

Background: CD4 and Tetherin are stabilized through intrachain and interchain disulfide bonds, respectively.
Results: CD4 and Tetherin retro-translocate from ER to cytosol with oxidized disulfide bridges as folded and multimeric molecules.
Conclusion: Cysteines reduction is not a prerequisite for ER to cytosol dislocation.
Significance: Our observations challenge the requirements of reduction and unfolding before dislocation.
Keywords: Biotin, Disulfide, ER-associated Degradation, ERAD, Oxidation-Reduction, BST-2, CD4, Biotinylation, Retro-translocation, Tetherin
Abstract
CD4 and BST-2/Tetherin are cellular membrane proteins targeted to degradation by the HIV-1 protein Vpu. In both cases proteasomal degradation following recruitment into the ERAD pathway has been described. CD4 is a type I transmembrane glycoprotein, with four extracellular immunoglobulin-like domains containing three intrachain disulfide bridges. BST-2/Tetherin is an atypical type II transmembrane glycoprotein with an N-terminal transmembrane domain and a C-terminal glycophosphatidylinositol anchor, which dimerizes through three interchain bridges. We investigated spontaneous and Vpu-induced retro-translocation of CD4 and BST-2/Tetherin using our novel biotinylation technique in living cells to determine ER-to-cytosol retro-translocation of proteins. We found that CD4 retro-translocates with oxidized intrachain disulfide bridges, and only upon proteasomal inhibition does it accumulate in the cytosol as already reduced and deglycosylated molecules. Similarly, BST-2/Tetherin is first exposed to the cytosol as a dimeric oxidized complex and then becomes deglycosylated and reduced to monomers. These results raise questions on the required features of the putative retro-translocon, suggesting alternative retro-translocation mechanisms for membrane proteins in which complete cysteine reduction and unfolding are not always strictly required before ER to cytosol dislocation.
Introduction
The transport of proteins across lipid bilayers is crucial for allowing cells to perform a large variety of different tasks necessary to support cell life and proliferation. In eukaryotes approximately one third (1) of all synthesized polypeptides are inserted co-translationally into the lumen of the endoplasmic reticulum (ER)4 to be either secreted or anchored on the cell surface or on inner membranes.
The ER resident protein Sec61 is necessary to promote insertion of nascent polypeptide chains into the ER. Sec61 is the main component of the translocon involved in the formation of the import channel and directly interacting with the ribosome (2).
As important as the import of proteins into the ER is the opposite process of retro-translocation from the ER to the cytosol of misfolded or unfolded proteins, which is necessary to maintain a correct cellular homeostasis. The ER-associated degradation (ERAD) pathway operates as a quality control mechanism to dispose of such unwanted molecules through proteasomal degradation (3). Retro-translocation, however, also operates in cases of exit from the ER not directly involved in ERAD, as observed for calreticulin (4), SV40 virus (5), and some bacterial and plant toxins (6–9).
Although ERAD has been widely studied both in yeast and mammalian cells (3, 10), a comprehensive picture of how the retro-translocation step works is still missing, particularly in relation to the requirements of the substrate and the structure of the molecular complex that actually drives dislocation. Here we report on the mechanism of retro-translocation of two widely studied proteins, CD4 and BST-2/Tetherin, using a novel technique to detect retro-translocation of defined proteins, based on the specific biotinylation in living cells only of molecules that from the ER reach the cytosolic compartment (11).
CD4, a classical functional marker of a subpopulation of T cells and the cellular receptor of HIV-1 retrovirus, is a type I transmembrane glycoprotein with four extracellular immunoglobulin (Ig) domains, three of which possess a canonical intramolecular disulfide bridge (12). Tetherin, however, is a cellular viral restriction factor that is able to antagonize the spreading of enveloped viruses (including HIV-1) by tethering newly formed viral particles to the plasma membrane of infected cells (13–16). Structurally, Tetherin is a type II transmembrane protein with a glycosylphosphatidylinositol (GPI) anchor at its C terminus that forms covalent dimers by three interchain disulfide bridges between two parallel monomers (17, 18).
The HIV-1 accessory protein Vpu is able to induce degradation of these two membrane proteins. Whereas degradation of CD4 employs a pathway that leads to the cytoplasmic proteasome (19–21), for Tetherin the evidences are more controversial (22–26).
Our results show that both CD4 and Tetherin are dislocated with intact disulfide bridges and hence as partially folded species. We also demonstrate that, although Tetherin can be in part degraded following the autophagy/lysosomal pathway, most of it is retro-translocated and degraded by the cytoplasmic proteasome. These data offer new insights on the mechanism of dislocation and raise questions on the required features of the putative retro-translocon.
EXPERIMENTAL PROCEDURES
Constructs
CD4 cDNA was modified by inserting at the N terminus the coding sequences for a secretion signal, a tag (SV5 tag, GKPIPNPLLGLD), and the biotin acceptor peptide (GLNDIFEAQKIEWHE (27)), then cloned into pcDNA3 vector (Invitrogen), yielding BAP-CD4. To obtain a CD4 with membrane-proximal BAP (18 amino acids from the transmembrane domain, CD4-BAP), the BAP tag was introduced immediately after Ser-382. A codon-optimized version of gp160 was cloned into pcDNA3 and used in all experiments with CD4. Human Tetherin was tagged with SV5 and BAP immediately upstream of the GPI anchor signal (Tetherin-BAP) and cloned into pcDNA3. Tetherin mutagenized in the two N-glycosylation sites (N65A,N92A) was obtained by site directed mutagenesis (QuikChange Site-directed Mutagenesis kit; Stratagene). Tetherin ectodomain, including amino acids 45–160 of human Tetherin (28) and containing the added SV5 and BAP tags, was amplified and cloned into pcDNA3 (cyt-ecto). Tetherin cyt-ecto cDNA was modified by inserting at its N terminus the coding sequence for a secretion signal to obtain a secreted version of Tetherin ectodomain (sec-ecto).
Tetherin with the membrane-proximal BAP (12 amino acids from the transmembrane domain, BAP-Tetherin) was obtained by inserting the BAP tag after Ala-48. Vpu from viral clone NL4-3 was expressed from a pcDNA3 vector containing an N-terminal leader peptide followed by SV5 tag.
Cell Culture and Transfection
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen), supplemented with 10% fetal calf serum (FCS; Invitrogen). Cells were transfected in 6-well plates (about 5 × 105 cells/well) by the standard calcium phosphate technique. When indicated, the Vpu-expressing plasmid was co-transfected. pEGFP-N1 plasmid was co-transfected as a loading and transfection control. Approximately 18 h after transfection, medium was discarded and replaced with 2 ml of medium supplemented with 0.1 mm biotin (Sigma), and cells were further incubated for at least 7–8 h. When indicated, after 4 h of incubation with biotin, the proteasome inhibitor MG132 (Sigma) was added in serum-free medium at a concentration of 10 μm for 4 h or at 5 μm for 12 h. Chloroquine (Sigma) was used at 50 μm for 4 h. The PNGase inhibitor Z-VAD(OMe)-fmk (IMGENEX) was used for 4 h at 100 μm.
Cell Extract Preparation, Gel Retardation Assay, and Western Blotting
Transfected HEK293T cells were washed with 20 mm N-ethylmaleimide (NEM) (Fluka) in PBS, pH 6.8, to remove free biotin and lysed with 100 μl/well SDS-lysis buffer (100 mm Tris-HCl, pH 6.8, 6% SDS, 20 mm NEM) and sonicated. For gel retardation assays samples were first boiled in SDS-gel loading buffer (25 mm Tris-HCl, pH 6.8, 1% SDS, 10% glycerol, 175 mm β-mercaptoethanol) and then incubated with 1 μg of streptavidin (StrAv) (Sigma) for 30 min at room temperature before loading. Gels were blotted onto PVDF membranes (Millipore), reacted with anti-SV5 mAb followed by incubation with an HRP-labeled anti-mouse whole IgG antibody (Jackson) for ECL detection. Quantification of bands was performed with the image processing software ImageJ v1.43 (National Institutes of Health) or using UVItec Alliance detection system.
