

Background: Tip110 synergizes with HIV-1 Tat protein and transactivates Tat-mediated HIV-1 LTR promoter, but the underlying mechanisms have not been understood.
Results: Tip110 bound to unphosphorylated RNAPII and led to increased P-TEFb recruitment and RNAPII phosphorylation, enhancing Tat-mediated transcription elongation of the HIV-1 LTR.
Conclusion: Tip110 is involved in transcription regulation of the HIV-1 LTR promoter.
Significance: These findings provide additional insights into HIV-1 transcription.
Keywords: HIV-1, Phosphorylation, Promoters, RNA Polymerase II, Transcription, Viral Replication, Long Terminal Repeat, Tip110, Transcription Elongation, Transcription Initiation
Abstract
Transcription plays an important role in both HIV-1 gene expression and replication and mandates complicated but coordinated interactions between the host and virus. Our previous studies have shown that an HIV-1 Tat-interacting protein of 110 kDa, Tip110, binds to and enhances Tat function in Tat-mediated HIV-1 gene transcription and replication (Liu, Y., Li, J., Kim, B. O., Pace, B. S., and He, J. J. (2002) HIV-1 Tat protein-mediated transactivation of the HIV-1 long terminal repeat promoter is potentiated by a novel nuclear Tat-interacting protein of 110 kDa, Tip110. J. Biol. Chem. 277, 23854–23863). However, the underlying molecular mechanisms by which this takes place were not understood. In this study, we demonstrated that Tip110 bound to unphosphorylated RNA polymerase II (RNAPII) in a direct and specific manner. In addition, we detected Tip110 at the HIV-1 long terminal repeat (LTR) promoter and found that Tip110 expression was associated with increased phosphorylation of serine 2 of the heptapeptide repeats within the RNAPII C-terminal domain and increased recruitment of positive transcription elongation factor b to the LTR promoter. Consistent with these findings, we showed that Tip110 interaction with Tat directly enhanced transcription elongation of the LTR promoter. Taken together, these findings have provided additional and mechanistic evidence to support Tip110 function in HIV-1 transcription.
Introduction
Initiation of transcription from the long terminal repeat (LTR)3 promoter of HIV-1 is a limiting step for HIV-1 expression and results from interactions between the virally encoded transactivator, Tat, and a number of cellular factors (1–5). Prior to Tat translation, general transcription factors, including TFIIB, TFIID, and TFIII, help to position RNA polymerase II (RNAPII) at the transcriptional start site. At this point, general transcription factor TFIIH associates with CDK7 to form a transcription initiation complex at the HIV-1 5′-LTR, which phosphorylates serine 5 of the heptapeptide repeat Tyr-Ser-Pro-Thr-Ser-Pro-Ser within the C-terminal domain (CTD) of RNAPII (6–8). RNAPII is then released from the preinitiation complex and becomes competent to initiate transcription. Compared with transcription initiation, RNAPII performs transcriptional elongation less efficiently (9). This is mainly due to the presence of the negative factors negative elongation factor (NELF) and DRB sensitivity-inducing factor (DSIF) (10, 11). In addition, a fraction of positive transcription factor b (P-TEFb) is inactive as it becomes bound to 7S RNA (12–15). When Tat is translated, it binds to cyclin T1 and recruits the P-TEFb complex, consisting of cyclin T1 and CDK9, to the transactivation response RNA element (TAR). These interactions lead to the phosphorylation of serine 2 of the heptapeptide repeats within the RNAPII CTD by CDK9, which as a result increases the rigidity of the CTD and the affinity of human capping enzymes and acts as a scaffold for the splicing and polyadenylation process. This in turn increases transcription elongation, 5′-end capping, and histone methylation at the HIV-1 LTR promoter (16, 17). At later stages of elongation, Tat associates with RNAPII rather than with TAR RNA, indicating that the Tat·P-TEFb·TAR complex is disrupted during transcription (18–20).
Besides P-TEFb, Tat-mediated transactivation also requires interaction between Tat and other cellular factors. Several studies have demonstrated that a number of splicing-associated or RNA-binding factors may be involved in HIV-1 LTR transcription initiation and elongation, indicating a link between HIV-1 transcription and pre-mRNA splicing. For example, human splicing factor SKIP, which associates with P-TEFb, has a dual function in Tat-mediated transcription elongation and HIV-1 pre-mRNA splicing (21). Another example is splicing-associated protein, Tat-SF1, which interacts with large RNAPII elongation-splicing complexes as well as the Tat·P-TEFb complex and stimulates HIV-1 transcription (22–24).
Tat-interacting protein of 110 kDa (Tip110), also known as SART3/p110, was initially identified as a nuclear protein in the mRNA splicing process during the study of human U6 snRNA capping enzyme (25). It was later reported to be a human homolog of yeast protein Prp24 and associates only transiently with U6 and U4/U6 snRNP during the recycling phase of the spliceosome cycle (26, 27). Human Tip110 was found to encode a tumor rejection antigen in various cancers; it is expressed in a number of cancer cell lines as well as the majority of cancer tissues (28–30). In addition, Tip110 also binds to the androgen receptor through the nuclear receptor box and functions as a repressor of androgen receptor transcription activation through interference with the complex formation between the androgen receptor and androgen receptor-responsive element (31).
We have previously reported that Tip110 is important for HIV-1 gene expression and virus replication by interacting with Tat and enhancing Tat-mediated transactivation activity (32). However, the molecular mechanisms underlying this process were not understood. In the current study, we characterized Tip110 interaction with RNAPII and P-TEFb and its effects on RNAPII phosphorylation and HIV-1 transcription initiation and elongation. We showed that Tip110 bound to unphosphorylated RNAPII and increased P-TEFb recruitment to the TAR·Tat·P-TEFb transcription complex, which was associated with increased phosphorylation of serine 2 of the heptapeptide repeats within the RNAPII CTD and enhanced Tat-mediated transcription elongation of the LTR promoter.
MATERIALS AND METHODS
Cell Lines and Cell Transfections
293T cells were purchased from the American Tissue Culture Collection, and U373MAGI and CEM-GFP cells were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. The cells were maintained in either Dulbecco's modified Eagle's medium (293T and U373MAGI), or RPMI 1640 medium (CEM-GFP), with 10% fetal bovine serum and incubated at 37 °C with 5% CO2. Transfections were carried out using the standard calcium phosphate precipitation method. pcDNA3 was used to equalize the total amount of DNA, whereas pEGFP was used to ensure that transfection efficiencies were comparable among all transfections.
Plasmids
Construction of pTip110.His, pGEX-Tip110, pTip110.HA, Tip110 deletion mutants (ΔRRM, ΔNLS, and ΔCT), pTat.Myc, and pLTR-Luc plasmid was described previously (32). GST-CTD plasmid was a gift from Dr. David Price (University of Iowa, Iowa City, IA). HIV-1 double guanine (G)-less plasmid was obtained from Dr. Carlos Sune (Instituto de Parasitologia y Biomedicina). Tip110 ΔNT mutant was constructed using pTip110.His as the template and primers 5′-CCG AAT TCA CCA TGG CTG CCG TAG ATG TGG AG-3′ and 5′-CCC GCT CGA GTC AAT GAT GAT GAT GAT GAT GCT TTC TCA GAA ACA GCT TGG C-3′. The PCR product was digested with EcoRI and SacI and inserted into pcDNA3.
Whole Cell Lysate Preparation, Immunoprecipitation, and Western Blot Analysis
Unless stated otherwise, cells were harvested 72 h post-transfection in a cell lysis buffer (50 mm Tris·HCl, pH 8.0, 0.5% Nonidet P-40, 2 mm EDTA, 137 mm NaCl, 10% glycerol, 0.5% sodium deoxycholate, 0.2% sodium azide, 0.004% sodium fluoride, 1× protease inhibitor mixture, 1 mm sodium orthovanadate, pH 7.25). After incubation on ice for 20 min, whole cell extracts were obtained by centrifugation at 15,000 × g for 10 min. Whole cell extracts of an equal amount of protein were separated by 8–12% SDS-PAGE and then electrotransferred to HyBond-P membrane (Amersham Biosciences). The membrane was probed with primary antibodies and appropriate peroxidase-labeled secondary antibody and then visualized with a homemade ECL system. For immunoprecipitation, whole cell extracts of 500 μg of protein were first precleaned using 20 μl of protein A-agarose beads (Millipore) and then incubated by rotation with 1 μg of antibody and 60 μl of protein A-agarose beads at 4 °C overnight. The beads were recovered by centrifugation and then washed with a washing buffer (50 mm Tris·HCl, pH 8.0, 0.5% Nonidet P-40, 2 mm EDTA, 0.4 m NaCl, 10% glycerol) four times. The beads were suspended in a SDS-PAGE sample buffer and used for SDS-PAGE and Western blot analysis.
