

Background: The target capture is a critical step for the selection of integration site.
Results: Mos1 transposon excision occurred before target capture. The TA dinucleotide present in the target and bent targets are important for this step.
Conclusion: Target capture mechanism and distribution of mariner elements are linked.
Significance: New insights for modeling Mos1 target capture complex and better understanding of mariner transposition cycle.
Keywords: DNA-Protein Interaction, Enzyme Mechanisms, Gene Transposable Elements, Genome Structure, Genomic Instability, Target Capture, Transposition
Abstract
DNA transposition contributes to genomic plasticity. Target capture is a key step in the transposition process, because it contributes to the selection of new insertion sites. Nothing or little is known about how eukaryotic mariner DNA transposons trigger this step. In the case of Mos1, biochemistry and crystallography have deciphered several inverted terminal repeat-transposase complexes that are intermediates during transposition. However, the target capture complex is still unknown. Here, we show that the preintegration complex (i.e., the excised transposon) is the only complex able to capture a target DNA. Mos1 transposase does not support target commitment, which has been proposed to explain Mos1 random genomic integrations within host genomes. We demonstrate that the TA dinucleotide used as the target is crucial both to target recognition and in the chemistry of the strand transfer reaction. Bent DNA molecules are better targets for the capture when the target DNA is nicked two nucleotides apart from the TA. They improve strand transfer when the target DNA contains a mismatch near the TA dinucleotide.
Introduction
Transposable elements are prominent in the make up of a major fraction of many eukaryotic genomes. They contribute to genome plasticity in many ways. They act as insertional mutagens, alter the regulation of gene expression, and provide coding information for the emergence of new functions (1–4). The study of their transposition pathways may shed light onto their impact on the dynamics of eukaryotic genome evolution. Among these transposons, Itm elements (to which belong mariner elements) are one of the most widespread groups of transposons. They transpose via a DNA intermediate, using a cut and paste mechanism (5). Elements of mariner are discrete DNA segments containing an ORF coding a single protein, the transposase, surrounded by inverted terminal repeats (ITRs).4 The active transposase is a homodimer that binds to one ITR. The second ITR is recruited to form a paired end complex, which is the catalytic complex where the strand transfer reactions promoting excision take place. The excision product containing the two cleaved ITR with a transposase dimer is known as a preintegration complex (PIC), in reference to the equivalent complex described for HIV integrase (6). Paired end complex assembly and excision of mariner are now well understood (7–10). In contrast, little is known about the subsequent step, namely the target capture. The target DNA is thought to be associated with the PIC in the so-called target capture complex (TCC) that drives the transposon to integrate at its new site in the genome.
Target capture has been extensively studied for prokaryotic model elements (Tn5, Tn7, and Tn10), the Mu phage, and two eukaryotic transposons (RAG1/2 and Himar1). All the related transposases display an RNase H-like catalytic domain that contains a DD(E/D) catalytic triad (11). However, these transposases are of different origins: Tn5 and Tn10 belong to the IS10 superfamily, and Tn7 is the prototype of a single family. Concerning the eukaryotic transposases, RAG1/2 belongs to the Transib family, whereas Himar1 belongs to the mariner family. These different origins result in differences in the transposition mechanisms. For instance, hairpins are produced during the excision at the ends of Tn5 and Tn10 but on the flanking DNA for RAG1/2. In contrast, mariner transposition does not require any hairpin (12). This suggests that the paired end and preintegration complexes are organized differently depending on the transposase involved. This is not the only difference existing between these well studied transposons. The target structure appears crucial in the recognition and/or integration of various elements (Tn7, Tn10, RAG1/2 transposases, and HIV-1 integrase) (13–17), whereas the only determinant for mariner target recognition and strand transfer is the presence of a TA dinucleotide, which is duplicated upon integration (18, 19). Moreover, some elements have specific (attTn7 for Tn7) (13) or preferred (HisG1 for Tn10, G(CT)(CT)(CT)(AT)(AG)(AG)(AG)C for Tn5) (20, 21) targets, whereas mariner elements display an essentially random integration pattern. However, it has been shown that Mos1 may have preferred integration spots such as the one in the chloramphenicol resistance gene of Tn9 (22). No structural features explaining the preferential integration of Mos1 have yet been identified (23). In addition, contrarily to RAG1/2 that creates a 5-bp duplication upon insertion (24), mariner elements generate a 2-bp duplication.
All these remarks suggest that target capture complexes are organized differently depending on the transposase involved and result in different integration pathways. An example is the timing of the target capture during various transposition cycles. Tn7 binds its specific target attTn7 before excising (13), whereas Mu captures its target DNA at different points of the reaction pathway (24, 25). In contrast, Tn5, Tn10, and RAG1/2 bind their target after excision (24, 26–29), and Himar1 could perform target capture both before and after excision (29). It has been demonstrated that Tn10, Himar1, and RAG1/2 capture their target in a two-step procedure. First, a labile interaction occurs between the target and the excised element. Second, the complex is stabilized, so that the target cannot be replaced by another DNA, resulting in what is known as “target commitment” (26, 28, 29).
The variety of the target capture mechanisms makes it necessary to describe them for each transposon family. In particular, within the mariner family, the fact that the first model to be studied, namely Himar1 (29), displayed a target capture contrasting with other transposases (Tn5, Tn10, and RAG1/2), leaves open the question of the chronology of events (excision/target capture/integration or target capture/excision/integration) for the other mariner elements. In this perspective, the Mos1 element is of particular interest, because it is the only eukaryotic transposon for which the complete transposition cycle can be reconstituted step by step in vitro and for which crystallographic data of the excised element (precleaved ITRs with two transposases) are available (9). Based on the crystal structure, a target capture complex has been recently proposed to account for the fact that a channel exists between the two transposases that might provide a niche to the target DNA (9). However, there is no biochemical evidence to support this model.
Here, we confirm that the organization of the Mos1 target capture complex is consistent with that predicted by cut and paste models of transposition. We show that target capture takes place only after Mos1 element has been fully excised. The TA dinucleotide present in the target is of particular importance not only for the strand transfer, but also for the target capture itself. DNA bending of the target significantly improves the efficiency of capture, highlighting the role of the +2 position relative to the TA dinucleotide. The various models previously published for Mos1 are discussed in the light of our findings.
EXPERIMENTAL PROCEDURES
Proteins
The MBP-MOS1 protein was produced using the pVL1392 baculovirus transfer vector and the BaculoGoldTM baculovirus expression system (BD Biosciences) and then extracted from baculovirus-infected Sf21 cells. The fusion protein was purified on a amylose resin (New England Biolabs) as previously described (30) (supplemental Fig. S1).
ITR and Target DNA
The sequences of the oligonucleotides used in the study are shown in supplemental Table S1. The precleaved ITR (PC-ITR) is 50 bp long (also used as a short ITR), and the long PC-ITR is 70 bp long. The uncleaved ITR (UC-ITR) and the prenicked ITR (PN-ITR) are 70 bp long. The TA targets are 30 bp long (also used as a short target) and 50 bp long (used as long target). GC target and targets containing mismatch and nick are 30 bp long. Oligonucleotides were provided by Eurofins MWG Biotech (Germany) or Eurogentec (Belgium) and purified by PAGE. Double-stranded DNA of ITRs and targets were obtained by annealing equimolar amounts of different complementary oligonucleotides. The wild type TA target was obtained by annealing T1 and T2. Targets exhibiting a nick at the integration site obtained by annealing T1, T3, and T3′ (N+1 target); T1, T4, and T4′ (N+2 target); T1, T5, and T5′ (N+3 target); and T3′, T4′, and T5′ were phosphorylated at the 5′ position using standard procedures. The targets exhibiting a mismatch were obtained by annealing T1 and T6 (M+1 target), T1 and T7 (M+2 target), and T1 and T8 (M+3 target). The GC target was obtained by annealing T9 and T10. Long targets were obtained by annealing T11 and T12 or T13 and T14. All targets were filled in with [α-32P]dATP by Klenow exo-minus in presence of dTTP. PC-ITR was obtained by annealing I4 and I5, UC-ITR was obtained by annealing I1 and I2, and PN-ITR was obtained by annealing I2, I3, and I4. The transferred strand (I2 or I5) was 5′ end-labeled with [γ-32P]ATP by T4 polynucleotide kinase prior to annealing.
