier playing an active role in intestinal homeostasis and inflammation. Intestinal barrier function is dysregulated during inflammatory bowel disease (IBD), with epithelial cells playing a significant part in generating an inflammatory milieu through the release of signals that attract leukocytes to the intestinal lamina propria. However, it is increasingly appreciated that the intestinal epithelium mediates a counterbalancing response to drive resolution. Drawing analogies with neuronal development, where the balance of chemoattractive and chemorepellent signals is key to directed neuronal movement it has been postulated that such secreted cues play a role in leukocyte migration. Netrin-1 is one of the best-described neuronal guidance molecules, which has been shown to play a significant role in directed migration of leukocytes. Prior to our study the potential role of netrin-1 in IBD was poorly characterized. We defined netrin-1 as an intestinal epithelial-derived protein capable of limiting neutrophil recruitment to attenuate acute colitis. Our study highlights that the intestinal epithelium releases factors during acute inflammation that are responsible for fine-tuning the immune response. Exploration of these epithelial-mediated protective mechanisms will shed light on the complexity of the intestinal epithelial barrier in health and disease.
Keywords: neuronal guidance molecule, intestinal epithelial cells, leukocyte migration, inflammatory bowel disease, apoptosis
Introduction
An epidemiological study published within the last year demonstrated that IBD, including Crohn disease (CD) and ulcerative colitis (UC) is significantly increasing in both incidence and prevalence worldwide, with North America having one of the highest reported prevalence rates.1 Substantial evidence supports the hypothesis that in a genetically susceptible host an inappropriate immune response to endogenous microbiota or self-antigen promotes a persistent and pathogenic inflammation in the intestinal mucosa leading to IBD.2 Mucosal inflammation as experienced during IBD is characterized by disrupted intestinal epithelial barrier function and inappropriate leukocyte accumulation in the intestinal lamina propria.2 Significant efforts are being made to understand the mechanisms of epithelial barrier and immunological dysfunction during IBD. Initially considered as a mere physical barrier to the intestinal luminal contents, the intestinal epithelial cell layer is now known to be a dynamic force that directs mucosal immune responses.3,4 Much information exists on the role that intestinal epithelial cells play in driving recruitment of leukocytes to the inflamed intestine through release of chemoattractants (chemokines).5 However, there is increasing evidence that intestinal epithelial cells can mediate a counterbalancing response to attenuate inflammation.3,6 Hypoxia driven signaling pathways have been highlighted as one such epithelial-mediated protective response in the intestine.7-12 Original studies hypothesized that soluble anti-inflammatory factors were released by hypoxic epithelial cells. One such factor identified by these studies was the neuronal guidance molecule, netrin-1.13 Netrin-1 is member of a class of secreted guidance molecules initially recognized as being essential for neuronal development.14 Novel studies point to an exciting additional role for netrin-1 in attenuating inflammatory responses in peripheral organs.13,15-18 We hypothesized the hypoxic intestinal epithelium was a source of netrin-1 which could have immunomodulatory effects in a model of IBD. Our investigations revealed that netrin-1 is a key mediator acting to balance the acute inflammatory response during intestinal inflammation as observed in IBD.12
Netrin-1 during Intestinal Homeostasis and Inflammation: An Epithelial Response
It is now evident that netrin-1 is part of an evolutionary conserved integrated network of signaling proteins that act simultaneously as chemoattractant or chemorepellent signals to control neuronal migration and development.19 Considering the parallels between axonal guidance and leukocyte migration an exciting body of work demonstrates that netrin-1 plays a guiding role to modulate functional immune responses.13,15-17,20 During IBD coordinated leukocyte migration is a key feature of disease pathogenesis.5 We therefore sought to investigate if netrin-1 might play a role in IBD.
