

Abstract
Pseudomonas syringae is a phylogenetically diverse species of Gram-negative bacterial plant pathogens responsible for crop diseases around the world. The HrpL sigma factor drives expression of the major P. syringae virulence regulon. HrpL controls expression of the genes encoding the structural and functional components of the type III secretion system (T3SS) and the type three secreted effector proteins (T3E) that are collectively essential for virulence. HrpL also regulates expression of an under-explored suite of non-type III effector genes (non-T3E), including toxin production systems and operons not previously associated with virulence. We implemented and refined genome-wide transcriptional analysis methods using cDNA-derived high-throughput sequencing (RNA-seq) data to characterize the HrpL regulon from six isolates of P. syringae spanning the diversity of the species. Our transcriptomes, mapped onto both complete and draft genomes, significantly extend earlier studies. We confirmed HrpL-regulation for a majority of previously defined T3E genes in these six strains. We identified two new T3E families from P. syringae pv. oryzae 1_6, a strain within the relatively underexplored phylogenetic Multi-Locus Sequence Typing (MLST) group IV. The HrpL regulons varied among strains in gene number and content across both their T3E and non-T3E gene suites. Strains within MLST group II consistently express the lowest number of HrpL-regulated genes. We identified events leading to recruitment into, and loss from, the HrpL regulon. These included gene gain and loss, and loss of HrpL regulation caused by group-specific cis element mutations in otherwise conserved genes. Novel non-T3E HrpL-regulated genes include an operon that we show is required for full virulence of P. syringae pv. phaseolicola 1448A on French bean. We highlight the power of integrating genomic, transcriptomic, and phylogenetic information to drive concise functional experimentation and to derive better insight into the evolution of virulence across an evolutionarily diverse pathogen species.
Author Summary
Pseudomonas syringae are environmentally ubiquitous bacteria of wide phylogenetic distribution, which can cause disease on a broad range of plant species. Pathogenicity requires the master regulator HrpL. HrpL controls the activation of virulence factor genes, including those encoding the type III secretion system which facilitates translocation of bacterial proteins into host cells. Here we overlaid transcriptome profiling of genes onto their phylogenetic distribution by characterizing the HrpL regulon across six diverse strains of P. syringae. We identified novel putative virulence factors, discovered two novel effector families, and functionally characterized an operon most likely involved in secondary metabolism that we show is required for virulence. We demonstrated that the size and composition of the HrpL regulon varies among strains, and explored how genes are recruited into, or lost from, the virulence regulon. Overall, our work widens the understanding of P. syringae pathogenicity and presents an experimental paradigm extensible to other pathogenic bacterial species.
Introduction
Many Gram-negative bacteria attach to host cells and translocate effector proteins into them via type III secretion systems (T3SS). Such systems are necessary for pathogenesis, are horizontally transferred across species, and are accompanied by dynamically evolving repertoires of type III effector (T3Es) genes [1], [2]. The T3SS is essential for Pseudomonas syringae pathogens to thrive in plant tissues. P. syringae represents an excellent example of the plasticity of T3E repertoires [3]. Despite a collectively broad host range for the species, individual isolates of P. syringae typically display pathogenic potential on a limited set of plants and either elicit immune responses, or simply fail to thrive on other plant species. Strains can be isolated from diseased plants, as epiphytes from healthy plants [4], and from various environmental sources [5].
The hrp/hrc group I T3SS is essential for P. syringae pathogens to cause disease on plants [1], [6]. The genes that encode the hrp/hrc T3SS and accessory proteins are clustered in a conserved pathogenicity island in P. syringae [7]. The genes for the associated T3Es can be scattered across the genome, often in association with mobile elements indicative of horizontal transmission [8]–[10]. Each strain's T3E repertoire ranges from 15–30 genes sampled from at least 57 different families and these collectively modify host cell biology to suppress immune response and favor bacterial proliferation and dispersion. However, the action of individual T3E proteins can be recognized by plant host disease resistance proteins, and this triggers immune responses sufficient to limit pathogen growth [11]. The conflicting selective pressures to retain a collection of T3E sufficient to suppress host defenses without triggering effector-specific immune responses [11] drives diversity in the suites of T3Es in plant pathogenic P. syringae isolates [3].
Transition from saprophytic to epiphytic or pathogenic lifestyle requires significant transcriptional reprogramming. Expression of genes encoding the P. syringae T3SS structural components and the associated T3E suite is controlled by the ECF-type sigma factor HrpL [12]–[14]. The expression of hrpL is induced in bacteria that encounter the leaf environment [13]. Subsequently, HrpL binds to promoters carrying a “hrp-box” consensus sequence to up-regulate the expression of the corresponding gene(s) [12]–[15].
Previous studies in P. syringae identified proteins that are neither T3Es nor structural components of the T3SS (hereafter, non-T3Es), but are HrpL-regulated [3], [16]–[19]. Non-T3Es coordinately regulated with the T3SS and its substrates were also found in other T3SS-expressing plant pathogens such as Erwinia amylovora [20], Ralstonia solanacearum [21], Xanthomonas campestris pv. vesicatoria [22], [23] and Pectobacterium carotovora [24]. Some HrpL-regulated non-T3E genes affect virulence on host plants in the well-studied strain P. syringae pv. tomato DC3000 (Pto DC3000); these include the corR regulator of coronatine toxin production [18], [25]. Notably, CorR expression is not HrpL-regulated in other strains, such as P. syringae pv. glycinea PG4180 [26].
Multi-Locus Sequence Typing (MLST) separates plant pathogenic P. syringae into at least 5 distinct phylogenetic groups [3], [27]. The fifth group, represented initially by P. syrinage pv. maculicola ES4326, was recently renamed P. cannabina pv. alisalensis ES4326 [28]. Many P. syringae genome sequences are now available, including three closed genomes from isolates representing major pathogen clades [29]–[31], and ∼120 additional draft sequences. Newly sequenced genomes also trace P. syringae disease outbreaks across the globe and over time [3], [32]–[39] attesting to the continued importance of the species. Recently, isolation and sequencing of saprophytic and epiphytic strains provided insight into a subgroup from group II that carries a non-canonical T3SS [40]. To date, transcriptome analyses using high throughput short read cDNA sequencing (RNA-seq) have been applied only to Pto DC3000, providing a well-curated reference gene annotation, but not specifically informing studies of the HrpL regulon [41]–[44].
In this study, we defined the HrpL regulon of six distinct strains of P. syringae with complete or draft genomes using RNA-seq coupled with the GENE-counter software package [45]–[47]. We sought primarily to compare the diversity of non-T3E HrpL-regulated genes between strains and secondarily to determine if there were additional type III effectors not found in our DNA-based analyses [3]. We detect non-T3E genes regulated directly or indirectly by HrpL. Those directly regulated by HrpL are distributed throughout the P. syringae clades in a mosaic pattern. However, most are either absent or not HrpL-regulated in MLST group II. We demonstrate that a novel cluster of non-T3E genes is required for P. syringae pv. phaseolicola 1448A virulence. We also identified two novel T3E families from a previously understudied clade. Our study reveals the mechanisms for gene recruitment into, and loss from, the key virulence regulon in P. syringae, and provides a roadmap for future functional studies.
Results
The HrpL regulons of six phylogenetically diverse P. syringae isolates are defined by RNA-seq
We defined the HrpL regulons of P. syringae pv. phaseolicola strain 1448A (Pph 1448A), P. syringae pv. lachrymans strain 107 (Pla 107) representing MLST group III; P. syringae pv. syringae strain B728a (PsyB728a), P. syringae pv. japonica strain MAFF 301072 PT (Pja) representing MLST group II; P. syringae pv. tomato strain DC3000 (Pto DC3000) representing MLST group I and P. syringae pv. oryzae strain 1_6 (Por), belonging to the relatively poorly studied clade, MLST group IV [3], [27]. The native hrpL gene from each isolate was cloned downstream of an arabinose-inducible promoter for controlled, high-level expression in the strain of origin. Isogenic strains carrying either the appropriate hrpL construct, or an empty vector (EV) as negative control, were grown with arabinose to induce the expression of the cloned hrpL gene in a minimal medium [19]. Expression of the native hrpL was repressed by addition of peptone to the media [48]. Figure S1 depicts our experimental pipeline and control validation.
We generated Illumina cDNA libraries from two biological replicates of each strain. Because our goal was to compare transcript abundance more than to improve annotation of transcribed genes, we used a simple cDNA method to minimize the RNA processing steps where transcripts could be lost. Therefore, we did not enrich for 5′ ends or distinguish transcript orientation. Transcript abundance was compared between isogenic HrpL and EV samples using GENE-counter [45]. Similar to other RNA-seq analysis methods like EdgeR or DESeq [49], [50], GENE-counter determines differential expression. While EdgeR and DESeq use the standard negative binomial distribution, GENE-counter relies on the negative binomial p distribution which better accounts for the over-dispersion observed in mRNA-seq data [51]–[53]. We bootstrapped the GENE-counter output for each isolate (Materials and Methods) to control for noise introduced by sample normalization. Between 1.6 and 5.6 million unambiguous reads per sample (mapping to only one location in the reference genome) were used for our analyses (Table 1). The sequencing depth ranged from 9.5 to 16.2 times the genome size, with the exception of the Psy B728a samples, which we sequenced to higher coverage (Table 1). On average 93.5% of the total number of annotated coding genes in a genome were covered by at least one read in at least one sample (Table 1). Bootstrapped-GENE-counter analysis established a median read count for every sample, a median q-value and a B-value, for every gene covered by at least one read in one biological replicate (Table S1). Genes not covered by any unambiguous reads are not represented in our GENE-counter output. The B-value represents the percentage of bootstraps in which a particular gene was called differentially expressed.
Table 1. Summary of Illumina RNA-seq data.
| Pph 1448A | Pla1 07_ Draft | Psy B728a | Pja_ Draft | Por_ Draft | Pto DC3000 | Pto DC3000_ Draft | |
| Technology | GAII | GAII | Hi-seq | GAII | GAII | GAII | GAII |
| read length (nt) | 36 | 36 | 50 | 36 | 36 | 36 | 36 |
| Average # of reads used for analysis after bootstrapping | 2,376,423 | 1,613,243 | 5,675,482 | 2,506,746 | 2,652,065 | 1,971,190 | 1,836,589 |
| Average # of nt used for analysis after bootstrapping | 85,551,228 | 58,076,775 | 283,774,100 | 90,242,856 | 95,474,340 | 70,962,840 | 66,117,204 |
| Genome size (bp) | 6,112,448 | 6,030,058 | 6,093,698 | 6,932,599 | 5,886,178 | 6,538,260 | 6,924,419 |
| Sequencing depth* | 14.0 | 9.6 | 46.6 | 13.0 | 16.2 | 10.9 | 9.5 |
| # coding genes in genome | 5,172 | 6,744 | 5,089 | 9,534 | 6,329 | 5,619 | 5,618 |
| # coding genes covered by reads | 5,047 | 6,362 | 5,061 | 7,414 | 5,951 | 5,317 | 5,573 |
The sequencing depth was defined as the number of nt used for the analysis divided by the size of the genome.
We further considered only genes with B-values≥50%. Like all “significance thresholds” the B-value cut-off is somewhat subjective. We selected a B-value of 50% to apply to all genomes because this threshold captured 95% of the known HrpL-regulated genes identified in our control genome, Pto DC3000, with a median q-value greater than 0.05. We identified between 59 to 192 genes differentially expressed across the strains (Table S1). For all strains, the large majority of the differentially expressed genes were up-regulated (between 53 and 180 genes, Table 2). These genes mainly encode T3SS components and known T3Es. Surprisingly, we identified few HrpL-down-regulated genes (Table S2): ranging from none in Pph 1448A to 45 in Pla 107. Genes called down-regulated in our analysis had relatively low q-values, reflecting low differences in read coverage between HrpL and EV samples. Lan et al. 2006 and Ferreira et al. 2006 identified down-regulated genes in their microarray studies for Pto DC3000. However, almost no overlap was found between the list of down-regulated genes from previous studies and ours, indicating that the down-regulated genes identified are most likely neither biologically, nor statistically robust, and thus unlikely to be biologically relevant. In contrast, there was stronger overlap between our HrpL-induced genes and those shared between these earlier studies (see below). Down-regulated genes were therefore not further analyzed. Finally, we manually inspected and curated all genes with B-values greater than or equal to 50% to define the HrpL regulon for each strain (Table 2, Table S3; Table S4; Materials and Methods).