Immunoprecipitations of CD4 and Tetherin were carried out by incubating lysates, diluted three times in TNN buffer (100 mm Tris-HCl, pH 8.0, 250 mm NaCl, 0.5% Nonidet P-40, 20 mm NEM, 1% protease inhibitors mixture (Sigma)) with anti-SV5 and protein A-agarose (Repligen) for 2 h at 4 °C. Immunoprecipitated proteins were eluted from agarose beads by boiling in SDS buffer. For immunoprecipitation of plasma membrane fractions, intact cells were first incubated with anti-SV5 for 1 h on ice, then washed with cold PBS and lysed in TNN buffer. 500 × g postnuclear supernatants were incubated with protein A-agarose for 30 min, and after washing, bound proteins were eluted by boiling in SDS buffer. Biotinylated material in the purified fraction was detected in Western blots with anti-SV5 or streptavidin-HRP (Jackson). Where indicated, cellular lysates or eluted material was treated with peptide N-glycosidase F (PNGase-F) or endoglycosidase H (Endo-H) (New England Biolabs) according to the manufacturer's instructions. For two-dimensional gel analysis, samples were first run on a nonreducing SDS-PAGE gel. The lane of interest was then cut and incubated with reducing loading buffer for 15 min at room temperature before being run in a second gel under reducing conditions. Where indicated, WB of rabbit anti-EGFP (Invitrogen) was used as loading and transfection control. In WB of cell fractionation experiments rabbit anti-actin (Sigma), mouse anti-calnexin (BD Bioscience), and rabbit anti-Derlin1 (Sigma) were used according to the manufacturers' instructions.
Trypsin Sensitivity Assay and Cell Fractionation
Microsome-containing lysates were obtained by resuspending cells in a fractionation buffer containing 20 mm Tris-HCl, pH 7.4, 250 mm sucrose, and 30 mm NEM, followed by 12 passages through a 1-inch 23-gauge needle and two centrifugations at 500 × g and 1000 × g for 5 min at 4 °C. For the trypsin sensitivity assay recovered supernatants were incubated with 1 μg of trypsin (Sigma) for 1 h at 37 °C. When indicated, Nonidet P-40 was added at 0.5% final concentration. For cell fractionation, 1000 × g supernatants were further centrifuged at 100,000 × g for 1 h at 4 °C. Supernatants represented cytosolic material and pellets the microsomal ER fraction. After a delicate wash in fractionation buffer, pellets were resuspended in the same buffer enriched with 1.2% SDS.
[35S]Methionine Labeling
Cells were first starved for 30 min in methionine/cysteine-free medium, supplemented with 10% dialyzed FCS and 0.1 mm biotin, then labeled for 10 or 15 min, as indicated, with 200 μCi/ml [35S]methionine/cysteine (PerkinElmer) and chased for 120 min in biotin-containing complete medium. Cells were lysed in 100 μl of SDS-lysis buffer, diluted with 400 μl of TNN, and sonicated or digested with DNaseI (Promega) for 1 h at 37 °C. SV5-tagged proteins were immunoprecipitated with anti-SV5 and protein A-agarose and eluted by boiling in SDS-lysis buffer, and samples were resolved on a nonreducing or reducing 10% SDS-PAGE. Purification of biotinylated material was performed with StrAv-coated magnetic beads (Dynabeads; Invitrogen) and the elution obtained by boiling in SDS buffer. Gels were fixed in 10% acetic acid, 10% methanol and incubated for 20 min in Amplify fluorographic enhancer (GE Healthcare), dried, and exposed for autoradiography on Kodak BioMax XAR films.
RESULTS
Biotinylation of Dislocated CD4 and Tetherin
We have used our recently described method of biotinylation in living cells (11) to investigate retro-translocation of CD4 and Tetherin induced by HIV-1 Vpu. In this technique, cytosolic expression of the biotin-ligase BirA causes specific monobiotinylation of cytosolically located protein substrates tagged with the 15-amino acid-long biotin acceptor peptide BAP (GLNDIFEAQKIEWHE(27)). The BAP tag was fused to ER luminal positions in both proteins, namely at the N terminus for CD4 and in the C-terminal part, just upstream of the GPI anchor signal, for Tetherin (Fig. 1). With this BAP tag configuration, only molecules that have reached the cytosolic compartment will be labeled by biotinylation. A second tag (SV5, 12 amino acids long) was also included next to BAP to favor recognition. The addition of the tags did not alter proper folding because both proteins were displayed on the cell surface as revealed by cytofluorometry with anti-SV5.5 A functional Tetherin tagged in the same position has been reported previously (15). Vpu was also SV5-tagged at its N terminus by fusing a leader peptide followed by the SV5 tag sequence.
FIGURE 1.
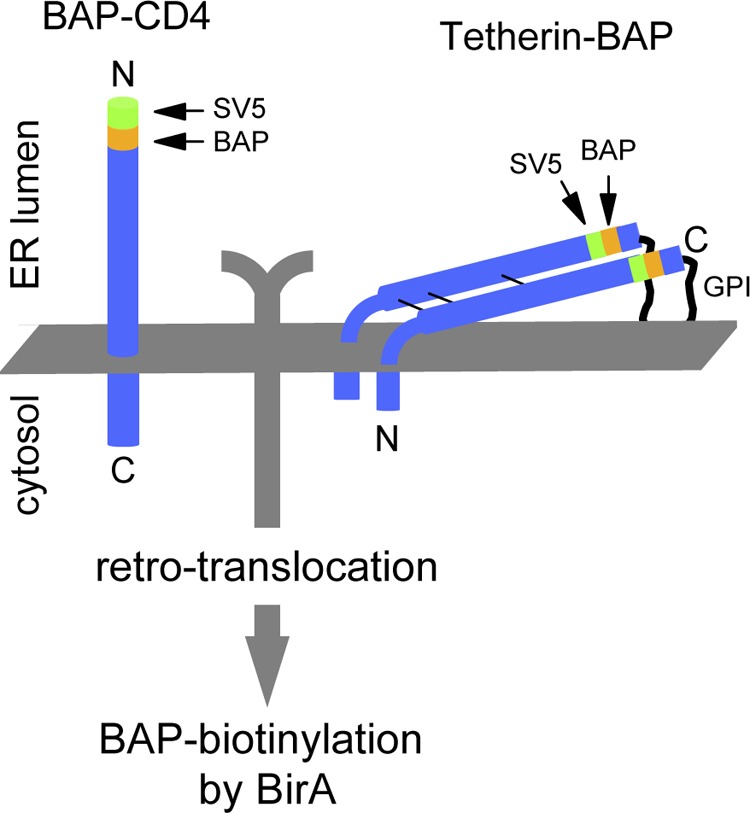
Scheme of CD4 and Tetherin tagged with BAP in ER-luminal positions. The 11-amino acid-long SV5 tag is also shown. Only retro-translocated BAP-tagged molecules are biotinylated by cytosolic BirA (cyt-BirA).
CD4 and Tetherin were independently co-expressed in HEK293T cells with cytosolic localized BirA (cyt-BirA), both in the presence and absence of Vpu and, in the case of CD4, also with HIV-1 gp160 to obtain a stronger degradation effect (29). The degree of retro-translocation was determined by monitoring the proportion of biotinylated molecules in a Western blot retardation assay (WB-ra). In this assay denatured samples are run in the presence of StrAv, causing retardation only of biotinylated molecules (30). As demonstrated previously for other proteins (11), all of the biotinylated CD4 and Tetherin found in cells expressing cyt-BirA corresponds to molecules that have been retro-translocated to the cytosol.