Recombinant Protein Purification and GST Pull-down Assay
pGEX-Tip110 and pGST-CTD were first transformed into Escherichia coli BL21 cells. The culture was grown to an A600 of 0.6. At this point, 0.5 mm isopropyl-β-thiogalactoside was added, and the culture was incubated at 37 °C for an additional 3 h. The bacteria were first lysed with a lysis buffer (50 mm Tris-HCl, pH 7.9, 12.5 mm MgCl2, 0.5 mm EDTA, 100 mm KCl, 20% glycerol, 1 mm β-mercaptoethanol, 10 μm ZnCl2) and then disrupted in a French press. The supernatant was collected and incubated with glutathione-Sepharose 4B, and the protein was eluted and concentrated. GST-Tip110 protein was digested with thrombin at 30 °C overnight, and then Tip110 was purified by adding glutathione beads to remove the GST protein. For the GST pull-down assay, GST-CTD (6 μg) protein was first phosphorylated overnight at 30 °C with 6 μl of casein kinase I (New England Biolabs) and 1 mm ATP with the phosphorylation buffer provided with the enzyme (New England Biolabs). Meanwhile, 4 μg of GST-Tip110 protein was digested with 1 μl of thrombin at 30 °C overnight to remove the GST tag. Unphosphorylated or phosphorylated GST-CTD proteins were first immobilized onto 30 μl of glutathione beads at 4 °C for 2 h. Then the protein-bound beads were incubated with purified Tip110 protein in 500 μl of GST pull-down buffer (20 mm HEPES, pH 7.9, 150 mm NaCl, 0.5 mm EDTA, 10% glycerol, 0.1% Triton X-100, 1 mm DTT) at room temperature for 2 h. Subsequently, the protein-bound beads were washed with PBS five times, and the bound proteins were eluted from the beads by 4× SDS-PAGE sample buffer. The proteins were separated by SDS-PAGE and analyzed by immunoblotting.
ChIP Assay
Cells (5 × 107) were first cross-linked with 1% formaldehyde at room temperature for 20 min; the cross-linking was terminated by the addition of glycine to a final concentration of 0.125 m. The cells were washed by PBS three times, and the cell pellet was suspended in a cell lysis buffer (85 mm KCl, 0.5% Nonidet P-40, 5 mm HEPES, pH 8.0) and then incubated on ice for 10 min. The nuclei were recovered by centrifugation at 3,000 × g for 5 min and then suspended in a nuclear lysis buffer (10 mm EDTA, 1% SDS, 50 mm Tris·HCl, pH 8.1). The nuclei were incubated on ice for an additional 10 min, and the supernatants were collected by centrifugation at 15,000 × g for 10 min and saved as nuclear lysates. The nuclear lysates were then sonicated on ice with 10 pulses, each for 15 s, to generate chromatin DNA with an average size of 600 bp. The sonicated lysates were diluted 10-fold with a buffer (165 mm NaCl, 0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris·HCl, pH 8.0) and precleared with 30 μl of protein A-Sepharose beads. The lysates were first incubated with the indicated antibodies overnight, and then 60 μl of protein A-Sepharose beads were added, and the lysates were incubated for an additional 4 h. The immunocomplexes were washed twice with a low salt buffer (150 mm NaCl, 0.1% SDS, 1% Nonidet P-40, 1 mm EDTA, 50 mm Tris·HCl), twice with a high salt buffer (500 mm NaCl, 0.1% SDS, 1% Nonidet P-40, 1 mm EDTA, 50 mm Tris·HCl), twice with LiCl buffer (250 mm LiCl, 0.1% SDS, 1% Nonidet P-40, 1 mm EDTA, 50 mm Tris·HCl), and finally twice with TE buffer (0.25 mm EDTA, 10 mm Tris-HCl). The recovered beads were eluted with 120 μl of elution buffer (1% SDS, 100 mm NaHCO3), the supernatants were collected and incubated at 65 °C overnight to reverse the formaldehyde cross-linking. The DNA from the supernatants was recovered by phenol extraction followed by ethanol precipitation and analyzed using PCR with primers spanning the HIV-1 LTR promoter (5′-CAT CCG GAG TAC TTC AAG AAC TGC-3′ and 5′-GGC TTA AGC AGT GGG TTC CCT AG-3′) or GAPDH (5′-GAA GGTGAA GGT CGGAGT-3′ and 5′-GAA GAT GGT GAT GGG ATT TC-3′). The PCR program consisted of 35 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 30 s.
In Vitro Transcription Assay
An in vitro transcription assay was performed as described previously (33) with some minor modifications. Briefly, recombinant Tat protein and Tip110 protein were mixed in buffer D (20 mm HEPES, pH 7.9, 0.1 m KCl, 20% glycerol, 0.2 mm EDTA, 0.5 mm DTT) and added to 3.5 μl of nuclear extracts (Promega) on ice. Then 200 ng of linearized HIV-1 dG-less DNA, unlabeled NTP, 4 units of RNase inhibitor, and 400 mm sodium citrate were added with a total 25-μl transcription reaction. The mixture was incubated at 30 °C for 30 min, followed by treatment with 1 μl of DNase I to remove the DNA template. RNA transcripts were recovered by phenol extraction and ethanol precipitation and suspended in 10 μl of double-distilled H2O. Two microliters of the RNA sample was used for RT-PCR analysis. The primers used to amplify the short G-less transcripts were 5′-GGG TCT CTC TGG TTA GAC CAG ATC TGA GCC TGG GAG CTC-3′ and 5′-AAA ACC AAA CCC TGC GCT CCA TCG CCA-3′. The primers used to amplify the long G-less transcripts were 5′-GCG AGG CAT AAA GTT GCG TGT G-3′ and 5′-AGG AGG GAG AGG TGA GGA GAG GAT-3′. The levels of long and short transcripts were quantitated by RT-PCR, followed by the densitometric analysis of the RT-PCR products; the ratio of the PCR products derived from the long transcripts to those derived from the short transcripts was calculated and used to express the elongation efficiency.
RESULTS
Tip110 Did Not Complex with Cyclin T1 and CDK9
Our previous work has shown that Tip110 enhances HIV-1 replication by activating Tat-mediated LTR transcription (32). Because Tip110 and P-TEFb co-localize within nuclear speckle structures (35) and P-TEFb is responsible for HIV-1 transcription elongation, we first determined whether Tip110 interacted with CDK9 or cyclin T1 by immunoprecipitation and Western blot analysis. First, we examined the possibility with ectopically expressed Tip110 in 293T cells. The results indicated that no Tip110·CDK9 or Tip110·cyclin T1 complex formation took place (Fig. 1A, top). CDK9 and cyclin T1 antibodies were not the problem, because they were capable of immunoprecipitating each other (Fig. 1A, middle and bottom), confirming complex formation between them (4). We then examined whether endogenous Tip110 associated with P-TEFb by immunoprecipitation. We found that neither endogenous cyclin T1 nor CDK9 could be detected in the anti-Tip110 immunoprecipitates (Fig. 1B). Similarly, we did not detect Tip110 protein in either anti-CDK9 or anti-cyclin T1 immunoprecipitates (Fig. 1, C and D). Therefore, these results suggest that Tip110 did not associate with CDK9 or cyclin T1.
FIGURE 1.
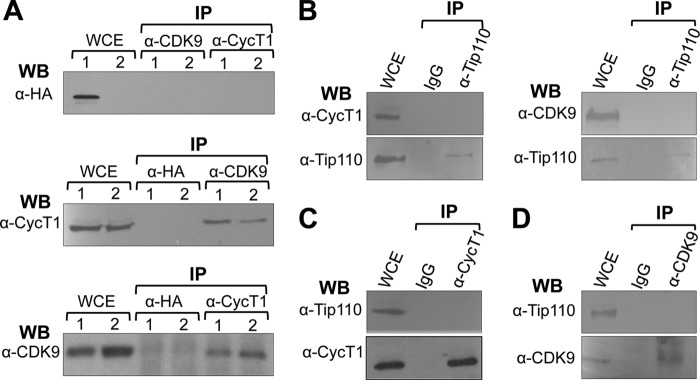
No complex formation between Tip110 and P-TEFb. A, 293T cells were transfected with pTip110.HA (lane 1) or pcDNA3 (lane 2). Whole cell extracts (WCE) were prepared and immunoprecipitated (IP) with α-HA, α-cyclin T1 (CycT1), or α-CDK9 antibody, followed by Western blot analysis (WB) with α-HA, α-CycT1, or α-CDK9 antibody. B, WCE were prepared from 293T cells and immunoprecipitated with α-Tip110 antibody or an isotype-matched IgG, followed by Western blot analysis with α-CycT1, α-CDK9, or α-Tip110 antibody. C, WCE were prepared from 293T cells and immunoprecipitated with α-CycT1 antibody or an isotype-matched IgG, followed by Western blot analysis with α-Tip110 or α-CycT1 antibody. D, WCE were prepared from 293T cells and immunoprecipitated with α-CDK9 antibody or an isotype-matched IgG, followed by Western blot analysis with α-Tip110 or α-CDK9 antibody.
