

Background: Insulin down-regulates GLUT4 by accelerating degradation in lysosomes.
Results: Insulin through H2O2 production dissociates retromer from LDM membrane in a protein kinase CK2-dependent manner.
Conclusion: Insulin switches GLUT4 traffic route toward lysosomes via retromer inhibition.
Significance: This revealed a unique oxidative stress-mediated insulin signal cascade that regulates the fate of GLUT4.
Keywords: Adipocyte, Glut4, Insulin, Membrane Trafficking, Signal Transduction, Retromer
Abstract
Although insulin acutely stimulates glucose uptake by promotion of GLUT4 translocation from intracellular compartments to the plasma membrane in adipocytes and muscles, long term insulin stimulation causes GLUT4 depletion that is particularly prominent in the insulin-responsive GLUT4 storage compartment. This effect is caused mainly by accelerated lysosomal degradation of GLUT4, although the mechanism is not fully defined. Here we show that insulin acutely induced dissociation of retromer components from the low density microsomal membranes of 3T3-L1 adipocytes that was accompanied by disruption of the interaction of Vps35 with sortilin. This insulin effect was dependent on the activity of protein kinase CK2 but not phosphatidylinositol 3-kinase or extracellular signal-regulated kinase 1/2. Knockdown of Vps26 decreased GLUT4 to a level comparable with that with insulin stimulation for 4 h. Vps35 with a mutation in the CK2 phosphorylation motif (Vps35-S7A) was resistant to insulin-induced dissociation from the low density microsomal membrane, and its overexpression attenuated GLUT4 down-regulation with insulin. Furthermore, insulin-generated hydrogen peroxide was an upstream mediator of the insulin action on retromer and GLUT4. These results suggested that insulin-generated oxidative stress switches the GLUT4 sorting direction to lysosomes through inhibition of the retromer function in a CK2-dependent manner.
Introduction
Although insulin acutely stimulates glucose uptake by promotion of translocation of the insulin-regulated glucose transporter GLUT4 from intracellular compartments to the plasma membrane in adipocytes and muscles (1, 2), long term insulin stimulation causes a depletion of GLUT4 protein (3–5), which is particularly prominent in the highly insulin-responsive GLUT4 storage compartment (GSC)2 (6, 7), and is thus accompanied by unresponsiveness to insulin of the glucose transport system (5). Although the precise mechanism of this downside insulin action is not fully understood, accelerated degradation of GLUT4 in the lysosomes may play a major role in its down-regulation with long term insulin stimulation because previous studies demonstrated that GLUT4 protein turnover is accelerated about 3-fold with insulin in 3T3-L1 adipocytes (4), and lysosome inhibitors effectively prevent insulin-induced GLUT4 depletion (7). Although insulin also inhibits transcription of GLUT4 (3, 5), this may contribute less to the insulin-evoked GLUT4 depletion especially in the early few hours of insulin stimulation because the turnover of GLUT4 protein is much slower (with half-lives of 50 and 15.4 h in the absence and presence of insulin, respectively) (4, 7) than the rate of GLUT4 loss with insulin stimulation (5). These observations suggested that insulin down-regulates GLUT4 mainly by promotion of its sorting toward the lysosomes.
Previous kinetic analyses have shown that insulin stimulation elicits entry of a significant number of GLUT4 from the relatively static insulin-responsive intracellular pool(s) into the endocytic recycling pathway (8–10). Although this shift in the subcellular traffic route of GLUT4 leads to a substantial increase in the number of GLUT4 at the cell surface (i.e. apparent “translocation”), the appearance of GLUT4 in the early endosomes, a major sorting center in the endocytic pathway, may possibly be relevant to promotion of its sorting to the lysosomes. Membrane proteins recycling from the endosome to the plasma membrane (e.g. the transferrin receptor and the LDL receptor) or those recycling between the endosome and the trans-Golgi network (TGN) (e.g. the mannose 6-phosphate receptor) would be delivered to the lysosomes when the sorting machinery on the early endosome is crippled (11), whereas non-recycling proteins (e.g. the EGF receptor) are transported all the way from the endosomes to the lysosomes for degradation after endocytosis. Thus, one possible mechanism for insulin-induced GLUT4 down-regulation would be that, under insulin stimulation, the endosomal GLUT4 sorting machinery may be functionally impaired, and GLUT4 not retrieved from the endosome-to-lysosome degradative flow would be transported to lysosomes with a consequent depletion of the transporter.
In the present study, to test this possibility, we investigated the role for the retromer complex in GLUT4 trafficking in 3T3-L1 adipocytes. Retromer is a key component of the endosomal protein sorting machinery that mediates the endosome-to-TGN retrieval of diverse functional transmembrane proteins (cargos) such as the cation-independent mannose 6-phosphate receptor, the Wnt-binding protein Wntless, and the sortilin family proteins (for reviews, see Refs. 12–15). The prototypical mammalian retromer (sorting nexin (SNX)-BAR retromer) is a heteropentameric complex comprising two functionally separate subcomplexes: the cargo-selective subcomplex composed of Vps26, Vps29, and Vps35 that recruits cargos via an association between Vps35 and a sorting motif within the cytoplasmic tail of cargo and the SNX-BAR dimer subcomplex composed of SNX1 and SNX2 or of SNX5 and SNX6 that drives membrane curvature by binding to the early endosomal phosphatidylinositol (PI) 3-phosphate resulting in the formation of membrane tubules.
We show herein that retromer components were present mainly in the low density microsomal (LDM) membranes of 3T3-L1 adipocytes and that retromer played a pivotal role in the regulation of GLUT4 protein level. Notably, insulin negatively regulated retromer function by disrupting its interaction with the membranes, which may be a possible mechanism of insulin-promoted GLUT4 sorting to the degradative pathway. Furthermore, the signaling mechanism of this insulin action was exceptionally unique in that it depends on insulin-generated oxidative stress, particularly hydrogen peroxide, as well as on the activity of protein kinase CK2 (formerly known as casein kinase 2) but not PI 3-kinase or extracellular signal-regulated kinase 1/2 (Erk1/2). Thus, this study revealed a unique oxidative stress-mediated insulin signal cascade that regulates the fate of GLUT4 by interfering with the retromer function.
EXPERIMENTAL PROCEDURES
Materials
Iodixanol, genistein, wortmannin, PD98059, SB203580, SP600125, 4,5,6,7-tetrabromobenzotriazole (TBB), and 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole, diphenyleneiodonium chloride, N-acetyl-l-cysteine were obtained from Sigma. LY294002 and hydrogen peroxide were from Wako Pure Chemical Industries (Osaka, Japan). Hydroxy-2-naphthalenylmethylphosphonic acid trisacetoxymethyl ester and microcystin-LR were from Enzo Life Sciences (Farmingdale, NY).