As shown in a Western blot retardation assay (Fig. 2A), a small fraction (approximately 5%) of biotinylated-CD4 (b-CD4) was detected in the absence of Vpu, corresponding to spontaneous retro-translocation. In contrast, when co-expressed with Vpu, a substantial proportion of biotinylated CD4 (approximately 30%) could be appreciated only after proteasome inhibition with MG132 because of the accumulation of retro-translocated molecules directed to proteasomal degradation. A band of deglycosylated CD4, generated by the activity of the cytosolic cellular PNGase, was also detected and, as expected for a retro-translocated protein, it was totally biotinylated (completely retarded by StrAv). Analysis of the glycosylation status of the b-CD4 fraction revealed both deglycosylated and glycosylated molecules. The latter were Endo-H-sensitive, therefore indicating that they were retro-translocated from the ER (Fig. 2B). The amount of deglycosylated CD4 accumulated in the presence of Vpu following MG132 treatment was not affected by the presence of BirA, ruling out interference of the BirA enzyme on retro-translocation (Fig. 2C). Further confirmation was obtained in [35S]methionine labeling experiments in cells treated with MG132. As shown in Fig. 2D, b-CD4 was already detected after 15-min pulse labeling in the presence of Vpu, whereas only marginal amount was present in the absence of Vpu. Also in this case, the b-CD4 population consisted of all the deglycosylated molecules and a small fraction of the glycosylated ones. Interestingly, a ladder of lower mobility bands was also detected. These bands, which likely correspond to a previously reported fraction containing CD4 species ubiquitinylated on residues of the cytosolic tail (19), were not retro-translocated as they did not get shifted by StrAv.
FIGURE 2.

Biotinylation of BAP-tagged CD4. A, WB-ra of BAP-tagged CD4. Extracts from HEK293T cells were co-transfected with BAP-CD4 and, where indicated, Vpu in the absence or presence of MG132 (5 μm for 12 h), and developed with anti-SV5. Biotinylated molecules appear as retarded bands when run with StrAv. B, glycosylation status of biotinylated CD4. WB shows b-CD4 from extracts of cells co-expressing Vpu in the presence of MG132 (5 μm for 12 h), immunoprecipitated with anti-SV5, treated with Endo-H or PNGase, and developed with HRP-conjugated StrAv. C, BirA lack of interference with CD4 retro-translocation. WB (developed with anti-SV5) shows BAP-CD4 from cells co-expressing Vpu, treated with MG132 (10 μm) for 4 h in the presence or absence of cyt-BirA. The deglycosylated band (de-glyc) is indicated. D, [35S]methionine labeling. Cells co-transfected with CD4 and cyt-BirA and with or without Vpu, were [35S]methionine-labeled for 15 min in the presence of MG132 (10 μm). CD4 was then immunoprecipitated (IP) with anti-SV5, run in a reducing PAGE retardation assay, and developed by autoradiography. Filled arrowheads indicate deglycosylated material, whereas open arrowheads indicate nonbiotinylated CD4 isoforms (observed in the presence of Vpu). E, trypsin-sensitivity assay. WB-ra of microsome-containing lysates (microsomes) derived from cells expressing BAP-CD4 and Vpu and treated with MG132 (5 μm) for 12 h before lysis, were incubated with (+) or without (−) trypsin for 1 h at 37 °C. Nonidet P-40 indicates the same microsomes-containing lysates treated with 0.5% Nonidet P-40 to make ER luminal proteins accessible to trypsin. Open arrowhead indicates CD4 with trypsin-digested cytosolic C-terminal tail; filled arrowhead indicates deglycosylated CD4. F, plasma membrane CD4. Membrane-displayed CD4 in MG132-treated cells was immunoprecipitated with anti-SV5 and analyzed in a WB-ra developed with anti-SV5. G, cellular fractionation. WB-ra shows total lysates of cells co-transfected with BAP-CD4 and cyt-BirA (tot) and of the pellet (ER) and supernatant (cytosolic, cyt) fractions obtained by ultracentrifugation. Calnexin and actin were used as ER and cytosol markers, respectively. WB of anti-EGFP was performed as loading and transfection control. Filled arrowheads indicate deglycosylated CD4.
Biotinylated CD4 corresponds to cytosolically localized molecules. In fact, trypsin sensitivity assays performed on microsome-containing lysates from MG132-treated cells showed that most of b-CD4 was sensitive to trypsin (a small fraction probably in an unaccessible complex was still present), whereas the nonbiotinylated material was trypsin-resistant, except for the predicted cleavage of the cytosolic C-terminal tail, which produced a band of higher mobility (Fig. 2E). However, the mature, membrane-exposed form of CD4 immunoprecipitated from cells first incubated with anti-SV5 and then washed and lysed, was found essentially not biotinylated, although a faint shifted band (representing a minimal proportion of the total) was detected (Fig. 2F). Interestingly, however, following cellular fractionation, most of b-CD4 was found associated with the microsomal-ER fraction with only a small part of the deglycosylated material recovered in the cytosol, indicating that despite exposure to the cytosol the dislocated material remains largely insoluble (Fig. 2G).
In the case of Tetherin, spontaneous retro-translocation (i.e. in the absence of Vpu) was already apparent both in control cells and, with increased amounts, in MG132-treated cells (Fig. 3A, first two lanes and fifth and sixth lanes). When co-expressed with Vpu the relative level of biotinylated Tetherin (b-Tetherin) compared with total Tetherin significantly increased both in untreated and MG132-treated cells (Fig. 3A, third and fourth lanes and seventh and eighth lanes). A complex pattern of bands was observed for Tetherin because it is a protein that undergoes several post-translational modifications, including two N-glycosylations (Asn-65 and Asn-92), the addition of a GPI anchor at the C terminus with the corresponding cleavage of the hydrophobic terminal sequence, and the formation of homodimers covalently stabilized by three interchain disulfide bonds (Cys-53, Cys-63, and Cys-91) between two parallel monomers. Of note, essentially all Tetherin was found processed for the cleavage of the GPI anchor signal.5 In the WBs shown (as in Fig. 3A) the upper set of bands labeled as Golgi/mem corresponds mostly to cell surface-exposed molecules. In fact, membrane-exposed Tetherin immunoprecipitated from the membrane of MG132-treated cells (reacted with anti-SV5 and then washed and lysed) was mostly not biotinylated, resistant to Endo-H and sensitive to PNGase (Fig. 3B). As shown in Fig. 3C, the same set of upper bands was sensitive to PNGase and resistant to Endo-H. In contrast, the group of bands showing an intermediate mobility (labeled ER-glyc in Fig. 3, A and C) represents a relevant fraction of Tetherin that, because of its sensitivity to Endo-H (Fig. 3C), corresponds to glycosylated molecules that have not yet trafficked through the Golgi. A significant part of this fraction was biotinylated; for instance, >70% in the presence of MG132 as shown in Fig. 3C (compare first and third lanes). Only upon MG132 treatment, however, a lower set of bands corresponding to fully biotinylated deglycosylated material (labeled de-glyc in Fig. 3, A and C) became apparent.
FIGURE 3.