Tip110 Bound to Unphosphorylated but Not Phosphorylated RNAPII
One of the essential events in HIV-1 LTR promoter transcriptional activation is phosphorylation within the heptapeptide repeats of the RNAPII CTD at serine 2 (CTDo2) and serine 5 (CTDo5). To investigate whether Tip110 directly interacted with RNAPII, 293T cells were transfected with pTip110.HA. An immunoprecipitation was performed for the unphosphorylated RNAPII (CTDa), serine 2-phosphorylated RNAPII (CTDo2), or serine 5-phosphorylated RNAPII (CTDo5) by three highly specific antibodies (8WG16 (CTDa), H5 (CTDo2), and H14 (CTDo5), respectively), followed by Western blot analysis using anti-HA antibody. The results demonstrated that only the 8WG16 immunocomplex contained Tip110 (Fig. 2A). This interaction was further confirmed by detecting CTDa in Tip110 immunoprecipitates (Fig. 2B), indicating that exogenous Tip110 only associated with CTDa. We further investigated whether endogenous Tip110 interacted with RNAPII by performing an immunoprecipitation of 293T cell lysates with anti-CTDa, anti-CTDo2, or anti-CTDo5 antibody, followed by Western blot analysis against Tip110 (Fig. 2, C–E, top panels). The results demonstrated that only the CTDa immunocomplex contained Tip110. This interaction was further confirmed by detecting unphosphorylated RNAPII in Tip110 immunoprecipitates (Fig. 2, C–E, bottom panels), indicating that endogenous Tip110 only interacted with CTDa but not CTDo2 and CTDo5.
FIGURE 2.

Complex formation between Tip110 and RNAPII. A and B, 293T cells were transfected with pTip110-HA (lane 1) or pcDNA3 (lane 2). WCE (500 μg) were prepared from 293T cells and immunoprecipitated (IP) with α-unphosphorylated CTD (CTDa), α-serine 2-phosphorylated CTD (CTDo2), or α-serine 5-phosphorylated CTD antibody (CTDo5), followed by Western blot analysis with α-HA (A), or immunoprecipitated with α-HA, followed by Western blot analysis with α-CDTa, α-CTDo2, or α-CTDo5 antibody (B). *, immunoreactive IgG. C, WCE were prepared from 293T cells and immunoprecipitated with α-CTDa antibody, α-Tip110 antibody, or an isotype-matched IgG, followed by Western blot analysis with α-Tip110 or α-CTDa antibody. D, WCE were prepared from 293T cells and immunoprecipitated with α-CTDo2 antibody, α-Tip110 antibody, or an isotype-matched IgG, followed by Western blot analysis with α-Tip110 or α-CTDo2 antibody. E, WCE were prepared from 293T cells and immunoprecipitated with α-CTDo5 antibody, α-Tip110 antibody, or an isotype-matched IgG, followed by Western blot analysis with α-Tip110 or α-CTDo5 antibody.
The results of these Western blots indicated that only CTDa, not CTDo2 and CTDo5, was detected to complex with Tip110 (Fig. 2). This difference could result from different expression levels of CTDa, CTDo2, and CTDo5 or from different efficiencies of the antibodies for these three RNAPII forms. To ascertain Tip110 interaction with CTDa, an in vitro GST pull-down assay was performed. First, recombinant GST-CTDa protein was purified from E. coli and then phosphorylated by casein kinase I in vitro. GST-CTDa and its phosphorylation by casein kinase I (GST-CTDo) were confirmed by Western blot analysis (Fig. 3A). Meanwhile, recombinant GST-Tip110 was expressed and purified (Fig. 3B, lane 2), treated with thrombin to remove the GST tag (Fig. 3B, lane 3), and then purified using the glutathione-beads to produce recombinant Tip110 protein (Fig. 3B, lane 4). Then recombinant proteins GST-CTDa or GST-CTDo and Tip110 were used in the GST pull-down assay. The proteins that remained on the beads (bound) as well as the proteins that were present in the supernatants (unbound) were analyzed by SDS-PAGE followed by Western blot analysis using anti-Tip110 or GST antibody. The results showed direct binding of Tip110 to CTDa but not to GST protein or CTDo (Fig. 3C). Taken together, these data demonstrated that Tip110 bound to CTDa in a direct manner but not CTDo2 or CTDo5.
FIGURE 3.
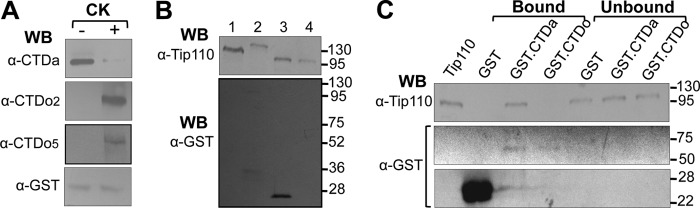
Direct binding of Tip110 to RNAPII CTDa. A, GST-CTDa protein was expressed and purified from E. coli and phosphorylated by casein kinase I (CK) in vitro. GST-CTDa and CK-phosphorylated GST-CTD (GST-CTDo) were confirmed by Western blot analysis with α-CTDo, α-CTDo2, α-CTDo5, or α-GST antibody. Western blot analysis against GST was included as a loading control. B, GST-Tip110 protein was expressed and purified from E. coli (lane 2). Its GST tag was removed by thrombin digestion (lane 3), followed by glutathione bead affinity purification to remove the GST tag (lane 4). All of those proteins were subjected to Western blot analysis with α-Tip110 antibody or α-GST antibody. WCE from 293T cells were included as a control (lane 1). C, GST-CTDa, CK-phosphorylated GST-CTD (GST-CTDo), and GST proteins were immobilized with 50 μl of glutathione beads and then incubated with purified Tip110 protein. The beads and the supernatants were separated by centrifugation and collected to represent bound and unbound fractions, respectively. The beads were then subject to repetitive washes to remove unbound proteins. Then the proteins bound on the beads and in the supernatants were detected by Western blot analysis with α-Tip110 antibody. Purified Tip110 protein was included as the control to indicate the correct protein size (lane 1). The membrane was stripped and reprobed with anti-GST antibody to ensure GST binding to the glutathione beads.
To determine which functional domains of Tip110 interacted with CTDa, we took advantage of a series of Tip110 mutants that contained the RRM domain (ΔRRM), C-terminal domain (ΔCT), NLS domain (ΔNLS), or N-terminal domain deletions (ΔNT) (32). Due to differences in antibody epitopes,4 the Tip110 mutants were divided into two groups for detection by Western blot analysis; ΔRRM, ΔNLS, and ΔCT mutants were detected by anti-Tip110 serum, and ΔNT was detected by a Tip110 monoclonal antibody. We first transfected 293T cells with the full-length Tip110 (WT) and each of the Tip110 mutants and confirmed their expression by Western blot analysis (Fig. 4, A and C). Then we performed immunoprecipitations with anti-CTDa antibody followed by Western blot analysis for Tip110. Mutants ΔCT, ΔNLS, and ΔRRM were all detected in CTDa immunoprecipitates, same as the binding with WT Tip110 (Fig. 4B). However, mutant ΔNT failed to be detected in the CTDa immunoprecipitates (Fig. 4D). To further confirm this finding, WT and ΔNT were co-transfected (WT + ΔNT) and analyzed similarly. The results showed that only WT and not ΔNT was detected in the CTDa immunoprecipitates of the co-transfection (Fig. 4D). These results suggest that the N-terminal domain of Tip110 is directly involved in Tip110-CTDa interactions.
FIGURE 4.
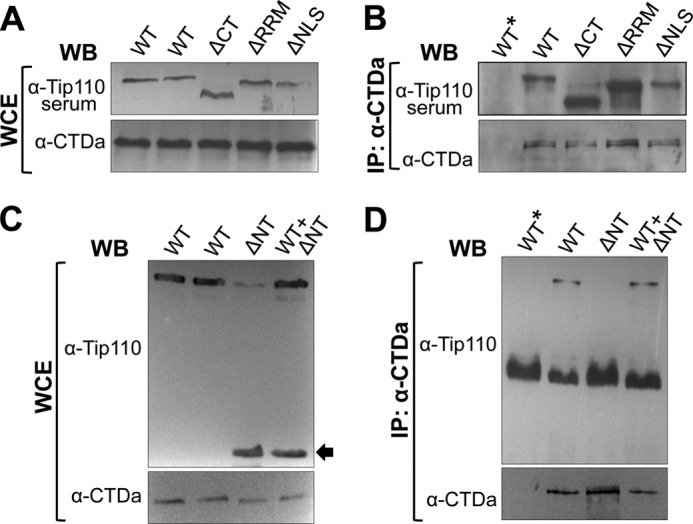
Requirement of the HAT domain for Tip110 binding to unphosphorylated RNAPII. 293T cells were transfected with Tip110 (WT), or each of the deletion mutants ΔCT, ΔRRM, and ΔNLS (A and B), ΔNT mutant, or WT + ΔNT (C and D). WCE were prepared from the transfected cells and directly analyzed by Western blot analysis with α-Tip110 serum and α-CTDa antibody (A) or α-Tip110 and α-CTDa antibody (C) or immunoprecipitated (IP) with α-CTDa antibody, followed by Western blot analysis with α-Tip110 serum and α-CTDa antibody (B) or α-Tip110 and α-CTDa antibody (D). WT*, an isotype-matched IgG control for immunoprecipitation. ΔNT is marked by an arrow (C).