In vitro and in vivo studies demonstrated that netrin-1 is an endogenous mediator released in the intestine to suppress inflammation during acute colitis.12 Mice with homozygous deficiency of netrin-1 do not live beyond postnatal day 3 due to improper axonal development,14 therefore we conducted our studies using mice heterozygous for netrin-1 expression (Ntn-1+/−).12 Initial findings revealed that in a model of acute colitis Ntn-1+/− mice experienced significantly exacerbated inflammation characteristic of acute disease compared with wildtype controls, including inflammatory cell influx and histological tissue damage.12 Netrin-1 expression was predominantly observed in intestinal epithelial cells and myenteric neural units of the colon.12 Netrin-1 has been observed to play an important role in the developing enteric nervous system, where it is expressed by the endoderm and outer gut mesenchyme in the fetal mouse gut.21,22 In line with our observations differentiated enteric nerves have been shown to express and synthesize netrin-1.23 There is evidence that the enteric nervous system is functionally altered during IBD and neuronal-derived molecules have been demonstrated to exert anti-inflammatory effects in models of IBD.24 Our studies revealed no differences in enteric neuronal anatomy in netrin-1 deficient mice during acute colitis, while netrin-1 expression in the colonic neural units remained consistent throughout the timecourse of colitis.12 To date there is no evidence that the enteric nervous system of netrin-1 deficient mice is functionally impaired. From our observations we conclude that neuronal derived netrin-1 was not the predominant source of netrin-1 during acute colitis. However, the possibility that neuronal derived netrin-1 is an immunomodulatory factor in the intestine remains to be fully examined.
In contrast, the colonic epithelium expresses a high level of netrin-1.12 It has been established that intestinal epithelial cells rapidly respond to environmental cues in order to control microbial invasion, intestinal barrier function and inflammation of the mucosa. This involves secretion of chemokines, cytokines and other factors that are responsible for controlling deleterious local responses.3-5 Our studies point to the release of netrin-1 by the intestinal epithelium as an endogenous protective response in acute colitis.12 In vitro studies revealed that cytokines relevant to the pathogenesis of IBD significantly increased netrin-1 expression in intestinal epithelial cells and epithelial netrin-1 expression was enhanced during the course of acute colitis.12 These findings are in line with observations in a study of IBD patients where netrin-1 expression in mucosal epithelial cells was observed to be dramatically increased above that of control patients.25 This group proceeded to demonstrate that enforced netrin-1 expression in the intestinal epithelium decreased epithelial cell apoptosis and predisposed mice to the development of colonic tumors.26 Increased epithelial cell apoptosis and dysregulated intestinal epithelial barrier function are hallmarks of IBD, as well as key features of IBD models.27,28 Treatments that prevent epithelial cell apoptosis have been demonstrated to protect mice from inflammation in acute colitis.29 In our studies mice treated with recombinant netrin-1 were protected during the course of acute colitis.12 Based on these findings, we considered the hypothesis that netrin-1 control of epithelial cell apoptosis was responsible for alterations in disease outcome in our model. Surprisingly, analysis revealed that despite demonstrating exacerbated inflammatory cell infiltrate and histological disease netrin-1 deficient mice did not have increased epithelial barrier dysfunction or epithelial cell apoptosis compared with wildtype controls during acute colitis.12 Similarly, administration of netrin-1 did not result in improved intestinal epithelial barrier function in vitro or in vivo. Despite evidence from cancer models, our studies led us to conclude that during acute inflammation as experienced in IBD, netrin-1 did not play a significant role in regulating intestinal epithelial cell apoptosis or barrier dysfunction. The exact reasons for this may be due to experimental design, choice of model and the timeline of disease progression. Original studies by Mazelin et al. utilized genetic overexpression of netrin-1 in the intestinal epithelium of mice.26 This approach demonstrated that persistent netrin-1 expression led to significant epithelial cell hyperplasia by 20 months. Crossing netrin-1 transgenic mice with the adenomatous polyposis coli (APC) mice, a common model of colorectal cancer, resulted in an increased frequency of high grade adenomas compared with controls. The authors concluded that chronic elevation of netrin-1 inhibited apoptotic signaling in the intestinal epithelium.26 These studies were followed up in the context of IBD associated colorectal cancer using the azoxymethane (AOM) plus dextran sulfate sodium (DSS) model.25 Using a method to block receptor binding of endogenous netrin-1 in AOM/DSS over a period of 10 weeks it was observed that blockade of netrin-1 signaling decreased the frequency of high grade adenomas.25 Neither study investigated the effect of netrin-1 signaling on epithelial cell apoptosis at acute timepoints. Both studies highlighted the need for chronic netrin-1 signaling to enhance epithelial cell survival and focused on timepoints of 10 or 20 weeks. Our study used a distinct model of direct epithelial injury that is associated with epithelial cell apoptosis and acute inflammation, namely DSS colitis. In addition, our animal studies were of considerably shorter duration to either cancer model used.25,26 These differences in model and timeline may in part account for our differential findings. In addition, the Mehlen group focused on the role of the netrin-1 receptors, deleted in colorectal cancer receptor (DCC) and the uncoordinated receptors (UNC5A-D).25,26 Both receptor groups have been identified to regulate cell survival upon netrin-1 binding.30 As yet the exact role for these two receptor families in IBD is unknown. Our study identified an alternative netrin-1 receptor, the A2B adenosine receptor (Adora2b) as the key receptor for mediating netrin-1 functional responses during DSS colitis (discussed below). Netrin-1 signaling through the Adora2b in our model of acute colitis may be an alternative reason why distinct netrin-1 effects were observed in our study. We cannot discount the possibility that netrin-1 may play a role in intestinal epithelial cell survival in a more chronic model of IBD. Taken together, our observation that netrin-1 did not affect intestinal epithelial cell apoptosis in acute colitis led us to consider other functions attributed to netrin-1 in peripheral organs.