Antibodies
Antibodies for GLUT4, insulin-regulated aminopeptidase, vesicle-associated membrane protein-2, β-tubulin, Syntaxin 6, mannose 6-phosphate receptor, and transferrin receptor were described previously (9, 16). Antibodies for Vps26, Vps29, and Vps35 were from Abcam (Cambridge, UK). Mouse antibodies for sortilin, SNX1, and SNX2 were from BD Transduction Laboratories. Antibodies for SNX5 and SNX6 were from Sigma. Antibodies for Akt, Rab7, Rab10, Rab11, pyruvate dehydrogenase, Na,K-ATPase, and early endosome antigen 1 were from Cell Signaling Technology (Danvers, MA). Anti-β subunit of CK2 (CK2β) antibody was from Merck Millipore.
Cell Culture and Differentiation
3T3-L1 cells provided by Howard Green (Harvard Medical School, Boston, MA) (17) were maintained in Dulbecco's modified Eagle's medium containing 4.5 g/liter d-glucose supplemented with 10% calf serum and differentiated into adipocytes in DMEM containing 1.0 g/liter d-glucose (DMEM-LG) supplemented with 10% fetal bovine serum (FBS) as described previously (7).
Subcellular Membrane Fractionation
The differential and sucrose density gradient centrifugations were carried out as described by Kono et al. (18). Briefly, cells were washed and homogenized in ice-cold STE buffer (250 mm sucrose, 10 mm Tris/Cl, pH 7.4, 1 mm EDTA) containing Complete protease inhibitor mixture and PhosSTOP phosphatase inhibitor mixture (Roche Diagnostics) with a Dounce tissue grinder. The homogenate was centrifuged for 2 min at 3,000 × g. The pellet and the fat fraction were discarded, and the infranatant solution was further centrifuged for 60 min at 150,000 × g. The pellet (crude microsomal fraction) was suspended in 0.5 ml of STE buffer containing Complete protease inhibitor mixture and PhosSTOP phosphatase inhibitor mixture and layered on top of a linear (15–45%, w/w) sucrose density gradient (∼14 × 80 mm in size) and centrifuged for 40 min at 160,000 × g. Fractions (0.5 cm in height) were collected from the top of the tube.
Iodixanol gradient centrifugation of the LDM fraction was carried out as described previously (7) except that all buffers contained Complete protease inhibitor mixture and PhosSTOP phosphatase inhibitor mixture. Fractions (0.3 ml) were collected starting from the bottom of the tube.
Immunoblotting
The amount of protein was analyzed by immunoblotting as described previously (16). The blots were visualized by using Amersham Biosciences ECL detection systems (GE Healthcare) and an LAS-4000 luminescent image analyzer (GE Healthcare). The intensity of the band was quantified with Multi Gauge software (Fuji Photo Film, Tokyo, Japan).
Transfection of Small Interfering RNA (siRNA) and Plasmid
siRNA duplexes targeting mouse Vps26 or the β subunit of CK2 mRNAs (Table 1) were purchased from Thermo Fisher Scientific Inc. (Waltham, MA) and Cosmo Bio Co. (Tokyo, Japan), respectively. The expression plasmid for mCherry-Vps35 fusion protein was prepared by insertion of the human Vps35 cDNA (from the Invitrogen MGC collection) into the pmCherry-C1 vector by using the In-Fusion Dry-Down PCR cloning kit (Takara Bio, Inc., Shiga, Japan). The Vps35-S7A mutation was introduced by using the QuikChange II site-directed mutagenesis kit (Agilent Technologies). The Vps26-myc-pcDNA3.1 expression plasmid was provided by Dr. Juan S. Bonifacino (National Institutes of Health, Bethesda, MD).
TABLE 1.
Target sequences for siRNA
| Target | Gene symbol | Target sequence of mRNA |
|---|---|---|
| Vps26 | Vps26a | AACACTATCTCTTCTATGA (113–131) |
| GAACTTTCACCAGCGGTTT (921–939) | ||
| GTAAGGTACTTTCTAAACC (808–826) | ||
| AGAAAGAGATCACAGGAAT (638–656) | ||
| CK2-B | Csnk2b | TCGCACAAATGTTGGAAAA (281–299) |
Differentiated 3T3-L1 adipocytes were dispersed with PBS containing 0.05% trypsin and 0.5 mg/ml collagenase (Type I; Worthington), washed three times with PBS, and resuspended in Electroporation Buffer (Bio-Rad) with siRNA or plasmid before electroporation in a 0.4-cm gap cuvette with Gene Pulser Xcell (Bio-Rad) set at 200 V and 500 microfarads. After electroporation, cells were resuspended in DMEM-LG containing 10% FBS and cultured for 24 h before assay.
Phosphoprotein Analysis
The phosphorylated proteins were analyzed by using the TALON phosphate metal affinity chromatography (PMAC) phosphoprotein enrichment kit (Clontech) according to the manufacturer's instructions. Briefly, the lysate of 3T3-L1 adipocytes was loaded onto the PMAC resin column. After washing the column, bound proteins were eluted, and the flow-through and the eluate fractions were subjected to immunoblotting.
Immunoprecipitation
Cells were washed two times with 2.5 ml of ice-cold PBS, scraped into 1 ml of solubilization buffer (150 mm NaCl, 1 mm EDTA, 1% (v/v) Nonidet P-40, 20 mm Hepes/Na, pH 7.6 containing protease inhibitor mixture and PhosSTOP phosphatase inhibitor mixture) and then kept in the ice for 30 min with brief vortexing every 5 min. The lysate was centrifuged at 20,000 × g for 30 min at 4 °C. The supernatant was precleared by incubation with 30 μl (bed volume) of Protein G-Sepharose (GE Healthcare) for 2 h at 4 °C and centrifuged at 5,000 × g for 3 min. The resultant supernatant was incubated with 1 μl of anti-Vps35 antibody and Protein G-Sepharose overnight. The beads were washed five times with 1 ml of solubilization buffer, and precipitated proteins were eluted with 100 μl of Laemmli sample buffer.
Measurement of Cytosolic Hydrogen Peroxide
3T3-L1 adipocytes were transfected with 30 μg of the pHyPer-Cyto vector (Evrogen, Moscow, Russia) by electroporation as described above and seeded on a 35-mm glass bottom culture dish. After incubation for 24 h, the medium was replaced with Hanks' balanced salt solution containing 138 mm NaCl, 5.4 mm KCl, 1.3 mm CaCl2, 0.5 mm MgCl2, 0.38 mm MgSO4, 0.44 mm KH2PO4, 0.34 mm Na2HPO4, 5.5 mm d-glucose, and 20 mm Hepes/NaOH, pH 7.4. For monitoring of the cytosolic concentration of hydrogen peroxide ([H2O2]c), cells were excited with a wavelength of 440 nm, and emission images were obtained at 1-min interval using the AQUACOSMOS/ASHURA fluorescence imaging system (Hamamatsu Photonics, Hamamatsu, Japan).
Statistics
Data were analyzed by Student's t test, and p < 0.05 was considered as statistically significant.