More P-TEFb Recruitment to the LTR Transcription Complex by Tip110 and Tat
Tip110 has been shown to directly interact with HIV-1 Tat protein (32). We next determined whether Tip110 interaction with Tat would recruit more P-TEFb to the HIV-1 LTR transcription complex. 293T cells were transfected with pLTR-Luc, pTat.Myc, and pTip110.HA. At 48 h post-transfection, we performed Western blot analysis to determine Tip110, Tat, CDK9, and cyclin T1 expression and found that neither Tip110 nor Tat altered the overall levels of CDK9 and cyclin T1 expression (Fig. 5A). An immunoprecipitation was performed for Tat, followed by Western blot analysis using cyclin T1 antibody. As expected, cyclin T1 was detected in anti-Myc immunoprecipitates, confirming cyclin T1/Tat interaction (Fig. 5B). Moreover, more cyclin T1 was detected in anti-Myc immunoprecipitates of cells co-expressing both Tat and Tip110 than in the cyclin T-Tat complex formed in the cells expressing Tat alone; the increase is estimated to be about 2.5-fold. Similarly, about 1.4-fold more CDK9 was detected in CTDa immunoprecipitates of cells co-expressing both Tat and Tip110 than in the CDK9-CTDa complex in the cells expressing Tat alone (Fig. 5C). To determine whether Tip110 binding to Tat/CTDa is directly involved in the increased recruitment, ΔNT that does not bind to CTDa (Fig. 4) or Tat (32) was transfected alone or with Tat; similar experiments were then performed (Fig. 5A). The results showed that unlike WT, ΔNT showed no changes in the levels of cyclin T1 and CDK9 in the transcription complex. The results were reproducible (Fig. 5D) and suggest that the interaction of Tip110 with Tat/CTDa led to the recruitment of more P-TEFb to the transcriptional complex.
FIGURE 5.

Increased recruitment of P-TEFb to Tat·RNAPII complex by Tip110. 293T cells were transfected with pLTR-Luc, pTat.Myc, and/or pTip110.HA or ΔNT mutant (marked by an asterisk) plasmids as indicated. pcDNA3 was added to equalize the total amounts of plasmid DNA transfected. WCE were prepared from the transfected cells and directly analyzed by Western blot analysis with α-Tip110, α-Myc, α-CycT1, α-CDK9, or α-β-actin antibody (A); immunoprecipitated (IP) with α-Myc antibody, followed by Western blot analysis with α-CycT1 and α-Myc antibody (B); or immunoprecipitated with α-CTDa antibody, followed by Western blot analysis with α-CDK9 or α-CTDa antibody (C). Rel, the relative changes of cyclin T1 (B) or CDK9 (C) induced by Tip110 expression over the recruitment of the cyclin T or CDK9 in the cells without exogenous Tip110. D, quantitative data were obtained from multiple independent experiments. Error bars, S.D.
Tip110 and Tat Enhanced RNAPII Phosphorylation
The most critical step during HIV-1 LTR promoter transactivation is the phosphorylation of RNAPII (36, 37). CDK7 phosphorylates serine 5 of RNAPII during the initiation of transcription, whereas CDK9 phosphorylates serine 2 during transcription elongation (4, 38). Because Tip110 could recruit more P-TEFb to the transcription complex, we next determined the effects of Tip110 and Tat expression on RNAPII phosphorylation. We transfected 293T cells with an increasing amount of pTip110.HA or pTat.Myc or the same amount of pTat.Myc with an increasing amount of Tip110.HA. After 48 h, Western blot analysis was performed against CTDa, CTDo2, and CTDo5. When cells were transfected with Tip110 alone, both CTDo2 and CTDo5 showed a 2-fold increase, whereas CTDa decreased (Fig. 6, A and D). When cells were transfected with Tat alone, CTDa showed a slight decrease, and CTDo5 exhibited little change, but CTDo2 showed a 1.5-fold increase (Fig. 6, B and E). In comparison, when cells were transfected with both Tat and Tip110, CTDo2 and CTDo5 both increased by 3-fold, whereas CTDa decreased (Fig. 6, C and F). These results indicate that expression of both Tip110 and Tat led to enhanced phosphorylation of RNAPII at both serine 2 and serine 5, which may account for Tip110 function in transcription activation. Expression analysis of P-TEFb (cyclin T1 and CDK9) or TFIIH (cyclin H and CDK7) confirmed that Tip110, Tat, or both did not significantly alter the expression of these proteins (data not shown), indicating that increased RNAPII phosphorylation by Tip110 or Tat was not the result of any changes in the expression levels of P-TEFb or TFIIH.
FIGURE 6.

Increased RNAPII CTD phosphorylation by Tip110 and Tat. 293T cells were transfected with an increasing amount of pTip110.HA (A), pTat.Myc (B), or pTat.Myc (0.5 μg) plus an increasing amount of pTip110.HA (C). ΔNT mutant (G; marked by an asterisk) was also included. pcDNA3 was added to equalize the total amounts of plasmid DNA transfected. Whole cell extracts were prepared 72 h post-transfection and subjected to Western blot analysis with α-Tip110, α-Myc, α-β-actin, α-CTDa, α-CTDo2, or α-CTDo5 antibody. The relative levels of CTDa, CTDo2, and CTDo5 in A, B, C, and G were quantitated by densitometry, and quantitative data from multiple independent experiments are presented in D, E, F, and H.
To further ascertain the effects of Tip110 interaction with Tat and CTDa on RNAPII phosphorylation, we took advantage of the ΔNT mutant that did not bind to CTDa (Fig. 4) and Tat (32) and performed similar experiments. Compared with the cells that were transfected with Tat alone, expression of ΔNT did not show considerable changes in RNAPII phosphorylation (Fig. 6, G and H).
Tip110 Was Present on the HIV-1 LTR Core Promoter
To determine whether Tip110 was recruited to the HIV-1 LTR promoter in vivo, ChIP experiments were carried out. The HIV-1 LTR core promoter and the primers for the ChIP assay are illustrated in Fig. 7A. For the ChIP assay with exogenous Tip110, 293T cells were transfected with the proviral plasmids pNL4-3 and pTip110.His, and Tip110 expression was confirmed (marked by an asterisk; Fig. 7B). Chromatin from transfected cells was isolated and immunoprecipitated with anti-His antibodies followed by PCR to amplify the HIV-1 LTR core promoter region. ChIP with no antibody (No Ab) and an isotype-matched IgG was included as negative controls, whereas ChIP with anti-CTDa antibody was included as a positive control. Moreover, PCR with primers spanning the GAPDH coding region was also performed as the other specificity control. Furthermore, PCR with ChIP input DNA was performed to ensure the quality and quantity of the DNA. The LTR promoter was amplified from the anti-His (Tip110) immunocomplex of cells that were transfected with pTip110.His (Fig. 7C, bottom panels). As expected, the LTR promoter was amplified from the anti-CTDa immunocomplex of cells that were transfected with pcDNA3 or pTip110.His. These results indicate that exogenous Tip110 is present at the HIV-1 LTR promoter in vivo.
FIGURE 7.

Presence of Tip110 at the HIV-1 LTR core promoter. A, schematic of the LTR core promoter and the primer locations for PCR amplification. B, Western blot analysis for Tip110 expression in 293T cells transfected with pNL4-3 and pTip110.His (293T*) or pcDNA3 (293T), U373MAGI containing the LTR promoter-driven β-galactosidase gene (MAGI), and CEM-GFP cells containing the LTR promoter-driven GFP gene (CEM.GFP). C and D, 293T cells transfected with the pNL4-3 and pTip110.His (C, bottom) or pcDNA3 (C, top), U373MAGI (D, top), and CEM-GFP cells (D, bottom) were subjected to the ChIP assay. Following chromatin cross-linking, shearing, and immunoprecipitation with α-CTDa, α-His, or α-Tip110 antibody, reverse cross-linking was performed, and the DNA was then purified and analyzed by PCR with the primer set specific for the HIV-1 LTR core promoter region, as shown in A. Input DNA and immunoprecipitated DNA without any antibody (No Ab) or with an isotype-matched IgG were included as the ChIP controls. In addition, PCR using a primer set specific for a GAPDH coding region was performed and also included as the control. E and F, Jurkat cells were transfected with Tip110.His or pcDNA3 and then infected with NL4-3 (equivalent to 20,000 cpm of reverse transcriptase, which gave rise to about 80% infection in 3 days determined by intracellular p24 staining). Cells were harvested for Western blot analysis for Tip110 expression (E) or ChIP assay as described above (F).