Leukocyte Reverse Migration Mediated by Netrin-1 during Acute Intestinal Inflammation
It is increasingly appreciated that leukocyte migration to sites of injury and inflammation is a complex phenomenon, regulated by a variety of signaling molecules and pathways.31 Chemoattractants responsible for leukocyte recruitment are known to be expressed by intestinal epithelial cells during inflammation that occurs in IBD.5 Much is known about chemokine-mediated leukocyte recruitment to the inflamed intestine, however the phenomenon of leukocyte egress from tissues is less well described.5,32 The existence of negative cues to reverse or prevent leukocyte recruitment are suggested by exciting studies performed in zebrafish, which demonstrate that neutrophils can retreat from sites of inflammation at the same rate that they are recruited to promote resolution of inflammation.33 Due to similarities with the developing nervous system where neuronal guidance molecules supply both attractive and repulsive cues to steer appropriate neuronal patterning it was hypothesized that these molecules may have a role to play in regulating leukocyte recruitment during an active immune response. Initial investigations with this in mind demonstrated that netrin-1 was one such molecule that could inhibit leukocyte migration during acute inflammation.15 Further studies in models of peripheral inflammation demonstrated that netrin-1 could inhibit neutrophil and macrophage migration to significantly reduce tissue inflammation and damage.13,16,17
During UC formation of neutrophil-filled crypt abscesses is a hallmark of disease.34,35 Neutrophil infiltration into the colonic lamina propria is also a key feature of the murine colitis model used in our studies.36 The definitive role of neutrophils during acute colitis is still debated. As discussed in a recent comprehensive review by Fournier and Parkos, neutrophils represent something of a “double-edged sword” in the context of intestinal inflammation.37 During acute intestinal injury neutrophils play a vital protective role and can assist in the resolution of inflammation. However, inappropriate persistence of activated neutrophils at the site of inflammation can be detrimental during intestinal injury and IBD. (reviewed in37) Loss of a neutrophilic response was observed to result in a worse outcome of experimental colitis.38,39 In contrast, suppressing neutrophil infiltration has been demonstrated to be an effective therapeutic intervention in a number of IBD models.12,40,41 These dichotomous results suggest that complete blockade of a neutrophil response is likely detrimental, however suppressing an over-active response could be of benefit in IBD. Given the importance of neutrophil recruitment in colitis coupled with studies demonstrating an inhibitory effect of netrin-1 on neutrophil and macrophage migration during acute inflammation prompted us to investigate the frequency of these cell types in our colitis studies. Detailed flow cytometric analysis demonstrated that netrin-1 deficient mice had a persistent and specific increase in neutrophil number within the colonic lamina propria during the course of colitis.12 This observation could be reversed by netrin-1 treatment. Our analysis revealed no significant effect on monocyte or macrophage recruitment to the intestine indicating that netrin-1 exhibited preference for limiting neutrophil recruitment to the inflamed colon. Many signaling molecules have been identified that are involved in leukocyte recruitment to the intestine during inflammation observed in IBD (Fig. 1). We have identified netrin-1 as an endogenously expressed guidance molecule that can limit neutrophil accumulation to attenuate acute mucosal inflammation as observed in IBD (Fig. 1). Importantly, netrin-1 treatment does not result in complete blockade of the neutrophilic response, an approach which can be detrimental in this model (discussed above). We suggest that netrin-1 expression acts as a braking mechanism to suppress an over-zealous neutrophil response as observed in IBD.