RESULTS
Prolonged Insulin Stimulation Down-regulates GLUT4
We first examined the dose responsiveness and the time course of insulin-induced GLUT4 down-regulation in 3T3-L1 adipocytes. As shown in Fig. 1A, stimulation of cells with insulin for 4 h elicited a dose-dependent decrease in GLUT4 protein. Under these conditions, insulin at 1 nm significantly decreased GLUT4, whereas the maximum effect (∼50% decrease) was obtained with 500 nm insulin. With 500 nm insulin, the GLUT4 amount dropped by more than 20% at 30 min, and the insulin effect reached almost maximum (by ∼50%) at 4 h (Fig. 1B). These results suggested that insulin turned on the GLUT4 down-regulation signal within 30 min, whereas the maximum effect was achieved with 4-h or longer stimulation.
FIGURE 1.
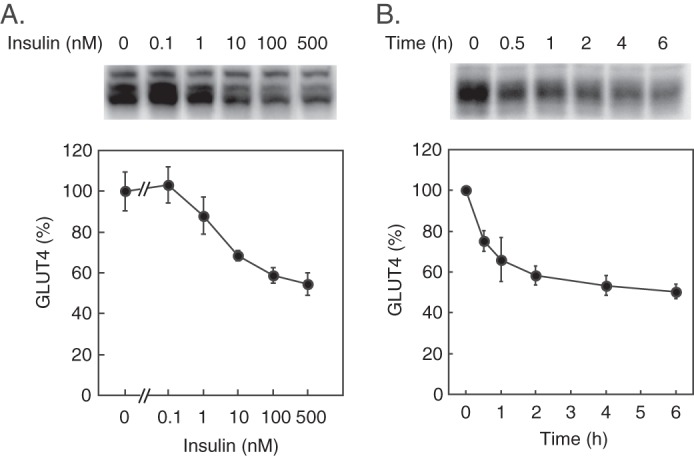
Effect of prolonged insulin stimulation on GLUT4. 3T3-L1 adipocytes were stimulated with the indicated concentrations of insulin for 4 h (A) or stimulated with 500 nm insulin for the indicated periods (B), and then the cellular amount of GLUT4 was assayed by immunoblotting. Representative immunoblots of GLUT4 (upper panels) and quantified GLUT4 amounts (lower panels) are shown. Results are the mean ± S.E. (n = 4). Error bars represent S.E.
Insulin Regulates Membrane Association of Retromer
To explore the role of retromer in GLUT4 trafficking, we next investigated the subcellular localization of the retromer components in 3T3-L1 adipocytes. Fractionation with a 15–45% (w/w) sucrose density gradient of the postnuclear membranes from 3T3-L1 adipocytes revealed that GLUT4 was localized mainly in the LDM fractions mostly coincident with Syntaxin 6, a TGN marker (Fig. 2A, Fractions 2–5) in the basal cells. Insulin stimulation increased GLUT4 in the fractions containing Na,K-ATPase (Fractions 7–9) and early endosome antigen 1 (Fractions 5–7), consistent with previous observations that GLUT4 enters the plasma membrane-endosome recycling pathway after insulin stimulation (8–10). Vps35 and SNX2, components of retromer, were broadly localized in the LDM fractions (Fractions 1–7) containing early endosome antigen 1 and Syntaxin 6 as well as GLUT4. Next, we further characterized the subcellular localization of retromer in the LDM membranes by iodixanol gradient centrifugation (7, 19). As shown in Fig. 2B, iodixanol gradient centrifugation separated two GLUT4-containing peaks: the GSC fraction (Fractions 3–6, referred to as Peak 1) and the endosome/TGN fraction (Fractions 9–12, referred to as Peak 2). Unexpectedly, Vps35, Vps26, and Vps29, the components of the cargo-selective subcomplex of retromer, were abundantly present in the GSC fraction in addition to the endosome/TGN fraction. By contrast, SNX1 and SNX2, the components of the SNX-BAR subcomplex, were mainly present in the endosome/TGN fraction (Peak 2) with minor localization in Peak 1, whereas SNX5 and SNX6 were detected only in Fraction 2 relevant to neither Peak 1 nor Peak 2. Rab7, which regulates retromer recruitment to endosomes (20, 21), was present in Peak 2 and the fractions lighter than Peak 2 corresponding to the lysosome-associated membrane protein 1-positive membranes (7). Rab10 and Rab11 were found mainly in Peak 2. Intriguingly, insulin stimulation for 30 min caused a decrease of the retromer components (Vps35, Vps26, Vps29, SNX1, and SNX2) in the LDM membranes without affecting the total cellular amounts (Fig. 2, B and C), indicating that insulin acutely dissociated the retromer components from the membranes. This was accompanied by the disruption of the interaction of Vps35 with sortilin, a retromer cargo, whereas the transferrin receptor was not co-immunoprecipitated with Vps35 (Fig. 2D). Because sortilin, a resident protein in the GLUT4-containing vesicles (22, 23), interacts with GLUT4 via its luminal Vps10p domain and plays an essential role in the biogenesis of the insulin-responsive GLUT4 pool (24, 25), these findings suggested that the insulin-induced retromer dissociation from the LDM membrane may affect subcellular trafficking of GLUT4.
FIGURE 2.

Subcellular localization of retromer components in 3T3-L1 adipocytes. A, the crude microsomal membranes of 3T3-L1 adipocytes stimulated without or with 100 nm insulin for 30 min were separated by 15–45% (w/w) sucrose density gradient centrifugation as described under “Experimental Procedures.” Fractions were collected and subjected to immunoblotting for GLUT4, Vps35, SNX2, and the indicated organelle marker proteins: Syntaxin 6 (the trans-Golgi network), early endosome antigen 1 (EEA1) (early endosome), Na,K-ATPase (the plasma membrane), and pyruvate dehydrogenase (PDH) (mitochondria). The top panel depicts the concentration (% (w/w)) of sucrose. B, the LDM membranes from 3T3-L1 adipocytes stimulated without or with 100 nm insulin for 30 min were analyzed by iodixanol gradient centrifugation. Fractions were subjected to immunoblotting for the indicated proteins. End, endosome. C, the amounts of retromer components in the LDM fraction (upper left panel) or the total lysate (upper right panel) of 3T3-L1 adipocytes stimulated without or with 100 nm insulin for 30 min were analyzed by immunoblotting. The lower panel shows the relative amount (percentage of basal) of retromer components in the LDM fraction of insulin-stimulated cells. Results are the mean ± S.E. (n = 3). Error bars represent S.E. D, the amounts of sortilin or the transferrin receptor (TfR) co-immunoprecipitated (IP) with Vps35 from the lysates of 3T3-L1 adipocytes stimulated without or with 100 nm insulin for 30 min were analyzed by immunoblotting.