Considering the unintegrated nature of the HIV proviral DNA and overexpression of Tip110 in the above experimental settings, we proceeded to determine whether endogenous Tip110 was present in the integrated HIV-1 promoter. We took advantage of U373MAGI cells that have an integrated HIV-1 LTR promoter-driven LacZ transgene cassette and CEM-GFP cells that have an integrated HIV-1 LTR promoter-driven GFP transgene cassette. Western blot analysis was first performed to confirm expression of endogenous Tip110 in these two cell lines (Fig. 7B). Then chromatin was isolated from these cells, immunoprecipitated, and subjected to PCR as above. The LTR promoter was clearly amplified from both CTDa and Tip110 immunocomplex prepared from both CEM-GFP and U373MAGI cells (Fig. 7D). Moreover, human CD4+ T lymphocyte Jurkat cells were transfected with pcDNA3 or pTip110.His (Fig. 7E) and then infected with HIV-1 NL4-3 viruses. A similar ChIP assay was performed. The LTR promoter was amplified from Tip110 immunocomplex prepared from cells that were transfected with pcDNA3 or pTip110.His, whereas the LTR promoter was only amplified from His immunocomplex prepared from pTip110.His-transfected but not pcDNA3-transfected cells (Fig. 7F). Collectively, these results suggest that endogenous Tip110 is also detectable at the integrated HIV-1 LTR core promoter in vivo.
Tip110 Expression Was Associated with P-TEFb Recruitment to the LTR Promoter and Phosphorylation of Serine 2 of the Heptapeptide of the RNAPII CTD
We then evaluated the relationship among Tip110 expression, P-TEFb recruitment to the LTR promoter, and RNAPII phosphorylation. 293T cells were first transfected with pLTR-Luc along with pTat.Myc, pTip110.HA, or both. Then a ChIP assay was performed using immunoprecipitates against Myc (Tat), HA (Tip110), cyclin T1, CDK9, CTDa, CTDo2, or CTDo5, followed by PCR to amplify the HIV-1 LTR region (Fig. 8A). Tat and Tip110 protein, included as positive controls, could be detected at the LTR region. No CDK9 or cyclin T1 was detected at the LTR promoter in the absence of Tat, but in the presence of Tip110 overexpression, increases in the levels of cyclin T1 and CDK9 recruited to the LTR promoter were observed. Meanwhile, CTDo2 was only observed in the LTR promoter in the presence of Tat expression and exhibited a 2-fold increase in the presence of Tip110, whereas CTDo5 was detected without Tat expression and showed little change in the presence of Tip110 and Tat. As expected, neither P-TEFb nor RNAPII could be detected at the LTR promoter when the immunoprecipitation was performed using mouse IgG as a control.
FIGURE 8.
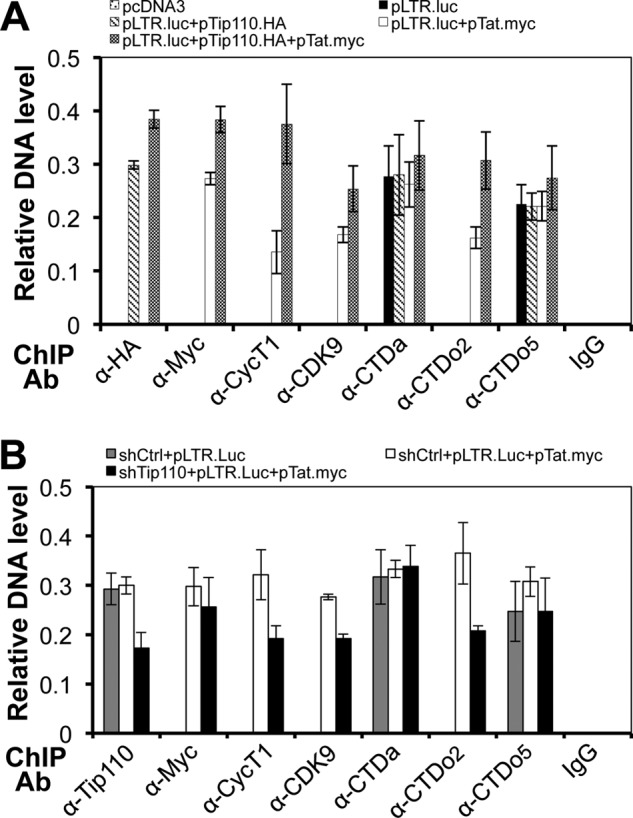
Recruitment of P-TEFb to the LTR promoter by Tip110. A, 293T cells were transfected with pLTR-Luc and pTip110.HA, pTat.Myc, or both. Chromatin from these cells was cross-linked, sheared, and immunoprecipitated with α-HA, α-Myc, α-CycT1, α-CDK9, α-CTDa, α-CTDo2, and α-CTDo5. After reverse cross-linking, the purified DNA was purified and analyzed by PCR with the primer set for the HIV-1 LTR promoter. PCR products were quantitated by densitometry and expressed as -fold increases over the input control. An isotype-matched IgG was included as a control. pcDNA3 was added to equalize the total amount of plasmid DNA transfected. B, 293T cells were first transfected with psh-Tip110 at day 0 and then transfected with pLTR-Luc and pTat.Myc plasmids at day 4. Cells were cultured for an additional 3 days and harvested for the ChIP assay as described above with the exception that α-Tip110 antibody was used in place of α-HA antibody for immunoprecipitation. pSIREN, the backbone vector for psh-Tip110, was included as a control for psh-Tip110. The quantitative data were derived from multiple independent experiments. Error bars, S.D.
We performed similar experiments in cells with down-modulated Tip110 expression using Tip110-specific shRNA. Tip110 siRNA expression led to drastic and lasting knockdown and subsequently cell death and was not used in these experiments (39, 40). Tip110-shRNA has been shown to reduce endogenous Tip110 expression at day 7, whereas its backbone vector pSIREN has no affect on Tip110 expression (see Fig. 9). We transfected Tip110-shRNA or its cognate control pSIREN into 293T cells at day 0, followed by transfection of pLTR-Luc and pTat.Myc at day 4. The cells were harvested at day 7 for ChIP analysis. We found that there was less CDK9 and cyclin T1 recruited to the LTR promoter, and only half of the CTDo2 was detected in the absence of Tip110 (Fig. 8B). Meanwhile, CTDa increased slightly, but CTDo5 decreased in the absence of Tip110. These data together suggest that Tip110 interaction with Tat enabled more P-TEFb to be recruited to the HIV-1 LTR promoter and further enhanced RNAPII serine 2 phosphorylation at the LTR promoter.
FIGURE 9.

Direct effects on HIV-1 transcription elongation by Tip110 and Tat. A, schematic of the HIV-1 LTR double G-less cassette used in the experiment. The transcript derived from this cassette contains two G-less regions, a short upstream G-less region (ST) and a long downstream G-less region (LT), which have been used to determine transcription initiation and transcription elongation, respectively. The primer locations to amplify the short and long transcripts are indicated. B, an in vitro transcription/elongation assay was performed with linearized double G-less plasmid and 0, 0.1, and 0.2 μg of recombinant Tat protein or 0.2 and 0.4 μg of recombinant Tip110 protein or 0.1 μg of recombinant Tat protein plus 0.2 and 0.4 μg of recombinant Tip110 protein. After DNase treatment, RNA was isolated and RT-PCR-amplified using primers specific for the short and long upstream G-less regions. C and D, 293T cells were first transfected with pSIREN or psh-Tip110 at day 0 and then transfected with the double G-less cassette and pTat.Myc plasmid at day 4 and again at day 7. Cells were harvested on day 7 and day 10 for whole cell extracts, followed by Western blot analysis with α-Tip110, α-Myc, or α-β-actin antibody (C), or for RNA extraction, followed by RT-PCR as above for the short and long upstream G-less regions (D). Elongation efficiency was calculated as the ratio of long to short upstream G-less region transcripts. E and F, HeLa nuclear extract was first added to α-Tip110 antibody, an isotype-matched IgG, or no antibody (No Ab) and then protein A beads. After a 2-h incubation, the protein A beads were recovered by brief centrifugation and discarded, whereas the supernatant was saved and subjected to Western blot analysis for Tip110 (E) or used in the in vitro transcription/elongation assay in the presence of recombinant Tat or Tat plus Tip110, followed by RNA isolation and RT-PCR as described above (F). The data were representative of three independent experiments.