Preintegration Complex Assembly
PICs were formed using 250 nm transposase (MBP-MOS1) and 250 nm PC-ITR in 10 mm Tris, pH 9, 50 mm NaCl, 0.5 mm DTT, 5% glycerol, and 5 mm MgCl2 in 20 μl. Reactions were carried out for 3 h at 30 °C. For PIC analyses, labeled ITRs were used instead of unlabeled ITRs, and the reaction products were analyzed by EMSA onto 6% native polyacrylamide gel in 0.25× TBE (Tris-borate-EDTA) buffer. The gel was dried and scanned. When specified, the PICs were assembled using UC-ITR or PN-ITR instead of PC-ITR.
Target Capture Assays
PICs were formed as described above using cold ITRs. After incubation, 250 nm of labeled targets were added with 5 mm EDTA. The addition of EDTA chelates MgCl2, blocking the strand transfer. The use of labeled target and EDTA allowed the detection of complexes if and only if the complexes contained a target and avoided strand transfer. For TCC assembly, reactions were carried out for another hour (30 °C). All the targets used contained a single dinucleotide TA, except for the GC target, which did not contain any TA. Complexes were analyzed by EMSA onto 6% native polyacrylamide gel in 0.25× TBE buffer. The gel was dried and scanned.
Stoichiometry of Target Capture Complex
Determination of the Number of Transposases in TCC
Cold PICs were prepared as described above but with an overnight incubation before proceeding to TCC assembly with a labeled target. The TCCs were then digested for 0–5 h by factor Xa (1 μg) in the presence of 5 mm CaCl2 at 30 °C. Reaction products were analyzed by EMSA onto 6% native polyacrylamide gel. The gel was dried and scanned.
Determination of the Number of Targets in TCC
Cold PICs were prepared as described above. Then labeled short (30 bp) and/or long (50 bp) targets were added before performing target capture. Reaction products were analyzed by EMSA onto 6% native polyacrylamide gel. The gel was dried and scanned.
Determination of the Number of ITRs in TCC
Cold PICs were prepared as described above using a mixture of equimolar unlabeled short (50 bp) and long (70 bp) PC-ITRs at a final concentration of 250 nm. Standard target capture (with a labeled target) assays were performed as described above. Reaction products were analyzed by EMSA onto 6% native polyacrylamide gel. The gel was dried and scanned.
Target Commitment
The TCCs were assembled as previously described using either short or long labeled targets as the first target. To find out whether the target could be exchanged, the second labeled target (short if the first was long and vice versa) was added at 250 nm. A control reaction was performed by adding long and short targets at the same time. Reactions were carried out for 1 h more at 30 °C. Complexes were analyzed by EMSA onto 6% native polyacrylamide gel. The gel was dried and scanned. Each point was repeated five times. The signal obtained for each target (long or short) was quantified using ImageQuant software. The percentage of each target was calculated as the signal obtained with the long (or short) target divided by the signal obtained with long and short targets. The percentage of target commitment was calculated as the percentage of the long or short target in a target commitment assay minus the percentage of the long or short target in the control reaction.
Integration Assays
Cold PICs were formed using PC-ITR at 30 °C but for 30 min. Labeled targets were then added with or without 5 mm EDTA for 3 h at 30 or 4 °C. Reactions were stopped by adding 0.1 mg/ml proteinase K and 0.1% SDS, incubated at 65 °C for 10 min, and incubated then at 37 °C for 30 min. DNA products were purified by phenol chloroform extraction and ethanol precipitation. Integration products were resuspended in a standard denaturing loading buffer, boiled for 5 min, and then loaded onto an 8% denaturing urea-acrylamide gel. The gel was dried and scanned. The integration rate is the ratio of the integration signal divided by the free target signal plus the integration signal. The integration efficiency found using the TA target was normalized to 100%. Each point was repeated five times. A labeled G+A ladder was obtained by annealing of GA1 (labeled at the 5′ end with [γ-32P]ATP by T4 polynucleotide kinase) to its complement GA2 (supplemental Table S1). This product was chemically cleaved at G and A using the Maxam and Gilbert standard procedure. G+A products were resuspended in denaturing loading buffer.
Integration products were analyzed by PCR, using F1 and F2 primers (supplemental Table S1). Integration products (3 μl) were amplified with GoTaq DNA polymerase (Promega) in conditions recommended by the manufacturer. 35 PCR cycles were carried out with an annealing temperature of 44 °C. PCR products were resolved on a 2% agarose gel containing ethidium bromide and purified using the Nucleospin extract II kit (Macherey-Nagel). PCR products were then cloned in the pGEM-T Easy System (Promega) and sequenced by Eurofins MWG Biotech (Germany).
Statistical Analyses
Quantification was done using ImageQuant software. Statistical analyses were performed using GraphPad Prism version 4.0c for Macintosh (GraphPad Software, La Jolla, CA). We used a Kruskal-Wallis one-way analysis of variance by ranks. This is a nonparametric method for testing whether samples originate from the same distribution or not. It is used to compare more than two samples that are not related. The null hypothesis is that the populations from which the samples originate all have the same median value. Analyses are done using an α level of 5% (α = 0.05). The calculated Kruskal-Wallis value is then compared with the critical value. If the Kruskal-Wallis test leads to significant results, then at least one of the samples is different from the other samples. The test does not identify where the differences lie, nor how many differences are actually present. To identify the differences, sample contrast determinations were carried out between individual sample pairs (or post hoc tests). Multiple comparisons were done using Dunn's test (pairwise comparisons) and the Bonferroni correction to find out whether the post hoc tests are significant.
RESULTS
The Target Capture Complex in the Mos1 System
According to the currently accepted models of mariner transposition, transpososome excision (i.e., the formation of the PIC) should precede the target capture (7). To set up a target capture assay, PICs were first formed using cold precleaved ITR (PC-ITR, mimicking the ITR extremities cleaved by transposase on the two strands) and the purified transposase MOS1 under catalytic conditions (Mg2+, 30 °C). The labeled target (containing a single TA) was added with EDTA after the PIC had been fully formed. In this situation, the observed complex will only correspond to the target capture complex and not to the strand transfer complex. The scheme of the assay is outlined in Fig. 1A. Formation of ITR-transposase complexes was controlled by the detection of SEC2 (single end complex 2) and PIC as already described (7) (Fig. 1B, lane 1). The use of equimolar amounts of unlabeled ITR, transposase, and labeled target gave rise to a complex detected by EMSA (Fig. 1B, lane 5). The only labeled DNA used in this assay is the target, allowing the detection of complexes if and only if a target molecule was present. The complex obtained in presence of EDTA and herein observed satisfied this condition. This complex should be considered as a TCC. As expected, this complex disappeared partially when equimolar herring sperm double-stranded DNA was added as competitor at the same time as the target, because this DNA could also be used as a target (Fig. 1B, lane 6). The band corresponding to TCC was expected to migrate above the PIC, because it contains an additional DNA fragment. However, the TCC has one surprising feature: it migrates slightly faster than the PIC (Fig. 1B). This discrepancy between the electrophoretic properties of TCC and PIC could result from the fact that the two complexes have different conformations (see “Bending DNA in the TCC”).
FIGURE 1.