Biotinylation of BAP-tagged Tetherin. A, WB-ra of BAP-tagged Tetherin. Extracts from HEK293T cells were co-transfected with Tetherin-BAP and, where indicated, Vpu in the absence or presence of MG132 (5 μm for 12 h) and developed with anti-SV5. Open and filled arrowheads indicate ER-like glycosylated (ER-glyc) and deglycosylated (de-glyc) Tetherin, respectively, whereas open and filled arrows indicate the corresponding biotinylated (retarded) fractions. B, plasma membrane displayed Tetherin in MG132-treated cells. Membrane-displayed Tetherin was immunoprecipitated with anti-SV5, and the resulting material was digested with Endo-H or PNGase and analyzed in a WB-ra developed with anti-SV5. Cells were co-transfected with Vpu. C, biotinylation of glycosylated and deglycosylated Tetherin. WB-ra shows lysates from cells expressing Tetherin treated with MG132 (5 μm for 12 h). Lysates were treated with Endo-H or PNGase, and the blot was developed with anti-SV5. Open and filled arrowheads and arrows are as in A. D, cyt-BirA biotinylating only retro-translocated molecules. WB-ra shows extracts from cells co-expressing Tetherin-BAP and a scFv BAP-tagged control protein, together with cyt-BirA and treated with MG132 (5 μm) for 12 h. Tetherin was detected with anti-SV5 and the scFv with the anti-roTag. E, cellular fractionation. WB-ra shows total lysates (tot) of cells co-transfected with Tetherin-BAP and cyt-BirA and treated with MG132 for 12 h before lysis and of the pellet (ER) and supernatant (cytosolic, cyt) fractions obtained by ultracentrifugation. Derlin1 and actin were used as ER and cytosolic markers, respectively. F, biotinylation as a non-postlysis event. Cells expressing only Tetherin-BAP (treated with 5 μm MG132 for 12 h) were mixed with cells co-transfected with cyt-BirA and a C-terminal BAP-tagged MHC-Iα (in its cytosolic tail) before mechanical lysis as performed in the cell fractionation experiments. Postnuclear supernatants corresponding to microsomal containing lysates were analyzed in WB-ra. Tetherin was revealed with anti-SV5 and MHC-Iα with anti-roTag. G, WB-ra shows lysates derived from Tetherin-BAP-transfected cells treated with MG132 (10 μm) or chloroquine (50 μm) for 4 h, as indicated. WB of anti-EGFP was used as loading and transfection control.
The cytosolic exposure of b-Tetherin was further demonstrated by co-expression (in cyt-BirA-expressing cells) of BAP-tagged Tetherin and a BAP-tagged control secretory protein (a scFv) that is very efficiently folded and secreted. Whereas Tetherin produced a biotinylated fraction, the control scFv did not (Fig. 3D).
As for CD4, cellular fractionation of Tetherin-expressing cells showed that most of the retro-translocated material was associated with the microsomal fraction with only a minor part of the deglycosylated molecules present in the soluble fraction (Fig. 3E). In these experiments the biotinylated material was not the consequence of postlysis activity of BirA because no biotinylation took place when cells expressing only BAP-tagged Tetherin or BirA were mixed before lysis, as shown in Fig. 3F. In contrast, a control protein (MHC-Iα) BAP-tagged in its cytosolic portion (11) co-expressed in the same BirA-expressing cells was fully biotinylated.
Controversial observations have been reported in relation to the degradation pathway followed by Tetherin, and a role has been proposed also for the lysosomal/autophagy pathway (22, 24, 26). To address this point, we analyzed lysates derived from cells co-expressing Tetherin and Vpu that were treated either with the proteasome inhibitor MG132 or with chloroquine, an inhibitor of autophagy and lysosomal degradation (31). Chloroquine treatment produced only a modest rescue of the total amount of intracellular protein (Fig. 3G). The fraction that increased upon chloroquine treatment was not biotinylated and not deglycosylated. This was expected because ER luminal molecules do not retro-translocate before lysosomal degradation, remaining in vesicular structures not accessible to cytosolic biotinylation and deglycosylation. In contrast, a strong increase was observed when the proteasome was inhibited, with the appearance of biotinylated and deglycosylated Tetherin species. These results indicate that Tetherin is preferentially targeted to the proteasome and only in part degraded through the lysosomal pathway. Altogether these data demonstrate that b-CD4 and b-Tetherin represent the respective fractions of retro-translocated molecules and that, for both molecules, the proteasome plays an important role in their degradation.
It has been previously proposed that for some ERAD substrates dislocation involves a step in which luminal domains are only partially exposed to the cytosolic side (32, 33). To investigate this point, we used two additional constructs in which the BAP tag was moved to positions on the luminal side proximal to the transmembrane domains of both CD4 and Tetherin (18 and 12 amino acids, respectively) (Fig. 4A). As shown in Fig. 4, B and C, membrane-proximal BAP-tagged CD4 and Tetherin behave in a way similar to molecules with the BAP in distal positions, regarding Vpu-induced degradation and stabilization by MG132. For the two proteins (in two different experiments) no differences were observed in the relative retro-translocation levels between molecules with distal and proximal BAP in the presence of MG132 (Fig. 4D). We thus concluded that, upon retro-translocation, the whole luminal domains of CD4 and Tetherin were completely exposed to the cytosolic environment.
FIGURE 4.
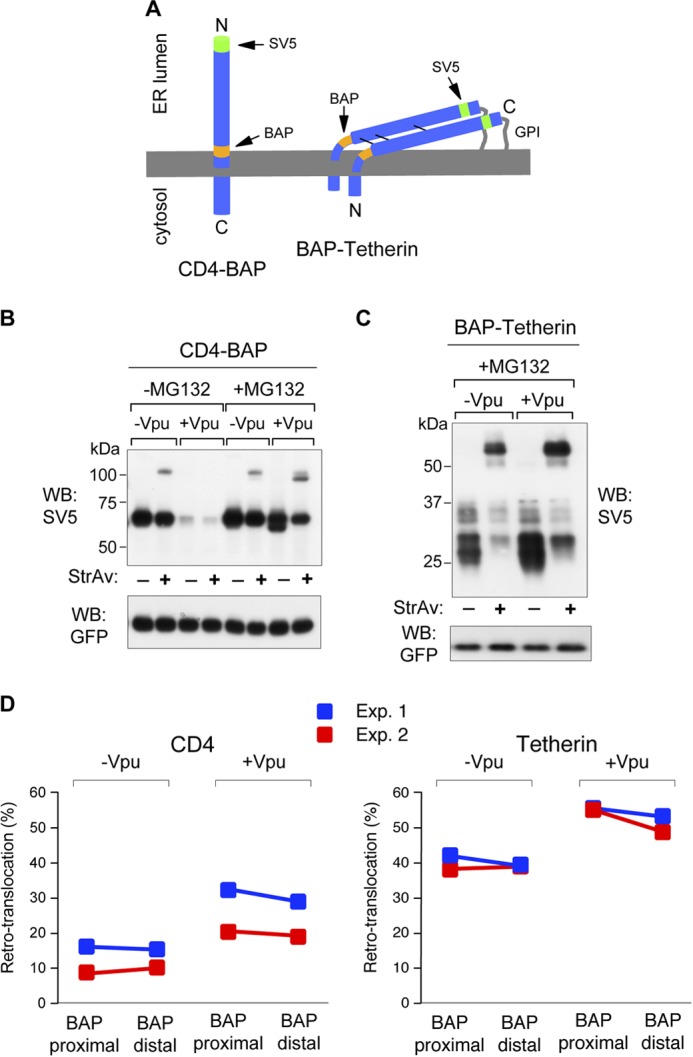
Cytosolic exposure of CD4 and Tetherin luminal domains. A, scheme of CD4 and Tetherin with the BAP tag positioned, in both cases, proximal to the transmembrane domains. The position of the SV5 tag is also shown. B, WB-ra of membrane-proximal BAP-tagged CD4 (CD4-BAP). Lysates were obtained from cells co-transfected with or without Vpu and in absence or presence of MG132 (5 μm for 12 h). C, WB-ra of membrane-proximal BAP-tagged Tetherin (BAP-Teth). Lysates were obtained from cells co-transfected with or without Vpu in the presence of MG132 (5 μm for 12 h.) D, quantification of retro-translocated fractions of membrane-proximal and membrane-distal BAP-tagged molecules. Comparison shows relative levels of retro-translocated CD4 (left panel) and Tetherin (right panel) for molecules with the BAP tag in membrane-distal or membrane-proximal position, co-expressed with or without Vpu (in all cases in the presence of MG132, 10 μm for 4 h), expressed as percentage of total protein. In both panels colors represent different experiments.