Tip110 and Tat Increased the LTR Transcription Elongation Efficiency
To determine if Tip110 interaction with Tat had direct effects on HIV-1 transcription, we adapted an in vitro guanine (G)-less transcription/elongation assay. This assay involves use of the template pHIVdG-less, which contains two G-less cassettes downstream from the HIV-1 LTR promoter (Fig. 9A) and allows synthesis of two RNaseT1-resistant transcripts: the promoter-proximal short transcript and the promoter-distal long transcript. Expression of these two transcripts has widely been used to determine transcription initiation and elongation, respectively (41). First, an in vitro transcription was set up to contain nuclear extract, the G-less DNA template, recombinant Tat, Tip110, or Tat and Tip110. After digestion of the template DNA with DNase I, synthesized RNA was recovered, and semiquantitative RT-PCR was performed to determine the relative levels of the short and long G-less transcripts using primers (Fig. 9A) that were specific for each of those two transcripts expected to give rise to PCR products of 300 (ST) and 520 bp (LT), respectively. As expected, Tat increased long transcript expression but had little effect on short transcript expression (Fig. 9B). Compared with Tat, Tip110 also slightly increased expression of the long transcripts and had little effect on the expression of short transcripts. Compared with Tat or Tip110 alone, the presence of both Tat and Tip110 showed considerably enhanced expression of the long transcripts but had no effect on the expression of short transcripts. These results indicate that the interaction of Tip110 with Tat led to a higher efficiency of elongation for RNAPII complexes formed on the LTR promoter.
To further characterize the significance of Tip110 in Tat-mediated LTR transcription elongation, we knocked down endogenous Tip110 and determined its effects on the LTR transcription. To achieve this, we knocked down endogenous Tip110 in 293T cells using Tip110-specific shRNA and performed an in vivo HIVdG-less assay in these cells. We transfected pHIVdG-less plasmid with pTat.Myc into 293T cells on day 0, 4, or 7 following transfection with either Tip110-specific shRNA or the backbone vector pSIREN. Cells were harvested at day 7 or 10 for Western blot or semiquantitative RT-PCR analysis. Western blot analysis demonstrated that endogenous Tip110 protein expression was down-modulated by pshTip110 at day 7 post-transfection but showed some recovery at day 10, and Tip110 expression showed no significant changes with pSIREN transfection (Fig. 9C). The RT-PCR analysis showed that the long transcripts showed considerable and consistent increases in cells that were transfected with Tat and pSIREN (Fig. 9D, left). In contrast, in cells that were transfected with Tat and pshTip110, the long transcripts showed similar increases at day 0 when Tip110 expression was not affected, considerable decreases at day 7 when Tip110 expression was knocked down, and considerable recovery at day 10 when Tip110 expression returned (Fig. 9D, right). The apparent interdependent relationship provides additional evidence to support the important roles of Tip110 in HIV-1 LTR transcription. Taken together, these results further demonstrate the specific role of Tip110 in regulation of the RNAPII elongation efficiency on the LTR promoter.
Last, we determined if direct depletion of endogenous Tip110 would impede Tat-mediated LTR transcription. To this end, nuclear extract was immunodepleted of endogenous Tip110 using an anti-Tip110 antibody before it was used in the in vitro HIVdG-less transcription assay as described above. Immunodepletion with an isotype-matched IgG and without any antibodies was also included as a control. Western blot analysis confirmed that immunodepletion with anti-Tip110 antibody removed more than half of the endogenous Tip110 from nuclear extract (Fig. 9E). The RT-PCR analysis showed that Tip110 depletion from nuclear extract led to a considerable decrease in synthesis of the long transcripts compared with the isotype-matched IgG-depleted nuclear extract (Fig. 9F). Interestingly, the addition of recombinant Tip110 back to the Tip110-depleted nuclear extract failed to restore synthesis of the long transcripts. The inability of recombinant Tip110 to restore the transcription elongation is probably due to removal of other transcription factors that are associated with Tip110 depletion, such as RNAPII itself.
DISCUSSION
Transcriptional activation of the HIV-1 LTR promoter is a complex event and requires the coordination of viral proteins and several cellular proteins. One of the key factors involved in this activation is the Tat protein, which enhances HIV-1 transcription by promoting the formation of transactivation complexes at the LTR promoter via protein-protein interactions. In addition to Tat protein, there are a large number of cellular factors also involved in transcription. They either function by removing the negative inhibitor that blocks RNAPII phosphorylation at the promoter region or by recruiting the elongation-competent RNAPII-containing complex (21, 42, 43). In this study, we focused on the Tip110 protein (Tat-interacting protein of 110 kDa). Studies from our group have shown that Tip110 functions synergistically with Tat in Tat-mediated transactivation of the HIV-1 LTR promoter, thereby increasing viral gene expression and virus production (32).
To understand the underlying molecular mechanisms of Tip110, we first examined its interaction with transcription factors. A large number of cellular factors are involved in Tat-mediated transcription; many recruit elongation-competent RNAPII-containing complexes and have been shown to interact with multiple transcription factors. For example, SKIP, Tat-SF1, and CA150 are reported to associate with Tat·P-TEFb in nuclear extracts and are present in large RNAPII elongation complexes (21, 23, 44). Tip110 was first identified as a Tat-interacting protein from a yeast two-hybrid screen of a human fetal brain cDNA library, using Tat as bait (32). We found that Tip110 bound to unphosphorylated RNAPII (CTDa) but not its phosphorylated form (CTDo) (Fig. 2). This direct and specific binding was further supported by the results of a GST pull-down assay (Fig. 3) and mutagenesis analysis (Fig. 4). Protein structure analysis predicted that the N-terminal two-thirds of the Tip110 protein contains seven HAT motifs. These HAT motifs provide a structural unit of two antiparallel α-helices that form functional TPR(s) and determine the specificity of protein-protein interactions (45). Besides binding to unphosphorylated RNAPII, this domain is also responsible for Tip110 interaction with Tat (32). This suggests that Tip110 forms a complex with Tat and unphosphorylated RNAPII through its N-terminal domain and may play a role in stabilizing the transactivation complex. In addition, the HAT domain of Tip110 interacts with a C-terminal region of the U4/U6 snRNP-specific 90-kDa protein that functions in the reassembly of the U4/U6 snRNP (26, 27). The HAT domain is also involved in Tip110 interaction with the splicing factor RNPS1 (46), suggesting that the HAT domain determines the specificity of Tip110 interactions with other proteins.
Although Tip110 and P-TEFb both co-localize in the nuclear speckle area (35), our data indicated that no interaction existed between them (Fig. 1). This phenomenon distinguishes Tip110 from other cellular transcription factors involved in the P-TEFb complex. For example, SKIP associates with P-TEFb and is recruited to the LTR promoter by Tat (21). ELL2 is another elongation factor in the P-TEFb complex, and Tat recruits more ELL2 to P-TEFb and assists in the stabilization of ELL2 to activate P-TEFb (47). Although there is no direct interaction between Tip110 and P-TEFb, Tip110 is capable of recruiting more P-TEFb to the transcription complex in the presence of Tat (Fig. 5). Therefore, we speculate that Tip110 might be first recruited to the unphosphorylated RNAPII on the LTR promoter in the absence of Tat and then assist in the recruitment of P-TEFb through Tat. After the RNAPII has been phosphorylated by P-TEFb and establishes transcription elongation, Tip110 would become disassociated from phosphorylated RNAPII and commence a new cycle of LTR transactivation.
Because P-TEFb is responsible for RNAPII serine 2 phosphorylation, we set out to investigate whether Tip110 or Tat would alter the RNAPII phosphorylation level. Our data showed that Tip110 protein alone could decrease the level of unphosphorylated RNAPII while increasing the level of both serine 2- and serine 5-phosphorylated RNAPII by 2-fold, suggesting that Tip110 is a weak transactivator of the HIV-1 LTR and leads to its basal level transcription. When Tip110 and Tat were co-expressed, an increase in serine 2 and serine 5 RNAPII phosphorylation levels coincided with a large decrease in unphosphorylated RNAPII (Fig. 6), which may account for the Tip110 function in Tat-mediated transcription. However, in these studies, we examined the overall RNAPII phosphorylation level in 293T cells in the presence of Tip110 and Tat, not the RNAPII specifically located on the LTR promoter. Several genes other than the HIV-1 LTR promoter are regulated at the stage of transcription elongation by P-TEFb, including hsp70 and proto-oncogenes c-myb, c-myc, and c-fos (48–50). For these genes, the RNAPII complexes are stalled in the 5′ region of the transcription unit, and P-TEFb recruitment is the key regulator that helps RNAPII to overcome this rate-limiting step. It is the TATA-box, not the TAR structure, that is important for the recruitment of P-TEFb (41). Therefore, the increased phosphorylated form of RNAPII may result from the recruitment of P-TEFb by Tip110 and Tat on these cellular genes. In order to examine RNAPII phosphorylation on the LTR promoter, one can label the 5′ end of the LTR-promoter templates with biotin and isolate LTR-bound preinitiation complexes to detect protein components associated with it (51). We would expect to see a greater increase in RNAPII phosphorylation located on the HIV-1 LTR promoter in the presence of Tip110 and Tat.