Table 2. Characterization of the HrpL-regulon across Pseudomonas syringae strains.
| Pph 1448A | Pla1 07_ Draft | Psy B728a | Pja_ Draft | Por_ Draft | Pto DC3000 | Pto DC3000_ Draft | |
| Genes up by RNA-seq raw | 88 | 167 | 63 | 78 | 232 | 133 | 131/124e |
| Genes up B-value≥50% | 76 | 108 | 53 | 69 | 180 | 115 | ND |
| Genes up after curationa | 71 | 90 | 51 | 46 | 114 | 110 | ND |
| % of known HrpL-genes found by RNA-seqb | 79% | NA | NA | NA | NA | 91% | 87% |
| # of genes expected to be HrpL-regulated by RNA-seqc | 58 | 62 | 45 | 38 | 82 | 96 | ND |
| # of putative novel virulence genes identified by RNA-seq | 13 | 28 | 6 | 8 | 32 | 14 | ND |
| # of missing genesd | 8 | NA | NA | NA | NA | 5 | ND |
| Genes down B-value≥50% | 0 | 45 | 5 | 12 | 12 | 38 | ND |
, See Materials and Methods.
, The number of known HrpL-regulated genes for Pto DC3000 was determined using data collected by [16], [17], [19], and defined as the number of genes found up-regulated in at least two studies. For Pph 1448A the number of known HrpL-regulated genes was determined according to data generated by [18], [19]; and defined as the number of genes found up-regulated in either study. See Table S5 for details.
, Genes expected to be HrpL-regulated were defined as genes being part of an operon known to be HrpL-regulated, genes found HrpL-dependent in other strains, or genes involved in coronatine synthesis. See Table S6 for details.
, Missing genes were defined as “ known HrpL-regulated” not found differentially expressed in our analysis.
, represent the number of genes found upregulated after genes split up between contigs were removed. NA, not applicable. ND, not determined.
Analysis of RNA-seq data is reproducible
To evaluate the reproducibility of our method, we compared the read coverage within and between biological samples for all Pto DC3000 genes covered by at least one read in our normalized GENE-counter data set. Biologically replicated samples had highly correlated results (R2 = 0.93 between EV replicates 1 and 2; R2 = 0.96 between conditional expression replicates HrpL1 and HrpL2, Figure 1A, lower panels). Comparing HrpL and EV replicates from two biological replicates, the majority of the data points correlate and cluster around the trend line (Figure 1A, upper panels). The outlier data points in red represent genes defined as differentially up-regulated by GENE-counter and having a B-value≥50%. We plotted the log of the median q-value of each Pto DC3000 gene defined to be differentially up-regulated (before manual curation) and their corresponding B-values ranked from smallest to largest (Figure 1B). As expected, genes with highly significant q-scores also have high B-values. Several genes not previously reported to be HrpL-regulated (marked in red) had more significant q-value scores (3.8E-02) than avrE (marked in blue), a well-characterized conserved HrpL-regulated type III effector [54].
Figure 1. Validation of our RNA-seq method.
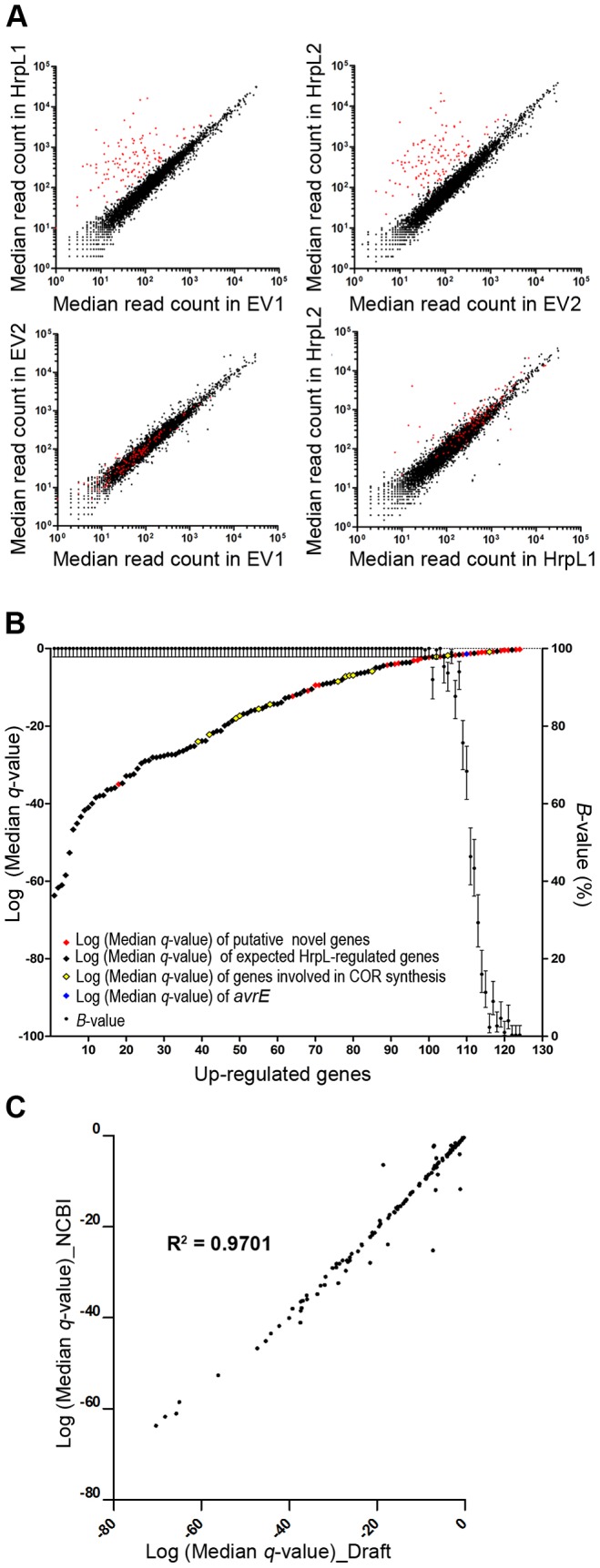
(A) Read coverage of Pto DC3000 genes within and between biological replicates displays high reproducibility. A graphical logarithmic representation of the median read counts of all genes covered by unique reads after bootstrapping of the data is presented. Top panels: comparison of Pto DC3000(pBAD::hrpL) and Pto DC3000(pBAD::EV) samples within (left) the first (HrpL1 or EV1), or the second (HrpL2 or EV2) (right) biological replicate. Bottom panels: comparison of Pto DC3000(pBAD::EV) samples (left), and Pto DC3000(pBAD::hrpL) samples (right) across two biological replicates. Red dots represent the logarithmic median read count of genes found to be significantly up-regulated in the presence of HrpL by GENE-counter with a B-value≥50%. (B) Graphical representation of GENE-counter data for Pto DC3000 bootstrapped 300 times. Genes identified as differentially up-regulated were plotted according to their Log (Median q-value). Data points were color-coded for putatively novel genes (genes not previously described as HrpL-regulated, in red), genes previously described as HrpL-regulated, hrp complex genes, type III effector genes (in black), and genes involved in coronatine synthesis (in yellow). avrE is marked in blue. (C) Draft and complete (NCBI) Pto DC3000 reference genomes yield highly similar RNA-seq results. Every gene found up-regulated with a B-value≥50 using both NCBI and draft genome as reference genomes was plotted according to its Log (median q-value).
We analyzed the same Pto DC3000 RNA-seq data set using either the complete Pto DC3000 genome sequence [30] or a draft Pto DC3000 genome sequence [36] as references. The draft genome sequence covers 85% of genes at over 90% of their length [36]. Using either the complete or the draft genome as a reference resulted in similar sequencing depths (Table 1). Using the draft genome as a reference, GENE-counter identified 124 HrpL-upregulated genes out of the 133 found using the complete Pto DC3000 genome (Table 2). Most of the genes that were not identified as differentially expressed using the draft genome were missing from the draft genome (data not shown). The high correlation between the Log(median q-value) of genes in the two data sets (Figure 1C) indicates that our method will effectively identify the majority of genes of the HrpL regulon from P. syringae isolates for which only a high quality draft genome is available.
RNA-seq successfully captures the HrpL regulon for Pto DC3000 and Pph 1448A
To further validate our pipeline to define HrpL–regulated genes, we compared our manually curated list of 110 Pto DC3000 HrpL-regulated genes (Table 2) to HrpL-regulated genes identified by three previous studies: one promoter probe study using an arabinose-inducible hrpL gene and two custom microarray analyses which compared expression between wild type and hrpL deletion mutant strains [16], [17], [19]. These studies produced largely overlapping, but not identical, lists of putatively HrpL-regulated genes (Table S5). Our Pto DC3000 HrpL-regulated gene set included 57 out of the 66 genes previously identified as HrpL-regulated in at least two of the previous studies (Table S5), even though our induction and analysis methods differed from these studies. 96 of the 110 genes we identified were also found to be HrpL-regulated in at least one of the previous studies [16], [17], [19] or were downstream genes in HrpL-regulated operons (Table 2). Overall, we found 91% of the previously identified HrpL-regulated genes in Pto DC3000. Our analysis also identified 14 novel HrpL-regulated genes (Table 2); six out of eight tested were confirmed to be HrpL-regulated using qRT-PCR (Table 3, see below).
Table 3. Real time RT-PCR analyses predominantly confirm RNA-seq data.
| Genes tested | Annotation | Median Q-value (Genecounter) | Putative hrp-box | pBAD system* # | Fold induction in pBAD system | Native system* ∧ | % expression in ΔhrpL vs. WT |
| PSPTO_4332 | hypothetical protein | 4.66E-13 | − | + | 15.3 | + | 3.8 |
| PSPTO_2130 | LuxR family DNA-binding response regulator | 3.45E-10 | + | + | 5.2 | + | 19.0 |
| PSPTO_3086 | transcriptional regulator | 3.75E-10 | − | + | 24.0 | + | 4.1 |
| PSPTO_2129 | sensory box histidine kinase/response regulator | 4.86E-05 | − | + | 3.4 | + | 21.3 |
| PSPTO_2208 | htpG heat shock protein 90 | 9.82E-05 | − | + | 2.7 | + | 53.6 |
| PSPTO_0871 | macrolide efflux protein | 1.51E-04 | + | + | 2.8 | ND | ND |
| PSPTO_1843 | aspartate kinase | 1.48E-03 | − | − | 0.9 | ND | ND |
| PSPTO_4716 | hypothetical protein | 1.89E-02 | − | − | 1.5 | ND | ND |
| PSPPH_A0112 | phosphoglycerate mutase family protein | 3.11E-11 | − | + | 4.8 | + | 20.5 |
| PSPPH_A0110 | hypothetical protein | 3.94E-10 | − | ND | ND | + | 53.6 |
| PSPPH_A0109 | sulfotransferase, putative | 7.96E-08 | − | + | 4.3 | + | 51.6 |
| PSPPH_1906 | LuxR family DNA-binding response regulator | 5.51E-07 | + | + | 4.9 | + | 4.1 |
| PSPPH_0762 | hypothetical protein | 4.45E-03 | − | + | 4.4 | + | 9.9 |
| PSPPH_A0106 | hypothetical protein | 7.49E-03 | − | ND | ND | + | 46.4 |
| PSPPH_A0108 | adenosylmethionine-8-amino-7-oxononanoate aminotransferase | 1.27E-02 | − | ND | ND | + | 36.7 |
| Psyr_0737 | putative transmembrane protein | 1.00E-46 | + | + | 23.3 | + | 8.0 |
| Psyr_0027 | hypothetical protein | 1.64E-14 | − | + | 4.3 | − | 84.5 |
| PORcurated_00518 | hypothetical protein | 2.49E-54 | + | + | 84.3 | + | 2.0 |
| PORcurated_04644 | Methyltransferase small domain | 1.16E-37 | + | + | 48.4 | + | 3.5 |
| PORcurated_03530 | hypothetical protein | 3.03E-30 | + | + | 19.5 | + | 34.2 |
| PORcurated_04640 | hypothetical protein | 1.72E-27 | + | + | 19.8 | + | 42.3 |
| PORcurated_04022 | Alkylated DNA repair protein | 4.24E-24 | − | +/− | 1.7 | ND | ND |
| PORcurated_04648 | hypothetical protein | 2.35E-14 | − | + | 4.1 | + | 41.7 |
| PORcurated_04371 | hypothetical protein | 3.39E-24 | + | + | 5.2 | + | 30.2 |
| PORcurated_04025 | hypothetical protein | 1.99E-11 | − | + | 3.4 | ND | ND |
| PORcurated_04024 | Domain of unknown function (DUF1883) | 1.06E-04 | − | − | 0.6 | ND | ND |
+ found up-regulated by qRT-PCR; − no up-regulation. ND, not determined. See Figure S2 and S3 for detailed qRT-PCR results.
expression compared between Ps (pBAD::EV) strain and Ps (pBAD::hrpL) strain grown in media containing arabinose.
expression compared between a wild type strain and an isogenic clean hrpL deletion mutant grown in MM media.