Organization of the Mos1 target capture complex. A, implemented protocol for TCC assembly. PIC was assembled with Tpase (gray ovals) and unlabeled PC-ITR (arrows) in catalytic conditions After assembly, labeled target (T*) was added with 5 mm EDTA to avoid strand transfer. TCCs were analyzed by EMSA. B, target capture assay. PIC assembly was monitored using labeled PC-ITRs (*) (lane 1). TCC assembly was monitored as described for A with labeled target (*) and unlabeled ITR (c) in the presence of transposase (+) (lane 5) and with competitor DNA (lane 6). The direct binding of MOS1 to the target was analyzed in the presence (+) or absence (−) of competitor DNA (lanes 3 and 4). Left side, complexes with labeled PC-ITR (SEC2, PIC). Right side, complexes with labeled targets (TCC and nonspecific binding complex (NSC)). Free DNAs (target, dimer of target (dtarget), and ITR) are indicated. C, PIC assembly was monitored using labeled PC-ITRs. TCC were performed with two labeled target: a short (S*) (30 bp) and a long target (L*) (50 bp), and analyzed by EMSA. D, determination of the number of target in the TCC. EMSAs were performed with short/long target combinations and a cold PC-ITR, as indicated. L, long target; S, short target. Labeled targets are indicated by asterisks. Targets present in the complexes are drawn on the right. E, determination of the number of transposase in TCC. For each condition, TCCs were assembled as described in A with labeled target (*), unlabeled ITR (c), and transposase. TCC assembly was performed without factor Xa treatment (0 h). After TCC assembly, TCCs were subjected to factor Xa cleavage for various times (1 h, 2 h 30 min, and 5 h) before EMSA. The proteins present in the various TCCs are drawn on the right. F, determination of the number of ITRs in the TCC. TCC assembly was performed with unlabeled (c) short (S)/long (L) ITR combinations and a labeled target (*) and analyzed by EMSA. The ITRs present in the complexes are drawn on the right. L/L, two long ITRs; S/L, one short and one long ITR; S/S, two short ITRs. G, kinetics of TCC formation. After PIC assembly as described in A, TCC formation was allowed to proceed for various times (0, 5, 30, 60, 120, and 180 min). The percentage of TCC formed was quantified (labeled target in TCC-labeled target in TCC + free target) and plotted as a function of time.
In the absence of ITR, a MOS1-labeled target complex is also detected. It corresponds to a nonspecific complex showing that the binding of MOS1 is not restricted to ITRs (Fig. 1B, lane 3). This binding is nonspecific because it decreased by addition of herring sperm double-stranded DNA (Fig. 1B, lane 4). This MOS1 target nonspecific complex migrates faster than the putative TCC and can be distinguished from TCC. To confirm that the complex observed with the labeled target should be a TCC, TCC was assembled with a short target (30 bp) and a long target (50 bp). As expected, when the target is longer, the TCC migrated slower than the complex containing a short target (Fig. 1C). This result showed that the observed complex corresponded to a TCC.
TCC is expected to contain a transposase dimer, a pair of ITRs, and a single target DNA molecule. We therefore determined the ITR-transposase target content of the putative Mos1 TCC. The number of targets was determined by a long/short target experiment. The PIC was assembled with cold ITRs, and then a mix of long or/and short targets was added. In the mix, either the short or the long target was labeled, as indicated (Fig. 1D). On the basis of what is known about TCC stoichiometry (a single target per complex), a single-band pattern was expected regardless of the conditions used. The resulting complexes were analyzed in EMSA. The same pattern was observed for reactions containing each combination of targets and corresponded to a single-band pattern. This result showed that the Mos1 TCC contained a single target DNA. Then the presence of a MOS1 dimer in the putative TCC was assayed by EMSA with a labeled target and unlabeled ITR. We took advantage of the cleavage site for the factor Xa protease between the MBP and MOS1 moieties in MBP-MOS1 fusion protein, as previously done to analyze the composition of the Mos1 PIC (7). If the TCC contains an MBP-MOS1 dimer, three bands would have been expected following factor Xa cleavage: a first band containing uncleaved MBP-MOS1, a second band containing both cleaved and uncleaved MOS1 monomers, and a third band containing two cleaved MOS1. Our data showed the sequential detection of each expected band. (Fig. 1E). Our data demonstrate that the TCC does contain a transposase dimer because it satisfies the conditions expected after factor Xa cleavage. Finally, The number of ITRs was determined using a long/short ITR experiment similar to that used to determine the number of ITR in the Mos1 PIC (7). The PIC was first assembled with short and/or long cold ITRs, and then a labeled target was added to allow TCC formation. With mixed ITRs, a three-band pattern would be expected if the TCC contains two ITRs (a band containing two short ITRs, a band containing one short and one long ITR, and a band containing two long ITRs). In contrast, a single-band pattern would be expected with mixed ITRs if the TCC contains only a single ITR. Because EMSA showed a three-band pattern with mixed ITRs (Fig. 1F), we concluded that the putative TCC contains two ITRs. As a conclusion, the putative TCC detected in Fig. 1B corresponds to a true TCC that contains a transposase dimer, a pair of ITRs, and a single target DNA. Finally, time course analyses showed that target capture was observed immediately after adding the target DNA, indicating a rapid capture mechanism. It reached its maximum after 1 h of incubation, suggesting a dynamic equilibrium between the partners (PIC on the one hand and the target on the other hand) (Fig. 1G).
Target capture efficiency of MOS1, as measured in our conditions (1.5%) could appear to be low compared with bacterial transposases (nearly 50%) (14), but we note that this value is highly correlated to the amount of preformed PIC (Fig. 1B, lane 6, and see below). This therefore suggested that the target capture is not a limiting step of the transposition cycle, which rather relies on the PIC assembly (7, 10).
Properties of the TCC
Most of our knowledge of the target capture mechanisms in eukaryotes is based on analogies with prokaryotic transposition. For Tn10 and RAG1/2, the excision precedes target capture, but for the mariner transposase Himar1, experiments exploring target commitment with uncleaved and precleaved ITR extremities suggest that target capture could occur before or after excision (28, 29). However, the assays used in these cases cannot separate the target capture step by itself from the integration step, thus preventing a fine analysis of the reaction. In contrast, our target capture assay allows the detection of the target capture complex in conditions preventing strand transfers, thus offering an unique opportunity to explore the sequence of events ranging from the assembly of the paired end complex to the strand transfer complex. To assess the need for ITRs cleavage to capture a target, UC-ITRs (mimicking ITRs present in genomic DNA) and PN-ITRs (mimicking ITRs cleaved at the nontransferred strand) (Fig. 2A) were used to prepare TCCs and were checked by EMSA (Fig. 2B). We first verified the ability of each ITR to promote PIC assembly in various conditions (noncatalytic conditions, Mg2+ at 4 °C; or catalytic conditions, Mg2+ at 30 °C) for 2 h. Under noncatalytic conditions, UC-ITRs (Fig. 2B, lane 1) and PN-ITRs (Fig. 2B, lane 5) only promoted the assembly of SEC2 (a single ITR and two transposases), and therefore no PIC was assembled (7), whereas in similar but noncatalytic conditions, PC-ITRs (mimicking ITRs cleaved at both strand) allowed the formation of a robust PIC (Fig. 2B, lanes 9). After the addition of a DNA target molecule, UC-ITRs and PN-ITRs (Fig. 2B, lanes 3 and 7) were unable to promote the assembly of a TCC. In similar conditions, PC-ITRs allowed the formation of a TCC (Fig. 2B, lane 11). Catalytic conditions (Mg2+ at 30 °C) allow the cleavage of two strands of the ITR (31, 32). In fact, PN-ITRs and UC-ITRs needed catalytic conditions (Mg2+ at 30 °C) to form a PIC, which was robust for PN-ITRs (Fig. 2B, lane 6), but in a lesser amount than that produced with PC-ITRs (Fig. 2B, lane 10), and remained faint for UC-ITRs (Fig. 2B, lane 2). For each ITR, in catalytic conditions, faint bands were observed above the PIC and could correspond to strand transfer complex and in noncatalytic conditions to oligomers between transposases and ITR as already observed (29, 31). In catalytic conditions, TCC was detected with each ITR and relied on PIC formation (Fig. 2B, lanes 4, 8, 11, and 12) because TCC was only observed in conditions allowing PIC assembly. Conversely, TCC was never observed when the PIC was absent (Fig. 2B, lanes 3 and 7). These results indicated that strand cleavage of PN-ITRs and UC-ITRs occurs during the formation of PIC, as previously shown (8, 10). They also indicated that the PIC is a more stable complex than the paired end complex (corresponding to the excision complex, still linked to flanking DNA), which cannot be detected in EMSA. Once the PIC is formed, the TCC can be assembled, if a target is provided to the reaction. The need for DNA cleavage was confirmed by replacing Mg2+ by Ca2+, because Ca2+ allows the second strand cleavage to occur, but not the cleavage of the first strand (26, 29). Under these conditions, a PIC was detected with PN-ITRs, but not with UC-ITRs, and TCC was observed only under conditions permitting PIC formation (supplemental Fig. S2). Altogether, these experiments support the hypothesis that MOS1 is able to capture a target only after cleavage.