HIV-1 transcription is regulated by the interplay between a combination of viral and cellular transcription factors with binding sites located within the HIV-1 LTR promoter. Using the ChIP assay, we first detected Tip110 associated with the LTR promoter expressed transiently by transfecting pNL4-3 into 293T cells (Fig. 7). To further examine whether Tip110 could be recruited to the integrated LTR promoter in the absence of Tat, we performed a ChIP assay with endogenous Tip110 using U373MAGI or CEM-GFP cells. In both cell lines, the LTR promoter is integrated into the chromosome with an inducible β-galactosidase reporter (U373MAGI) or GFP reporter gene (CEM-GFP) (34, 52, 53). The results showed that endogenous Tip110 could be detected on the integrated LTR promoter in the absence of Tat. Furthermore, similar results were obtained in the context of HIV-1 infection. Taken together, these results support the notion that Tip110 is probably recruited to the LTR promoter at the RNAPII-pausing step before Tat is translated.
The ChIP assay also demonstrated that approximately a 3-fold increase in cyclin T1 and a 1.5-fold increase in CDK9 were recruited to the LTR promoter in the presence of Tip110 and Tat (Fig. 8), which is consistent with the previous Western blot results (Fig. 5). We observed that Tip110 recruits more cyclin T1 than CDK9 into the transcription complex, and this is most likely because Tat directly interacts with Tip110 and mediates the recruitment of cyclin T1 by Tip110. We further examined RNAPII phosphorylation levels at the LTR promoter by performing a ChIP assay with 8WG16, H5, and H14 antibodies. Serine 2 phosphorylation of RNAPII only took place in the presence of Tat and increased with Tip110 overexpression, possibly as a result of the increased recruitment of P-TEFb by Tip110 and Tat. Unphosphorylated and serine 5-phosphorylated RNAPII were readily detected at the HIV-1 promoter in the absence of Tat, whereas Tip110 supplied along with Tat produced a slight effect on serine 5 phosphorylation, consistent with previous reports that TFIIH is recruited to the LTR promoter for RNAPII serine 5 phosphorylation in the absence of Tat (38). The increased recruitment of T-PEFb to the LTR promoter and increased CTDo2 (Fig. 8) were associated with increased transcription elongation of the LTR promoter (Fig. 9) in the presence of Tip110, providing additional evidence that Tip110 is an important factor for HIV-1 Tat-mediated transcription.
What might be the actual mechanism by which Tip110 facilitates Tat transactivation? First, overexpression of Tip110 significantly increased viral gene expression; this enhancement might due to the role of Tip110 in LTR transactivation by stimulating RNAPII phosphorylation with Tat. Second, Tip110 formed a complex with both Tat and unphosphorylated RNAPII CTD and was detected on the LTR promoter. Also, cyclin T1 and CDK9 levels were greatly increased within the transcriptional complex in the presence of Tip110 and Tat. On the basis of these observations, we propose a working model of the function of Tip110 in Tat-mediated LTR transcription (Fig. 10). Tip110 is first recruited to the LTR promoter independently of Tat. It forms a complex with unphosphorylated RNAPII and Tat and then recruits more cyclin T1 and CDK9 to the transcriptional complex, which would further enhance RNAPII serine 2 phosphorylation. When RNAPII serine 2 is fully phosphorylated, LTR transcription elongation commences, and the full-length mRNA is synthesized. At this stage, Tip110 becomes dissociated with the hyperphosphorylated RNAPII and is recycled to begin a new round of transactivation.
FIGURE 10.

A working model for Tip110 function in HIV-1 LTR promoter transactivation. In the absence of Tip110, Tat will bind to cyclin T1 and recruit the P-TEFb complex to the TAR RNA (1). These interactions lead to the phosphorylation of RNAPII serine 2 by CDK9 (2), and HIV-1 transcription elongation commences (3). In the presence of Tip110, Tip110 is first recruited to the LTR promoter and binds to unphosphorylated RNAPII (RNAPIIa) (4). Additional complex formation of Tip110 with Tat protein recruits more cyclin T1 and CDK9 to the transcription complex (5). As a result, more RNAPII serine 2 is phosphorylated (RNAPIIo), which facilitates the transcription elongation and synthesis of full-length HIV-1 mRNA. Tip110 then becomes disassociated from the hyperphosphorylated form of RNAPII and will start a new round of transactivation.
Acknowledgments
We thank Dr. David Price (University of Iowa) for the pGST-CTD plasmid and Dr. Carlos Sune (Instituto de Parasitologia y Biomedicina) for the pHIV-LTR-dG plasmid. We also thank Drs. Andy (Qigui) Yu, Mark Kaplan, Harikrishna Nakshatri, and Yuichiro Takagi for advice and input throughout the study.
This work was supported, in whole or in part, by National Institutes of Health, NINDS, Grant R01NS065785 and National Institutes of Health, NIMH, Grant R01092673 (to J. J. H.).
W. Zhao, Y. Liu, K. A. Timani, and J. J. He, unpublished data.
- LTR
- long terminal repeat
- RNAPII
- RNA polymerase II
- CTD
- C-terminal domain
- snRNP
- small nuclear ribonucleoprotein
- WCE
- whole cell extracts
- P-TEFb
- positive transcription factor b
- TAR
- transactivation response RNA element
- CycT1
- cyclin T1.
REFERENCES
- 1. Garber M. E., Mayall T. P., Suess E. M., Meisenhelder J., Thompson N. E., Jones K. A. (2000) CDK9 autophosphorylation regulates high-affinity binding of the human immunodeficiency virus type 1 tat-P-TEFb complex to TAR RNA. Mol. Cell. Biol. 20, 6958–6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peterlin B. M., Price D. H. (2006) Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 [DOI] [PubMed] [Google Scholar]
- 3. Price D. H. (2000) P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell. Biol. 20, 2629–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei P., Garber M. E., Fang S. M., Fischer W. H., Jones K. A. (1998) A Novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462 [DOI] [PubMed] [Google Scholar]
- 5. Lis J. (1998) Promoter-associated pausing in promoter architecture and postinitiation transcriptional regulation. Cold Spring Harb. Symp. Quant. Biol. 63, 347–356 [DOI] [PubMed] [Google Scholar]
- 6. Ramanathan Y., Rajpara S. M., Reza S. M., Lees E., Shuman S., Mathews M. B., Pe'ery T. (2001) Three RNA polymerase II carboxyl-terminal domain kinases display distinct substrate preferences. J. Biol. Chem. 276, 10913–10920 [DOI] [PubMed] [Google Scholar]
- 7. Roy R., Adamczewski J. P., Seroz T., Vermeulen W., Tassan J. P., Schaeffer L., Nigg E. A., Hoeijmakers J. H., Egly J. M. (1994) The MO15 cell cycle kinase is associated with the TFIIH transcription-DNA repair factor. Cell 79, 1093–1101 [DOI] [PubMed] [Google Scholar]
- 8. Trigon S., Serizawa H., Conaway J. W., Conaway R. C., Jackson S. P., Morange M. (1998) Characterization of the residues phosphorylated in vitro by different C-terminal domain kinases. J. Biol. Chem. 273, 6769–6775 [DOI] [PubMed] [Google Scholar]
- 9. Zawel L., Kumar K. P., Reinberg D. (1995) Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev. 9, 1479–1490 [DOI] [PubMed] [Google Scholar]
- 10. Wu C.-H., Yamaguchi Y., Benjamin L. R., Horvat-Gordon M., Washinsky J., Enerly E., Larsson J., Lambertsson A., Handa H., Gilmour D. (2003) NELF and DSIF cause promoter proximal pausing on the Hsp70 promoter in Drosophila. Genes Dev. 17, 1402–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu C.-H., Lee C., Fan R., Smith M. J., Yamaguchi Y., Handa H., Gilmour D. S. (2005) Molecular Characterization of Drosophila NELF. Nucleic Acids Res. 33, 1269–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barboric M., Kohoutek J., Price J. P., Blazek D., Price D. H., Peterlin B. M. (2005) Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 24, 4291–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barboric M., Yik J. H., Czudnochowski N., Yang Z., Chen R., Contreras X., Geyer M., Matija Peterlin B., Zhou Q. (2007) Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 35, 2003–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Q., Cooper J. J., Altwerger G. H., Feldkamp M. D., Shea M. A., Price D. H. (2007) HEXIM1 is a promiscuous double-stranded RNA-binding protein and interacts with RNAs in addition to 7SK in cultured cells. Nucleic Acids Res. 35, 2503–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sedore S. C., Byers S. A., Biglione S., Price J. P., Maury W. J., Price D. H. (2007) Manipulation of P-TEFb control machinery by HIV. Recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 35, 4347–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou M., Deng L., Kashanchi F., Brady J. N., Shatkin A. J., Kumar A. (2003) The Tat/TAR-dependent phosphorylation of RNA polymerase II C-terminal domain stimulates cotranscriptional capping of HIV-1 mRNA. Proc. Natl. Acad. Sci. U.S.A. 100, 12666–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou M., Deng L., Lacoste V., Park H. U., Pumfery A., Kashanchi F., Brady J. N., Kumar A. (2004) Coordination of transcription factor phosphorylation and histone methylation by the P-TEFb kinase during human immunodeficiency virus type 1 transcription. J. Virol. 78, 13522–13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerber M., Shilatifard A. (2003) Transcriptional elongation by RNA polymerase II and histone methylation. J. Biol. Chem. 278, 26303–26306 [DOI] [PubMed] [Google Scholar]