Retromer Regulates GLUT4 Amount
To clarify the role of retromer in GLUT4 trafficking, we knocked down Vps26 with siRNA. As depicted in Fig. 3A, Vps26 depletion decreased GLUT4 amount by ∼60%, which was comparable and not additive with the effect of insulin. Vps26 knockdown also decreased vesicle-associated membrane protein-2, a vesicle SNARE in the GLUT4-containing vesicles (26, 27), whereas sortilin was less sensitive to Vps26 knockdown, suggesting that vesicle-associated membrane protein-2 and sortilin may be differentially regulated by Vps26. On the other hand, overexpression of Vps26 caused an increase in the GLUT4 amount (Fig. 3B), although insulin still down-regulated GLUT4 to the same level as in control cells. Likewise, Vps35 overexpression had a similar effect on GLUT4 (data not shown). These data indicated that retromer plays a pivotal role in the maintenance of the GLUT4 amount in the basal state (without insulin stimulation) possibly by retrieving the glucose transporter from the endosome-to-lysosome degradative flow. These findings also suggested that insulin-induced retromer dissociation from the LDM membrane might be relevant to accelerated sorting of GLUT4 to the lysosomes.
FIGURE 3.
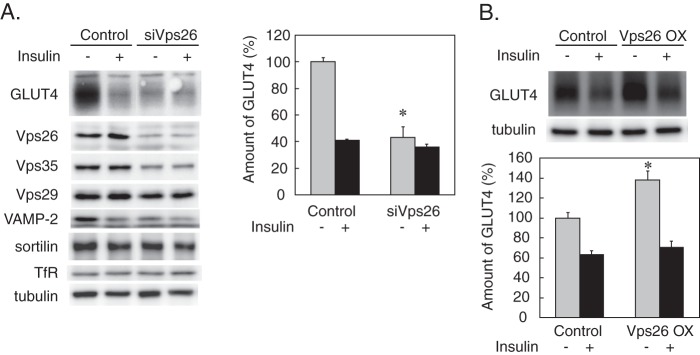
Effects of Vps26 knockdown and overexpression on the GLUT4 Amount. A, 3T3-L1 adipocytes were transfected with 20 nmol of non-silencing (Control) or Vps26-silencing siRNA (siVps26) by electroporation. After incubation for 24 h, cells were stimulated without or with 500 nm insulin for 4 h. The cells were lysed and subjected to immunoblotting for the indicated proteins. Representative immunoblot data (left panel) and the relative amounts of GLUT4 (right panel) shown as the percentage of the control value (control cells without insulin) are depicted. Results are the mean ± S.E. (n = 4). *, p < 0.05 (versus control). B, 3T3-L1 adipocytes were transfected by electroporation with 30 μg of pcDNA3.1 vector alone (Control) or containing Vps26-myc cDNA. After incubation for 24 h, cells were stimulated without or with 500 nm insulin for 4 h before immunoblotting for GLUT4. Representative immunoblot data (upper panel) and the relative amounts of GLUT4 shown as the percentage of the control value (control cells without insulin)) (lower panel) are depicted. Results are the mean ± S.E. (n = 4). *, p < 0.05 (versus control). Error bars represent S.E. OX, overexpression.
Insulin Action Depends on Protein Kinase CK2
In the next series of experiments, we investigated the mechanism of insulin-induced retromer dissociation and its relevance to GLUT4 down-regulation. To this end, we first tested the effects of inhibitors of various protein kinases involved in the insulin signaling pathways. As shown in Fig. 4, A and B, inhibition of the insulin receptor tyrosine kinase with genistein or hydroxy-2-naphthalenylmethylphosphonic acid trisacetoxymethyl ester significantly blocked insulin-induced GLUT4 depletion, whereas neither wortmannin nor PD98059 were effective, suggesting that the insulin effect on GLUT4 was dependent on the insulin receptor tyrosine kinase but was not mediated through PI 3-kinase or Erk1/2. In addition, the insulin effect was not affected with inhibitors of p38 mitogen-activated protein kinase (SB203580) or JNK (SP600125). Unexpectedly, LY294002, another widely used PI 3-kinase inhibitor, considerably blocked the insulin effect. Because LY294002 also inhibits protein kinase CK2 with potency similar to PI 3-kinase (IC50 values, 10 μm for PI 3-kinase and 6.9 μm for CK2) (28), we investigated the role for CK2 in the insulin action by using specific pharmacological inhibitors as well as by siRNA-mediated gene silencing of CK2. As shown in Fig. 4B, both TBB and 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole, specific inhibitors of CK2, significantly interfered with the insulin effect on GLUT4. TBB also blocked insulin-induced down-regulation of the insulin-regulated aminopeptidase, a GLUT4 vesicle-resident protein (Fig. 4C). In agreement, siRNA-mediated knockdown of the CK2β also prevented insulin-induced GLUT4 down-regulation (Fig. 4D). Furthermore, TBB but neither wortmannin nor PD98059 inhibited insulin-induced dissociation of Vps35 from the LDM membrane (Fig. 4, E and F), suggesting that CK2 regulates the Vps35 dissociation from the LDM membrane.
FIGURE 4.

Effects of protein kinase inhibitors on the insulin action. A and B, 3T3-L1 adipocytes were treated without (Control) or with the indicated inhibitors (genistein (Geni) (400 μm), hydroxy-2-naphthalenylmethylphosphonic acid (HNMPA) (150 μm), wortmannin (Wort) (100 nm), LY294002 (LY) (50 μm), PD98059 (PD) (20 μm), SB203580 (SB) (50 μm), SP600125 (SP) (20 μm), TBB (50 μm), or 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) (20 μm)) for 30 min before stimulation with insulin (500 nm) for 4 h. Cells were then lysed and subjected to immunoblotting. The representative immunoblot data (upper panels) and the relative amounts of GLUT4 (lower panels) are shown. Results are the means ± S.E. (n = 3). *, p < 0.05 (versus without insulin). C, cells were treated with the indicated concentrations of TBB for 30 min before stimulation with insulin (Ins) (500 nm) for 4 h. Then the cellular amounts of GLUT4 and insulin-regulated aminopeptidase (IRAP) were analyzed by immunoblotting. D, 3T3-L1 adipocytes were transfected by electroporation with siRNA (10 nmol) targeting the CK2β (siCK2) and cultured for 24 h before stimulation with insulin (500 nm) for 4 h. The amounts of CK2β, GLUT4, and tubulin were analyzed by immunoblotting. E, 3T3-L1 adipocytes were treated without or with TBB (50 μm) for 30 min before stimulation with 100 nm insulin for 30 min. Then the LDM fraction was prepared and subjected to iodixanol gradient centrifugation. The amounts of Vps35 in the fractions were analyzed by immunoblotting (IB). F, 3T3-L1 adipocytes were treated without (control) or with TBB (50 μm), wortmannin (Wort) (200 nm), or PD98059 (PD) (20 μm) for 30 min before stimulation with 100 nm insulin for 30 min. Then the LDM fractions were prepared and subjected to immunoblotting for Vps35. Representative immunoblot data (upper panel) and the relative amounts of Vps35 in the LDM fraction (lower panel) are shown. Results are the mean ± S.E. (n = 3). *, p < 0.05 (versus control value). Error bars represent S.E.