FIGURE 2.
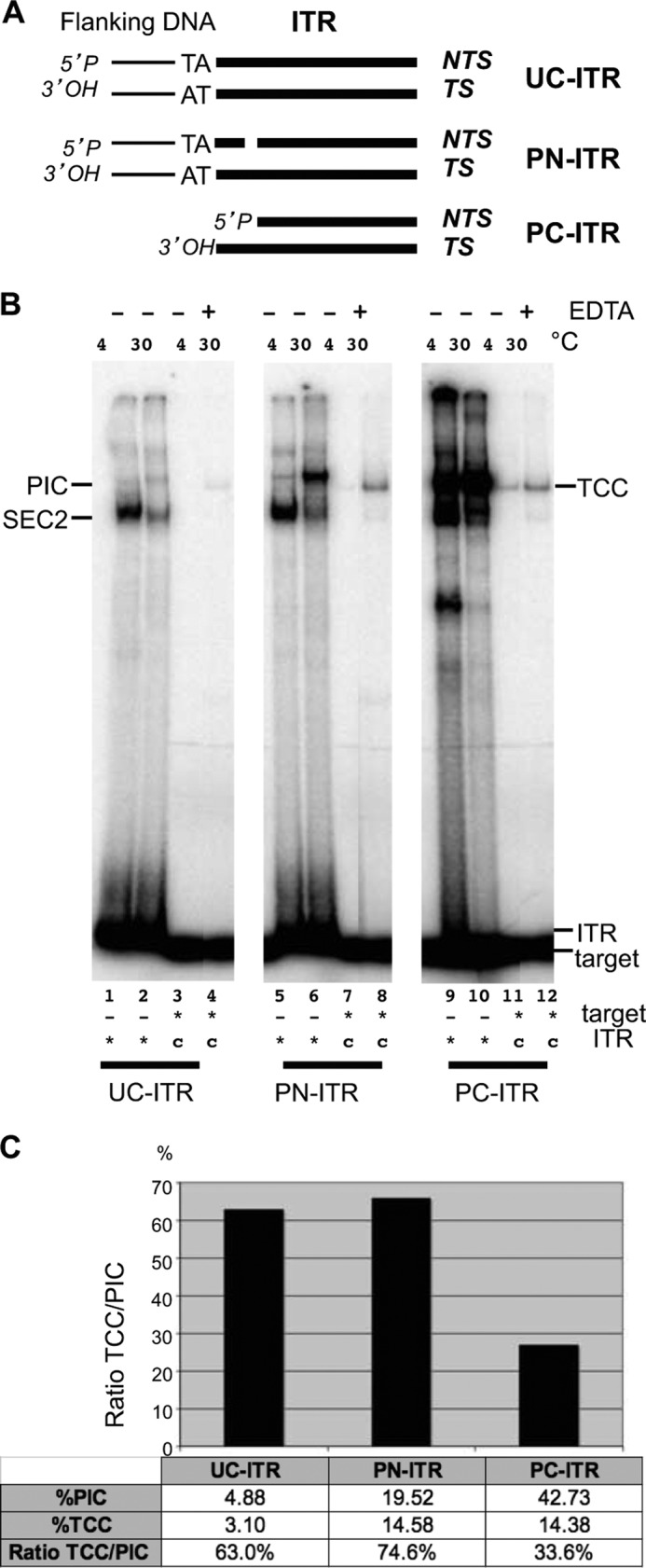
TCC assembly according to the ITR ends. A, diagram of the different types of ITR used. UC-ITR, PN-ITR, and nontransferred strand (NTS) cleaved 3 bp inside the ITR; PC-ITR, nontransferred strand, and transferred strand (TS) are cleaved. B, TCC assembly with various ITRs. For each ITR, PIC assembly was assayed using labeled ITRs (*) and Mg2+, at either 4 or 30 °C (as specified). TCC were assembled using cold PICs and a labeled target (*) without EDTA, at 4 °C when the PICs were preformed at 4 °C (lanes 3, 7, and 11) and with or without EDTA at 30 °C when the PICs were preformed at 30 °C (lanes 4, 8, and 12). C, for quantification, the percentage of PIC was obtained by dividing the amount of PIC by the amount of PIC + free ITR. The percentage of TCC was obtained by dividing the amount of TCC by the amount of TCC + free target. The ratio TCC/PIC was calculated for each ITR and plotted on a graph, and the values are indicated in the table.
Then we checked whether the PICs obtained from the three ITRs (UC-, PN-, and PC-ITR) were similarly efficient in capturing the target DNA. The percentage of PIC and TCC obtained at 30 °C was quantified for each type of ITRs and the TCC/PIC ratio was calculated for each ITR (Fig. 2C). The percentage of PIC formation is low with UC-ITRs and PN-ITRs, but the TCC/PIC ratio indicated that 60–70% of the formed PICs were proficient for target capture versus only 27% of the PICs formed with PC-ITRs. This suggests that the conformation of the PIC involving PC-ITRs is quite different from that of those involved UC-ITRs or PN-ITRs. We therefore assume that the conformational changes in the transposase-ITR complex between the first and second DNA strand cleavage (33) are essential to generate a target capture-competent PIC in which the 3′OH of the transferred strand is correctly exposed.
We then investigated the target commitment. We used two linear targets, short (30 bp) and long (50 bp), both containing a single TA to find out whether a target could be exchanged after its capture (Fig. 3A). As previously, the PIC was first assembled using PC-ITRs in the presence of Mg2+. A long labeled or a short labeled target was added concomitantly with EDTA, thus allowing TCC formation to occur but preventing strand transfer. After incubation with the first target for 1 h, the second labeled target (a short target if the first one was long and vice versa) was added. To estimate the influence of the target length on the target capture efficiency, TCC formation was achieved using a mix of short and long targets. An important bias was observed toward the binding of the long target, which was preferentially engaged in ∼70% of the TCCs (Fig. 3B, lane 3). To measure the target commitment, the amount of complex formed with the first added target was divided by the amount of complex formed with the same target when both targets were added together. An absence of target commitment would have resulted in the same ratio, irrespective of the probe (short or long) added first. Target commitment was assayed six times and was ∼16 ± 4% with respect to the second target (Fig. 3B). This difference was not statistically significant (Kruskal-Wallis test, p = 0.7). The same experiments were performed in the presence of Mg2+ that could help the protein conformation. Even if strand transfer was allowed, similar results were obtained, and no target commitment was detected (data not shown). This led to the conclusion that Mos1 is not sensitive to target commitment or, in other words, that the target DNA could be exchanged for another DNA until the TCC was converted to a strand transfer complex, as already observed when two different targets are added together (Fig. 1B, lane 6).
FIGURE 3.
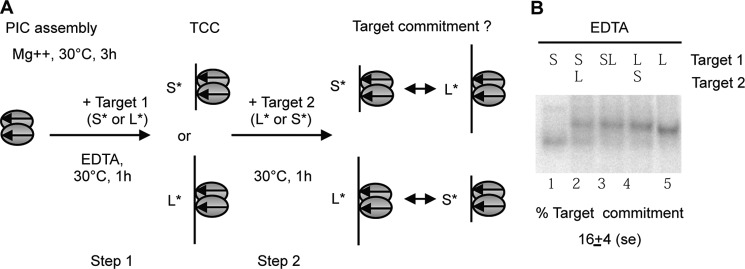
Target commitment. A, diagram of the two-step assay used to measure target commitment. Two labeled targets different in size (short (S*) or long (L*)) were used. PIC was first assembled with unlabeled PC-ITR. During this first step, TCC was allowed to proceed with the labeled target 1 (short (S*) or long (L*)). At step 2 of the assay, a second target of distinguishable size was added. After a further incubation for 1 h at 30 °C, the complexes obtained were analyzed by EMSA. To find out whether either of the two targets is preferred for TCC formation, a mix of both targets was incubated during a 2-h assay. The same molar concentration of targets was used in each assay. B, TCC formation was performed with short (S), long (L), or a mix of short and long targets (SL) in the same molar concentrations as controls. Target commitment was assayed as described in A (lanes 2–4). Experiments were repeated six times and quantified. The amount of complex formed with the first target is divided by the amount of the complex formed with the same target when both targets are added together. The averages of target commitment are indicated below the panel. The significance of the differences (lane 2 versus lane 3 and lane 4 versus lane 3) was assayed using a Kruskal-Wallis test and resulted in a p value of 0.7.