- 19. Shilatifard A., Conaway R. C., Conaway J. W. (2003) The RNA polymerase II elongation complex. Annu. Rev. Biochem. 72, 693–715 [DOI] [PubMed] [Google Scholar]
- 20. Sims R. J., 3rd, Belotserkovskaya R., Reinberg D. (2004) Elongation by RNA polymerase II. The Short and long of it. Genes Dev. 18, 2437–2468 [DOI] [PubMed] [Google Scholar]
- 21. Brès V., Gomes N., Pickle L., Jones K. A. (2005) A human splicing factor,SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1. Genes Dev. 19, 1211–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fong Y. W., Zhou Q. (2000) Relief of two built-In autoinhibitory mechanisms in P-TEFb is required for assembly of a multicomponent transcription elongation complex at the human immunodeficiency virus type 1 promoter. Mol. Cell. Biol. 20, 5897–5907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Q., Sharp P. A. (1996) Tat-SF1. Cofactor for stimulation of transcriptional elongation by HIV-1 Tat. Science 274, 605–610 [DOI] [PubMed] [Google Scholar]
- 24. Kameoka S., Duque P., Konarska M. M. (2004) p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. EMBO J. 23, 1782–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu J., Shimba S., Nomura N., Reddy R. (1998) Isolation and characterization of a new 110-kDa human nuclear RNA-binding protein (p110nrb). Biochim. Biophys. Acta 1399, 1–9 [DOI] [PubMed] [Google Scholar]
- 26. Bell M., Schreiner S., Damianov A., Reddy R., Bindereif A. (2002) P110, a novel human U6 snRNP protein and U4/U6 snRNP recycling factor. EMBO J. 21, 2724–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Medenbach J., Schreiner S., Liu S., Lührmann R., Bindereif A. (2004) Human U4/U6 snRNP recycling factor P110. Mutational analysis reveals the function of the tetratricopeptide repeat domain in recycling. Mol. Cell. Biol. 24, 7392–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawagoe N., Shintaku I., Yutani S., Etoh H., Matuoka K., Noda S., Itoh K. (2000) Expression of the {SART3} tumor rejection antigen in renal cell carcinoma. J. Urol. 164, 2090–2095 [PubMed] [Google Scholar]
- 29. Sasatomi T., Suefuji Y., Matsunaga K., Yamana H., Miyagi Y., Araki Y., Ogata Y., Itoh K., Shirouzu K. (2002) Expression of tumor rejection antigens in colorectal carcinomas. Cancer 94, 1636–1641 [DOI] [PubMed] [Google Scholar]
- 30. Yang D., Nakao M., Shichijo S., Sasatomi T., Takasu H., Matsumoto H., Mori K., Hayashi A., Yamana H., Shirouzu K., Itoh K. (1999) Identification of a gene Coding for a Protein Possessing Shared Tumor Epitopes Capable of Inducing HLA-A24-restricted Cytotoxic T Lymphocytes in Cancer Patients. Cancer Res. 59, 4056–4063 [PubMed] [Google Scholar]
- 31. Liu Y., Kim B. O., Kao C., Jung C., Dalton J. T., He J. J. (2004) Tip110, the human immunodeficiency virus type 1 (HIV-1) Tat-interacting protein of 110 kDa as a negative regulator of androgen receptor (AR) transcriptional activation. J. Biol. Chem. 279, 21766–21773 [DOI] [PubMed] [Google Scholar]
- 32. Liu Y., Li J., Kim B. O., Pace B. S., He J. J. (2002) HIV-1 Tat protein-mediated transactivation of the HIV-1 long terminal repeat promoter is potentiated by a novel nuclear Tat-interacting protein of 110 kDa, Tip110. J. Biol. Chem. 277, 23854–23863 [DOI] [PubMed] [Google Scholar]
- 33. Suñé C., Goldstrohm A. C., Peng J., Price D. H., Garcia-Blanco M. A. (2000) An in vitro transcription system that recapitulates equine infectious anemia virus tat-mediated inhibition of human immunodeficiency virus type 1 Tat activity demonstrates a role for positive transcription elongation factor b and associated proteins in the mechanism of Tat activation. Virology 274, 356–366 [DOI] [PubMed] [Google Scholar]
- 34. Brockman M. A., Tanzi G. O., Walker B. D., Allen T. M. (2006) Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J. Virol. Methods 131, 134–142 [DOI] [PubMed] [Google Scholar]
- 35. Herrmann C. H., Mancini M. A. (2001) The Cdk9 and cyclin T subunits of TAK/P-TEFb localize to splicing factor-rich nuclear speckle regions. J. Cell Sci. 114, 1491–1503 [DOI] [PubMed] [Google Scholar]
- 36. Zhu Y., Pe'ery T., Peng J., Ramanathan Y., Marshall N., Marshall T., Amendt B., Mathews M. B., Price D. H. (1997) Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 11, 2622–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim Y. K., Bourgeois C. F., Pearson R., Tyagi M., West M. J., Wong J., Wu S. Y., Chiang C. M., Karn J. (2006) Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 25, 3596–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen D., Zhou Q. (1999) Tat activates human immunodeficiency virus type 1 transcriptional elongation independent of TFIIH kinase. Mol. Cell. Biol. 19, 2863–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Y., Lee M. R., Timani K., He J. J., Broxmeyer H. E. (2012) Tip110 maintains expression of pluripotent factors in and pluripotency of human embryonic stem cells. Stem Cells Dev. 21, 829–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu Y., Timani K., Ou X., Broxmeyer H. E., He J. J. (2013) C-MYC controlled TIP110 protein expression regulates OCT4 mRNA splicing in human embryonic stem cells. Stem Cells Dev. 22, 689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montanuy I., Torremocha R., Hernández-Munain C., Suñé C. (2008) Promoter influences transcription elongation. TATA-box element mediates the assembly of processive transcription complexes responsive to cyclin-dependent kinase 9. J. Biol. Chem. 283, 7368–7378 [DOI] [PubMed] [Google Scholar]
- 42. Baillat D., Hakimi M. A., Näär A. M., Shilatifard A., Cooch N., Shiekhattar R. (2005) Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276 [DOI] [PubMed] [Google Scholar]
- 43. Vardabasso C., Manganaro L., Lusic M., Marcello A., Giacca M. (2008) The histone chaperone protein nucleosome assembly protein-1 (hNAP-1) binds HIV-1 Tat and promotes viral transcription. Retrovirology 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carty S. M., Goldstrohm A. C., Suñé C., Garcia-Blanco M. A., Greenleaf A. L. (2000) Protein-interaction modules that organize nuclear function. FF domains of CA150 bind the phosphoCTD of RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 97, 9015–9020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blatch G. L., Lässle M. (1999) The tetratricopeptide repeat. A structural motif mediating protein-protein interactions. BioEssays 21, 932–939 [DOI] [PubMed] [Google Scholar]
- 46. Harada K., Yamada A., Yang D., Itoh K., Shichijo S. (2001) Binding of a SART3 tumor-rejection antigen to a pre-mRNA splicing factor RNPS1. A possible regulation of splicing by a complex formation. Int. J. Cancer 93, 623–628 [DOI] [PubMed] [Google Scholar]
- 47. He N., Liu M., Hsu J., Xue Y., Chou S., Burlingame A., Krogan N. J., Alber T., Zhou Q. (2010) HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38, 428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rougvie A. E., Lis J. T. (1988) The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell 54, 795–804 [DOI] [PubMed] [Google Scholar]
- 49. Krumm A., Meulia T., Brunvand M., Groudine M. (1992) The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 6, 2201–2213 [DOI] [PubMed] [Google Scholar]
- 50. Roberts S., Purton T., Bentley D. L. (1992) A protein-binding site in the c-myc promoter functions as a terminator of RNA polymerase II transcription. Genes Dev. 6, 1562–1574 [DOI] [PubMed] [Google Scholar]
- 51. Zhou M., Halanski M. A., Radonovich M. F., Kashanchi F., Peng J., Price D. H., Brady J. N. (2000) Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol. Cell. Biol. 20, 5077–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gervaix A., West D., Leoni L. M., Richman D. D., Wong-Staal F., Corbeil J. (1997) A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. U.S.A. 94, 4653–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sundaravaradan V., Das S. R., Ramakrishnan R., Sehgal S., Gopalan S., Ahmad N., Jameel S. (2007) Role of HIV-1 subtype C envelope V3 to V5 regions in viral entry, coreceptor utilization and replication efficiency in primary T-lymphocytes and monocyte-derived macrophages. Virol. J. 4, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]