CD4 and Tetherin Retro-translocate with Oxidized Disulfide Bonds
Because both CD4 and Tetherin ectodomains were completely exposed in the retro-translocation intermediates detected, we next investigated the folding state of the retro-translocated fractions by analyzing the presence of disulfide bonds. To this end, total cellular extracts from cells co-expressing BirA, Vpu, and CD4 were prepared in the presence of NEM to avoid possible postlysis oxidation of reduced cysteines. NEM alkylation was effective because it caused a clear retardation in SDS-PAGE of the bands corresponding to glycosylated and deglycosylated CD4 in MG132-treated cells (Fig. 5A). This was expected because CD4 contains five cysteines in its cytosolic tail. However, the change in migration was different between the glycosylated and de-glycosylated fractions, suggesting a different oxidation status for the two populations. Similar samples were then analyzed in reducing and nonreducing PAGE (Fig. 5B). Under nonreducing conditions most of the glycosylated CD4 showed higher mobility than under reducing conditions because of the presence of oxidized intrachain disulfide bridges. Interestingly, part of this fraction was biotinylated, indicating that this retro-translocated glycosylated b-CD4 still contained oxidized disulfide bridges. In contrast, the biotinylated deglycosylated fraction did not show mobility differences in reducing and nonreducing conditions, indicating that disulfide bonds were already reduced (Fig. 5B). To further confirm the presence of retro-translocated molecules with oxidized disulfide bonds, CD4 was immunoprecipitated with anti-SV5 and directly analyzed in reducing and nonreducing WBs with HRP-conjugated StrAv to reveal only biotinylated molecules. As shown in Fig. 5C, whereas the deglycosylated fraction showed similar migration in reducing and nonreducing conditions (labeled de-glyc/red), the glycosylated b-CD4 clearly migrated with a lower mobility in reducing conditions (very close to the deglycosylated reduced fraction and labeled glyc/ox), thus demonstrating that glycosylated CD4 was retro-translocated with disulfide bonds still formed. The biotinylated upper band corresponds to the glycosylated fraction as confirmed by Endo-H digestion (Fig. 5D), which converted the upper band into a single one with identical mobility to the deglycosylated CD4 (detected only after MG132 treatment).
FIGURE 5.

Retro-translocation of CD4 with oxidized disulfide bonds. A, alkylation of CD4-free cysteines. Cells co-transfected with BirA, BAP-CD4, and Vpu were incubated for 5 min at 4 °C in PBS in the presence or absence of 20 mm NEM and rinsed in SDS-lysis buffer. Samples were then run in reducing SDS-PAGE, blotted, and developed with anti-SV5. B, nonreducing and reducing WB-ra of BAP-tagged CD4. Lysates from cells co-transfected with Vpu and treated with MG132 (5 μm for 12 h) were run in nonreducing (left) and reducing (right) conditions and developed with anti-SV5. Biotinylated and nonbiotinylated fractions of glycosylated and deglycosylated CD4 and their oxidation status are indicated with corresponding open and filled arrowheads and arrows. C, WB of biotinylated BAP-tagged CD4. CD4 was immunoprecipitated (IP) with anti-SV5 from cells expressing Vpu in the presence of MG132 (5 μm for 12 h), analyzed in nonreducing (left) and reducing (right) conditions, and developed with HRP-conjugated StrAv. CD4 glycosylation and oxidation status are indicated with open and filled arrows. D, ER-like glycosylation of biotinylated BAP-tagged CD4. WB shows reducing conditions of CD4 immunoprecipitated with anti-SV5 from cells expressing Vpu in the presence or absence of MG132 (5 μm for 12 h) and treated with or without Endo-H. Blot was developed with HRP-conjugated StrAv. E, [35S]methionine labeling of biotinylated CD4. Biotinylated CD4 fraction was immunoprecipitated from cells expressing Vpu after 2-h labeling with [35S]methionine in the presence of MG132 (10 μm), first with anti-SV5 and then with StrAv-conjugated beads, and analyzed in nonreducing (left) and reducing (right) conditions and developed by autoradiography. The glycosylation and oxidation status of biotinylated CD4 bands is indicated.
Biotinylated CD4 with oxidized disulfide bonds was also observed following [35S]methionine labeling (2 h) of cells in the presence of MG132. Analysis in nonreducing and reducing conditions of the [35S]methionine-labeled CD4, immunoprecipitated first with anti-SV5 and then with StrAv, showed the band of glycosylated CD4 band migrating with a reduced mobility under reducing conditions (Fig. 5E). Therefore, dislocated CD4 molecules become reduced and deglycosylated after reaching the cytosol.
Analysis of Tetherin in nonreducing WB-ra from cells not treated with MG132 showed that the biotinylated fraction corresponded only to disulfide stabilized dimers (Fig. 6A). This was independent of the presence or absence of Vpu, indicating that Tetherin has an intrinsic tendency to spontaneous retro-translocation despite the fact that, as expected, less Tetherin accumulated when co-expressed with Vpu. Following proteasome inhibition with MG132, the level of biotinylated dimers (both with and without Vpu) increased and, in addition, totally biotinylated monomers accumulated (Fig. 6A, first four lanes). Deglycosylated material was evident in both dimeric and monomeric fractions (Fig. 6A, arrowheads). For comparison, the same samples analyzed in reducing conditions are also shown (Fig. 6A). The presence of biotinylated dimers indicates that Tetherin retro-translocates with oxidized interchain disulfide bridges. To further confirm this observation, cellular extracts from cells treated with or without MG132 were analyzed in bidimensional nonreducing/reducing WBs developed with either anti-SV5 or HRP-conjugated StrAv (Fig. 6, B and C).
FIGURE 6.

Retro-translocation of dimeric Tetherin. A, nonreducing and reducing WB-ra of BAP-tagged Tetherin. Lysates from cells co-transfected with or without Vpu in the absence and presence of MG132 (5 μm for 12 h) were run in nonreducing (left) and reducing (right) conditions and developed with anti-SV5. Arrowheads indicate the deglycosylated fractions of Tetherin monomers and dimers. B and C, bidimensional (nonreducing/reducing) analysis of Tetherin. Lysates from cells co-expressing Tetherin and BirA treated (C) or not treated (B) with MG132 (5 μm for 12 h) were analyzed in a two-dimensional (nonreducing/reducing) WB developed with anti-SV5 or HRP-conjugated StrAv, as indicated. The uppermost panels show the same sample after the first (nonreducing) dimension. Vertical lanes show the same sample separated only through the reducing dimension. The different Tetherin glycosylation species of monomers and dimers are indicated. D, Z-VAD-fmk treatment confirming the presence of deglycosylated Tetherin dimers. Nonreducing WB of Tetherin from cells treated with MG132 (10 μm) and the PNGase inhibitor Z-VAD-fmk (100 μm) for 4 h is indicated. To better observe dimeric and monomeric glycosylation isoforms, diverse exposure times from the same gel are shown. Boxes highlight Tetherin molecules accumulated during proteasomal inhibition, which were sensitive to the concomitant presence of the PNGase inhibitor. E, [35S]methionine pulse-chase labeling. Cells co-transfected with Tetherin-BAP were labeled with a 15-min pulse of [35S]methionine and then chased for 2 h. MG132 (10 μm) was present from the beginning of the pulse. Tetherin was immunoprecipitated (IP) with anti-SV5, run in a nonreducing PAGE retardation assay, and developed by autoradiography. F, nonreducing WB-ra of cell extracts form cells expressing WT or N65A,N92A Tetherin mutant after MG132 treatment (10 μm, 4 h) is shown. The nonglycosylated monomer and the shifted biotinylated monomer and dimers are indicated. G, alkylation of Tetherin-free cysteines. Cells co-transfected with cyt-BirA and Tetherin-BAP were incubated 5 min at 4 °C in PBS in the presence or absence of 20 mm NEM and lysed in SDS-lysis buffer. Samples were run in nonreducing WB-ra and developed with anti-SV5. H, Tetherin ectodomain oxidation state. Extracts from cells transfected with full-length Tetherin (Teth), the cytosolically expressed Tetherin ectodomain (cyt-ecto), or the ER/secretory version (sec-ecto) of the same domain were analyzed in reducing and nonreducing WBs developed with anti-SV5.