Notably, four of the nine missing genes were not present in our laboratory strain, which has lost part of the Pto DC3000 plasmid A. One gene, shcA (PSPTO_5353) was found differentially expressed in our analysis but had a B-value less than 50%. Further, GENE-counter discards RNA-seq reads that map non-uniquely to more than one location in the genome, and HrpL-regulated duplicated genes account for three missing Pto DC3000 genes: T3E genes hopAM1-1(PSPTO_1022) and hopQ1-2 (PSPTO_4732), and the non-T3E gene plcA2 (PSPTO_B0005) (Table S5). Finally, hopK1 (PSPTO_0044), was covered by RNA-seq reads but the differences in expression in HrpL and EV treatments were not statistically significant (Table S1, S5).
Two previous studies focused on the identification of HrpL-regulated genes in Pph 1448A [18], [19] and identified 43 HrpL-regulated genes comparing expression between wild type and hrpL mutants. We identified 35 (∼80%). Four of the missing eight genes were covered by reads but not found significantly differentially expressed, hopAK1 (PSPPH_1424), a gene encoding a MarR transcriptional regulator (PSPPH_1519), avrRps4 (PSPPH_A0087), and hopAS1 (PSPPH_4736). Those four genes had a median read coverage ranging from 100 to 1000, indicating that the absence of differential expression in our analysis is not due to weak or undetectable levels of expression. One, PSPPH_2294 is a pseudogene. PSPPH_1525 encoding a putative effector related to Ralstonia Hpx30 [55], PSPPH_A0009 and A00075 encoding truncated hopW1 are duplicated and had very low to no read coverage (Table S5). Our GENE-counter analysis pipeline results are consistent with previous transcriptional studies, reinforcing the validity of our methods. Additionally, we identified robustly HrpL-induced genes that were not previously identified.
Quantitative RT-PCR analyses predominantly confirm our RNA-seq data
We identified between six and 32 genes previously not known to be HrpL-regulated in each strain with corresponding q-values ranging from E-02 to E-54 (Table 2, Table S3). Some of these are shared across strains. We could not identify a consensus upstream hrp-box in the promoters of several, and suggest that these could be indirectly activated by HrpL. We performed qRT-PCR using samples derived from strains expressing HrpL in the pBAD system and confirmed 19 of 23 tested (Figure S2). Additionally, we confirmed HrpL-dependent expression of 19 genes out of 20 tested, by comparing wild type expression with expression in a hrpL deletion mutant in hrpL-inducing minimal medium (Table 3, Figure S3). We observed a high correlation between RNA-seq data and either qRT-PCR profiling method, especially for genes with a q value>E-03 (Table 3, Figures S2, S3). In sum, we identified the majority of previously identified HrpL-regulated genes in two well-studied strains and we confirmed wild type HrpL regulation for nearly all of the newly identified members of this key virulence regulon.
RNA-seq identifies new T3E genes
Most of the known T3E and candidate T3E genes in our tested strains and those previously defined by similarity and/or functional criteria were included in the HrpL regulons we defined in our RNA-seq analyses (Figure S4). Most of strains used in this study had previously been screened for novel type III effector genes by functional translocation assays with the exception of Por and Pja [3], [19]. Therefore, we searched the Por and Pja HrpL regulons for potential novel effector genes based on the criteria of having an identifiable upstream hrp-box sequence and no homology to previously identified T3E families. We chose six Por genes (Porcurated_02784, 04644, 04640, 03530, 02145, and 04371) to investigate as potentially encoding novel T3Es. Pja also carries a gene homologous to Porcurated_04644; but only the Por allele was tested. All six putative T3E were tested for their ability to be translocated via a native T3SS using an established assay [56] (Materials and Methods) from Pto DC3000D28E, an “effector-less” Pto DC3000 strain [57]. Only Pto DC3000D28E carrying Por curated _02784-Δ79avrRpt2 or Por curated _04640-Δ79avrRpt2 triggered a Hypersensitive Response (HR) in Col-0 (Figure 2A). We verified that HA-tagged versions of all six T3E candidates were expressed in Pto DC3000D28E indicating that lack of HR in our translocation assay was unlikely due to a lack of protein accumulation (Figure 2B). No HR was observed in the rps2 mutant, indicating that the response was avrRpt2-specific and not the result of toxicity. These two new P. syringae effectors will henceforth be referred to as HopBH1Por and HopBI1Por according to proposed T3E naming guidelines [58].
Figure 2. Identification of a novel type III effector.

(A) Δ79avrRpt2 translocation assay. Four week old Col-0 and Col-0 rps2–101c (rps2) plants were hand inoculated with Pto DC300028E without plasmid or Pto DC300028E carrying either citrine (negative control), hopBA1 and its upstream region (positive control), IMG/ER Porcuarted_02784, 04644, 04640, 03530, 02145, and 04371 sequence and upstream sequence into Δ79avrRpt2 -fusion vector pJC532. Plants were scored and pictures were taken 24 h after inoculation for hypersensitive response (HR). (B) Western blotting analysis. Pto DC300028E without plasmid (negative control), Pto DC300028E carrying IMG/ER Porcurated_02784, 04644, 04640, 03530, 02145, and 04371 sequence and upstream region into HA-fusion vector pJC531 were grown on MM media for 5 hours. Culture aliquots were subjected to western blot using anti-HA antibody. CBB, Coomassie brilliant blue.
None of the 19 P. syringae strains for which we previously performed comparative genomic analysis encode either hopBH1 or hopBI1 [3]. However, each can be found in P. syringae strains isolated from various sources ranging from non-symptomatic plants to snow [33], [35], [40], [59]–[62] (Figure S5). Amino acid sequence alignments suggest that HopBH1 is a bi-modular effector exhibiting sequence conservation within its C-terminal domain and sequence diversity toward its N-terminal half (Figure S6). In the non-pathogenic strain Psy 642, the putative HopBH1 protein appears to have been disrupted by a frameshift mutation, leading to two putative open reading frames designated as ORF29-30 [40]. Phylogenetic analysis of strains carrying either hopBH1 and/or hopBI1 indicates that both effector genes occur with a mosaic distribution across the P. syringae, but are absent from the phylogenetic group III [3], [27] (Figure S5). Neither HopBH1 nor HopBI1 contain known protein folds, nor do they display sequence or structural homology to proteins of known function.
The HrpL regulons are diverse across isolates
The composition of the HrpL regulon across strains was surveyed by functional classification based on protein annotation and sequence homology determined by BLASTP (Table S6). As summarized in Figure 3 and Table S7, Pto DC3000 and Por possess the largest and most diverse HrpL regulons among the sampled strains, while the Group II strains Pja and Psy B728a have the smallest. We are confident that the less complex HrpL regulons are not a sampling artifact, because the data collected from Pja has a transcriptome depth similar to the other strains, and the Psy B728a HrpL regulon was sampled at relatively high depth compared to our other transcriptomes. We conclude that HrpL regulons vary in size and composition across the P. syringae phylogeny.
Figure 3. HrpL regulons across phylogenetically distinct strains vary in size and composition.

Each gene of every HrpL regulon (after manual curation) was categorized according to its putative function based on annotation, or annotation of its best hit after BLAST against the completely sequenced strains. For details on which category every gene was assigned refer to Table S7.
Recruitment of genes into and out of the HrpL regulon
We observed variable HrpL-dependent expression for several highly conserved non-T3E genes present in all six strains (Table S6). We identified polymorphisms in the hrp-box sequences from two of these (Figure 4A). In the first case, new HrpL-regulated genes we identified, PSPTO_2130, PSPPH_1906 and Lac107_00061530, are orthologs that encode a DNA-binding response regulator. HrpL-dependent induction was confirmed by qRT-PCR (Table 3, Figure 4B). Orthologous genes are also present in Pja, Psy B728a, Por (Pjap_00016990, Psyr_1940, and Porcurated_00527, respectively) but were not identified as differentially expressed (Table S1). PSPTO_2130 and all of its orthologs have conserved hrp-box motifs. However, the promoters of the orthologs from Por and all other group II strains contain single nucleotide polymorphisms (in red, Figure 4A) in the consensus hrp-box sequence. Our RNA-seq data suggested that expression of these polymorphic alleles was not HrpL-dependent, a finding confirmed by qRT-PCR performed with both of our HrpL-regulation experimental tests (Figure 4B, Figure S7A).
Figure 4. hrp-box mutations associated with differential HrpL-dependent up-regulation of PSPTO_2105, and 2130 orthologs.

(A) PSPTO_2105 and PSPTO_2130 hrp-box sequence variation across P. syringae strains as presented in [3]. In blue, canonical hrp-box nucleotides. In red, nucleotides of divergent hrp-box sequences. (B) HrpL-dependent expression of PSPTO_2130 orthologs across P. syringae strains. qRT-PCR analysis was performed on RNA samples derived from isogenic strains expressing, or lacking, an arabinose-inducible hrpL gene. Expression was normalized to gap-1. Relative expression: each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD. (C) HrpL-dependent expression of PSPTO_2105 orthologs across P. syringae strains, as above. Each experiment was repeated twice.
PSPTO_2130 and its orthologs are part of a putative operon composed of four genes, PSPTO_2128-2131 (Figure S8A). Unusually, the hrp-box sequences were located within the first ORF of the putative operons of PSPTO_2130 and its orthologs. We monitored HrpL-dependent expression using qRT-PCR of all genes from PSPTO_2131 to 2128 from three strains (Figure S8B, C, D). In none of these strains was the first gene of the operon, containing the putative hrp-box, differentially expressed. By contrast, HrpL-dependent expression was observed for genes downstream of the predicted hrp-box, including coding sequences, PSPTO_2130 and PSPPH_1906, in all but the group II reference strain Psy B728a (Figure S8). Deletion mutants in Pto DC300 and Pph 1448A of PSPTO_2130 and PSPPH_1906 did not display any growth defect on Arabidopsis accession Col-0 or French bean cultivar Tendergreen (susceptible to Pto DC3000 and Pph 1448A, respectively) (data not shown). Thus, the role of PSPTO_2130 and its orthologs in virulence remains unclear.
In the second case, PSPTO_2105 and its orthologs, which encode a putative ApbE-family protein, are highly conserved across P. syringae and are HrpL-regulated in Pph 1448A, Pla 107, PtoDC3000 and Por but not in the group II strains Psy B728a or Pja (Table S5, S6). qRT-PCR (Figure 4C, Figure S7B) support our RNA-seq data. PSPTO_2105 is required for full virulence of Pto DC3000 on Arabidopsis [18]. We also observed significantly reduced virulence when we tested two independent deletion mutants of the Pph 1448A ortholog PSPPH_1855 for growth on the native host, French beans (Figure S9). Every group II strain analyzed has variations in the otherwise well conserved hrp-box sequence in at least two positions (Figure 4A). Collectively, these data demonstrate that promoter erosion within the hrp-box is a mechanism to remove genes from the virulence regulon.
HrpL regulons of isolates from phylogenetic group II contain fewer non-T3E genes
Both Psy B728a and Pja appear to have relatively small HrpL regulons; both belong to the MLST group II. To address whether this was a general feature of group II strains, and to address the distribution of the genes that we identified experimentally across the phylogeny, we extended our investigation of non-T3E HrpL regulon diversity to BLAST homology searches of 44 sequenced Pseudomonas spp. strains [3], [35], [63]. Our non-T3E gene search set included genes likely to be directly HrpL-regulated, derived from either previous studies [19], [64] or this study. From our study, these included genes we experimentally confirmed for HrpL-dependent expression, genes that encoded proteins found not to be translocated, or genes unlikely to encode a translocated product by annotation. We removed T3SS genes and known T3Es (Figure 5). Most of the directly HrpL-regulated non-T3E genes we identified are absent from group II strains, but distributed across strains from groups I and III. Some are present in the previously described group IV and V, as well as the novel MLST groups VII, IX, X (Berge et al., personal communication, Figure S5) for which we had limited sampling. Further, the promoters of group II homologs of Por curated_02977, 01635 are divergent, and lack canonical hrp-boxes (data not shown). Thus, not only do group II strains possess lower numbers of known T3E genes on average than the other phylogroups, group II strains also possess fewer non-T3E genes in their HrpL regulon suggesting a potential shift in virulence mechanisms of this clade [3].
Figure 5. Strains of phylogenetic group II have the fewest non-effector genes in their HrpL regulons.