Vps35 Phosphorylation Regulates Membrane Association
Next, we attempted to identify the CK2 substrate involved in the insulin action on retromer. As shown in Fig. 5A, insulin dissociated Vps35 from the LDM fraction only in the presence of microcystin-LR, an inhibitor of serine/threonine protein phosphatases 1 and 2A (PP1 and PP2A) (29), in the homogenization buffer but not in its absence, suggesting that phosphorylation at serine/threonine residue(s) of either retromer components or its cargo proteins is involved in the Vps35 dissociation from the LDM membrane. This result led us to analyze the phosphorylation state of the retromer components by using PMAC resin (from Clontech), which selectively binds proteins with a phosphate group on serine, threonine, or tyrosine. The lysates of 3T3-L1 adipocytes stimulated without or with insulin for 30 min were loaded on the resin and eluted. As shown in Fig. 5B, insulin stimulation increased Akt bound to the resin (i.e. in the eluate fraction), indicating that this method was useful to detect the phosphorylation state of insulin target proteins. Among the retromer components, Vps35 was present in both the flow-through and the eluate fractions, whereas other retromer components were mainly present in the flow-through fraction, although a very small amount of SNX2 was detected in the eluate fraction. Insulin stimulation increased Vps35 bound to the resin without affecting other components, suggesting that Vps35 is a putative target of insulin action. Mouse Vps35 possesses 12 CK2 phosphorylation consensus sequences ((S/T)XX(D/E) where S/T denotes the phosphorylated serine or threonine and X represents any non-basic residue (30)) among which the serine residue at position 7 (S7PQD) was actually phosphorylated in the phosphoprotein database at PhosphoSitePlus. To examine whether phosphorylation at this serine was relevant to the Vps35 interaction with the membrane, we overexpressed a Vps35 mutant in which serine 7 was replaced with alanine (Vps35-S7A). Also, the mCherry fluorescent protein was fused with this mutant to distinguish it from endogenous Vps35. As shown in Fig. 5C, mCherry-Vps35-S7A but not mCherry-Vps35 (wild type) was resistant to insulin-induced dissociation from the LDM membrane. Additionally, overexpression of this mutant interfered with insulin-induced GLUT4 down-regulation. These data indicated that phosphorylation of serine 7 of Vps35 is necessary for its dissociation from the LDM membrane.
FIGURE 5.
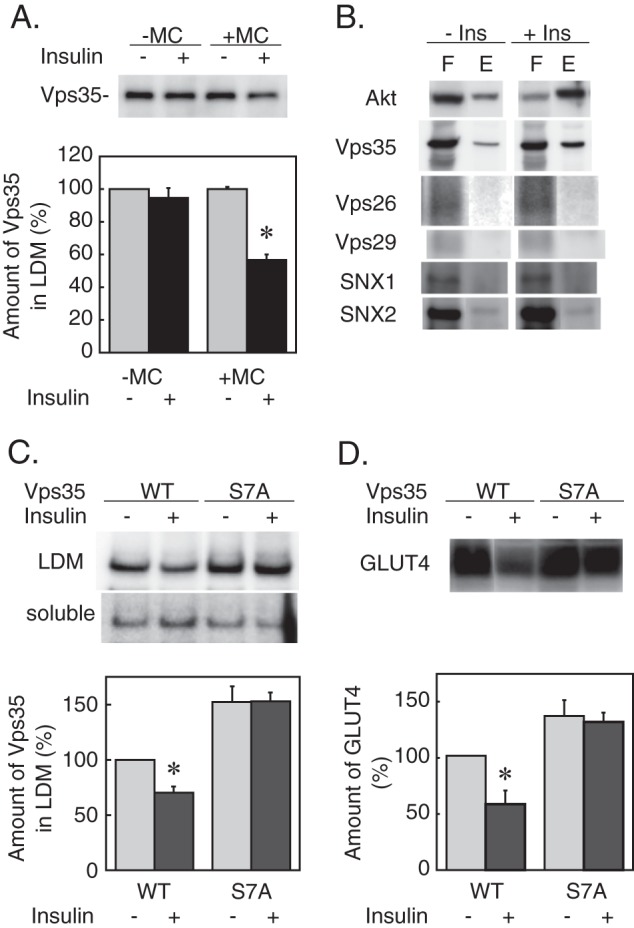
Role of Vps35 phosphorylation in the insulin action. A, 3T3-L1 adipocytes were stimulated without or with insulin (100 nm) for 30 min. Then cells were homogenized in STE buffer containing no (−MC) or 2 μm microcystin-LR (+MC). The LDM fractions were prepared and subjected to immunoblotting for Vps35. Representative immunoblot data (upper panel) and the relative amounts of Vps35 in the LDM fraction (lower panel) are shown. Results are the mean ± S.E. (n = 4). *, p < 0.05 (versus without insulin). B, the lysates of 3T3-L1 adipocytes stimulated without or with insulin (Ins) (100 nm) for 30 min were loaded on a PMAC resin column, washed, and eluted as described under “Experimental Procedures.” The flow-through (F) and eluate (E) fractions were analyzed by immunoblotting for the indicated proteins. C, 3T3-L1 adipocytes were transfected by electroporation with the expression plasmids (30 μg each) encoding wild-type (WT) or S7A mutant Vps35 fused with mCherry and cultured for 24 h before stimulation with insulin (100 nm) for 30 min. Then the LDM and soluble fractions were prepared and subjected to immunoblotting for Vps35. The upper panel shows the representative immunoblot data of mCherry-Vps35 in the LDM and the soluble fractions. The lower panel shows the relative amount of mCherry-Vps35 in the LDM fraction as the percentage of the control value (wild-type Vps35 without insulin). Results are the means ± S.E. (n = 3). *, p < 0.05 (versus without insulin). D, 3T3-L1 adipocytes were transfected with the expression plasmids as described in C and cultured for 24 h before stimulation with 500 nm insulin for 4 h. The amount of GLUT4 was analyzed by immunoblotting. The upper panel shows the representative immunoblot data of GLUT4. The lower panel shows the relative amount of GLUT4 as the percentage of the control value (wild-type Vps35 without insulin). Results are the means ± S.E. (n = 3). *, p < 0.05 (versus without insulin). Error bars represent S.E.