In the absence of MG132 two main isoforms of Tetherin dimers are present, which correspond to the ER-like and Golgi/membrane-glycosylated forms (Fig. 6B, upper panel, labeled as ER-glyc and Golgi/mem). Only the lower spot (ER-glyc) is abundantly biotinylated (Fig. 6B, lower panel). These results are consistent with the previous data shown in Figs. 3A and 6A. When the same experiment was performed in the presence of MG132, both dimers and monomers were found (Fig. 6C, upper panel). In addition to the same two protein populations (ER-glyc and Golgi/mem), of which only the ER-like glycosylated one was biotinylated, other isoforms of higher mobility corresponding to deglycosylated molecules were also found biotinylated (Fig. 6C, lower panel). These biotinylated and deglycosylated isoforms were present both in monomers and dimers, indicating that Tetherin dislocates as disulfide-bridged dimers, which then become deglycosylated by the cytosolic PNGase. This deglycosylation represents a second independent criterion (in addition to biotinylation) supporting the dislocation of Tetherin molecules in the form of dimers. In fact, the fastest migrating band in the dimeric population corresponds to deglycosylated molecules, as shown by the effect of the PNGase inhibitor Z-VAD-fmk in cells treated with MG132 (Fig. 6D). Additional evidence of retro-translocation of dimeric Tetherin was obtained from [35S]methionine pulse-chase labeling experiments (Fig. 6E). After a 15-min pulse, all labeled Tetherin already appeared as a dimer with an ER-like glycosylation, and a very small biotinylated fraction could be appreciated. After a 2-h chase, instead, part of the material was converted to the more mature Golgi/membrane isoforms (slower mobility), and an increased level of retro-translocated (biotinylated) dimer was also apparent (from 11% after the pulse to approximately 30–35% after the chase). As expected, the biotinylated band was derived from the ER-like glycosylation isoform. The small amount of monomer, which appeared only during the chase, was also completely biotinylated.
N-Glycosylation does not appear to be involved in maintaining the dimeric structure during retro-translocation. A Tetherin with the two glycosylation sites mutated to alanine (N65A,N92A) and located very close to two of the three cysteines (Cys-63 and Cys-91) involved in interchain SS bonds, still retro-translocated as dimer (Fig. 6F).
Effective alkylation of reduced cysteines in Tetherin is shown in Fig. 6G. The presence of NEM caused different migration of monomeric isoforms (compare Fig. 6G, lanes 1 and 3) and also prevented the formation of spurious heterodimers visible in the upper part of the gel (Fig. 6G, lanes 3 and 4). Therefore, biotinylated homodimers (lanes 2 and 4) represent true retro-translocated material and are not the consequence of postlysis artifacts. Further confirmation of dimer retro-translocation, as opposed to reoxidation after dislocation to the cytosol, was obtained by cytosolic expression of the Tetherin ectodomain (cyt-ecto, corresponding to residues 45–160), which was found totally reduced in the monomeric form, whereas the same ectodomain targeted to the lumen through a secretory leader peptide (sec-ecto) was found completely dimerized (Fig. 6H).
Taken together, these results indicate that Tetherin retro-translocation initiates by dislocating disulfide-bonded stabilized dimers, which at a later stage become deglycosylated, reduced to monomers, and finally degraded. Thus, both CD4 and Tetherin retro-translocate from the ER to the cytosol with intrachain and interchain disulfide bridges still formed, respectively, and not as completely unfolded molecules.
DISCUSSION
The way in which secretory and membrane proteins that have entered the ERAD pathway cross the membrane barrier to be transported to the cytosol still remains obscure. Retro-translocation itself is frequently described as a step involving the unfolding of the polypeptide chain, followed by the actual extraction, probably through some sort of dislocation channel. Many hypotheses from different groups have indicated the translocon Sec61 (34, 35), the Derlin family proteins (36, 37), TRAM1 (38), and BAP31 (39) as possible components of this macromolecular structure. On the cytosolic side, the AAA ATPase VCP/p97 has been widely reported as necessary to provide energy for the entire process (40–42).
The problem of the folding state of molecules that have entered the ERAD pathway, including the presence of disulfide bridges, is still controversial and remains an important issue (3, 32, 43–46). We took advantage of our recently described method that allows precise and specific detection of molecules retro-translocated from the ER to the cytosol (11, 47) to address the redox state of disulfide bridges in the retro-translocated fractions of CD4 and Tetherin. Biotinylation in living cells has also been used recently to study spontaneous retro-translocation of MHC class I molecules and to determine cytosolic/luminal localization of prion protein isoforms (48, 49).
When co-expressed with HIV-1 Vpu, CD4, and Tetherin are induced to enter the ERAD pathway (19, 25). For both proteins we showed that biotinylation identifies the population of retro-translocated molecules and demonstrated that Tetherin preferentially follows the retro-translocation/proteasome pathway, even in the absence of Vpu. More interestingly, both proteins are actually retro-translocated with disulfide bridges still formed. Whereas for CD4 the three disulfide bonds are intrachain, within each of the three of four extracellular Ig domains, for Tetherin three interchain disulfide bonds are formed between two parallel monomers. Hence, CD4 and Tetherin exit the ER as a partially folded molecule and as a dimer, respectively. This observation is particularly remarkable in the case of Tetherin also because of its double unusual N-terminal (transmembrane domain) and C-terminal (GPI) anchorage to the membrane.
The folding status of retro-translocated molecules remains controversial. There are few examples reported in literature of artificial substrates that seem to be dislocated as folded molecules (50, 51). These evidences seem to suggest, in a very indirect way, that unfolding is not a prerequisite for crossing the ER lipid bilayer. It has recently been shown that the very large SV40 viral particle (30–40 nm in diameter), which dislocates from the ER to the cytosol to subsequently enter the nucleus, exits the ER as a complex assembly assisted by two ERAD-involved proteins, BiP and BAP31 (5). A role for Derlin2 has been also established during infection by mouse polyomavirus (52).
CD4 and Tetherin are N-glycosylated. We found that for both proteins, a fraction of the biotinylated molecules retain their glycosylation state, in agreement with previous findings (11, 53) and consistent with the fact that the deglycosylating activity in mammalian cells is provided by the cytosolic PNGase (54). This confirms that deglycosylation appears to be the rate-limiting step of the process because deglycosylated biotinylated material only accumulates upon proteasomal inhibition, whereas glycosylated biotinylated molecules are also detected without inhibition. In addition, we observed dislocated CD4 molecules that were still glycosylated and with disulfide bridges formed, whereas all of the deglycosylated isoforms were found completely reduced, indicating that, in addition to deglycosylation, the reduction of these molecules also takes place after dislocation. In the particular case of Tetherin, biotinylated dimers were in part also deglycosylated, indicating that PNGase digestion can precede reduction.
However, not all proteins may behave like CD4 and Tetherin. For example, we have observed reduction prior to dislocation when the MHC-Iα chain is directed to degradation by the cytomegalovirus immunoevasin US2,5 suggesting that different requirements may apply to different proteins. To this respect, a crucial role for the activity of the ER-resident oxidoreductases protein disulfide isomerase and ERdj5 was reported for the MHC-Iα chain (55) and for other ERAD substrates such as BACE457 and BACE457Δ (56), the null Hong Kong α1-antitrypsin and the J chain of mouse IgM (57, 58). Indeed, a role for the redox potential during ERAD has been proposed (55, 59).
An interesting case is represented by the conditional nonsecretory mouse Ig κ-light chain NS1 for which the evidence indicates that, in the presence of MG132, one of the two intrachain disulfide bonds (in the CL domain) remains oxidized whereas the one in the VL domain is already reduced (32). In this case, a model of partial dislocation was proposed with the reduced domain exposed to the cytosolic side and the oxidized one still in the ER lumen until its final extraction and degradation (32). In light of the results here presented, however, an alternative model compatible with complete dislocation of both domains can be envisaged.