“Non-effector” genes are listed across the top; Pseudomonas genomes, color-coded by phylogenetic group, on the left, according to Figure S5 (Pseudomonas UB246 was not included, because it belongs to a divergent pseudomonad lineage). Only the first gene of an operon is represented. Dark blue box indicates presence of full-length ORFs (with at least 80% nucleotide identity and 40% coverage), by similarity search as well as the presence of a hrp-box in the 500 bp upstream region. Grey boxes indicate that the corresponding gene was present, but the presence of putative hrp-boxes could not be determined, due to incomplete sequencing of the upstream region. Yellow boxes indicate that genes are present but either no hrp-boxes were detected in the upstream region or hrp-boxes are presumably not functional (because divergent from the hrp-box for which HrpL-dependent expression was confirmed). White boxes indicate absence of genes based on homology searches. The light blue boxes indicate the presence of a divergent ORF with upstream hrp-box sequence present. S, indicates that the gene is present as a single gene. O, indicates that the gene is present in an operon. + or − indicate that the gene was differentially expressed or not in our RNA-seq data. ND indicates not determined: PSPTO_A00030 is absent from our Pto DC3000 laboratory strain, and a homologous sequence of Psyr_0737 is present in Pja genome but was not annotated as an ORF.
Recruitment of a novel gene cluster into an avrD-containing virulence operon in Pph 1448A
Both Pph 1448A and Pla 107 contain avrD, a gene required for synthesis of syringolides, small molecules sufficient for HR on soybean cultivars expressing the Rpg4 disease resistance gene [65]–[67]. avrD is a non-T3E gene, as defined above (Figure 5), and its expression in E. coli is sufficient for production of syringolides [65]. RNA-seq analysis identified a series of orthologous, HrpL-regulated genes directly downstream of avrD in both Pph 1448A and Pla 107 (Table S3, S6). In Pph 1448A, those genes are arranged in two clusters composed of PSPPH_A0112-A0110 and PSPPH_A0109-A0106, which are flanked by transposable elements (Figure 6A). While most of these genes seem to encode hypothetical proteins, PSPPH_A0112, A0109, A0108, A0107 encode putative enzymes: a phosphoglycerate mutase, a sulfotransferase, an amino transferase, and an oxidoreductase respectively. We confirmed the HrpL-dependent expression of PSPPH_A0112, A0110, A0109, and A0107 (Table 3, Figure S2, and S3). This operon is typically found as a presence/absence polymorphism; when present, it is almost always downstream from avrD (Figure 6B). PSPPH_A0111 corresponds to a 99 bp sequence present in Pla 107, P. syringae pv. mori (Pmo), P. syringae pv. glycinea R4 (Pgy R4), P. syringae pv. tomato T1 (Pto T1), and P. syringae pv. actinidiae (Pan) but not annotated as an ORF, thus it is not represented in the graphical representation of the conserved neighborhood region (Figure 6B). In P. syringae CC1629, this putative operon appears to have been disrupted by insertion of a transposable element. In P. syringae pv aesculi 0893_23 (Pae) this locus is not entirely sequenced. To determine whether avrD is part of an operon with PSPPH_A0112-A0106, we used RT-PCR to confirm that the intragenic regions between avrD/PSPPH_A0112 and between PSPPH_A0110/PSPPH_A0109 were transcribed in wild type Pph 1448A but either very weakly or not at all in the ΔhrpL mutant (Figure 6C).
Figure 6. A novel HrpL-regulated virulence operon linked to avrD in Pph 1448A identified by RNA-seq analysis.

(A) Color coded genomic context of avrD and downstream genes from PSPPH_A0106 to A0112. Grey arrows represent transposases. White arrows represent additional ORFs that are not necessary related. (B) Graphical representation of avrD operon according to IMG/ER conserved neighborhood region search with the 45 Pseudomonas strains presented in Figure 5 and S5. Grey, white, black, and colored arrows as in A. Brackets represent breaks and end of contigs or scaffolds. Not to scale. (C) PCR was performed using primers spanning intragenic regions between PSPPH_A0109 and A0110 (top panels), or PSPPH_A0112 and avrD (middle panels) on cDNA prepared from Pph 1448A (Pph) or an isogenic Pph 1448AΔhrpL mutant (ΔhrpL) grown in MM media. Total RNA was subject to reverse transcriptase (+RT), or without reverse transcriptase (−RT, as negative control). gDNA, DW indicate respectively that genomic DNA or distilled water were used as template for positive and negative controls of amplification. Equal loading was controlled by monitoring gap-1 amplification across samples (bottom panels). (D) Two week old bean cv. Tendergreen beans were dip inoculated with wild type Pph 1448A (Pph), two independent clean deletion avrD mutants (ΔavrD #1, ΔavrD #2), and two independent clean deletion PSPPH_A0107 mutants (ΔPSPPH_A0107 #1, ΔPSPPH_A0107 #2) at OD600 = 0.001. Bacterial growth of each strain was determined after 3.5 dpi. Letters represent significant differences with P<0.05 according to Tukey's highly significant difference and error bars display standard deviation. This experiment was repeated at least twice.
We generated two independent deletion mutants for avrD and PSPPH_A0107 (ΔavrD #1 and 2, ΔPSPPH_A0107 # 1 and 2, respectively) and tested their growth on French bean cv. Tendergreen (Figure 6D). All mutants displayed reduced growth compared to wild type Pph 1448A (Pph), indicating that both avrD and PSPPH_A0107 are required for full virulence on cv. Tendergreen. We confirmed that the HrpL-dependent expression of several downstream genes was not disrupted by mutations (Figure S10). However, PSPPH_A0112, A0107 and A0106 were consistently slightly up-regulated in avrD mutants compared to the wild type. The intact remaining hrp-box is closer to PSPPH_A0112-A0106 in the avrD mutants, which could account for increased transcript levels. Additionally, these data could explain why the avrD mutants displayed a reduced growth defect compared to the ΔPSPPH_A0107 mutants (Figure 6D). We speculate that these non-T3E genes are involved in the synthesis of a secondary metabolite(s) required for virulence of Pph 1448A.
Discussion
P. syringae is a broadly distributed and agronomically important pathogen of many plant species. Full virulence for many strains within this species requires expression of genes induced by the sigma factor HrpL, but the HrpL regulon has only been systematically surveyed using microarrays in Pto DC3000 [16], [17] and to a limited extent by promoter probe studies in a few strains [3], [19]. Using RNA-seq, we successfully defined HrpL regulons across six phylogenetically diverse strains. We benchmarked our data set with previous transcriptional studies of two reference genomes [16]–[19] (Table 2) and with qRT-PCR analysis (Table 3, Figure S2, Figure S3). Our approach allowed us to efficiently define the HrpL regulon of multiple strains, even those for which only draft genome sequence is available. We found a plethora of non-T3E genes in these regulons and experimentally verified both newly identified T3Es and non-T3E virulence factors. Additionally, we identified a variety of mechanisms that could drive recruitment into and loss from, the main virulence regulon of P. syringae.
Identification of two novel HrpL-regulated type III effectors in Por
We identified HopBH1Por and HopBI1Por, defining two novel effector families. Both have a mosaic phylogenetic distribution across P. syringae [35], [40], [63] (and an unpublished strain, TLP2, JGI taxon ID: 2507262033). Both are present in CC1513 and CC1629, two other strains belonging to the MLST group IV. They appear to be absent from sequenced MLST group III strains. HopBH1 has a bi-modular structure. The ∼170 amino-acid N-terminus is divergent compared to the relatively well conserved ∼250 amino acid C-terminal domain across HopBH1 alleles (Figure S6). The HopBH1 C-terminal domain is 50% identical to a protein from P. fluorescens SS101 which lacks a putative hrp-box or a T3SS secretion competent N-terminal sequence [68], suggesting that it may have been recruited as an effector by N-terminal assortment [69]. Several putative proteins present in Pantoea, Serratia, Burkholderia species, as well as Myxobacteria, display ∼50% identity with the HopBH1 C-terminal domain. Remarkably, about 150 amino acids of the HopBH1 C-terminal domain also shares 40% identity with part of the ∼1000 amino acid long P. savastanoi pv. savastanoi NCPPB3335 HrpK. Notably, this hrpK gene (PSA3335_2516) is from a rhizobia-like type III secretion and is different from the hrpK(Pto) (PSA3335_1389) of canonical T3SS conserved in plant pathogenic P. syringae [70]. HopBI appears to be confined to Pseudomonas. Neither HopBH1, nor HopBI1 display similarity to known-effectors. Their virulence functions remain to be determined.
HrpL regulons include diverse non-T3E genes some of which are known virulence factors
Although analysis of type III virulence systems focuses mainly on the characterization and function of T3SS and T3E proteins, several non-T3E genes are co-regulated with the T3SS. They encode hypothetical proteins, transporters, or enzymes likely involved in secondary metabolism (Figure 5). In contrast to T3E genes, for which functional redundancy is predominant and generation of multiple effector mutants is often required to affect virulence [54], [57], [71], [72], several single knockout mutants of non-T3E HrpL-regulated genes in Pto DC3000 and Pph 1448A displayed reduced virulence on Arabidopsis and beans [18], [73]. In general, little is known about the non-T3E genes in HrpL regulons, but homology provides reasonable scenarios for several that we identified, and we functionally validated others (below).
Among our collection of diverse HrpL-regulated, non-T3E genes, none are present in the HrpL regulon of all six strains tested, and nearly all are distributed in a mosaic pattern among the genomes of available strains (Figure 5).
PSPTO_0370 and orthologs encode a MATE efflux transporter present in an operon with iaaL which is involved in auxin conjugation to IAA-Lys [74]. Porcurated_02977 encodes a putative indole-3-glycerol phosphate synthase. Both potentially alter auxin signaling and could interfere with the balance between immune response and growth and development [75].
Several other putative transporters were identified as HrpL-regulated. PSPTO_2691 encodes a putative membrane protein TerC; PSPTO_0871 a putative macrolide efflux protein; Porcurated_01635 a putative threonine efflux protein; and PSPTO_0838 a putative major facilitator family transporter. Co-regulation of putative transporters with the T3SS suggests that promotion of nutrient acquisition, export of secondary metabolites, or detoxification of plant-encoded antimicrobials are important features of the virulence regulon.
PSPTO_0834, encoding a putative alcohol dehydrogenase, is the first gene of a putative operon comprising five genes (up to PSPTO_0838). This operon includes genes of unknown function, genes encoding a putative bifunctional deaminase-reductase enzyme and a transporter. The function of this operon remains unknown but at least PSPTO_0834 is required for full virulence of Pto DC3000 on Arabidopsis [18].
The PSPTO_0873-0875 putative operon is widely distributed across Pseudomonas and Erwinia species and also present in Pantoea stewartii pv. stewartii DC283. In Erwinia and P. stewartii, this operon is physically linked to the T3SS and is HrpL-regulated. PSPTO_0873 is a putative amidinotransferase that makes ornithine and homo-arginine from arginine and lysine. Ornithine or homo-arginine may be then incorporated into a tri- or di-peptide natural product generated by the rest of this operon. Most interestingly, hsvC, hsvB, hsvA from Erwinia amylovora, corresponding to PSPTO_0873-0875, are required for full virulence on apple shoots [76].
PSPTO_2105 and orthologs encode a protein similar to ApbE from Salmonella typhimurium involved in thiamine synthesis. ApbE was identified through the analysis of several mutants defective in thiamine biosynthesis, and was implicated in iron-sulfur cluster biosynthesis/repair, as well as FAD binding [77]–[79] suggesting a role during oxidative stress [78]. PSPTO_2105 is required for full virulence of Pto DC3000 on Arabidopsis [18]. We extend this finding by showing that the PSPPH_1855 ortholog of PSPTO_2105 is required for full virulence of Pph 1448A on French bean (Figure S9).
PSPTO_2130 and orthologs encode LuxR family DNA-binding response regulators that may be involved in regulation of regulons downstream of HrpL. Our deletion mutants of this gene in Pto DC3000 and Pph 1448A, or of the entire Pto DC3000 operon, did not alter growth on Arabidopsis or French bean cv. Tendergreen, respectively (data not shown), undermining the probability of a necessary function during plant colonization in our experimental conditions. However this operon is conserved across Pseudomonas, and PFLU_2937, the ortholog of PSPTO_2129 from P. fluorescence SBW25, was identified as a plant-induced gene [80]. It therefore remains plausible that this operon is involved in plant association.
Porcurated_04644 appears to encode a putative RNA N-methyltransferase, while the hypothetical protein Porcurated_03530 has homology to FliB which, in Salmonella, is responsible for methylation of flagellin [81]. We speculate that both may be involved in modification of conserved molecules known to induce host defense responses [82]–[84].
avrD is widely distributed across bacteria and is involved in the synthesis of syringolides [85]. Syringolides are elicitors of cell death in soybean expressing the Rpg4 disease resistance gene [86], [87]. The putative function of avrD is discussed below.