Insulin-generated Hydrogen Peroxide Regulates Retromer Function and GLUT4 Level
As shown above, the putative insulin signal(s) that regulates the retromer localization and the GLUT4 amount is unique in that it depended neither on PI 3-kinase nor on Erk1/2. Previous studies have demonstrated that insulin stimulates the generation of reactive oxygen species, mainly H2O2 (31, 32), via the activation of Nox4-based NADPH oxidase (33, 34) in adipocytes, the mechanism of which is independent of PI 3-kinase (34). Although such oxidative stress plays a facilitative role in the transduction of insulin signals (35–37), prolonged exposure to oxidative stress impairs insulin-induced GLUT4 translocation accompanied by de novo protein synthesis-independent reduction in the GLUT4 protein amount (38, 39). These observations led us to investigate the role of insulin-generated H2O2 in the regulation of retromer and GLUT4. Thus, we monitored the generation of H2O2 in 3T3-L1 adipocytes by using HyPer, an H2O2-sensitive fluorescent protein with submicromolar affinity to H2O2 that is insensitive to other oxidants (40). As shown in Fig. 6A, the addition of exogenous H2O2 increased the intensity of HyPer fluorescence in a dose-dependent manner. Insulin also elicited a rapid and sustained (over 150 min) elevation of the HyPer fluorescence intensity that was accompanied by prolonged phosphorylation of Vps35 (Fig. 6A). As depicted in Fig. 6, B and C, insulin-induced GLUT4 down-regulation was considerably attenuated with diphenyleneiodonium chloride, an inhibitor of NADPH oxidases, or N-acetyl-l-cysteine, an antioxidant, whereas the addition of either H2O2 or glucose oxidase caused GLUT4 depletion, indicating a pivotal role of NADPH oxidase-generated H2O2 in GLUT4 down-regulation. The addition of H2O2 also caused dissociation of retromer components (Vps35 and Vps29) from the LDM membranes (Fig. 6D) that was associated with disruption of the Vps35 interaction with sortilin (Fig. 6E), suggesting that H2O2 is a mediator in the insulin regulation of retromer. We also investigated the role for CK2 in this H2O2-mediated insulin signaling cascade. As shown in Fig. 6F, TBB did not inhibit the insulin-induced H2O2 generation, but it inhibited H2O2-mediated GLUT4 down-regulation, suggesting that CK2 lies downstream of H2O2 in the insulin signaling cascade.
FIGURE 6.

Role of hydrogen peroxide in GLUT4 down-regulation. A, 3T3-L1 adipocytes were transfected with the pHyPer-Cyto expression vector (30 μg) by electroporation and cultured on a 35-mm glass bottom dish for 24 h before monitoring the [H2O2]c. Cells were stimulated with the indicated concentrations of exogenous H2O2 (upper left panel) or insulin (upper right and lower left panels) at the time points depicted with arrows. The data are shown as the means ± S.E. (n = 6). Lower right panel, cells were stimulated with insulin (500 nm) in DMEM-LG for the indicated periods, and the amounts of Vps35 bound (Eluate) and unbound (Flow-through) to PMAC resin were analyzed. B, 3T3-L1 adipocytes pretreated for 15 min with diphenyleneiodonium chloride (DPI) (left panel) or N-acetyl-l-cysteine (NAC) (right panel) were stimulated with 500 nm insulin for 4 h, and GLUT4 amounts were analyzed by immunoblotting. C, 3T3-L1 adipocytes were stimulated with insulin (Ins) (500 nm) or the indicated concentrations of H2O2 (left panel) or glucose oxidase (GO) (right panel) for 4 h, and the GLUT4 amounts were analyzed by immunoblotting. D, the LDM membranes prepared from 3T3-L1 adipocytes treated without or with 100 nm insulin or 10 mm H2O2 for 30 min were subjected to iodixanol gradient centrifugation, and the amounts of Vps35 and Vps29 in the fractions were analyzed by immunoblotting. E, the amounts of sortilin co-immunoprecipitated (IP) with Vps35 from the lysates of 3T3-L1 adipocytes stimulated without (Cont) or with 100 nm insulin or 10 mm H2O2 for 30 min were analyzed by immunoblotting. F, left panel, 3T3-L1 adipocytes expressing HyPer were stimulated with insulin (100 nm) in the presence of TBB (50 μm), and the [H2O2]c was monitored as described in A. The data are shown as the means ± S.E. (n = 6). Right panel, 3T3-L1 adipocytes were stimulated with 10 mm H2O2 for 4 h in the absence or the presence of TBB (50 μm), and the GLUT4 amounts were measured by immunoblotting. Error bars represent S.E. Cont, control; IB, immunoblotting.
DISCUSSION
In the present study, we studied the mechanism of GLUT4 down-regulation by prolonged insulin stimulation and have provided evidence that retromer plays a pivotal role in the regulation of GLUT4 amount and is a critical functional component in this downside effect of insulin. This was supported by the following observations. First, manipulation of the retromer function by knockdown of Vps26 reduced GLUT4 to the level induced by 4-h insulin stimulation, whereas overexpression of Vps26 caused an increase in GLUT4. Second, insulin acutely interfered with the membrane interaction of retromer, and this was accompanied by disruption of the Vps35 interaction with its cargo protein sortilin. Thus, retromer plays an indispensable role in the maintenance of GLUT4 amount, and its function is negatively regulated by insulin. Given that retromer mediates the retrograde transport of cargo proteins from endosome to TGN, thus retrieving them from the endosome-to-lysosome degradative flow, the insulin interference with the retromer function may cause switching of the GLUT4 traffic route toward the lysosomes with consequent acceleration of GLUT4 degradation. This model is consistent with previous observations that insulin accelerates GLUT4 turnover (4) and that prolonged insulin stimulation reduces GLUT4 by a de novo protein synthesis-independent mechanism (38). On the other hand, because TGN is the most likely compartment for the generation of insulin-sensitive GLUT4 pool(s) (41, 42), retromer-mediated retrograde transport of GLUT4 may be critical for the generation of an insulin-sensitive pool (43), and insulin-elicited switching of GLUT4 sorting at the endosomes may partly explain the inverse correlation between GLUT4 turnover and its targeting to GSC or biogenesis of the insulin-responsive GLUT4 vesicles (7, 24). A recent proteomic analysis of GLUT4 vesicles (44) has shown that the insulin-responsive GLUT4 vesicles contain retromer components such as Vps35 and Vps26 as well as retromer cargo proteins including the mannose 6-phosphate receptor, sortilin, and sortilin-related LDL receptor relative 11 (SorLA). Because the latter two proteins contain the luminal Vps10p domain that can serve as the binding site for GLUT4 (25), they are possible candidates for mediating retromer-dependent sorting of GLUT4.
Another important finding in the present study is that protein kinase CK2, but not PI 3-kinase or Erk1/2, plays a crucial role in the insulin signaling cascade to retromer inhibition as well as GLUT4 down-regulation. This was demonstrated by using pharmacological inhibitors and RNAi-based knockdown of CK2. In addition, by using the Vps35-S7A mutant, we showed that CK2-mediated phosphorylation of Vps35 is a possible mechanism for the insulin-induced disruption of the Vps35 interaction with the LDM membrane. Furthermore, by real time monitoring of the HyPer fluorescence, we showed that insulin stimulates rapid and sustained generation of H2O2, which is a possible candidate for the upstream mediator in the CK2 regulation of Vps35, although the mechanism of insulin regulation of CK2 activity is unclear at present. The basal activity of CK2 is very high, and there has been no known intracellular signal molecule that activates CK2 (45). In this regard, our data indicated that a microcystin-LR-sensitive protein phosphatase (i.e. PP1 or PP2A) antagonized the Vps35 dissociation from the LDM membrane. Thus, it is possible that H2O2 may facilitate CK2-mediated Vps35 phosphorylation by inhibiting a putative redox-sensitive serine/threonine phosphatase(s) such as PP2A (46, 47) that may dephosphorylate Vps35. Alternatively, because H2O2 inactivates redox-sensitive protein tyrosine phosphatases such as PTP1B (48, 49), it is also possible that H2O2 may inactivate PP2A through phosphorylation of its catalytic subunit at tyrosine 307, which negatively regulates the PP2A activity (50). Also, we cannot exclude the possibility that H2O2 may regulate CK2 activity through subcellular localization or interaction with other regulatory molecules.