The fact that some proteins retro-translocate with both carbohydrate chains attached and intact disulfide bonds poses a limit to an exit mechanism based on a simple protein channel because of the high steric hindrance imposed by the sugar moieties and the globular or multimeric structure of the oxidized polypeptide. Whereas the CD4 species to be dislocated is still represented by a monomeric chain, for Tetherin there are two transmembrane domains and two GPI anchors to deal with, thus making the whole process far more complex.
In any event, the minimal width of CD4 with disulfide bridges formed, and without considering the sugar moieties, is approximately 30 Å (60, 61), and similar values have been estimated for the width of Tetherin dimers in the coiled-coil region (residues 68–138) (62). The Sec61 channel is only 15–25 Å wide and during synthesis can accommodate the transport of nonglycosylated unfolded polypeptides of 5–8 Å (63), suggesting that a pore of at least four times as wide is needed. It has been proposed but not proven completely that the Sec61 channel could form the retro-translocation pore by dynamically interacting with different molecular partners and thereby changing its permeability properties (64). Pores of larger size such as the ones formed by perforin (130–300 Å wide) (65, 66), which allow the movement of granzymes of 45–50 Å width (65), are large enough to compromise tight control of the transported material (67).
An alternative model for type I transmembrane proteins that does not consider dislocation as a pore-driven process has been suggested (68). The model postulated that extraction of the substrate could arise from the rearrangement of the ER membrane lipids leading to the formation of bicellular structures released in the cytosol. Recently, however, new experimental data have challenged this view (69).
In conclusion, we provide direct evidence that proteins can retro-translocate from the ER to the cytosol with folded oxidized disulfide bridges. Our results therefore seriously challenge the prevalent view of a retro-translocation step involving transport of already fully denatured polypeptides and impose important structural constraints to the molecular complex participating in the extraction phase.
G. Petris and O. R. Burrone, unpublished observation.
- ER
- endoplasmic reticulum
- BAP
- biotin acceptor peptide
- BirA
- Escherichia coli biotin ligase
- cyt-
- cytosolic
- Endo-H
- endoglycosidase H
- ERAD
- endoplasmic reticulum-associated degradation
- fmk
- fluoromethyl ketone
- GPI
- glycosylphosphatidylinositol
- NEM
- N-ethylmaleimide
- PNGase-F
- peptide N-glycosidase F
- sec-
- secretory
- StrAv
- streptavidin
- WB-ra
- Western blot retardation assay
- Z
- benzyloxycarbonyl.
REFERENCES
- 1. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 2. Rapoport T. A. (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- 3. Vembar S. S., Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Afshar N., Black B. E., Paschal B. M. (2005) Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol. Cell. Biol. 25, 8844–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Geiger R., Andritschke D., Friebe S., Herzog F., Luisoni S., Heger T., Helenius A. (2011) BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 13, 1305–1314 [DOI] [PubMed] [Google Scholar]
- 6. Li S., Spooner R. A., Hampton R. Y., Lord J. M., Roberts L. M. (2012) Cytosolic entry of Shiga-like toxin a chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS One 7, e41119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lord J. M., Roberts L. M., Lencer W. I. (2005) Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr. Top. Microbiol. Immunol. 300, 149–168 [DOI] [PubMed] [Google Scholar]
- 8. Sokołowska I., Wälchli S., Wȩgrzyn G., Sandvig K., Słominska-Wojewódzka M. (2011) A single point mutation in ricin A-chain increases toxin degradation and inhibits EDEM1-dependent ER retrotranslocation. Biochem. J. 436, 371–385 [DOI] [PubMed] [Google Scholar]
- 9. Teter K., Allyn R. L., Jobling M. G., Holmes R. K. (2002) Transfer of the cholera toxin A1 polypeptide from the endoplasmic reticulum to the cytosol is a rapid process facilitated by the endoplasmic reticulum-associated degradation pathway. Infect. Immun. 70, 6166–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernasconi R., Molinari M. (2011) ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 23, 176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petris G., Vecchi L., Bestagno M., Burrone O. R. (2011) Efficient detection of proteins retro-translocated from the ER to the cytosol by in vivo biotinylation. PLoS One 6, e23712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brady R. L., Barclay A. N. (1996) The structure of CD4. Curr. Top Microbiol. Immunol. 205, 1–18 [DOI] [PubMed] [Google Scholar]
- 13. Jouvenet N., Neil S. J., Zhadina M., Zang T., Kratovac Z., Lee Y., McNatt M., Hatziioannou T., Bieniasz P. D. (2009) Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83, 1837–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neil S. J., Zang T., Bieniasz P. D. (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430 [DOI] [PubMed] [Google Scholar]
- 15. Perez-Caballero D., Zang T., Ebrahimi A., McNatt M. W., Gregory D. A., Johnson M. C., Bieniasz P. D. (2009) Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139, 499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakuma T., Noda T., Urata S., Kawaoka Y., Yasuda J. (2009) Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83, 2382–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishikawa J., Kaisho T., Tomizawa H., Lee B. O., Kobune Y., Inazawa J., Oritani K., Itoh M., Ochi T., Ishihara K. (1995) Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 26, 527–534 [DOI] [PubMed] [Google Scholar]
- 18. Kupzig S., Korolchuk V., Rollason R., Sugden A., Wilde A., Banting G. (2003) Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4, 694–709 [DOI] [PubMed] [Google Scholar]
- 19. Magadán J. G., Pérez-Victoria F. J., Sougrat R., Ye Y., Strebel K., Bonifacino J. S. (2010) Multilayered mechanism of CD4 down-regulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 6, e1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Margottin F., Bour S. P., Durand H., Selig L., Benichou S., Richard V., Thomas D., Strebel K., Benarous R. (1998) A novel human WD protein, h-βTrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1, 565–574 [DOI] [PubMed] [Google Scholar]
- 21. Schubert U., Antón L. C., Bacík I., Cox J. H., Bour S., Bennink J. R., Orlowski M., Strebel K., Yewdell J. W. (1998) CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 72, 2280–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Douglas J. L., Viswanathan K., McCarroll M. N., Gustin J. K., Früh K., Moses A. V. (2009) Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a βTrCP-dependent mechanism. J. Virol. 83, 7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goffinet C., Allespach I., Homann S., Tervo H. M., Habermann A., Rupp D., Oberbremer L., Kern C., Tibroni N., Welsch S., Krijnse-Locker J., Banting G., Kräusslich H. G., Fackler O. T., Keppler O. T. (2009) HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 5, 285–297 [DOI] [PubMed] [Google Scholar]
- 24. Iwabu Y., Fujita H., Kinomoto M., Kaneko K., Ishizaka Y., Tanaka Y., Sata T., Tokunaga K. (2009) HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284, 35060–35072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mangeat B., Gers-Huber G., Lehmann M., Zufferey M., Luban J., Piguet V. (2009) HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its β-TrCP2-dependent degradation. PLoS Pathog. 5, e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitchell R. S., Katsura C., Skasko M. A., Fitzpatrick K., Lau D., Ruiz A., Stephens E. B., Margottin-Goguet F., Benarous R., Guatelli J. C. (2009) Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-TrCP and endolysosomal trafficking. PLoS Pathog. 5, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beckett D., Kovaleva E., Schatz P. J. (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8, 921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swiecki M., Scheaffer S. M., Allaire M., Fremont D. H., Colonna M., Brett T. J. (2011) Structural and biophysical analysis of BST-2/tetherin ectodomains reveals an evolutionary conserved design to inhibit virus release. J. Biol. Chem. 286, 2987–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Willey R. L., Maldarelli F., Martin M. A., Strebel K. (1992) Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J. Virol. 66, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Predonzani A., Arnoldi F., López-Requena A., Burrone O. R. (2008) In vivo site-specific biotinylation of proteins within the secretory pathway using a single vector system. BMC Biotechnol. 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoon Y. H., Cho K. S., Hwang J. J., Lee S. J., Choi J. A., Koh J. Y. (2010) Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 51, 6030–6037 [DOI] [PubMed] [Google Scholar]