The group II strains have smaller HrpL regulons
One of our most striking comparative observations is the relatively small size and diversity of the HrpL regulons of the phylogenetic group II strains PsyB728a and Pja. We observed that most of the non-T3E genes known to be HrpL-regulated in other strains are not present, or lack HrpL-regulation in group II strains, underpinning the conclusion that the limited regulon observed for Psy B728a and Pja can most likely be generalized to all group II strains (Figure 5). They also contain fewer T3Es than the other clades [3]. The group II strains carry genes for phytotoxins not shared by other P. syringae groups. Expression of these phytotoxins is not regulated by HrpL, and could compensate for missing T3E functions, making a smaller T3E repertoire sufficient to suppress plant defenses [3].
Modes of recruitment of non-T3E genes into, and out of, the HrpL regulon
Turnover within the HrpL regulon is known to be influenced by gene gain and loss, mediated by association of genes within the regulon with mobile elements and horizontal gene transfer (data not shown, Figure 6 A, B). However, we also observed that all the group II strains analyzed here have polymorphisms in the hrp-box sequence that correlated with the loss of HrpL-dependent regulation of PSPTO_2105 and orthologs (likely encoding AbpE). Several different polymorphisms within the hrp-box were observed, suggesting independent mutational events (Figure 4). Additionally, the group II strain orthologs of PSPTO_2130 (LuxR family), carry nucleotide polymorphisms in the consensus hrp-box, and are not HrpL-regulated (Figure 4). Orthologous genes from Por also display nucleotide variation in this hrp-box, also leading to loss of HrpL-regulation. The substitution patterns of these alterations suggest multiple, independent losses of HrpL-regulation. PSPTO_2130 and its orthologs are part of an operon where the consensus hrp-box is embedded within the first ORF in this operon (Figure S8) and is thus likely to be constrained by the genetic code. Interestingly, PSPTO_2130 and its orthologs have variation in the second half of the hrp box where CCAC is replaced by TCAC. This hrp-box motif, while uncommon, is also found in PSPTO_0370, PORcurated_01251 (hopAO1Por), and Pjap_00002060 (hopC1Pja), each of which we defined as HrpL-regulated.
The promoter erosion we observe could be driven by negative host selection pressure, or weak selection for maintenance of HrpL regulon membership combined with subsequent drift. Similarly, reversion of at least the SNPs could quickly recruit genes back into the HrpL regulon. Because the ORFs have not accumulated stop mutations, these promoter mutations are either relatively recent or there is active maintenance of the ORF sequence, perhaps for expression under different conditions.
Horizontal transfer or other types of recombination could explain how 5′ regions diverge and how these regions and associated genes are recruited in to the HrpL regulon. Porcurated_02977, 01635, and 04371, encode an indole-3-glycerol phosphate synthase, a putative threonine efflux transporter and a hypothetical protein, respectively, that are HrpL-regulated. Similar genes are present in Pja and Psy B728a but are not HrpL-regulated (Figure 5). Putative hrp-boxes can be identified in all three Por genes, but not for the corresponding genes in Pja and Psy B728a. These genes are not syntenic (data not shown). They display high similarity in their coding sequence (data not shown); however their corresponding 5′ upstream regions are highly divergent. This could be the result of horizontal transfer, though there is no obvious footprint of mobile element DNA, or independent recombination events.
Lastly, loss of transcription termination regulation could lead to read-through transcription, and thus provide a mechanism for recruitment of non-T3E genes into the HrpL regulon. This mechanism was first highlighted by the recruitment into the HrpL regulon of the corR gene which was recombined downstream of the hrp-box associated hopAQ1 gene, in Pto DC3000 [25]. We observed that several genes found differentially expressed in our analysis were located downstream of HrpL-regulated T3E genes (Table S3) and could potentially be recruited into the HrpL regulon via loss of transcription termination regulation and subsequent transcriptional read-through.
Recruitment of a novel gene cluster into an avrD-containing virulence operon in Pph 1448A
We identified a cluster of HrpL-regulated genes, PSPPH_A0106-A0112, downstream from avrD that were recruited into a novel HrpL-regulated operon transcribed from the avrD promoter. These genes are flanked in Pla 107 and Pph 1448A, by transposable elements, suggesting that they could be acquired by horizontal gene transfer (Figure 6). Deletion mutants of either PSPPH_A0107 or avrD resulted in reduced virulence on French bean. The slightly reduced virulence we observed is in conflict with observations that allelic replacement of avrD by the nptII gene did not result in any growth defect in completive index assays [72]. This discrepancy could be explained by transcription from the nptII promoter in the previous work, or by the use of different growth assays, time points, and bean cultivars.
The PSPPH_A0106-A0112 operon is most likely involved in small molecule(s) synthesis promoting bacterial growth on host plants. Component(s) synthesized by the products of this operon and their effect on plants remain to be determined. However, since syringolides can be made from AvrD-expressing E. coli, and since the PSPPH_A0106-A0112 operon is not present in E. coli, we speculate that that the PSPPH_A0106-A0112 operon is not required for syringolide production. When present, AvrD shares no less than 84% amino acid identity across P. syringae strains. Genes encoding an AvrD-like protein with about 30% identity are widely distributed among bacteria, including Bacillus, Streptomyces and Vibrio. In general, these avrD-like genes are not found as singletons, but instead are linked to genes encoding various enzymes not related to any of the PSPPH_A0112-A0106 genes. In Streptomyces coelicolor A3(2), AvrD is part of an mmy operon responsible for synthesis of methylenomycin [88]. The PSPPH_A0110 to PSPPH_A0107 locus and to some extent the PSPPH_A0106 genes have similarity to genes in operons from Xanthomonas, Acidovorax, Pectobacterium and Ralstonia. Only the Ralstonia solanacearum PSI07 megaplasmid, carries both an avrD-like gene and a PSPPH_A0110-A0106 cluster of genes, but they are not contiguous on this plasmid. PSPPH_A0112 is mainly limited to P. syringae, but shares some homology with HMPREF9336_00100 (29% amino acid identity) found in Segniliparus rugosus ATCC BAA-974, an opportunistic pathogen associated with mammalian lung disease [89]. HMPREF9336_00100 and an avrD-like gene are linked in Segniliparus rugosus, being separated by only two genes and encoded on the same strand. We additionally observed that this operon has been disrupted by insertion of a transposable element in P. syringae CC1629, reminiscent of transposon disruptions of T3E genes commonly observed across the P. syringae phylogeny [3].
hrpL is widely distributed, and tightly linked in all hrp/hrc group I T3SS [1] and the non-canonical T3SS found in some P. syringae, as well as the T3SS of P. viridiflava, P. fluorescens, Erwinia, Pantoea stewartii, and Dickeya. It is the key virulence regulator in most if not all of these species. Our work highlights the advantages of integrating next generation transcriptional and genomic data to better understand the role of non-T3E HrpL regulon genes in plant-pathogen interactions. Our approach is readily applied to strains with sequenced genomes and broad phylogenetic sampling [63] to better understand P. syringae virulence mechanisms and their evolution.
Materials and Methods
Bacterial strains and growth conditions
For maintenance and transformation, P. syringae were grown in King's B media (KB) at 28°C. E. coli DH5α was grown in Luria-Bertani (LB) media at 37°C. Antibiotics were used at the following concentrations: 50 µg/ml rifampicin, 25 µg/ml kanamycin, 10 µg/ml tetracycline, and 25 µg/ml gentamycin, according to vector selection. Strains used or analyzed in this study are listed with their abbreviation in Table S8.
Cloning and plasmids
Native hrpL from the various P. syringae were PCR amplified using LA-Taq (TaKaRa) and oligonucleotides listed in Table S9 containing XbaI and Hind III sites, then cloned into pTOPO-TA (Invitrogen). The pTOPO-TA::hrpL was sequenced, digested with XbaI and HindIII, and cloned into NheI/HindIII-digested pCF340 (Newman and Fuqua, 1999) and designated pBAD::hrpL.
Porcurated_02784, 04644, 03530, 01245, 04371 and their respective upstream region containing the hrp-box were PCR amplified using Pfx (Invitrogen) and primers described in Table S9. Resulting PCR fragments were cloned into pENTR-D-TOPO (Invitrogen) and sequenced. Porcurated_04644 was amplified similarly using primers containing attB1/attB2 sites and cloned into the pDONR 207 vector (Invitrogen). All resulting constructs were sub-cloned into either the gateway-compatible pJC532 vector containing the in-frame Δ79avrRpt2 sequence for translocation assays or the pJC531 vector containing an in-frame HA sequence [3] to check for protein expression. All vectors used in this study were transformed into P. syringae strains using tri-parental mating with an E. coli helper strain containing pRK2013.
Preparation of samples for RNA-seq analysis
Pseudomonas strains containing pBAD::hrpL native or pBAD::EV were grown overnight at 28°C, in KB media supplemented with tetracycline, then sub-cultured in fresh media at OD600 = 0.2, and grown until OD600 = 0.4–0.5. Bacteria were washed twice with 10 mM MgCl2 and resuspended in minimal medium [48] (MM is 50 mMKPO4 pH 5.7, 7.6 mM (NH4)2SO4, 1.7 mM MgCl2, 1.7 mM NaCl) containing 10 mM mannitol and supplemented with 1% glycerol and 0.1% peptone which suppresses hrpL induction. Bacteria were then inoculated in supplemented minimal media at OD600 = 0.2, and incubated shaking for 30 min at 28°C. Expression of hrpL was induced by addition of 200 mM L-arabinose. Aliquots of cell culture were taken 1, 3, 5 hours post-induction and treated with RNAprotect reagent (Qiagen). RNA isolation was performed by using the RNeasy minikit (Qiagen). Isolated total RNA was treated twice with TURBO DNase (Ambion). Total RNAs derived from each time point were pooled at a 1 to 3 ratio. 10 µg of pooled total RNA was depleted of 16S and 23S ribosomal RNA using RiboMinus (Invitrogen). cDNA were prepared from ∼1 µg of ribosomal depleted RNA, using random hexamer primers and Superscript II reverse transcriptase (Invitrogen). Second strand cDNA was prepared using DNA polymerase I and Ribonuclease H (Invitrogen). Double stranded cDNA was purified using Qiaquick spin columns (Qiagen) and eluted with EB buffer. Double stranded cDNA was sheared using a Covaris Disruptor. Library was prepared according to the manufacturer's protocol (Illumina). Sequencing of the library was performed according the manufacturer's protocol on either Illumina GAII including single-end, 36 cycles or Illumina HiSeq 2000 including single-end, 70 cycles.
Sequence mapping and analysis
We analyzed our RNA reads using the GENE-counter pipeline. For the Pto DC3000, Psy B728A, and Pph 1448A datasets, we used the publically available genomes provided by NCBI, along with the transcriptome constructed by NCBI's gene prediction pipeline. For the Por, Pjap and Pla107 dataset, we used in-house assembly for the genome and used JGI's Integrated Microbial Genomes – Expert Review gene prediction pipeline for the transcriptome. All ribosomal RNA genes were excluded from the transcriptome file for all datasets. Transcriptome sequences for each strain were blasted against their corresponding genome and GFF files were constructed from the Blast reports using an in-house script. We processed the RNA reads and aligned the reads using the default parameters of GENE-counter's CASHX read mapping algorithm. Reads mapping to multiple genomic locations were excluded. Annotated genes were included in the analysis only if at least one read in one sample matched that gene which can lead to highly duplicated genes not being considered. The false discovery rate cutoff for determining differential expression was set to 0.05. We made a small modification to GENE-counter's findDGE.pl script that allowed for random seeding during the sample depth normalization process. By repeating the normalization process 300 times we generated B-values to measure and control for normalization effects. The GenBank accession (http://www.ncbi.nlm.nih.gov/) and Gold ID (http://img.jgi.doe.gov/cgi-bin/w/main.cgi) of the genomes used in this study are CP000058-CP000060, Gi04410, CP000075, Gi07003, Gi03478, and AE016853-AE016855. RNA-seq data have been deposited in NCBI Gene Expression Omnibus and will be accessible through GEO Series accession number GSE46930 (http://www.ncbi.nlm.nih.gov/geo/).