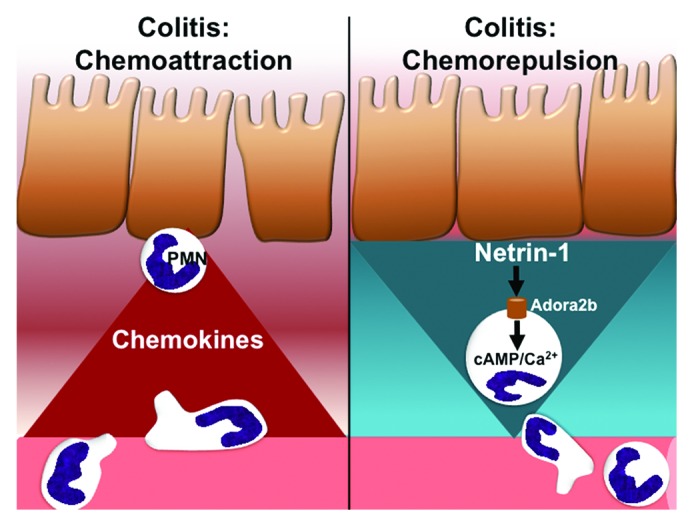
Figure 1. Balance between chemoattraction and repulsion mediated by epithelial cells during intestinal inflammation. The chemokines known to regulate neutrophil (PMN) migration into the intestinal lamina propria during colitis are well established. Our findings in an acute murine model of colitis present the neuronal guidance molecule netrin-1 as a chemorepulsive factor released by intestinal epithelial cells that limits PMN recruitment to the inflamed intestine. Initial studies point to netrin-1 signaling through the A2B adenosine receptor (Adora2b) as the mechanism by which netrin-1 mediates its therapeutic effects. Adora2b signaling can induce intracellular signaling molecules that have been demonstrated to alter PMN migration. Our study reveals that the intestinal epithelium can also be a source of chemorepulsive cues during acute colitis.
Recent studies in atherosclerotic mouse models have pointed to a pathogenic role for netrin-1 guidance of macrophages.20 This study implicated macrophage-derived netrin-1 as a retention signal for macrophages in the atherosclerotic plaque.20 These differential findings to ours and other acute models point to a tissue specific and disease contextual role for netrin-1. Future studies would need to focus on a potential role for netrin-1 in the context of chronic intestinal inflammation, as IBD is a relapsing, remitting, long-term disease. Chronic inflammation observed in IBD and murine models is heavily reliant on lymphocytes,6,42 therefore the tissue source and cell target for netrin-1 may be different in the context of chronic intestinal inflammation to that observed during acute colitis. The deleterious role of leukocyte-derived netrin-1 in atherosclerosis indicates that netrin-1 may have a distinct function in chronic models of IBD to that of acute colitis.
Netrin-1 Signaling Pathways in Acute Intestinal Inflammation
An important aspect of netrin-1 function is the signaling cascade that it can induce. Netrin-1 can bind to a number of cell surface receptors, including the deleted in colorectal cancer receptor (DCC), the uncoordinated receptors (UNC5A-D), neogenin and the A2B adenosine receptor (Adora2b).43 Signaling via the majority of the proposed netrin-1 receptors has yet to be investigated in IBD. Of note the Adora2b has been demonstrated to be a protective signaling pathway during acute tissue inflammation,44-49 including colitis.50 Considering our investigations demonstrated that netrin-1 mediated protection was driven by inhibition of neutrophil recruitment we proceeded to investigate netrin-1 signaling through receptors known to be expressed on this cell type. Previous findings have demonstrated that UNC5b is expressed on multiple leukocyte populations and have implicated netrin-1 signaling through this receptor in inhibition of granulocyte and lymphocyte migration.15,20 Similarly, studies have demonstrated that the Adora2b is expressed on neutrophils and signaling through this receptor inhibits neutrophil migration in vitro and in vivo.13,44,51,52 Following a previously described protocol53 we utilized antibody inhibition of the UNC5b receptor in our model of colitis. This proved unsuccessful in altering the protective function of netrin-1 during disease. In contrast, mice deficient in Adora2b could not be rescued from colitis progression by netrin-1 treatment, thereby implicating this receptor in netrin-1 mediated repression of neutrophil recruitment during acute intestinal inflammation.12 Since its identification as a netrin-1 receptor the exact signaling mechanism by which Adora2b functions as a netrin-1 receptor has yet to be defined.54 It is intriguing that studies link Adora2b signaling with both induction of cyclic adenosine monophosphate (cAMP) and also calcium mobilization.55 The balance between these two intracellular signaling molecules has been implicated in determining directional migration in response to cues such as netrin-1.31 We would predict that netrin-1 induces intracellular signaling pathways downstream of the Adora2b to inhibit neutrophil migration. Future studies will seek to identify the specific molecular signature induced by netrin-1 to regulate reverse leukocyte migration during IBD.
