

Background: Nicotinic acetylcholine receptor trafficking is governed by compartment-specific molecular signals.
Results: Novel motifs in the muscle β/δ subunit cytoplasmic loops mediate Golgi retention and recovery from the plasma membrane.
Conclusion: The β/δ motifs regulate surface trafficking of assembled receptor and retain unassembled subunit loops.
Significance: This work identifies a novel Golgi-based regulatory step in nicotinic receptor trafficking.
Keywords: Golgi, Nicotinic Acetylcholine Receptors, Protein Motifs, Skeletal Muscle, Trafficking, Neuromuscular Junction
Abstract
The molecular determinants that govern nicotinic acetylcholine receptor (AChR) assembly and trafficking are poorly defined, and those identified operate largely during initial receptor biogenesis in the endoplasmic reticulum. To identify determinants that regulate later trafficking steps, we performed an unbiased screen using chimeric proteins consisting of CD4 fused to the muscle AChR subunit cytoplasmic loops. In C2 mouse muscle cells, we found that CD4-β and δ subunit loops were expressed at very low levels on the cell surface, whereas the other subunit loops were robustly expressed on the plasma membrane. The low surface expression of CD4-β and δ loops was due to their pronounced retention in the Golgi apparatus and also to their rapid internalization from the plasma membrane. Both retention and recovery were mediated by the proximal 25–28 amino acids in each loop and were dependent on an ordered sequence of charged and hydrophobic residues. Indeed, βK353L and δK351L mutations increased surface trafficking of the CD4-subunit loops by >6-fold and also decreased their internalization from the plasma membrane. Similarly, combined βK353L and δK351L mutations increased the surface levels of assembled AChR expressed in HEK cells to 138% of wild-type levels. This was due to increased trafficking to the plasma membrane and not decreased AChR turnover. These findings identify novel Golgi retention signals in the β and δ subunit loops that regulate surface trafficking of assembled AChR and may help prevent surface expression of unassembled subunits. Together, these results define molecular determinants that govern a Golgi-based regulatory step in nicotinic AChR trafficking.
Introduction
Nicotinic acetylcholine receptors (nAChRs)2 are pentameric, ligand-gated ion channels belonging to the superfamily that includes GABA, glycine, and 5-hydroxytryptamine receptors. In the peripheral nervous system, nAChRs mediate fast synaptic transmission at neuromuscular and autonomic ganglionic synapses, and their dysfunction results in human neurological disorders including myasthenia gravis (1) and congenital myasthenic syndrome (2). In the CNS, nAChRs modulate transmitter release and neuronal excitability and have been implicated in a variety of neurological disorders including autosomal dominant nocturnal frontal lobe epilepsy, Alzheimer disease, Parkinson disease, and schizophrenia, as well as in nicotine addiction (for review, see Refs. 3–5).
Nicotinic AChRs occur in multiple subtypes formed from different combinations of subunits that convey different pharmacological and physiological properties. The muscle AChR is a hetero-pentamer composed of 2 α, and one β, δ, and γ (fetal) or ϵ (adult) subunits. Neuronal receptors are either homo-pentamers composed of α7, α8, or α9 subunits or hetero-pentamers composed of α2-α6 and β2-β4 subunits. All subunits are homologous and have a large extracellular N-terminal domain, four transmembrane domains, and a large cytoplasmic loop between transmembrane domains 3 and 4 that is thought to contain determinants for trafficking and targeting of the receptor (3, 6).