Altogether, we propose a working model deduced from the present study for insulin-induced switching of GLUT4 traffic route (Fig. 7). In the model, insulin stimulates H2O2 generation via activation of the NADPH oxidase (possibly Nox4) by a yet undefined mechanism. The generated H2O2 promotes CK2-mediated Vps35 phosphorylation by inactivating a microcystin-LR-sensitive serine/threonine phosphatase, causing disruption of the retromer interaction with cargo proteins such as sortilin, thus switching GLUT4 sorting to lysosomes. Apparently, a few missing links are present in this model among which are (a) the postreceptor mechanism of insulin activation of NADPH oxidase, (b) the mechanism of H2O2 regulation of CK2 activity, and (c) the entity of the cargo protein that connects GLUT4 and retromer in the endosome-to-TGN retrograde transport. Further work will be necessary to elucidate these points.
FIGURE 7.
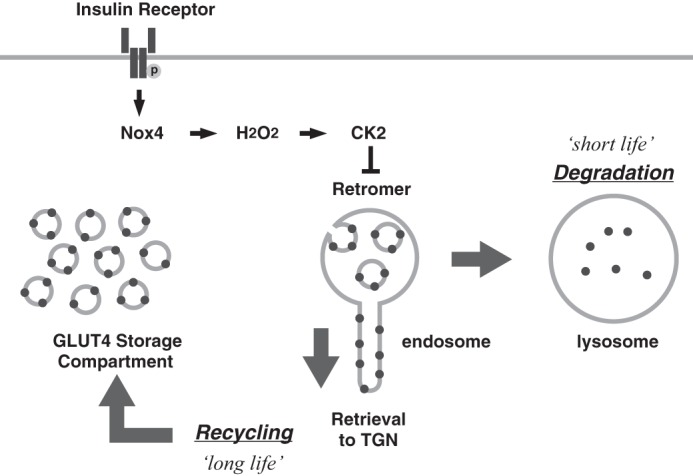
A model for insulin-induced switching of GLUT4 traffic route from “recycling” to “degradation.” Insulin receptor tyrosine kinase activation stimulates Nox4-mediated H2O2 generation, which may increase the apparent activity of CK2 by inhibition of counteracting redox-sensitive phosphatase(s). CK2-induced Vps35 phosphorylation may dissociate retromer from the LDM membrane, leading to switching of GLUT4 sorting at endosomes from “recycling to TGN” to “lysosomal degradation.” This may cause shortening of the GLUT4 half-life and depletion of GLUT4 in GSC.
Finally, although the present study provides a novel insight into the mechanism of the downside action of insulin, it is unclear whether retromer plays any physiological role in the insulin action(s) such as GLUT4 translocation. In this regard, we unexpectedly found the cargo-selective components (Vps35, Vps26, and Vps29) in the GSC fraction without the SNX-BAR components (SNX1 and SNX2), which were mainly present in the endosome/TGN fraction (Fig. 2B). The reason for this discrepancy is unknown, but it may reflect the absence of PI 3-phosphate or the presence of as yet unidentified SNX protein(s) in the GSC fraction. In light of recent studies showing that non-prototypical retromer is involved in the endosome-to-plasma membrane traffic of G-protein-coupled receptors (14, 15), it would be intriguing to investigate the role of GSC-associated retromer components in the physiological action of insulin on subcellular trafficking of GLUT4.
Acknowledgments
We thank Howard Green (Harvard University) for providing 3T3-L1 cells and Juan S. Bonifacino (National Institutes of Health) for the Vps26-myc-pcDNA3.1 expression plasmid.
This work was supported by the Global Centers of Excellence Program “Signal Transduction in the Regulatory System and Its Disorders” and Grant-in-aid 23591295 from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to H. S.). Parts of this study were presented in abstract form at the 69th Scientific Sessions of the American Diabetes Association, New Orleans, LA, June 5–9, 2009.
- GSC
- GLUT4 storage compartment
- TGN
- trans-Golgi network
- SNX
- sorting nexin
- BAR
- Bin-Amphiphysin-Rvs
- LDM
- low density microsome
- TBB
- 4,5,6,7-tetrabromobenzotriazole
- PDH
- pyruvate dehydrogenase
- PI
- phosphatidylinositol
- PMAC
- phosphate metal affinity chromatography
- PP
- protein phosphatase
- CK2β
- β subunit of CK2.
REFERENCES
- 1. Cushman S. W., Wardzala L. J. (1980) Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 255, 4758–4762 [PubMed] [Google Scholar]
- 2. Suzuki K., Kono T. (1980) Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc. Natl. Acad. Sci. U.S.A. 77, 2542–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flores-Riveros J. R., McLenithan J. C., Ezaki O., Lane M. D. (1993) Insulin down-regulates expression of the insulin-responsive glucose transporter (GLUT4) gene: effects on transcription and mRNA turnover. Proc. Natl. Acad. Sci. U.S.A. 90, 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sargeant R. J., Pâquet M. R. (1993) Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. Biochem. J. 290, 913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomson M. J., Williams M. G., Frost S. C. (1997) Development of insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 272, 7759–7764 [DOI] [PubMed] [Google Scholar]
- 6. Maier V. H., Gould G. W. (2000) Long-term insulin treatment of 3T3-L1 adipocytes results in mis-targeting of GLUT4: implications for insulin-stimulated glucose transport. Diabetologia 43, 1273–1281 [DOI] [PubMed] [Google Scholar]
- 7. Liu L. B., Omata W., Kojima I., Shibata H. (2007) The SUMO conjugating enzyme Ubc9 is a regulator of GLUT4 turnover and targeting to the insulin-responsive storage compartment in 3T3-L1 adipocytes. Diabetes 56, 1977–1985 [DOI] [PubMed] [Google Scholar]
- 8. Slot J. W., Geuze H. J., Gigengack S., Lienhard G. E., James D. E. (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113, 123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shibata H., Suzuki Y., Omata W., Tanaka S., Kojima I. (1995) Dissection of GLUT4 recycling pathway into exocytosis and endocytosis in rat adipocytes. Evidence that GTP-binding proteins are involved in both processes. J. Biol. Chem. 270, 11489–11495 [DOI] [PubMed] [Google Scholar]
- 10. Govers R., Coster A. C., James D. E. (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell. Biol. 24, 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arighi C. N., Hartnell L. M., Aguilar R. C., Haft C. R., Bonifacino J. S. (2004) Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 165, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonifacino J. S., Hurley J. H. (2008) Retromer. Curr Opin. Cell Biol. 20, 427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGough I. J., Cullen P. J. (2011) Recent advances in retromer biology. Traffic 12, 963–971 [DOI] [PubMed] [Google Scholar]
- 14. Seaman M. N. (2012) The retromer complex—endosomal protein recycling and beyond. J. Cell Sci. 125, 4693–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cullen P. J., Korswagen H. C. (2012) Sorting nexins provide diversity for retromer-dependent trafficking events. Nat. Cell Biol. 14, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu L. B., Omata W., Kojima I., Shibata H. (2003) Insulin recruits GLUT4 from distinct compartments via distinct traffic pathways with differential microtubule dependence in rat adipocytes. J. Biol. Chem. 278, 30157–30169 [DOI] [PubMed] [Google Scholar]