Manual curation of data set and designation of HrpL regulon
First, protein sequences of genes found up-regulated in our analysis with B-values≥50% were used to search each genome used in this study with BlastP to identify genes split up in different contigs/scaffolds. Possible duplication was ruled out by comparing the size of the query to the size of the subject sequence (of complete genomes, principally). Putative sequencing errors leading to stop codons and discontinuous ORFs, led to consecutive queries matching the same subject sequence. Only the entry with the most significant q-value was kept. Secondly, genes encoding open reading frames shorter than 60 amino acids were excluded from our data set. Thirdly, loci of genes not previously found HrpL-dependent were assessed for linkage to genes with a hrp-box. As previously described [41], [90], we observed potential transcriptional read-through artifacts for which directly HrpL-targeted genes led to apparent up-regulation of adjacent genes. Therefore, genes found differentially expressed adjacent to a HrpL-regulated gene, but on the opposite DNA strand were considered to be putative transcriptional read-through and removed from our analysis. Genes encoded on the same strand as the HrpL-regulated gene were kept. Fourth, genes with a hrp-box embedded within their ORF on either sense or anti-sense strands were not included. Adjacent genes encoded on the same strand as the manually predicted hrp-box were included in the defined HrpL regulon, but genes on the opposite strand of the hrp-box were excluded. All genes removed from the HrpL regulons after manual curation are listed Table S4.
Quantitative RT-PCR
For native gene expression, bacteria were grown for 4 hours in KB media from OD600 = 0.2, washed twice with sterile 10 mM MgCl2 and transferred into MM minimum media containing 10 mM mannitol for PtoDC3000, PsyB728a, Pla107, Pja, Por strains or MM minimum media containing 10 mM fructose for Pph 1448A strain. Cells were collected after 5 hours of incubation shaking at 28°C and treated with RNAprotect reagent (Qiagen). Total RNA derived from cells grown in MM media or arabinose inducing media (as above) was extracted using the RNeasy minikit (Qiagen), DNase treated twice (Ambion Turbo DNase), and cleaned up with Qiagen RNeasy Mini kit. Reverse transcription was performed using SuperScript II (Invitrogen) with 2 µg total RNA. Diluted cDNA was analyzed with SYBR green (Applied Biosystem) using the Opticon 2 System (BioRad). Primers used are listed in Table S9.
Δ79avrRpt2-based translocation assay
Four week old Col-0 and Col-0 rps2–101c (rps2) plants were hand inoculated with Pto DC300028E [57] carrying Δ79avrRpt2 fusion clones at OD600 = 0.1. Plants were scored for Hypersensitive Response (HR) and pictures were taken 24 h after inoculation.
Generation of P. syringae knockout mutants
Knockout constructs were generated using MTN1907, a modified version of pLVC-D which allows for SacB counter-selection [3], [91]. To create Pph 1448AΔPSPPH_A0107, Pph1448AΔPSPPH_A0113 mutants, 5′ and 3′ regions flanking the gene of interest were amplified using Pfx (Invitrogen) and combined by overlap extension PCR (Table S9), then cloned into pENTR-D-TOPO and sequenced. To generate the Pph 1448AΔPSPPH_1855, PsyB728aΔhrpL and PorΔhrpL mutants, nucleotide sequences corresponding to the fused flanking regions of each gene were synthesized including Gateway recombination sites and cloned in the pUC17 vector (GenScript). All five clones were recombined into MTN1907 and transformed into either Pph1448A, PsyB728a or Por by tri-parental mating. After selection on tetracycline plates, merodiploids resulting from homologous recombination were verified by PCR. Two independent merodiploids carrying either a 3′ or 5′ insertion were grown on KB agar containing 5% sucrose to select for the loss of sacB via a second recombination event. Putative clean-deletion mutants were verified by PCR using flanking primers and gene specific primers.
Bacterial growth on French bean
Before inoculation, Pph 1448A and mutants were grown overnight and sub-cultured from OD600 = 0.2 for 4 hours in KB media, then washed twice with 10 mM MgCl2. Two week old French bean cv. Tendergreen improved (Livingston Seed Co.) were dip inoculated with freshly grown bacteria at OD600 = 0.001 bacteria in 10 mM MgCl2 and 0.04% Silwet L-77. Four plants were dip inoculated for each strain. Three days and an half after inoculation leaf discs were cored (12 to 16 replicates, each 4 cores), ground in 10 mM MgCl2, serially diluted and plated on KB/50 µg/ml rifampicin and bacteria counted. Each set of mutants were tested side by side with the wild type strain at least 3 times.
Supporting Information
Graphical representation of our experimental pipeline. Isogenic P. syringae strains carrying either pBAD::EV or pBAD::hrpL were grown on MM media supplemented with arabinose and collected 1, 3 and 5 hours post induction. RNA was extracted for each time point and cDNA prepared to confirm induction of hrpL and hrcC for P. syringae pBAD::hrpL. Total RNA for each time point was pooled equally. Pooled RNA for each strain was subjected to rRNA removal and double stranded cDNA prepared (Materials and Methods). Illumina libraries were prepared according to manufacturer's protocol and sequenced. Resulting reads were used to run GENE-counter. After 300 bootstraps of GENE-counter, genes from each sample were assigned a median read count, a median p and q-value as well as a B-value.
(TIF)
Detailed results of qRT-PCRs described in Table 3 from samples derived from the pBAD system. (A) For Pto DC3000 genes, (B) for Pph 1448A genes, (C) for Psy B728a genes, (D) for Por genes. cDNA was prepared from the same total RNA used to generate our RNA-seq data for all strains except Pto DC3000. For Pto DC3000, cDNAs were prepared from an independent biological replicate. Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD.
(TIF)
Detailed results of qRT-PCRs described in Table 3 from samples derived from isogenic strains grown in MM media. (A) For Pto DC3000 genes, (B) for Pph 1448A genes, (C) for Psy B728a genes, (D) for Por genes. cDNA was prepared from wild type strains and corresponding isogenic ΔhrpL mutants grown in MM media for 5 hours. Expression was normalized to gap-1. For determination of the relative % expression, each wild type strain sample was set to 100% and ΔhrpL samples normalized accordingly. Error bars represent SD.
(TIF)
The majority of effector genes are found up-regulated in our analysis. Effector suites present in each strain are listed. In bold, effector genes found up-regulated by RNA-seq. Underlined, effector genes HrpL-dependent according to Pseudomonas syringae Genome Resources, (PPI database http://www.pseudomonas-syringae.org/) but not found up-regulated in our analysis. In grey, effector genes previously identified according to a combination of homology and functional criteria described in Chang et al., 2005 and Baltrus et al., 2011 but not found to be HrpL-regulated in these strains in any experiment, to our knowledge. ‘ indicates insertion or truncation according to PPI database. The new Por type III effectors defined in this study are listed in red.
(TIF)
hopBH1 and hopBI1 are both present across phylogenetically diverse strains of P. syringae. (A) Bayesian phylogenetic tree of 45 Pseudomonas strains [3], [40], [63] based on seven conserved loci as described in [3]. Bayesian posterior probabilities are displayed on the phylogeny only at nodes where these values are <0.95. Each phylogenetic group (defined according to Berge et al., personal communication and [27]) is color coded. (B) Distribution of hopBH1 and hopBI1 across the 45 Pseudomonas strains. Dark blue boxes indicate presence of corresponding full length ORF. Light Blue box indicates truncated ORF. White boxes indicate absence of corresponding ORF in the sequenced genome.
(TIF)
Amino-acid sequence alignment of HopBH1. Alignment performed using clustal W with sequences from all P. syringae strains known to date to contain hopBH1.
(TIF)
Expression of PSPTO_2105 and 2130 or their orthologs under native conditions supports our results obtained with arabinose-inducible hrpL system. (A) Relative expression of PSPTO_2130 and its orthologs. (B) Relative expression of PSPTO_2105 and its orthologs. qRT-PCR analysis was performed on RNA samples derived from wild type strains and the cognate isogenic ΔhrpL mutant grown in MM media. Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD.
(TIF)
HrpL-dependent up-regulation of genes downstream of PSPTO_2130 and its orthologs in Pto DC3000, Pph 1448A, but not Psy B728a. (A), Graphical representation of PSPTO_2131-PSPTO-2128 operon. qRT-PCR analysis was performed on RNA samples derived from (B) Pto DC3000(pBAD::hrpL) and Pto DC3000(pBAD::EV) (C) Pph 1448A(pBAD::hrpL) and Pph 1448A(pBAD::EV); and (D) Psy B728a(pBAD::hrpL) and Psy B728a(pBAD::EV). ORF nomenclature for operons from Pph 1448A and Psy B728a in C and D, respectively, is listed directly under the corresponding ORF numbers in Pto DC3000 in B. Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD. qRT-PCR analysis was performed on RNA samples derived from (E) Pto DC3000 and Pto DC3000 ΔhrpL (F) Pph 1448A and Pph 1448A ΔhrpL, For determination of the relative % expression, each wild type strain sample was set to 100% and ΔhrpL samples normalized accordingly. Error bars represent SD.
(TIF)
Pph 1448A mutants deleted in thiamine biosynthesis lipoprotein gene display reduced growth on Tendergreen beans. Two week old bean cv. Tendergreen beans were dip inoculated with wild type Pph 1448A (Pph) or two independent mutants with a clean deletion of PSPPH_1855 (PphΔ1855 #1, PphΔ1855 #2), at OD600 = 0.001. Bacterial growth of each strain was determined after 3.5 dpi. Error bars represent SD. This experiment was repeated twice with similar results.
(TIF)
Determination of the relative expression of avrD PSPPH_A0112, _A0107, _A0106 in various Pph 1448A mutants. qRT-PCR was performed on cDNA derived from wild type Pph 1448A (Pph), Pph 1448AΔhrpL, two independent clean deletion avrD mutants (ΔavrD #1, ΔavrD #2), and two independent clean deletion PSPPH_A0107 mutants (ΔA0107 #1, ΔA0107 #2). Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD. This experiment was repeated 3 times with similar results.
(TIF)
Raw bootstrapped GENE-counter results for each strain.
(XLSX)
Genes found down-regulated by RNA-seq.
(XLSX)
Our defined HrpL regulons across various P. syringae after manual curation.
(XLSX)
List of genes excluded from analysis after manual curation.
(XLSX)
List of “confirmed” HrpL-regulated genes for Pto DC3000 and Pph 1448A according to previous studies.
(XLSX)
BlastP search results of the product of each HrpL regulon gene across the strains tested in this study.
(XLSX)
Details on the attribution of the functional classification for each HrpL regulon gene.
(XLSX)
List of Pseudomonas strains used or analyzed in this study.
(XLSX)
List of primers and sequences used in this study.
(XLSX)
Acknowledgments
We thank Barclay McGhee and Dr. Yijian He for technical assistance.