Conclusion
As our knowledge of the intestinal epithelial barrier increases it has become evident that intestinal epithelial cells are capable of active involvement in mucosal immune regulation. Analogous to the field of neuronal development it is tempting to suggest that intestinal epithelial secretion of guidance cues such as netrin-1 plays a fundamental role in directing the local immune response. Increasing our knowledge of the potential expression and function of these cues in the intestine may open up avenues of potential therapeutic intervention in IBD. It could be speculated that tipping the balance of mediators within the intestine toward a chemorepellent signature may be beneficial in reducing pathogenic leukocyte recruitment in IBD. We conclude from our studies that netrin-1 is a novel endogenous response generated within the intestinal epithelium to dampen neutrophil recruitment in acute colitis (Fig. 1). We would postulate that netrin-1 may have a beneficial function in chronic models of intestinal inflammation. However, recent findings in models of atherosclerosis identify leukocytes themselves as a source of chemorepellent cues to mediate leukocyte tissue retention. Therefore, future studies in IBD would need to consider the tissue source and dynamic expression of these guidance cues during the timecourse of disease to identify appropriate modalities of therapeutic intervention.
Glossary
Abbreviations:
- Adora2b
A2B adenosine receptor
- AOM
azoxymethane
- cAMP
cyclic adenosine monophosphate
- CD
Crohn disease
- DCC
deleted in colorectal cancer
- DSS
dextran sulfate sodium
- IBD
inflammatory bowel disease
- Ntn-1
netrin-1
- UC
ulcerative colitis
- UNC
uncoordinated
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/tissuebarriers/article/24957
References
- 1.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54, e42, quiz e30. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Scharl M, Rogler G. Inflammatory bowel disease pathogenesis: what is new? Curr Opin Gastroenterol. 2012;28:301–9. doi: 10.1097/MOG.0b013e328353e61e. [DOI] [PubMed] [Google Scholar]
- 3.Glover LE, Colgan SP. Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology. 2011;140:1748–55. doi: 10.1053/j.gastro.2011.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4607–14. doi: 10.1073/pnas.1000092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papadakis KA. Chemokines in inflammatory bowel disease. Curr Allergy Asthma Rep. 2004;4:83–9. doi: 10.1007/s11882-004-0048-7. [DOI] [PubMed] [Google Scholar]
- 6.Collins CB, Aherne CM, Kominsky D, McNamee EN, Lebsack MD, Eltzschig H, et al. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and T regulatory cells. Gastroenterology. 2011;141:1821–31. doi: 10.1053/j.gastro.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. 2011;186:4367–74. doi: 10.4049/jimmunol.0903617. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–18. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 9.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glover LE, Irizarry K, Scully M, Campbell EL, Bowers BE, Aherne CM, et al. IFN-γ attenuates hypoxia-inducible factor (HIF) activity in intestinal epithelial cells through transcriptional repression of HIF-1β. J Immunol. 2011;186:1790–8. doi: 10.4049/jimmunol.1001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aherne CM, Collins CB, Masterson JC, Tizzano M, Boyle TA, Westrich JA, et al. Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut. 2012;61:695–705. doi: 10.1136/gutjnl-2011-300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, et al. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994;78:425–35. doi: 10.1016/0092-8674(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 15.Ly NP, Komatsuzaki K, Fraser IP, Tseng AA, Prodhan P, Moore KJ, et al. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14729–34. doi: 10.1073/pnas.0506233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirakaj V, Thix CA, Laucher S, Mielke C, Morote-Garcia JC, Schmit MA, et al. Netrin-1 dampens pulmonary inflammation during acute lung injury. Am J Respir Crit Care Med. 2010;181:815–24. doi: 10.1164/rccm.200905-0717OC. [DOI] [PubMed] [Google Scholar]