- 17. Djian P., Phillips M., Green H. (1985) The activation of specific gene transcription in the adipose conversion of 3T3 cells. J. Cell. Physiol. 124, 554–556 [DOI] [PubMed] [Google Scholar]
- 18. Kono T., Robinson F. W., Sarver J. A., Vega F. V., Pointer R. H. (1977) Actions of insulin in fat cells. Effects of low temperature, uncouplers of oxidative phosphorylation, and respiratory inhibitors. J. Biol. Chem. 252, 2226–2233 [PubMed] [Google Scholar]
- 19. Hashiramoto M., James D. E. (2000) Characterization of insulin-responsive GLUT4 storage vesicles isolated from 3T3-L1 adipocytes. Mol. Cell. Biol. 20, 416–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rojas R., van Vlijmen T., Mardones G. A., Prabhu Y., Rojas A. L., Mohammed S., Heck A. J., Raposo G., van der Sluijs P., Bonifacino J. S. (2008) Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 183, 513–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seaman M. N., Harbour M. E., Tattersall D., Read E., Bright N. (2009) Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J. Cell Sci. 122, 2371–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin B. Z., Pilch P. F., Kandror K. V. (1997) Sortilin is a major protein component of Glut4-containing vesicles. J. Biol. Chem. 272, 24145–24147 [DOI] [PubMed] [Google Scholar]
- 23. Morris N. J., Ross S. A., Lane W. S., Moestrup S. K., Petersen C. M., Keller S. R., Lienhard G. E. (1998) Sortilin is the major 110-kDa protein in GLUT4 vesicles from adipocytes. J. Biol. Chem. 273, 3582–3587 [DOI] [PubMed] [Google Scholar]
- 24. Shi J., Kandror K. V. (2005) Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev. Cell 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 25. Shi J., Kandror K. V. (2007) The luminal Vps10p domain of sortilin plays the predominant role in targeting to insulin-responsive Glut4-containing vesicles. J. Biol. Chem. 282, 9008–9016 [DOI] [PubMed] [Google Scholar]
- 26. Cheatham B., Volchuk A., Kahn C. R., Wang L., Rhodes C. J., Klip A. (1996) Insulin-stimulated translocation of GLUT4 glucose transporters requires SNARE-complex proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 15169–15173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tamori Y., Hashiramoto M., Araki S., Kamata Y., Takahashi M., Kozaki S., Kasuga M. (1996) Cleavage of vesicle-associated membrane protein (VAMP)-2 and cellubrevin on GLUT4-containing vesicles inhibits the translocation of GLUT4 in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 220, 740–745 [DOI] [PubMed] [Google Scholar]
- 28. Davies S. P., Reddy H., Caivano M., Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. MacKintosh C., Beattie K. A., Klumpp S., Cohen P., Codd G. A. (1990) Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 264, 187–192 [DOI] [PubMed] [Google Scholar]
- 30. Allende J. E., Allende C. C. (1995) Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J. 9, 313–323 [DOI] [PubMed] [Google Scholar]
- 31. Czech M. P., Lawrence J. C., Jr., Lynn W. S. (1974) Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proc. Natl. Acad. Sci. U.S.A. 71, 4173–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. May J. M., de Haën C. (1979) Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 254, 2214–2220 [PubMed] [Google Scholar]
- 33. Krieger-Brauer H. I., Kather H. (1995) Antagonistic effects of different members of the fibroblast and platelet-derived growth factor families on adipose conversion and NADPH-dependent H2O2 generation in 3T3 L1-cells. Biochem. J. 307, 549–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahadev K., Motoshima H., Wu X., Ruddy J. M., Arnold R. S., Cheng G., Lambeth J. D., Goldstein B. J. (2004) The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 24, 1844–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mahadev K., Wu X., Zilbering A., Zhu L., Lawrence J. T., Goldstein B. J. (2001) Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J. Biol. Chem. 276, 48662–48669 [DOI] [PubMed] [Google Scholar]
- 36. Mahadev K., Zilbering A., Zhu L., Goldstein B. J. (2001) Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 276, 21938–21942 [DOI] [PubMed] [Google Scholar]
- 37. Seo J. H., Ahn Y., Lee S. R., Yeol Yeo C., Chung Hur K. (2005) The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol. Biol. Cell 16, 348–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rudich A., Tirosh A., Potashnik R., Hemi R., Kanety H., Bashan N. (1998) Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 47, 1562–1569 [DOI] [PubMed] [Google Scholar]
- 39. Tirosh A., Potashnik R., Bashan N., Rudich A. (1999) Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J. Biol. Chem. 274, 10595–10602 [DOI] [PubMed] [Google Scholar]
- 40. Belousov V. V., Fradkov A. F., Lukyanov K. A., Staroverov D. B., Shakhbazov K. S., Terskikh A. V., Lukyanov S. (2006) Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 3, 281–286 [DOI] [PubMed] [Google Scholar]
- 41. Bogan J. S., Kandror K. V. (2010) Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Biol. 22, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kandror K. V., Pilch P. F. (2011) The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic 12, 665–671 [DOI] [PubMed] [Google Scholar]
- 43. Hatakeyama H., Kanzaki M. (2011) Molecular basis of insulin-responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic 12, 1805–1820 [DOI] [PubMed] [Google Scholar]
- 44. Jedrychowski M. P., Gartner C. A., Gygi S. P., Zhou L., Herz J., Kandror K. V., Pilch P. F. (2010) Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J. Biol. Chem. 285, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Litchfield D. W. (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J. 369, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guy G. R., Cairns J., Ng S. B., Tan Y. H. (1993) Inactivation of a redox-sensitive protein phosphatase during the early events of tumor necrosis factor/interleukin-1 signal transduction. J. Biol. Chem. 268, 2141–2148 [PubMed] [Google Scholar]
- 47. Rao R. K., Clayton L. W. (2002) Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem. Biophys. Res. Commun. 293, 610–616 [DOI] [PubMed] [Google Scholar]
- 48. Denu J. M., Tanner K. G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 [DOI] [PubMed] [Google Scholar]
- 49. Lee S. R., Kwon K. S., Kim S. R., Rhee S. G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 [DOI] [PubMed] [Google Scholar]
- 50. Chen J., Martin B. L., Brautigan D. L. (1992) Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264 [DOI] [PubMed] [Google Scholar]