Funding Statement
This work was supported by NIH training grant Predoctoral Training Program in Bioinformatics and Computational Biology T32 GM067553-06 (SY, CDJ, JLD) and R01-GM066025 (CDJ, JLD, SRG). JSC was supported by the National Research Initiative Competitive Grants Program Grant no. 2008-35600-04691 (JHC). JLD is an HHMI Investigator and this work was funded in part by the Howard Hughes Medical Institute and the Gordon and Betty Moore Foundation (via Grant GBMF3030). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Troisfontaines P, Cornelis GR (2005) Type III secretion: more systems than you think. Physiology (Bethesda) 20: 326–339. [DOI] [PubMed] [Google Scholar]
- 2. Naum M, Brown EW, Mason-Gamer RJ (2009) Phylogenetic evidence for extensive horizontal gene transfer of type III secretion system genes among enterobacterial plant pathogens. Microbiology 155: 3187–3199. [DOI] [PubMed] [Google Scholar]
- 3. Baltrus DA, Nishimura MT, Romanchuk A, Chang JH, Mukhtar MS, et al. (2011) Dynamic Evolution of Pathogenicity Revealed by Sequencing and Comparative Genomics of 19 Pseudomonas syringae Isolates. PLoS Pathog 7: e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirano SS, Upper CD (2000) Bacteria in the leaf ecosystem with emphasis on Pseudomonas syringae-a pathogen, ice nucleus, and epiphyte. Microbiol Mol Biol Rev 64: 624–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morris CE, Monteil CL, Berge O (2013) The Life History of Pseudomonas syringae: Linking Agriculture to Earth System Processes. Annu Rev Phytopathol 51: 85–104. [DOI] [PubMed] [Google Scholar]
- 6. O'Brien HE, Desveaux D, Guttman DS (2011) Next-generation genomics of Pseudomonas syringae . Curr Opin Microbiol 14: 24–30. [DOI] [PubMed] [Google Scholar]
- 7. Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, et al. (2000) The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc Natl Acad Sci U S A 97: 4856–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stavrinides J, McCann HC, Guttman DS (2008) Host-pathogen interplay and the evolution of bacterial effectors. Cell Microbiol 10: 285–292. [DOI] [PubMed] [Google Scholar]
- 9. Rohmer L, Guttman DS, Dangl JL (2004) Diverse evolutionary mechanisms shape the type III effector virulence factor repertoire in the plant pathogen Pseudomonas syringae . Genetics 167: 1341–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindeberg M, Myers CR, Collmer A, Schneider DJ (2008) Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol Plant Microbe Interact 21: 685–700. [DOI] [PubMed] [Google Scholar]
- 11. Jones JD, Dangl JL (2006) The plant immune system. Nature 444: 323–329. [DOI] [PubMed] [Google Scholar]
- 12. Xiao Y, Heu S, Yi J, Lu Y, Hutcheson SW (1994) Identification of a putative alternate sigma factor and characterization of a multicomponent regulatory cascade controlling the expression of Pseudomonas syringae pv. syringae Pss61 hrp and hrmA genes. J Bacteriol 176: 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang X, Xiao Y, Zhou JM (2006) Regulation of the type III secretion system in phytopathogenic bacteria. Mol Plant Microbe Interact 19: 1159–1166. [DOI] [PubMed] [Google Scholar]
- 14. Xiao Y, Hutcheson SW (1994) A single promoter sequence recognized by a newly identified alternate sigma factor directs expression of pathogenicity and host range determinants in Pseudomonas syringae . J Bacteriol 176: 3089–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fouts DE, Abramovitch RB, Alfano JR, Baldo AM, Buell CR, et al. (2002) Genomewide identification of Pseudomonas syringae pv. tomato DC3000 promoters controlled by the HrpL alternative sigma factor. Proc Natl Acad Sci U S A 99: 2275–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lan L, Deng X, Zhou J, Tang X (2006) Genome-wide gene expression analysis of Pseudomonas syringae pv. tomato DC3000 reveals overlapping and distinct pathways regulated by hrpL and hrpRS . Mol Plant Microbe Interact 19: 976–987. [DOI] [PubMed] [Google Scholar]
- 17. Ferreira AO, Myers CR, Gordon JS, Martin GB, Vencato M, et al. (2006) Whole-genome expression profiling defines the HrpL regulon of Pseudomonas syringae pv. tomato DC3000, allows de novo reconstruction of the Hrp cis clement, and identifies novel coregulated genes. Mol Plant Microbe Interact 19: 1167–1179. [DOI] [PubMed] [Google Scholar]
- 18. Vencato M, Tian F, Alfano JR, Buell CR, Cartinhour S, et al. (2006) Bioinformatics-enabled identification of the HrpL regulon and type III secretion system effector proteins of Pseudomonas syringae pv. phaseolicola 1448A. Mol Plant Microbe Interact 19: 1193–1206. [DOI] [PubMed] [Google Scholar]
- 19. Chang JH, Urbach JM, Law TF, Arnold LW, Hu A, et al. (2005) A high-throughput, near-saturating screen for type III effector genes from Pseudomonas syringae . Proc Natl Acad Sci U S A 102: 2549–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McNally RR, Toth IK, Cock PJ, Pritchard L, Hedley PE, et al. (2012) Genetic characterization of the HrpL regulon of the fire blight pathogen Erwinia amylovora reveals novel virulence factors. Mol Plant Pathol 13: 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Occhialini A, Cunnac S, Reymond N, Genin S, Boucher C (2005) Genome-wide analysis of gene expression in Ralstonia solanacearum reveals that the hrpB gene acts as a regulatory switch controlling multiple virulence pathways. Mol Plant Microbe Interact 18: 938–949. [DOI] [PubMed] [Google Scholar]
- 22. Noel L, Thieme F, Nennstiel D, Bonas U (2001) cDNA-AFLP analysis unravels a genome-wide hrpG-regulon in the plant pathogen Xanthomonas campestris pv. vesicatoria . Mol Microbiol 41: 1271–1281. [DOI] [PubMed] [Google Scholar]
- 23. Schmidtke C, Findeiss S, Sharma CM, Kuhfuss J, Hoffmann S, et al. (2012) Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res 40: 2020–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hogan CS, Mole BM, Grant SR, Willis DK, Charkowski AO (2013) The Type III Secreted Effector DspE Is Required Early in Solanum tuberosum Leaf Infection by Pectobacterium carotovorum to Cause Cell Death, and Requires Wx(3-6)D/E Motifs. PLoS One 8: e65534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sreedharan A, Penaloza-Vazquez A, Kunkel BN, Bender CL (2006) CorR regulates multiple components of virulence in Pseudomonas syringae pv. tomato DC3000. Mol Plant Microbe Interact 19: 768–779. [DOI] [PubMed] [Google Scholar]
- 26. Ullrich M, Penaloza-Vazquez A, Bailey AM, Bender CL (1995) A modified two-component regulatory system is involved in temperature-dependent biosynthesis of the Pseudomonas syringae phytotoxin coronatine. J Bacteriol 177: 6160–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hwang MS, Morgan RL, Sarkar SF, Wang PW, Guttman DS (2005) Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae . Appl Environ Microbiol 71: 5182–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bull CT, Manceau C, Lydon J, Kong H, Vinatzer BA, et al. (2010) Pseudomonas cannabina pv. cannabina pv. nov., and Pseudomonas cannabina pv. alisalensis (Cintas Koike and Bull, 2000) comb. nov., are members of the emended species Pseudomonas cannabina (ex Sutic & Dowson 1959) Gardan, Shafik, Belouin, Brosch, Grimont & Grimont 1999. Syst Appl Microbiol 33: 105–115. [DOI] [PubMed] [Google Scholar]
- 29. Joardar V, Lindeberg M, Jackson RW, Selengut J, Dodson R, et al. (2005) Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J Bacteriol 187: 6488–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buell CR, Joardar V, Lindeberg M, Selengut J, Paulsen IT, et al. (2003) The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc Natl Acad Sci U S A 100: 10181–10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feil H, Feil WS, Chain P, Larimer F, DiBartolo G, et al. (2005) Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc Natl Acad Sci U S A 102: 11064–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Butler MI, Stockwell PA, Black MA, Day RC, Lamont IL, et al. (2013) Pseudomonas syringae pv. actinidiae from recent outbreaks of kiwifruit bacterial canker belong to different clones that originated in China. PLoS One 8: e57464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Brien HE, Thakur S, Gong Y, Fung P, Zhang J, et al. (2012) Extensive remodeling of the Pseudomonas syringae pv. avellanae type III secretome associated with two independent host shifts onto hazelnut. BMC Microbiol 12: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Green S, Studholme DJ, Laue BE, Dorati F, Lovell H, et al. (2010) Comparative genome analysis provides insights into the evolution and adaptation of Pseudomonas syringae pv. aesculi on Aesculus hippocastanum . PLoS One 5: e10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sarris PF, Trantas EA, Baltrus DA, Bull CT, Wechter WP, et al. (2013) Comparative genomics of multiple strains of Pseudomonas cannabina pv. alisalensis, a potential model pathogen of both monocots and dicots. PLoS One 8: e59366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reinhardt JA, Baltrus DA, Nishimura MT, Jeck WR, Jones CD, et al. (2009) De novo assembly using low-coverage short read sequence data from the rice pathogen Pseudomonas syringae pv. oryzae . Genome Res 19: 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cai R, Lewis J, Yan S, Liu H, Clarke CR, et al. (2011) The plant pathogen Pseudomonas syringae pv. tomato is genetically monomorphic and under strong selection to evade tomato immunity. PLoS Pathog 7: e1002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Studholme DJ, Ibanez SG, MacLean D, Dangl JL, Chang JH, et al. (2009) A draft genome sequence and functional screen reveals the repertoire of type III secreted proteins of Pseudomonas syringae pathovar tabaci 11528. BMC Genomics 10: 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCann HC, Rikkerink EHA, Bertels F, Fiers M, Lu A, et al. (2013) Genomic Analysis of the Kiwifruit Pathogen Pseudomonas syringae pv. actinidiae Provides Insight into the Origins of an Emergent Plant Disease. PLoS Pathog 9: e1003503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clarke CR, Cai R, Studholme DJ, Guttman DS, Vinatzer BA (2010) Pseudomonas syringae strains naturally lacking the classical P. syringae hrp/hrc Locus are common leaf colonizers equipped with an atypical type III secretion system. Mol Plant Microbe Interact 23: 198–210. [DOI] [PubMed] [Google Scholar]
- 41. Filiatrault MJ, Stodghill PV, Myers CR, Bronstein PA, Butcher BG, et al. (2011) Genome-wide identification of transcriptional start sites in the plant pathogen Pseudomonas syringae pv. tomato str. DC3000. PLoS One 6: e29335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Filiatrault MJ, Stodghill PV, Bronstein PA, Moll S, Lindeberg M, et al. (2010) Transcriptome analysis of Pseudomonas syringae identifies new genes, noncoding RNAs, and antisense activity. J Bacteriol 192: 2359–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greenwald JW, Greenwald CJ, Philmus BJ, Begley TP, Gross DC (2012) RNA-seq analysis reveals that an ECF sigma factor, AcsS, regulates achromobactin biosynthesis in Pseudomonas syringae pv. syringae B728a. PLoS One 7: e34804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hockett KL, Burch AY, Lindow SE (2013) Thermo-regulation of genes mediating motility and plant interactions in Pseudomonas syringae . PLoS One 8: e59850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cumbie JS, Kimbrel JA, Di Y, Schafer DW, Wilhelm LJ, et al. (2011) GENE-counter: a computational pipeline for the analysis of RNA-Seq data for gene expression differences. PLoS One 6: e25279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bushley KE, Raja R, Jaiswal P, Cumbie JS, Nonogaki M, et al. (2013) The genome of Tolypocladium inflatum: evolution, organization, and expression of the cyclosporin biosynthetic gene cluster. PLoS Genet 9: e1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Filichkin SA, Mockler TC (2012) Unproductive alternative splicing and nonsense mRNAs: a widespread phenomenon among plant circadian clock genes. Biol Direct 7: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huynh TV, Dahlbeck D, Staskawicz BJ (1989) Bacterial blight of soybean: regulation of a pathogen gene determining host cultivar specificity. Science 245: 1374–1377. [DOI] [PubMed] [Google Scholar]
- 49. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Greene W (2008) Functional forms for the negative binomial model for count data. Economics Letters 99: 585–590. [Google Scholar]
- 52. Greene W (2005) Functional Form and Heterogeneity in Models for Count Data. Foundations and Trends® in Econometrics 1: 113–218. [Google Scholar]
- 53. Di Y, Schafer D, Cumbie J, Chang J (2011) The NBP Negative Binomial Model for Assessing Differential Gene Expression from RNA-Seq. Stat Appl Genet Mol Biol 10 Article 24. [Google Scholar]
- 54. Kvitko BH, Park DH, Velasquez AC, Wei CF, Russell AB, et al. (2009) Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog 5: e1000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mukaihara T, Tamura N (2009) Identification of novel Ralstonia solanacearum type III effector proteins through translocation analysis of hrpB-regulated gene products. Microbiology 155: 2235–2244. [DOI] [PubMed] [Google Scholar]
- 56. Guttman DS, Vinatzer BA, Sarkar SF, Ranall MV, Kettler G, et al. (2002) A functional screen for the type III (Hrp) secretome of the plant pathogen Pseudomonas syringae . Science 295: 1722–1726. [DOI] [PubMed] [Google Scholar]
- 57. Cunnac S, Chakravarthy S, Kvitko BH, Russell AB, Martin GB, et al. (2011) Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae . Proc Natl Acad Sci U S A 108: 2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lindeberg M, Stavrinides J, Chang JH, Alfano JR, Collmer A, et al. (2005) Proposed guidelines for a unified nomenclature and phylogenetic analysis of type III Hop effector proteins in the plant pathogen Pseudomonas syringae . Mol Plant Microbe Interact 18: 275–282. [DOI] [PubMed] [Google Scholar]