Activity of the TCC
To check the reactivity of the Mos1 TCC, i.e., its ability to promote strand transfer, integration assays were performed using a labeled target under various conditions. PIC was formed with unlabeled ITRs. After PIC assembly, a target labeled on a single strand was added, and reactions were conducted in various conditions: noncatalytic (4 °C and Mg2+ or 30 °C and EDTA) or catalytic (30 °C and Mg2+). In catalytic conditions, the expected integration product would contain the ITR transferred strand inserted into the target DNA at the TA dinucleotide, giving rise to an expected integration product of 66 bp (Fig. 4A). Integration reactions were analyzed on a denaturing gel (Fig. 4B). Noncatalytic conditions make it impossible to detect integration products (Fig. 4B, lanes 1 and 3). Catalytic conditions make it possible to detect two integration products into the labeled target that differ in size by one base (Fig. 4B, lane 2). The largest had the size expected for integration at the TA dinucleotide. The smallest (65 bp) could result from the integration of an ITR into a shorter target, because of the labeling method used (Klenow filling). To ascertain that the integrations did occur at the TA dinucleotide, integration products were amplified by PCR, cloned, and sequenced. As expected, we found that the integrations were localized at the TA dinucleotide (Fig. 4C), confirming that the Mos1 TCC is able to promote bona fide integrations. Moreover, using a target labeled on both strands allowed us to detect concerted integration, which mimics transposition (supplemental Fig. S3). The percentage of integration is quite low, but the percentage of integration is correlated with the percentage of target capture (Fig. 1F).
FIGURE 4.
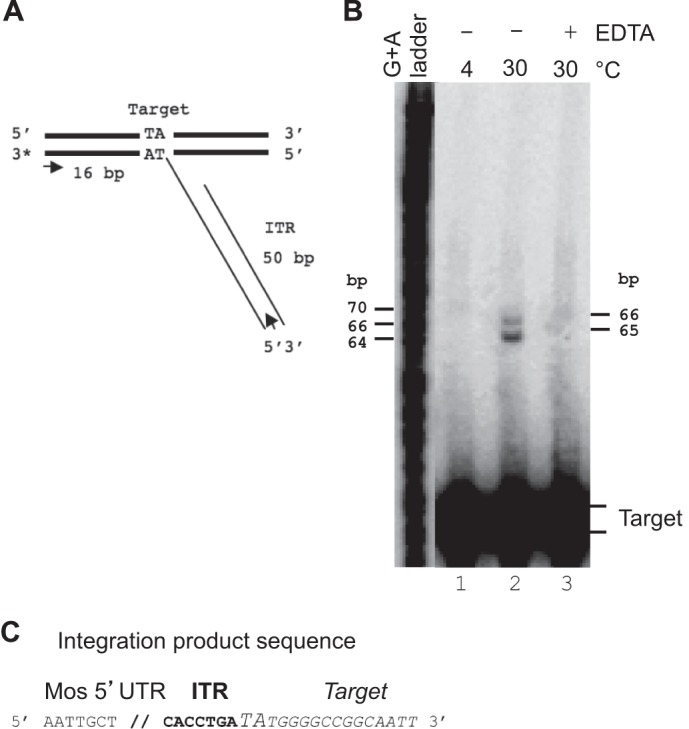
Integration assays. A, diagram showing the expected integration of the PC-ITR into the labeled target. The positions of the PCR primers used to amplify the integration products are indicated by arrows. B, TCCs were allowed to proceed as described for Fig. 1A, under various conditions: 4 °C with Mg2+ (lane 1), 30 °C with Mg2+ (lane 2), and 30 °C with EDTA (lane 3). Integration products were recovered and loaded onto a denaturing gel. A G+A ladder was used to calculate the sizes of the integration products, which are indicated on the right. C, three products were sequenced after PCR amplification. They all contained the ITR (bold type) integrated in the target DNA (italic type) at the TA dinucleotide (oversized uppercase type), as expected.
Integration at a TA dinucleotide (which is duplicated upon integration) is a common feature of several DNA transposons in both eukaryotes and prokaryotes. The TA dinucleotide, present on each side of the transposon, has been demonstrated to be essential for mariner excision (10), but it was not clear whether the TA dinucleotide present in the target is essential for target capture and/or strand transfer reactions. To address this issue, a TCC was assembled using a target containing no TA (GC target), under noncatalytic conditions, and compared with a TCC assembled with the TA target (Fig. 5A). EMSA showed that the TCC could be assembled with a target without TA, but that the percentage of target capture obtained with the GC target was 70% lower than that obtained with the TA target, showing the TA dinucleotide is important for target capture. The strand transfer into a labeled GC target was then assayed. No discrete band was detected under catalytic conditions, showing that the GC target does not support specific strand transfer (Fig. 5B). No integration product could be amplified by PCR from the smear detected in the gel (Fig. 5B), strongly suggesting that no conventional integration had occurred in the GC target. Because no integration was observed with GC target, target capture assays were also done in the presence of Mg2+ to evaluate its role on GC target capture. The efficiency of GC target in presence of Mg2 is similar to that observed in absence of Mg2 (data not shown). Taken together, our results provide evidence about the role of the target TA dinucleotide, which is a key feature for both target capture and integration.
FIGURE 5.
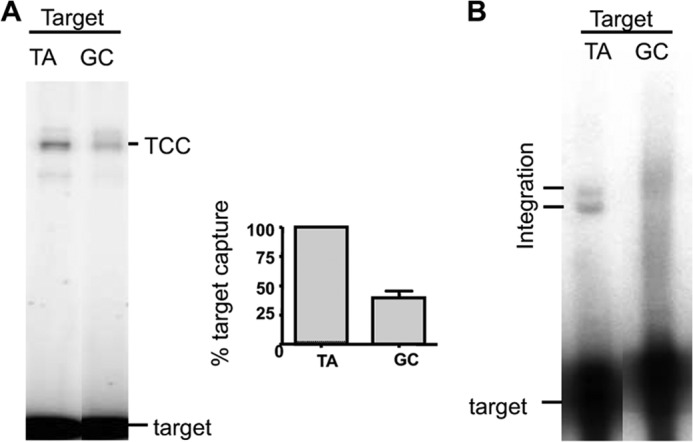
Role of the TA dinucleotide at the insertion site. A, TCC assembly was done as indicated in Fig. 1A using a TA target or a GC target (which contains no TA dinucleotide) and analyzed by EMSA. The analysis was repeated five times, and the percentage of TCC for each target was obtained by dividing the amount of TCC by the amount of TCC + free target. The normalized results were plotted on a graph. B, integration assays were performed as described for Fig. 4, using cold PC-ITR and a labeled TA target or a labeled GC target. Integration products were analyzed onto denaturing gel.
Richardson et al. (9) have proposed an involvement of the MOS1 Arg186 residue in the target capture. They assume that the two Arg186 side chains play a key role in binding the symmetrical TA target DNA sequence, possibly by recognition of the TA base pair or the TA dinucleotide. We test the ability of a R186A mutant to promote PIC and/or TCC assembly in EMSA (Fig. 6). We confirm that the R186A mutant allows PIC formation. TCC is also observed with this mutant, sustaining the fact that Arg186 is a residue involved in strand transfer rather than in target capture.
FIGURE 6.

Checking the role of Arg186 in target capture. PIC assembly was controlled using a labeled PC-ITR for each transposase (WT and R186A) as indicated. TCCs were assembled as described for Fig. 1.