The function of nAChRs in muscle or neurons depends on their appropriate assembly and trafficking to the plasma membrane. Individual subunits are synthesized and assembled in an ordered fashion into receptor in the endoplasmic reticulum (ER). This is a surprisingly inefficient process, with only 10–30% of the synthesized subunits being incorporated into surface nAChR in muscle or neurons (6, 7). Only pentameric receptor is exported from the ER, however, and unassembled subunits and partially assembled intermediates are retained and degraded by the ER-associated degradation (ERAD) machinery (8). This discrimination process is accomplished by trafficking signals in the AChR subunits that prevent surface expression of assembly intermediates, which could decrease the pool of subunit available for assembly and uncouple transmitter binding from channel function (9). Most notably, a retention motif located in the first transmembrane domain of each subunit retains unassembled subunits in the ER but becomes buried in receptor pentamers, allowing export (10). Additional signals in the major cytoplasmic loop, including a dibasic retention motif (11) and paired hydrophobic residues that are required for export (12), likely provide additional quality control. The ER components that bind these motifs are not yet identified, although the sorting receptor Rer1 has been implicated in retention of unassembled α subunits (9), and several other ER proteins have been identified that promote expression of different nAChR subtypes, including RIC-3 (13), VILIP-1 (14) and UBXD4 (15).
After ER export, AChRs are trafficked to the Golgi complex for further processing, including modification of N-linked oligosaccharides on the γ and δ subunits to more complex forms (16), and then are sorted and delivered to the plasma membrane. The molecular determinants in the AChR that govern these later steps in the secretory pathway are unknown, but several findings suggest that Golgi trafficking is also regulated and may contribute to quality control. First, mutation of the dibasic motif in the α1 subunit cytoplasmic loop allows some unassembled subunits to be trafficked from the ER to the Golgi, but they are then retained in the Golgi by an unknown mechanism (11). Second, in Caenorhabditis elegans, the Golgi-resident protein, unc-50, is required for trafficking of one subtype of nAChR to the neuromuscular junction (17). Third, a significant intracellular pool of mature, assembled AChR has been detected in both muscle cells and neurons (3, 18). Consequently, the Golgi complex could constitute a second checkpoint in AChR biogenesis and trafficking, which further monitors the composition and processing of nAChRs and regulates their delivery to the plasma membrane.
Here, using the mouse muscle AChR as a model, we performed an unbiased screen to identify molecular determinants for post-ER trafficking of the AChR. We identify a novel signal in the β and δ subunit loops that mediates Golgi retention, and mutation of this motif permits surface expression of unassembled subunit loops and increases surface levels of assembled receptor. We propose that this Golgi retention motif regulates AChR trafficking to the plasma membrane and that it may also contribute to quality control along with ER-based mechanisms.
EXPERIMENTAL PROCEDURES
CD4-Subunit Loop Constructs
Chimeric constructs consisting of mouse CD4 extracellular and transmembrane domains fused with the major cytoplasmic loop of each mouse nAChR subunit were generated as described previously (19). Briefly, a BglII site was introduced by site-directed mutagenesis at the end of the transmembrane domain of mouse CD4. The intracellular domain of CD4 was then excised, and PCR fragments comprising each of the acetylcholine receptor subunit loops or intracellular loop regions were ligated into this site. Mutations in the intracellular loops were introduced using the QuikChange Lightning site-directed mutagenesis kit (Stratagene). All CD4-subunit loop chimeras were expressed using the pcDNA3 vector and confirmed by sequencing.
Cell Culture, Transfection, and Immunostaining
C2 mouse muscle cells were maintained in growth medium (DMEM supplemented with 20% fetal bovine serum, 0.5% chick embryo extract, 2 mm l-glutamine, and 200 units/ml penicillin-streptomycin) at 37 °C and 8% CO2. For immunostaining experiments, myoblasts were grown on 8-well chamber slides (Nalgene Nunc, Inc.) and transfected at ∼60–70% confluence using FuGENE (Roche Applied Science). Upon reaching confluence the cells were incubated with fusion medium (DMEM supplemented with 5% horse serum and 2 mm l-glutamine) and allowed to differentiate into myotubes for 3–4 days prior to analysis (19).