- 59. Lindow SE (1985) Ecology of Pseudomonas syringae relevant to the field use of Ice - deletion mutants constructed in vitro for plant frost control. Engineered Organisms in the Environment: Scientific Issues 23–35. [Google Scholar]
- 60. Mohr TJ, Liu H, Yan S, Morris CE, Castillo JA, et al. (2008) Naturally occurring nonpathogenic isolates of the plant pathogen Pseudomonas syringae lack a type III secretion system and effector gene orthologues. J Bacteriol 190: 2858–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morris CE, Sands DC, Vanneste JL, Montarry J, Oakley B, et al. (2010) Inferring the evolutionary history of the plant pathogen Pseudomonas syringae from its biogeography in headwaters of rivers in North America, Europe, and New Zealand. MBio 1: e00107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morris CE, Sands DC, Vinatzer BA, Glaux C, Guilbaud C, et al. (2008) The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J 2: 321–334. [DOI] [PubMed] [Google Scholar]
- 63. Baltrus DA, Yourstone S, Lind A, Guilbaud C, Sands DC, et al. (2013) Draft Genomes for a Phylogenetically Diverse Suite of Pseudomonas syringae Isolates From Multiple Source Populations. J Bacteriol in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schechter LM, Vencato M, Jordan KL, Schneider SE, Schneider DJ, et al. (2006) Multiple approaches to a complete inventory of Pseudomonas syringae pv. tomato DC3000 type III secretion system effector proteins. Mol Plant Microbe Interact 19: 1180–1192. [DOI] [PubMed] [Google Scholar]
- 65. Keen NT, Tamaki S, Kobayashi D, Gerhold D, Stayton M, et al. (1990) Bacteria expressing avirulence gene D produce a specific elicitor of the soybean hypersensitive reaction. Mol Plant Microbe Interact 3: 112–121. [Google Scholar]
- 66. Kobayashi DY, Tamaki SJ, Keen NT (1989) Cloned avirulence genes from the tomato pathogen Pseudomonas syringae pv. tomato confer cultivar specificity on soybean. Proc Natl Acad Sci U S A 86: 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kobayashi DY, Tamaki SJ, Keen NT (1990) Molecular characterization of avirulence gene D from Pseudomonas syringae pv. tomato . Mol Plant Microbe Interact 3: 94–102. [DOI] [PubMed] [Google Scholar]
- 68. Loper JE, Hassan KA, Mavrodi DV, Davis EW 2nd, Lim CK, et al. (2012) Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet 8: e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stavrinides J, Ma W, Guttman DS (2006) Terminal reassortment drives the quantum evolution of type III effectors in bacterial pathogens. PLoS Pathog 2: e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gazi AD, Sarris PF, Fadouloglou VE, Charova SN, Mathioudakis N, et al. (2012) Phylogenetic analysis of a gene cluster encoding an additional, rhizobial-like type III secretion system that is narrowly distributed among Pseudomonas syringae strains. BMC Microbiol 12: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Badel JL, Shimizu R, Oh HS, Collmer A (2006) A Pseudomonas syringae pv. tomato avrE1/hopM1 mutant is severely reduced in growth and lesion formation in tomato. Mol Plant Microbe Interact 19: 99–111. [DOI] [PubMed] [Google Scholar]
- 72. Macho AP, Zumaquero A, Gonzalez-Plaza JJ, Ortiz-Martin I, Rufian JS, et al. (2012) Genetic analysis of the individual contribution to virulence of the type III effector inventory of Pseudomonas syringae pv. phaseolicola . PLoS One 7: e35871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Melotto M, Underwood W, Koczan J, Nomura K, He SY (2006) Plant stomata function in innate immunity against bacterial invasion. Cell 126: 969–980. [DOI] [PubMed] [Google Scholar]
- 74. Glass NL, Kosuge T (1986) Cloning of the gene for indoleacetic acid-lysine synthetase from Pseudomonas syringae subsp. savastanoi . J Bacteriol 166: 598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Denance N, Sanchez-Vallet A, Goffner D, Molina A (2013) Disease resistance or growth: the role of plant hormones in balancing immune responses and fitness costs. Front Plant Sci 4: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Oh CS, Kim JF, Beer SV (2005) The Hrp pathogenicity island of Erwinia amylovora and identification of three novel genes required for systemic infectiondouble dagger. Mol Plant Pathol 6: 125–138. [DOI] [PubMed] [Google Scholar]
- 77. Beck BJ, Downs DM (1998) The apbE gene encodes a lipoprotein involved in thiamine synthesis in Salmonella typhimurium . J Bacteriol 180: 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Skovran E, Lauhon CT, Downs DM (2004) Lack of YggX results in chronic oxidative stress and uncovers subtle defects in Fe-S cluster metabolism in Salmonella enterica . J Bacteriol 186: 7626–7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Boyd JM, Endrizzi JA, Hamilton TL, Christopherson MR, Mulder DW, et al. (2011) FAD binding by ApbE protein from Salmonella enterica: a new class of FAD-binding proteins. J Bacteriol 193: 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, et al. (2009) Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens . Genome Biol 10: R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Burnens AP, Stanley J, Sack R, Hunziker P, Brodard I, et al. (1997) The flagellin N-methylase gene fliB and an adjacent serovar-specific IS200 element in Salmonella typhimurium . Microbiology 143 Pt 5: 1539–1547. [DOI] [PubMed] [Google Scholar]
- 82. Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, et al. (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337: 1111–1115. [DOI] [PubMed] [Google Scholar]
- 83. Zipfel C, Robatzek S, Navarro L, Oakeley EJ, Jones JD, et al. (2004) Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 428: 764–767. [DOI] [PubMed] [Google Scholar]
- 84. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, et al. (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410: 1099–1103. [DOI] [PubMed] [Google Scholar]
- 85. Murillo J, Shen H, Gerhold D, Sharma A, Cooksey DA, et al. (1994) Characterization of pPT23B, the plasmid involved in syringolide production by Pseudomonas syringae pv. tomato PT23. Plasmid 31: 275–287. [DOI] [PubMed] [Google Scholar]
- 86. Atkinson MM, Midland SL, Sims JJ, Keen NT (1996) Syringolide 1 Triggers Ca2+ Influx, K+ Efflux, and Extracellular Alkalization in Soybean Cells Carrying the Disease-Resistance Gene Rpg4 . Plant Physiol 112: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yucel I, Midland SL, Sims JJ, Keen NT (1994) Class I and class II avrD alleles direct the production of different products in gram-negative bacteria. Mol Plant Microbe Interact 7: 148–150. [DOI] [PubMed] [Google Scholar]
- 88. Gust B, Chandra G, Jakimowicz D, Yuqing T, Bruton CJ, et al. (2004) Lambda red-mediated genetic manipulation of antibiotic-producing Streptomyces . Adv Appl Microbiol 54: 107–128. [DOI] [PubMed] [Google Scholar]
- 89. Earl AM, Desjardins CA, Fitzgerald MG, Arachchi HM, Zeng Q, et al. (2011) High quality draft genome sequence of Segniliparus rugosus CDC 945(T) = (ATCC BAA-974(T)). Stand Genomic Sci 5: 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wernisch L, Kendall SL, Soneji S, Wietzorrek A, Parish T, et al. (2003) Analysis of whole-genome microarray replicates using mixed models. Bioinformatics 19: 53–61. [DOI] [PubMed] [Google Scholar]
- 91. Marco ML, Legac J, Lindow SE (2005) Pseudomonas syringae genes induced during colonization of leaf surfaces. Environ Microbiol 7: 1379–1391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Graphical representation of our experimental pipeline. Isogenic P. syringae strains carrying either pBAD::EV or pBAD::hrpL were grown on MM media supplemented with arabinose and collected 1, 3 and 5 hours post induction. RNA was extracted for each time point and cDNA prepared to confirm induction of hrpL and hrcC for P. syringae pBAD::hrpL. Total RNA for each time point was pooled equally. Pooled RNA for each strain was subjected to rRNA removal and double stranded cDNA prepared (Materials and Methods). Illumina libraries were prepared according to manufacturer's protocol and sequenced. Resulting reads were used to run GENE-counter. After 300 bootstraps of GENE-counter, genes from each sample were assigned a median read count, a median p and q-value as well as a B-value.
(TIF)
Detailed results of qRT-PCRs described in Table 3 from samples derived from the pBAD system. (A) For Pto DC3000 genes, (B) for Pph 1448A genes, (C) for Psy B728a genes, (D) for Por genes. cDNA was prepared from the same total RNA used to generate our RNA-seq data for all strains except Pto DC3000. For Pto DC3000, cDNAs were prepared from an independent biological replicate. Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD.
(TIF)
Detailed results of qRT-PCRs described in Table 3 from samples derived from isogenic strains grown in MM media. (A) For Pto DC3000 genes, (B) for Pph 1448A genes, (C) for Psy B728a genes, (D) for Por genes. cDNA was prepared from wild type strains and corresponding isogenic ΔhrpL mutants grown in MM media for 5 hours. Expression was normalized to gap-1. For determination of the relative % expression, each wild type strain sample was set to 100% and ΔhrpL samples normalized accordingly. Error bars represent SD.
(TIF)
The majority of effector genes are found up-regulated in our analysis. Effector suites present in each strain are listed. In bold, effector genes found up-regulated by RNA-seq. Underlined, effector genes HrpL-dependent according to Pseudomonas syringae Genome Resources, (PPI database http://www.pseudomonas-syringae.org/) but not found up-regulated in our analysis. In grey, effector genes previously identified according to a combination of homology and functional criteria described in Chang et al., 2005 and Baltrus et al., 2011 but not found to be HrpL-regulated in these strains in any experiment, to our knowledge. ‘ indicates insertion or truncation according to PPI database. The new Por type III effectors defined in this study are listed in red.
(TIF)
hopBH1 and hopBI1 are both present across phylogenetically diverse strains of P. syringae. (A) Bayesian phylogenetic tree of 45 Pseudomonas strains [3], [40], [63] based on seven conserved loci as described in [3]. Bayesian posterior probabilities are displayed on the phylogeny only at nodes where these values are <0.95. Each phylogenetic group (defined according to Berge et al., personal communication and [27]) is color coded. (B) Distribution of hopBH1 and hopBI1 across the 45 Pseudomonas strains. Dark blue boxes indicate presence of corresponding full length ORF. Light Blue box indicates truncated ORF. White boxes indicate absence of corresponding ORF in the sequenced genome.
(TIF)
Amino-acid sequence alignment of HopBH1. Alignment performed using clustal W with sequences from all P. syringae strains known to date to contain hopBH1.
(TIF)
Expression of PSPTO_2105 and 2130 or their orthologs under native conditions supports our results obtained with arabinose-inducible hrpL system. (A) Relative expression of PSPTO_2130 and its orthologs. (B) Relative expression of PSPTO_2105 and its orthologs. qRT-PCR analysis was performed on RNA samples derived from wild type strains and the cognate isogenic ΔhrpL mutant grown in MM media. Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD.
(TIF)
HrpL-dependent up-regulation of genes downstream of PSPTO_2130 and its orthologs in Pto DC3000, Pph 1448A, but not Psy B728a. (A), Graphical representation of PSPTO_2131-PSPTO-2128 operon. qRT-PCR analysis was performed on RNA samples derived from (B) Pto DC3000(pBAD::hrpL) and Pto DC3000(pBAD::EV) (C) Pph 1448A(pBAD::hrpL) and Pph 1448A(pBAD::EV); and (D) Psy B728a(pBAD::hrpL) and Psy B728a(pBAD::EV). ORF nomenclature for operons from Pph 1448A and Psy B728a in C and D, respectively, is listed directly under the corresponding ORF numbers in Pto DC3000 in B. Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD. qRT-PCR analysis was performed on RNA samples derived from (E) Pto DC3000 and Pto DC3000 ΔhrpL (F) Pph 1448A and Pph 1448A ΔhrpL, For determination of the relative % expression, each wild type strain sample was set to 100% and ΔhrpL samples normalized accordingly. Error bars represent SD.
(TIF)
Pph 1448A mutants deleted in thiamine biosynthesis lipoprotein gene display reduced growth on Tendergreen beans. Two week old bean cv. Tendergreen beans were dip inoculated with wild type Pph 1448A (Pph) or two independent mutants with a clean deletion of PSPPH_1855 (PphΔ1855 #1, PphΔ1855 #2), at OD600 = 0.001. Bacterial growth of each strain was determined after 3.5 dpi. Error bars represent SD. This experiment was repeated twice with similar results.
(TIF)
Determination of the relative expression of avrD PSPPH_A0112, _A0107, _A0106 in various Pph 1448A mutants. qRT-PCR was performed on cDNA derived from wild type Pph 1448A (Pph), Pph 1448AΔhrpL, two independent clean deletion avrD mutants (ΔavrD #1, ΔavrD #2), and two independent clean deletion PSPPH_A0107 mutants (ΔA0107 #1, ΔA0107 #2). Expression was normalized to gap-1. For determination of the relative expression, each EV sample was set to 1 and HrpL samples normalized to the corresponding EV samples. Error bars represent SD. This experiment was repeated 3 times with similar results.
(TIF)
Raw bootstrapped GENE-counter results for each strain.
(XLSX)
Genes found down-regulated by RNA-seq.
(XLSX)
Our defined HrpL regulons across various P. syringae after manual curation.
(XLSX)
List of genes excluded from analysis after manual curation.
(XLSX)
List of “confirmed” HrpL-regulated genes for Pto DC3000 and Pph 1448A according to previous studies.
(XLSX)
BlastP search results of the product of each HrpL regulon gene across the strains tested in this study.
(XLSX)
Details on the attribution of the functional classification for each HrpL regulon gene.
(XLSX)
List of Pseudomonas strains used or analyzed in this study.
(XLSX)
List of primers and sequences used in this study.
(XLSX)