- 17.Grenz A, Dalton JH, Bauerle JD, Badulak A, Ridyard D, Gandjeva A, et al. Partial netrin-1 deficiency aggravates acute kidney injury. PLoS One. 2011;6:e14812. doi: 10.1371/journal.pone.0014812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mirakaj V, Gatidou D, Pötzsch C, König K, Rosenberger P. Netrin-1 signaling dampens inflammatory peritonitis. J Immunol. 2011;186:549–55. doi: 10.4049/jimmunol.1002671. [DOI] [PubMed] [Google Scholar]
- 19.Kolodkin AL, Tessier-Lavigne M. Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol. 2011;3:3. doi: 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Gils JM, Derby MC, Fernandes LR, Ramkhelawon B, Ray TD, Rayner KJ, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–43. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang Y, Liu MT, Gershon MD. Netrins and DCC in the guidance of migrating neural crest-derived cells in the developing bowel and pancreas. Dev Biol. 2003;258:364–84. doi: 10.1016/S0012-1606(03)00136-2. [DOI] [PubMed] [Google Scholar]
- 22.Ratcliffe EM, Setru SU, Chen JJ, Li ZS, D’Autréaux F, Gershon MD. Netrin/DCC-mediated attraction of vagal sensory axons to the fetal mouse gut. J Comp Neurol. 2006;498:567–80. doi: 10.1002/cne.21027. [DOI] [PubMed] [Google Scholar]
- 23.Ratcliffe EM, Fan L, Mohammed TJ, Anderson M, Chalazonitis A, Gershon MD. Enteric neurons synthesize netrins and are essential for the development of the vagal sensory innervation of the fetal gut. Dev Neurobiol. 2011;71:362–73. doi: 10.1002/dneu.20869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakhan SE, Kirchgessner A. Neuroinflammation in inflammatory bowel disease. J Neuroinflammation. 2010;7:37. doi: 10.1186/1742-2094-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paradisi A, Maisse C, Coissieux MM, Gadot N, Lépinasse F, Delloye-Bourgeois C, et al. Netrin-1 up-regulation in inflammatory bowel diseases is required for colorectal cancer progression. Proc Natl Acad Sci U S A. 2009;106:17146–51. doi: 10.1073/pnas.0901767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, et al. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature. 2004;431:80–4. doi: 10.1038/nature02788. [DOI] [PubMed] [Google Scholar]
- 27.Schulzke JD, Bojarski C, Zeissig S, Heller F, Gitter AH, Fromm M. Disrupted barrier function through epithelial cell apoptosis. Ann N Y Acad Sci. 2006;1072:288–99. doi: 10.1196/annals.1326.027. [DOI] [PubMed] [Google Scholar]
- 28.Araki Y, Mukaisyo K, Sugihara H, Fujiyama Y, Hattori T. Increased apoptosis and decreased proliferation of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol Rep. 2010;24:869–74. doi: 10.3892/or.2010.869. [DOI] [PubMed] [Google Scholar]
- 29.Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology. 2010;139:2093–101. doi: 10.1053/j.gastro.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 30.Cirulli V, Yebra M. Netrins: beyond the brain. Nat Rev Mol Cell Biol. 2007;8:296–306. doi: 10.1038/nrm2142. [DOI] [PubMed] [Google Scholar]
- 31.Huttenlocher A, Poznansky MC. Reverse leukocyte migration can be attractive or repulsive. Trends Cell Biol. 2008;18:298–306. doi: 10.1016/j.tcb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadakis KA, Prehn J, Zhu D, Landers C, Gaiennie J, Fleshner PR, et al. Expression and regulation of the chemokine receptor CXCR3 on lymphocytes from normal and inflammatory bowel disease mucosa. Inflamm Bowel Dis. 2004;10:778–88. doi: 10.1097/00054725-200411000-00013. [DOI] [PubMed] [Google Scholar]
- 33.Mathias JR, Perrin BJ, Liu TX, Kanki J, Look AT, Huttenlocher A. Resolution of inflammation by retrograde chemotaxis of neutrophils in transgenic zebrafish. J Leukoc Biol. 2006;80:1281–8. doi: 10.1189/jlb.0506346. [DOI] [PubMed] [Google Scholar]