Bending DNA in the TCC
We had observed a distinctive mobility profile of TCC, indicating possible conformational changes of DNA within this complex. The use of nicked and mismatched targets with Tn10 transposase and RAG1/2 have shown that target capture was enhanced when using these modified targets (15, 16). Indeed, the DNA target within the TCC can be markedly bent. This was shown to be a major parameter during TCC assembly for various transposable elements (13–17). We therefore checked the role of the conformational flexibility of the double-stranded DNA molecule by introducing a discontinuity into one of the strands. A nick in duplex DNA increases the conformational flexibility of the DNA, thus reducing the free energy cost of forming a bend. Another way to bend a double-stranded DNA is to introduce a mismatch at one point of the sequence. In this case, the mismatch will force the DNA to adopt a bend-constraint structure. We therefore tested whether the presence of a nick or a mismatch positioned at different positions relative to the TA dinucleotide could influence target capture. TCCs were formed with modified labeled targets (Fig. 7A) and analyzed by EMSA (Fig. 7B). Similar levels of TCC were detected for targets containing a mismatch at positions +1 and +2 when compared with the wild type TA target, but a lesser amount of TCC was detected with M+3 (Fig. 7B). On the other hand, we observed that TCC assembly was greater with all three nicked targets than with the wild type, the most efficient being the target nicked at +2 relative to the TA (Fig. 7B). These experiments were repeated five times and quantified, confirming that the presence of a mismatch at position +1 or +2 had no effect on TCC assembly (Fig. 7C). In contrast, the target containing a mismatch at position +3 was captured statistically significantly less efficiently (p < 0.05), whereas the presence of a nick at position +1, +2, or +3 increased target capture. The increase in target capture was statistically significantly different in the target with a nick at +2 from the wild type target (p < 0.001). We then assayed the six modified targets for their ability to support integration (Fig. 7D). For the mismatched targets, the percentage of integration depended on the percentage of target capture, except for the M+2 target (Fig. 7, D and F). In our assays, integration was the result of both target capture and strand transfer. To assess the efficiency of strand transfer independently of that of target capture, the integration was normalized relative to the percentage of TCC formed with each target. Normalization showed that mismatched targets enhanced strand transfer, with the most marked effect being associated with the M+2 target because integration/TCC ratio is 310.5 with this target, whereas the same ratio is only 151 and 169.8 with M+1 and M+3 target, respectively (Fig. 7F). This showed that a mismatch at M+2 position enhanced the integration step 3-fold. Sequencing the subsequent integration products showed that integration of the ITR occurred at the TA dinucleotide, whatever the position of the mismatch, indicating that the position of the induced bend did not affect the integration reaction. Similar experiments were done with nicked targets (Fig. 7D). Normalization revealed that the three nicked targets all behaved in the same way as the wild type target toward strand transfer, i.e., the percentage of integration depends of the percentage of target capture (integration/TCC percentage of ∼130%) (Fig. 7F), and sequencing the integration products showed that the ITR was integrated at the TA dinucleotide.
FIGURE 7.

Target capture assays using modified targets. A, the sequence of the wild type TA target (bold type) and targets with one mismatch (M+1, M+2, and M+3) or one nick (N+1, N+2, and N+3) are given. The mismatches are boxed, and arrows indicate the nicks. B, TCCs were assayed under standard conditions (Fig. 1A), using either mismatched targets (left panel) or nicked targets (right panel). The wild type TA target (WT) is used as a reference. The resulting complexes were analyzed in EMSA. C, experiments were repeated at least three times. For each target, the relative percentage of TCC was calculated and plotted as histogram bars. The significance of the differences between the different modified targets and the TA target was assayed using a Kruskal-Wallis test. Significant p values are indicated. ***, p < 0.001; *, p < 0.05. D, integration assays were performed as in Fig. 4, using either mismatched targets (left panel) or nicked targets (right panel). The results for the nicked targets were obtained using targets labeled on both strands and compared with a similarly labeled WT target. E, experiments were repeated at least three times, quantified, and plotted as histogram bars. The significance of the differences between each of the modified targets and the WT target was assayed using a Kruskal-Wallis test. Significant p values are indicated. ***, p < 0.001. F, the percentages of TCC and integration obtained in C and E, respectively, were reported for each target. To assess the strand transfer efficiency, regardless of the target capture efficiency, the integration was normalized according to the percentage of TCC formed with each target (integration percentages divided by TCC percentages).
In conclusion, we showed that modified targets had different effects on the integration step: nicked targets improved the target capture but did not influence integration. The strand transfer detected on the opposite strand was as effective as with an unmodified target. In contrast, mismatched targets improved strand transfer, without having any effect on target capture. In both cases, the target modified at position +2 relative to the TA dinucleotide had the greatest effect (N+2 for target capture and M+2 for integration).
DISCUSSION
The insertion sites of transposons have a major impact on genome structure and gene expression regulation (1, 2). One way to know how insertion sites are selected is to study target capture, a critical albeit poorly known step in the transposition cycle of eukaryotic DNA transposons. To obtain information about the nature and dynamics of TCC, we developed a target capture assay using Mos1 as a model. This model might also help to better understand the mechanism of transposition of transposons of biotechnological interest (Sleeping Beauty and PiggyBac) for which biochemical approaches are not available. Our assay is based on a preformed PIC that is converted into a TCC by the recruitment of a DNA molecule. This assay was used to demonstrate that Mos1 proceeds to target capture only once the transposon has been excised from its donor site. In this respect, the transposition cycle of Mos1 (an eukaryotic transposon) is similar to that described for Tn5 (26), Tn10 (27) (prokaryotic transposons), and RAG1/2 (24). HIV integrase proceeds similarly, because the 3′ processing of viral ends must be done before target capture (34). In contrast, Himar1 transposases can capture target after or before end cleavage. This apparent discrepancy between HIMAR1 and MOS1 probably results from the assays used. Indeed, the timing of HIMAR1 target capture was tested in the presence of Mg2+, thus allowing end cleavage during the TCC assays (29). In contrast, we used EDTA (which prevents end cleavage) to control the nature (cleaved versus un-cleaved) of the ITR ends. We thus propose that all mariner elements proceed to target capture only once the transposon has been excised, in agreement with prokaryotic DNA transposons and RAG1/2.
Tn10 and Tn5 have been useful models for understanding the molecular mechanisms of target capture. These elements perform integration by resolving a hairpin present at the extremities of the excised element. This step results in blunt DNA ends that need a profound rearrangement when target capture occurs, resulting in the 3′OH of the transferred strand being better exposed (35). In the case of mariner elements, this rearrangement is not necessary, because the transferred strand is directly produced with a 3-bp overhang by the excision (32, 36) that actually precedes target capture. However, conformational changes have to occur, between the first and second DNA strand cleavage, to allow the second DNA strand cleavage to occur, thus generating a target capture-competent complex. These conformational changes have been recently documented for Mos1 by solution scattering methods and PIC crystallography (9, 33). SEC2 presented first an elongated form, the two monomers interacting by the first helix turn helix domain. One monomer had to rotate for the recruitment of the second ITR (giving a compact SEC2), conducting to the compact crossed architecture observed in the PIC. These conformational changes may account for the fact that uncleaved and prenicked ITRs are more efficient in promoting target capture than precleaved ITRs. We assume that both of these ITRs, which need to be cleaved to allow PIC assembly, support conformational changes that lead to a target capture-competent complex. In contrast, the precleaved ITRs lead to less efficient target capture complexes. This suggests that this complex does not have the same conformation as the complex obtained with ITR cleaved by the transposase.
Elements of mariner do not display integration proximate to the excision site (“local hopping”) in vivo. This is consistent with the fact the complete excision of the transposon precedes its reintegration. However, autointegration (i.e., the integration of a single ITR into the sequence of the transposon itself) has been detected in vitro for both Hsmar1 (10) (32, 37) and Mos1, suggesting that partial local hopping may occur (38). Taken together, these observations imply that a more sophisticated mechanism prevents autointegrations and/or proximate integrations in vivo. One such mechanism could rely on the fact that the transposable element is mostly or totally insensitive to target commitment, excluding neighboring sequences as targets. In this case, the target capture complex would remain labile until the strand transfer reaction is completed. The element could “explore” several targets within the whole genome before being integrated at any given location, particularly at a distant location from the excision site. The lack of target commitment that we have demonstrated for Mos1 is consistent with the biology of mariner elements. It may account for the random distribution of mariner elements seen in natural populations (39, 40) and for the results obtained in transgenesis assays using Mos1 as a vector (41, 42). One of the consequences of the excision/target capture pathway that we elucidated in the present study is a possible risk of losing the excised element before it is integrated. However, the biochemistry of transposition makes this unlikely, because excision is the limiting step of the reaction (at least in vitro). This risk still exists even if double-stranded breaks are efficiently repaired using the sister chromosome. It may contribute to the stochastic loss of mariner elements that has been commonly observed (43). This can be perceived as the price to be paid for random distribution.