To assay cellular localization, C2 myotubes transfected with each CD4-subunit chimera were fixed in 2% paraformaldehyde/PBS, blocked, and incubated with anti-CD4 antibody H129.19 (BD Biosciences) and Alexa Fluor 488-conjugated anti-rat secondary antibody to label surface CD4 chimeras. The cells were then washed, permeabilized with 0.5% Triton X-100 for 10 min, blocked, and incubated with anti-CD4 antibody and Alexa Fluor 594-conjugated secondary antibody to label intracellular chimeras. In control immunostaining and on-cell Western experiments, we did not detect intracellular proteins without this permeabilization step.
To define the cellular compartment involved in retention, transfected myotubes were permeabilized and co-labeled for CD4 (rat anti-CD4 (H129.19) or goat anti-rmCD4 (R&D Systems) and specific markers for the ER (mouse anti-protein disulfide isomerase, clone 1D3, Enzo Life Sciences, Cambridge, MA) and Golgi compartments (rabbit anti-giantin, clone ab24586, Abcam, Farmingdale, NY). Laser scanning confocal microscopy was performed using an Olympus Fluoview Scanning Confocal Microscope (Olympus America, Center Valley, PA). Digital images were processed with Adobe Photoshop (Adobe Systems Inc., San Jose, CA).
To assay internalization from the plasma membrane, we transfected C2 muscle cells in duplicate with each CD4 subunit loop chimera, and after 3-day expression, incubated the live cells with rat anti-CD4 antibody (H129.19) at 4 °C for 10 min. After washing in growth media to remove unbound antibody, one set of cultures was fixed immediately with 2% paraformaldehyde, blocked, and incubated with Alexa Fluor 594-anti-rat secondary antibody to label surface CD4 chimeras. The second set was incubated for 15 min at 37 °C to allow endocytosis to occur before fixing, permeabilizing with 0.5% Triton X-100, and labeling with Alexa Fluor 594-secondary antibody; this labeled both surface and internalized chimeras. To confirm internalization, transfected myotubes were labeled sequentially for surface and internalized chimeras. After labeling with CD4 antibody at 4 °C and incubation at 37 °C for 15 min, the cells were fixed with 2% paraformaldehyde and incubated with Alexa Fluor 488-secondary antibody to detect surface chimeras. Then, the cells were washed, permeabilized with 0.5% Triton-X, and incubated with Alexa Fluor 594-secondary antibody to detect internalized chimeras. In addition, we performed acid stripping experiments, where the transfected myotubes were labeled with anti-CD4 antibody as above, but were then treated with 0.5 m NaCl/0.2 m acetic acid (pH 2.5) for 4 min at 4 °C prior to fixation to remove surface-bound antibody (20, 21).
The labeled culture slides were viewed with a Zeiss Axioplan 2 IE fluorescence microscope and Plan-Apo 40× objective, and quantified in two ways. First, in random fields, we counted the number of transfected myotubes that showed mostly punctuate intracellular staining rather than diffuse surface staining and expressed this as a percentage of total transfected myotubes (20 random fields/experiment). This included myotubes with both high and low expression levels of the CD4-subunit loops. Second, digital images were acquired using an Axiocam MRM camera and Axiovision software, with automatic exposure times. Then, in blinded fashion, we counted the number of puncta within an 8 × 8-μm region centered in the brightest area of each transfected myotube. Objects were defined as puncta by their circular shape and intensity compared with the surrounding region and size (<1 μm). Data were collected from 2–5 independent experiments for each CD4 chimeric construct.
On-cell Western Assays and Immunoblotting
C2 muscle cells were grown on 8-well chamber slides and transfected in duplicate with each CD4-subunit loop chimera. After 3–4 days for expression the cells were fixed, blocked, and incubated with anti-CD4 antibody, either with or without permeabilization, to detect total and surface CD4-subunit loops, respectively. Bound antibodies were detected using IRDye-conjugated anti-rat secondary antibodies and an Odyssey Imaging System (LI-COR). Total signal intensity was measured for each well and the percentage of surface (nonpermeabilized) versus total (permeabilized) expression calculated for each CD4-subunit loop chimera. Data were collected from 3–6 independent experiments for each construct.