- 34.Lampinen M, Sangfelt P, Taha Y, Carlson M. Accumulation, activation, and survival of neutrophils in ulcerative colitis: regulation by locally produced factors in the colon and impact of steroid treatment. Int J Colorectal Dis. 2008;23:939–46. doi: 10.1007/s00384-008-0509-x. [DOI] [PubMed] [Google Scholar]
- 35.Hermanowicz A, Gibson PR, Jewell DP. The role of phagocytes in inflammatory bowel disease. Clin Sci (Lond) 1985;69:241–9. doi: 10.1042/cs0690241. [DOI] [PubMed] [Google Scholar]
- 36.Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5:354–66. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 38.Zhang R, Ito S, Nishio N, Cheng Z, Suzuki H, Isobe K. Up-regulation of Gr1+CD11b+ population in spleen of dextran sulfate sodium administered mice works to repair colitis. Inflamm Allergy Drug Targets. 2011;10:39–46. doi: 10.2174/187152811794352114. [DOI] [PubMed] [Google Scholar]
- 39.Kühl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–92. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 40.Farooq SM, Stillie R, Svensson M, Svanborg C, Strieter RM, Stadnyk AW. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2009;329:123–9. doi: 10.1124/jpet.108.145862. [DOI] [PubMed] [Google Scholar]
- 41.Bento AF, Leite DF, Claudino RF, Hara DB, Leal PC, Calixto JB. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J Leukoc Biol. 2008;84:1213–21. doi: 10.1189/jlb.0408231. [DOI] [PubMed] [Google Scholar]
- 42.Collins CB, Aherne CM, McNamee EN, Lebsack MD, Eltzschig H, Jedlicka P, et al. Flt3 ligand expands CD103⁺ dendritic cells and FoxP3⁺ T regulatory cells, and attenuates Crohn’s-like murine ileitis. Gut. 2012;61:1154–62. doi: 10.1136/gutjnl-2011-300820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore SW, Tessier-Lavigne M, Kennedy TE. Netrins and their receptors. Adv Exp Med Biol. 2007;621:17–31. doi: 10.1007/978-0-387-76715-4_2. [DOI] [PubMed] [Google Scholar]
- 44.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–35. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–15. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ehrentraut H, Westrich JA, Eltzschig HK, Clambey ET. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS One. 2012;7:e32416. doi: 10.1371/journal.pone.0032416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grenz A, Kim JH, Bauerle JD, Tak E, Eltzschig HK, Clambey ET. Adora2b adenosine receptor signaling protects during acute kidney injury via inhibition of neutrophil-dependent TNF-α release. J Immunol. 2012;189:4566–73. doi: 10.4049/jimmunol.1201651. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, et al. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hart ML, Jacobi B, Schittenhelm J, Henn M, Eltzschig HK. Cutting Edge: A2B Adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J Immunol. 2009;182:3965–8. doi: 10.4049/jimmunol.0802193. [DOI] [PubMed] [Google Scholar]
- 50.Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009;182:4957–64. doi: 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wakai A, Wang JH, Winter DC, Street JT, O’Sullivan RG, Redmond HP. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock. 2001;15:297–301. doi: 10.1097/00024382-200115040-00008. [DOI] [PubMed] [Google Scholar]
- 52.Koeppen M, Harter PN, Bonney S, Bonney M, Reithel S, Zachskorn C, et al. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology. 2012;116:1245–57. doi: 10.1097/ALN.0b013e318255793c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tadagavadi RK, Wang W, Ramesh G. Netrin-1 regulates Th1/Th2/Th17 cytokine production and inflammation through UNC5B receptor and protects kidney against ischemia-reperfusion injury. J Immunol. 2010;185:3750–8. doi: 10.4049/jimmunol.1000435. [DOI] [PubMed] [Google Scholar]
- 54.Corset V, Nguyen-Ba-Charvet KT, Forcet C, Moyse E, Chédotal A, Mehlen P. Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature. 2000;407:747–50. doi: 10.1038/35037600. [DOI] [PubMed] [Google Scholar]
- 55.Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta. 2011;1808:1329–39. doi: 10.1016/j.bbamem.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]