Two recent publications have proposed a TCC model derived from the Mos1 PIC. The first model deals with the fact that this PIC has a channel between the catalytic domains, in which the modeled target DNA is straight and approximately perpendicular to both ITRs. This model predicts that the Arg186 MOS1 residue will bind to the target through the target TA dinucleotide, supporting subsequent integration (9). The assay that we have developed allows us to discriminate between Mos1 target capture and strand transfer reactions. Hence, we have demonstrated that Arg186 is not involved in target capture and that it only plays a role in strand transfer reactions. Montaño and Rice (44) have proposed an alternative model for the Mos1 TCC, in which the target DNA is strongly bent. This has been shown to occur in the intasome of the prototype foamy virus and in the Mu transpososome (6, 45). The Mu transpososome displays enhanced capture efficiency when mismatched targets are provided (46). We checked modified targets for their efficiency in the capture by Mos1.
We have shown that nicks and mismatches influence integration, but in two different ways. Nicks enhance target capture up to 5-fold, indicating that the capture of the target is more efficient on a flexible target DNA molecule. Similar findings of enhanced target capture with nicked and mismatched targets have been observed with Tn10 transposase and RAG1/2 (15, 16). Our results are consistent with the Mos1 TCC model of Montaño and Rice (44). This suggests that target bending could serve to position the scissile phosphate in the active site.
The target bending could also induce a local DNA deformation that would confer a high energy conformation to the scissile phosphate that is necessary for a nucleophilic attack and subsequent strand transfers, as detected with Tn10 (15). Such severe deformation of the target and expansion of the major groove that makes it possible to position the scissile phosphate at the active sites have been detected in the prototype foamy virus and Mu TCCs (6, 47). This interpretation was confirmed after using mismatched targets for which strand transfer reactions are strongly enhanced (up to 3-fold). Our results indicate that target DNA bends need to be strictly localized. In fact, the position of the mismatch is a main feature determining strand transfer efficiency. Using both nicked and mismatched targets highlighted the significance of the position +2 relative to the TA dinucleotide.
During the past decade, all studies have failed to identify determinants of mariner integration other than the TA dinucleotide. Used as the target and duplicated upon the integration of all mariner elements, the TA dinucleotide was only known for its involvement in the excision process (10). We demonstrated here for the first time that the TA dinucleotide plays a crucial role in target capture and DNA strand transfer. Previous studies revealed that the local DNA sequences immediately surrounding the TA dinucleotide confer little target specificity, although they show a slight preference for TA dinucleotides that are flanked by A-T base pairs (10, 22, 23). It is tempting to imagine that the bending of the TA-rich targets is energetically more favorable for setting up the strand transfer reactions. This hypothesis is sustained by the lack of efficient strand transfer with TA-free targets. Accordingly, the target binding to TA dinucleotide is poorly specific, because displacement is observed with any sequence, but all containing TA. In addition, the target length, at least in vitro, influences the target capture efficiency, but the length is probably not the major factor, which is rather the target bendability, with respect with its length. Recently, it has been shown that the topology of the target affects integration, because negatively supercoiled DNA is a better target. One hypothesis accounting for this result is that supercoiling could underwind DNA, thus increasing its bendability (19). This hypothesis fits in with our results.
Mos1 has proved to be a valuable model for studying critical steps in transposition, especially because it offers a unique possibility to decipher in vitro key features of target selection. Other actors of the dynamics of transposition have been discovered in the course of in vivo studies. This is the case of HMGB1 during Sleeping Beauty transposition (48) or of nucleosomes positioning along the HIV integrasome (18). This indicates that in vitro biochemical results are relevant to the more complex in vivo situations. In this context, it would be interesting to investigate the local deformation of the target DNA in vivo. In fact, alterations of the DNA/chromatin structure have been shown to be essential to initiate replication and gene transcription. Interestingly, both replication and gene transcription involve TA-rich regions, which, in turn, have been shown to be essential for mariner transposition. This raises the issue of whether promoters and replication origins could be good mariner targets. Elements of mariner do not appear to have an insertion bias for active genes, suggesting that transcription is not a major factor in target capture nor in the subsequent integration. A possible relationship between mariner transposition and replication, especially at the level of its initiation (replication origins), remains an open issue. Eukaryotic genomes contain tens of thousands of replication origins distributed along the chromosomes in a way that matches the distribution of mariner elements. Moreover, the amplification of cut and paste transposons implies that replication and transposition have to be coupled. This correlation is in accordance with our hypothesis of target choice in AT-rich regions. This could provide new insights into the mechanisms that govern the dynamics of the transposition of DNA transposons into host genomes.
Acknowledgments
We thank G. Carpentier for the construction of baculovirus expressing the MBP-MOS. We also thank P. Gaudray for helpful discussions. The autoradiograms were recovered using the PPF genome facilities (UFR Sciences & Techniques, University of Tours). Dr. M. Ghosh revised the English text.
This work was supported by the Région Centre (InhDDE Project), the University of Tours, and the Centre National de la Recherche Scientifique.

This article contains supplemental Table S1 and Figs. S1–S3.
- ITR
- inverted terminal repeat
- M
- mismatched
- MBP
- maltose-binding protein
- N
- nicked
- PC-ITR
- precleaved ITR
- PN-ITR
- prenicked ITR
- PIC
- preintegration complex
- SEC
- single end complex
- TCC
- target capture complex
- UC-ITR
- uncleaved ITR.