- 32. Okuda-Shimizu Y., Hendershot L. M. (2007) Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell 28, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shamu C. E., Flierman D., Ploegh H. L., Rapoport T. A., Chau V. (2001) Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol. Biol. Cell 12, 2546–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmitz A., Herrgen H., Winkeler A., Herzog V. (2000) Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 148, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wiertz E. J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T. R., Rapoport T. A., Ploegh H. L. (1996) Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432–438 [DOI] [PubMed] [Google Scholar]
- 36. Lilley B. N., Ploegh H. L. (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429, 834–840 [DOI] [PubMed] [Google Scholar]
- 37. Sun F., Zhang R., Gong X., Geng X., Drain P. F., Frizzell R. A. (2006) Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 281, 36856–36863 [DOI] [PubMed] [Google Scholar]
- 38. Ng C. L., Oresic K., Tortorella D. (2010) TRAM1 is involved in disposal of ER membrane degradation substrates. Exp. Cell Res. 316, 2113–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang B., Heath-Engel H., Zhang D., Nguyen N., Thomas D. Y., Hanrahan J. W., Shore G. C. (2008) BAP31 interacts with Sec61 translocons and promotes retrotranslocation of CFTRΔF508 via the derlin-1 complex. Cell 133, 1080–1092 [DOI] [PubMed] [Google Scholar]
- 40. Carlson E. J., Pitonzo D., Skach W. R. (2006) p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 25, 4557–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Elkabetz Y., Shapira I., Rabinovich E., Bar-Nun S. (2004) Distinct steps in dislocation of luminal endoplasmic reticulum-associated degradation substrates: roles of endoplasmic reticulum-bound p97/Cdc48p and proteasome. J. Biol. Chem. 279, 3980–3989 [DOI] [PubMed] [Google Scholar]
- 42. Ye Y., Meyer H. H., Rapoport T. A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 43. Hampton R. Y., Sommer T. (2012) Finding the will and the way of ERAD substrate retrotranslocation. Curr. Opin. Cell Biol. 24, 460–466 [DOI] [PubMed] [Google Scholar]
- 44. Bagola K., Mehnert M., Jarosch E., Sommer T. (2011) Protein dislocation from the ER. Biochim. Biophys. Acta 1808, 925–936 [DOI] [PubMed] [Google Scholar]
- 45. Feige M. J., Hendershot L. M. (2011) Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 23, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Claessen J. H., Kundrat L., Ploegh H. L. (2012) Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 22, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vecchi L., Petris G., Bestagno M., Burrone O. R. (2012) Selective targeting of proteins within secretory pathway for endoplasmic reticulum-associated degradation. J. Biol. Chem. 287, 20007–20015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., Yu Y. Y., Myers N., Hansen T. H. (2013) Decoupling the role of ubiquitination for the dislocation versus degradation of major histocompatibility complex (MHC) class I proteins during endoplasmic reticulum-associated degradation (ERAD). J. Biol. Chem. 288, 23295–23306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emerman A. B., Zhang Z. R., Chakrabarti O., Hegde R. S. (2010) Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Mol. Biol. Cell 21, 4325–4337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fiebiger E., Story C., Ploegh H. L., Tortorella D. (2002) Visualization of the ER-to-cytosol dislocation reaction of a type I membrane protein. EMBO J. 21, 1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tirosh B., Furman M. H., Tortorella D., Ploegh H. L. (2003) Protein unfolding is not a prerequisite for endoplasmic reticulum-to-cytosol dislocation. J. Biol. Chem. 278, 6664–6672 [DOI] [PubMed] [Google Scholar]
- 52. Lilley B. N., Gilbert J. M., Ploegh H. L., Benjamin T. L. (2006) Murine polyomavirus requires the endoplasmic reticulum protein Derlin-2 to initiate infection. J. Virol. 80, 8739–8744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blom D., Hirsch C., Stern P., Tortorella D., Ploegh H. L. (2004) A glycosylated type I membrane protein becomes cytosolic when peptide N-glycanase is compromised. EMBO J. 23, 650–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Misaghi S., Pacold M. E., Blom D., Ploegh H. L., Korbel G. A. (2004) Using a small molecule inhibitor of peptide N-glycanase to probe its role in glycoprotein turnover. Chem. Biol. 11, 1677–1687 [DOI] [PubMed] [Google Scholar]
- 55. Tortorella D., Story C. M., Huppa J. B., Wiertz E. J., Jones T. R., Bacik I., Bennink J. R., Yewdell J. W., Ploegh H. L. (1998) Dislocation of type I membrane proteins from the ER to the cytosol is sensitive to changes in redox potential. J. Cell Biol. 142, 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Molinari M., Galli C., Piccaluga V., Pieren M., Paganetti P. (2002) Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J. Cell Biol. 158, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hagiwara M., Maegawa K., Suzuki M., Ushioda R., Araki K., Matsumoto Y., Hoseki J., Nagata K., Inaba K. (2011) Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 41, 432–444 [DOI] [PubMed] [Google Scholar]
- 58. Ushioda R., Hoseki J., Araki K., Jansen G., Thomas D. Y., Nagata K. (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572 [DOI] [PubMed] [Google Scholar]
- 59. Hosokawa N., Wada I., Natsuka Y., Nagata K. (2006) EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded α1-antitrypsin. Genes Cells 11, 465–476 [DOI] [PubMed] [Google Scholar]
- 60. Rudd P. M., Wormald M. R., Stanfield R. L., Huang M., Mattsson N., Speir J. A., DiGennaro J. A., Fetrow J. S., Dwek R. A., Wilson I. A. (1999) Roles for glycosylation of cell surface receptors involved in cellular immune recognition. J. Mol. Biol. 293, 351–366 [DOI] [PubMed] [Google Scholar]
- 61. Wu H., Kwong P. D., Hendrickson W. A. (1997) Dimeric association and segmental variability in the structure of human CD4. Nature 387, 527–530 [DOI] [PubMed] [Google Scholar]
- 62. Hinz A., Miguet N., Natrajan G., Usami Y., Yamanaka H., Renesto P., Hartlieb B., McCarthy A. A., Simorre J. P., Göttlinger H., Weissenhorn W. (2010) Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 7, 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Van den Berg B., Clemons W. M., Jr., Collinson I., Modis Y., Hartmann E., Harrison S. C., Rapoport T. A. (2004) X-ray structure of a protein-conducting channel. Nature 427, 36–44 [DOI] [PubMed] [Google Scholar]
- 64. Kalies K. U., Allan S., Sergeyenko T., Kröger H., Römisch K. (2005) The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 24, 2284–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Law R. H., Lukoyanova N., Voskoboinik I., Caradoc-Davies T. T., Baran K., Dunstone M. A., D'Angelo M. E., Orlova E. V., Coulibaly F., Verschoor S., Browne K. A., Ciccone A., Kuiper M. J., Bird P. I., Trapani J. A., Saibil H. R., Whisstock J. C. (2010) The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 468, 447–451 [DOI] [PubMed] [Google Scholar]
- 66. Praper T., Sonnen A., Viero G., Kladnik A., Froelich C. J., Anderluh G., Dalla Serra M., Gilbert R. J. (2011) Human perforin employs different avenues to damage membranes. J. Biol. Chem. 286, 2946–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu C. C., Walsh C. M., Young J. D. (1995) Perforin: structure and function. Immunol. Today 16, 194–201 [DOI] [PubMed] [Google Scholar]
- 68. Ploegh H. L. (2007) A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 448, 435–438 [DOI] [PubMed] [Google Scholar]
- 69. Olzmann J. A., Kopito R. R. (2011) Lipid droplet formation is dispensable for endoplasmic reticulum-associated degradation. J. Biol. Chem. 286, 27872–27874 [DOI] [PMC free article] [PubMed] [Google Scholar]