To assay surface and intracellular pools of AChR, HEK cells were grown on 10-cm dishes and transfected with wild-type or mutant AChR subunits using the CaP method. After 1 day for expression, the cells were incubated live with biotinylated α-bungarotoxin (α-BuTx) for 45 min to label surface AChR, and then washed, collected, and extracted in buffer containing 0.5% Triton X-100, 25 mm Tris, 25 mm glycine, 150 mm NaCl, 5 mm EDTA, and Halt protease inhibitor mixture (ThermoScientific). First, biotin-α-BuTx-labeled surface receptor was isolated from the extracts using streptavidin beads (Invitrogen). Then, unlabeled intracellular AChR was isolated from the remaining supernatant by reincubation with biotin-α-BuTx and pulldown on streptavidin beads. The samples were separated on 10% polyacrylamide gels (14 × 14 cm) and immunoblotted with anti-β subunit antibody (mAb148). Bound antibodies were detected using IRDye-conjugated anti-rat secondary antibody, imaged with an Odyssey Imaging System, and band intensities were quantified using ImageStudio (LI-COR). The percentage of receptor in the surface and intracellular pools was calculated from 4 independent experiments.
Assays of AChR Surface Levels and Turnover
Heterologous COS and HEK cells were maintained in growth medium (DMEM-HI supplemented with 10% FBS and 100 units/ml penicillin/streptomycin) at 37 °C and 5% CO2. To assay levels of surface AChR, HEK cells were grown on 6-well plates and transfected with pcDNA3 plasmids encoding the mouse AChR subunits using X-tremeGene (Roche Applied Science). After 1 day for expression, the cells were labeled with 10 nm 125I-labeled-α-BuTx (PerkinElmer Life Sciences) for 45 min. Nonspecific binding was determined by treating myotubes with 1 μm cold α-BuTx for 30 min prior to incubation with 125I-α-BuTx. Cells were then washed three times with growth medium to remove unbound 125I-α-BuTx, solubilized in 0.1 n NaOH, and the 125I-α-BuTx bound to surface AChR was measured with a Packard gamma counter. Background counts were subtracted from the experimental counts, and values are reported as a percentage of the total surface counts for cells transfected with wild-type AChR.
For receptor turnover experiments, COS cells were grown on 6-cm dishes and transfected in triplicate with wild-type or mutant AChR. Surface AChR was labeled with 125I-α-BuTx (as above), and its degradation was followed over time as described in Refs. 22–24. Briefly, as 125I-α-BuTx-labeled AChR degrades, free 125I accumulates in the culture medium. This was measured in samples collected at 5, 19.5, and 27.5 h, along with the amount of labeled receptor remaining on the cells at 27.5 h. The total amount of labeled AChR at the beginning of the experiment was calculated by adding the medium and cell counts, and the percentage remaining at each time point was graphed on a semilog plot. Degradation curves were fitted by linear regression, and half-lives were calculated for each experiment and then averaged (n = 5 independent experiments). Receptor turnover experiments were also performed on HEK cells, with similar results.