REFERENCES
- 1. Babatz T. D., Burns K. H. (2013) Functional impact of the human mobilome. Curr. Opin. Genet. Dev. 23, 264–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kunarso G., Chia N.-Y., Jeyakani J., Hwang C., Lu X., Chan Y.-S., Ng H.-H., Bourque G. (2010) Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634 [DOI] [PubMed] [Google Scholar]
- 3. Cordaux R., Udit S., Batzer M. A., Feschotte C. (2006) Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl. Acad. Sci. U.S.A. 103, 8101–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chalopin D., Galiana D., Volff J.-N. (2012) Genetic innovation in vertebrates. Gypsy integrase genes and other genes derived from transposable elements. Int. J. Evol. Biol. 2012, 724519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Plasterk R. H., van Luenen H. G. (2002) in Mobile DNA II (Craig N. L., Craigie R., Gellert M., Lambowitz A. L., eds) pp. 519–532, ASM Press, Washington, D. C [Google Scholar]
- 6. Maertens G. N., Hare S., Cherepanov P. (2010) The mechanism of retroviral integration from x-ray structures of its key intermediates. Nature 468, 326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaillet J., Genty M., Cambefort J., Rouault J.-D., Augé-Gouillou C. (2012) Regulation of mariner transposition. The peculiar case of Mos1. PLoS One 7, e43365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carpentier G., Jaillet J., Pflieger A., Adet J., Renault S., Augé-Gouillou C. (2011) Transposase-transposase interactions in MOS1 complexes. A biochemical approach. J. Mol. Biol. 405, 892–908 [DOI] [PubMed] [Google Scholar]
- 9. Richardson J. M., Colloms S. D., Finnegan D. J., Walkinshaw M. D. (2009) Molecular architecture of the Mos1 paired-end complex. The structural basis of DNA transposition in a eukaryote. Cell 138, 1096–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Claeys Bouuaert C., Chalmers R. (2010) Transposition of the human Hsmar1 transposon. Rate-limiting steps and the importance of the flanking TA dinucleotide in second strand cleavage. Nucleic Acids Res. 38, 190–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nowotny M. (2009) Retroviral integrase superfamily. The structural perspective. EMBO Rep. 10, 144–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Claeys Bouuaert C., Chalmers R. M. (2010) Gene therapy vectors. The prospects and potentials of the cut-and-paste transposons. Genetica 138, 473–484 [DOI] [PubMed] [Google Scholar]
- 13. Kuduvalli P. N., Rao J. E., Craig N. L. (2001) Target DNA structure plays a critical role in Tn7 transposition. EMBO J. 20, 924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pribil P. A., Haniford D. B. (2003) Target DNA bending is an important specificity determinant in target site selection in Tn10 transposition. J. Mol. Biol. 330, 247–259 [DOI] [PubMed] [Google Scholar]
- 15. Pribil P. A., Wardle S. J., Haniford D. B. (2004) Enhancement and rescue of target capture in Tn10 transposition by site-specific modifications in target DNA. Mol. Microbiol. 52, 1173–1186 [DOI] [PubMed] [Google Scholar]
- 16. Tsai C.-L., Chatterji M., Schatz D. G. (2003) DNA mismatches and GC-rich motifs target transposition by the RAG1/RAG2 transposase. Nucleic Acids Res. 31, 6180–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Müller H. P., Varmus H. E. (1994) DNA bending creates favored sites for retroviral integration. An explanation for preferred insertion sites in nucleosomes. EMBO J. 13, 4704–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lohe A. R., De Aguiar D., Hartl D. L. (1997) Mutations in the mariner transposase. The D,D(35)E consensus sequence is nonfunctional. Proc. Natl. Acad. Sci. U.S.A. 94, 1293–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Claeys Bouuaert C., Chalmers R. (2013) Hsmar1 transposition is sensitive to the topology of the transposon donor and the target. PLoS One 8, e53690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benjamin H. W., Kleckner N. (1989) Intramolecular transposition by Tn10. Cell 59, 373–383 [DOI] [PubMed] [Google Scholar]
- 21. Shevchenko Y., Bouffard G. G., Butterfield Y. S., Blakesley R. W., Hartley J. L., Young A. C., Marra M. A., Jones S. J., Touchman J. W., Green E. D. (2002) Systematic sequencing of cDNA clones using the transposon Tn5. Nucleic Acids Res. 30, 2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crénès G., Ivo D., Hérisson J., Dion S., Renault S., Bigot Y., Petit A. (2009) The bacterial Tn9 chloramphenicol resistance gene. An attractive DNA segment for Mos1 mariner insertions. Mol. Genet. Genomics 281, 315–328 [DOI] [PubMed] [Google Scholar]
- 23. Crénès G., Moundras C., Demattei M.-V., Bigot Y., Petit A., Renault S. (2010) Target site selection by the mariner-like element, Mos1. Genetica 138, 509–517 [DOI] [PubMed] [Google Scholar]
- 24. Matthews A. G., Elkin S. K., Oettinger M. A. (2004) Ordered DNA release and target capture in RAG transposition. EMBO J. 23, 1198–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naigamwalla D. Z., Chaconas G. (1997) A new set of Mu DNA transposition intermediates. Alternate pathways of target capture preceding strand transfer. EMBO J. 16, 5227–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakai J., Kleckner N. (1997) The Tn10 synaptic complex can capture a target DNA only after transposon excision. Cell 89, 205–214 [DOI] [PubMed] [Google Scholar]
- 27. Gradman R. J., Ptacin J. L., Bhasin A., Reznikoff W. S., Goryshin I. Y. (2008) A bifunctional DNA binding region in Tn5 transposase. Mol. Microbiol. 67, 528–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neiditch M. B., Lee G. S., Landree M. A., Roth D. B. (2001) RAG transposase can capture and commit to target DNA before or after donor cleavage. Mol. Cell Biol. 21, 4302–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lipkow K., Buisine N., Chalmers R. (2004) Promiscuous target interactions in the mariner transposon Himar1. J. Biol. Chem. 279, 48569–48575 [DOI] [PubMed] [Google Scholar]
- 30. Bouchet N., Jaillet J., Gabant G., Brillet B., Briseño-Roa L., Cadene M., Augé-Gouillou C. (2013) cAMP protein kinase phosphorylates the Mos1 transposase and regulates its activity. Evidences from mass spectrometry and biochemical analyses. Nucleic Acids Res. 10.1093/nar/gkt874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Augé-Gouillou C., Brillet B., Hamelin M.-H., Bigot Y. (2005) Assembly of the mariner Mos1 synaptic complex. Mol. Cell Biol. 25, 2861–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dawson A., Finnegan D. J. (2003) Excision of the Drosophila mariner transposon Mos1. Comparison with bacterial transposition and V(D)J recombination. Mol. Cell 11, 225–235 [DOI] [PubMed] [Google Scholar]
- 33. Cuypers M. G., Trubitsyna M., Callow P., Forsyth V. T., Richardson J. M. (2013) Solution conformations of early intermediates in Mos1 transposition. Nucleic Acids Res. 41, 2020–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krishnan L., Engelman A. (2012) Retroviral integrase proteins and HIV-1 DNA integration. J. Biol. Chem. 287, 40858–40866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klenchin V. A., Czyz A., Goryshin I. Y., Gradman R., Lovell S., Rayment I., Reznikoff W. S. (2008) Phosphate coordination and movement of DNA in the Tn5 synaptic complex. Role of the (R)YREK motif. Nucleic Acids Res. 36, 5855–5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miskey C., Papp B., Mátés L., Sinzelle L., Keller H., Izsvák Z., Ivics Z. (2007) The ancient mariner sails again. Transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol. Cell Biol. 27, 4589–4600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Claeys Bouuaert C., Liu D., Chalmers R. (2011) A simple topological filter in a eukaryotic transposon as a mechanism to suppress genome instability. Mol. Cell Biol. 31, 317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sinzelle L., Jégot G., Brillet B., Rouleux-Bonnin F., Bigot Y., Augé-Gouillou C. (2008) Factors acting on Mos1 transposition efficiency. BMC Mol. Biol 9, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bryan G., Garza D., Hartl D. (1990) Insertion and excision of the transposable element mariner in Drosophila. Genetics 125, 103–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lohe A. R., Timmons C., Beerman I., Lozovskaya E. R., Hartl D. L. (2000) Self-inflicted wounds, template-directed gap repair and a recombination hotspot. Effects of the mariner transposase. Genetics 154, 647–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang W., Swevers L., Iatrou K. (2000) Mariner (Mos1) transposase and genomic integration of foreign gene sequences in Bombyx mori cells. Insect Mol. Biol. 9, 145–155 [DOI] [PubMed] [Google Scholar]
- 42. Vallin E., Gallagher J., Granger L., Martin E., Belougne J., Maurizio J., Duverger Y., Scaglione S., Borrel C., Cortier E., Abouzid K., Carre-Pierrat M., Gieseler K., Ségalat L., Kuwabara P. E., Ewbank J. J. (2012) A genome-wide collection of Mos1 transposon insertion mutants for the C. elegans research community. PLoS One 7, e30482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lohe A. R., Moriyama E. N., Lidholm D. A., Hartl D. L. (1995) Horizontal transmission, vertical inactivation, and stochastic loss of mariner-like transposable elements. Mol. Biol. Evol. 12, 62–72 [DOI] [PubMed] [Google Scholar]
- 44. Montaño S. P., Rice P. A. (2011) Moving DNA around. DNA transposition and retroviral integration. Curr. Opin. Struct. Biol. 21, 370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Montaño S. P., Pigli Y. Z., Rice P. A. (2012) The Mu transpososome structure sheds light on DDE recombinase evolution. Nature 491, 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yanagihara K., Mizuuchi K. (2002) Mismatch-targeted transposition of Mu. A new strategy to map genetic polymorphism. Proc. Natl. Acad. Sci. U.S.A. 99, 11317–11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cherepanov P., Maertens G. N., Hare S. (2011) Structural insights into the retroviral DNA integration apparatus. Curr. Opin. Struct. Biol. 21, 249–256 [DOI] [PubMed] [Google Scholar]
- 48. Zayed H., Izsvák Z., Khare D., Heinemann U., Ivics Z. (2003) The DNA-bending protein HMGB1 is a cellular cofactor of Sleeping Beauty transposition. Nucleic Acids Res. 31, 2313–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]