RESULTS
To identify molecular determinants for AChR trafficking we performed an unbiased screen using chimeric constructs consisting of mouse CD4 extracellular and transmembrane domains fused with the major cytoplasmic loop of each mouse muscle nAChR subunit (19) (Fig. 1A). The CD4-loop chimeras do not assemble with endogenous AChR subunits in muscle cells (19) and lack a previously identified ER retention motif in the first transmembrane domain of each subunit (10). Thus, they allow us to test each subunit cytoplasmic loop for molecular signals that act in later, post-ER trafficking steps. For this, we transfected mouse C2 muscle cells with each CD4-subunit loop chimera. After 3 days for expression, the differentiated myotubes were fixed with 2% paraformaldehyde and immunostained for surface CD4 chimeras with anti-CD4 antibody and Alexa Fluor 488-conjugated secondary antibody; the cells were then permeabilized with detergent and immunostained for intracellular chimeras with anti-CD4 antibody and Alexa Fluor 594-secondary antibody. Whereas CD4 and CD4-α, γ, and ϵ loops were robustly expressed on the myotube surface, we found that CD4-β and δ loops were mostly retained intracellularly and detectable only after permeabilization (Fig. 1B). To confirm this we used an on-cell Western blot assay (see “Experimental Procedures”) to quantify surface versus total expression for each CD4-subunit loop chimera (Fig. 1C). This revealed that a high percentage of CD4 and CD4-ϵ loop (>70%) and moderate percentage of CD4-α and γ loops (30–37%) were trafficked to the cell surface. In contrast, only 6% of CD4-β and 0.3% of CD4-δ loop were detectable on the plasma membrane. This suggests that the β and δ subunit cytoplasmic loops contain retention signals that largely prevent their surface expression.
FIGURE 1.

Cellular localization of CD4-AChR subunit loops in muscle cells. A, schematic shows the subunit composition of muscle AChR, the topology of each subunit, and the CD4-subunit cytoplasmic loop chimeras used to screen for post-ER trafficking signals. B, C2 myotubes transfected with CD4-subunit loops were fixed and immunostained for surface CD4 chimeras with anti-CD4 antibody and Alexa Fluor 488-conjugated secondary antibody (green). The cells were then permeabilized and immunostained for intracellular chimeras with anti-CD4 and Alexa Fluor 594 secondary antibody (red). CD4 and CD4-α and ϵ loops were expressed on the cell surface, whereas CD4-β and δ loops were retained intracellularly. C, quantification of surface expression of each CD4-subunit loop was done by on-cell Western blot assays. A high percentage of CD4 and CD4-ϵ loops and moderate percentage of CD4-α and γ loops were expressed on the cell surface. In contrast, only 6% of CD4-β and 0.3% of CD4-δ loops were detectable on the plasma membrane; thus, they are almost exclusively localized in the intracellular compartment.**, p < 0.01 compared with CD4 and CD4-ϵ; #, p < 0.05 versus CD4-α; ANOVA with Tukey's post hoc test. Error bars, S.E.
In addition, we found that the intracellular localization of CD4-β and δ loops differed from that of the other CD4-subunit loops. CD4 and CD4-α, γ, ϵ loops exhibited a relatively diffuse intracellular distribution, consistent with normal trafficking through the ER and Golgi to the cell surface (Fig. 1B). In contrast CD4-β and δ loops showed a more punctate, restricted localization. To identify this cellular compartment we performed immunofluorescence microscopy using a laser scanning confocal microscope (Fig. 2). Both CD4-β and δ-loop chimeras localized to discrete, punctate/reticular, cytoplasmic structures that tended to encircle the nucleus (Fig. 2, A and B), a morphology and distribution characteristic of the Golgi complex in differentiated muscle fibers as well as cultured myotubes (16, 25). Indeed, double labeling with antibodies to the Golgi scaffolding protein giantin (Fig. 2A) revealed extensive overlap between CD4-β and δ loops and the Golgi marker, but no overlap for the CD4 control. In addition, we observed minimal overlap between CD4-β and δ loop chimeras and the endoplasmic reticulum marker protein disulfide isomerase (Fig. 2B). CD4-δ loop had a slightly more diverse subcellular distribution to CD4-β loop, in that it also localized to small puncta that did not co-localize with Golgi markers. From these results we conclude that CD4-β and δ loop chimeras localize principally to the Golgi complex and not the ER. Thus, the low surface expression of CD4-β and δ loops is due, at least in part, to selective retention in the Golgi apparatus, identifying novel trafficking signals specific to the β and δ cytoplasmic loops.
FIGURE 2.

CD4-β and δ cytoplasmic loops are retained mainly in the Golgi complex. A, C2 myotubes transfected with CD4 and CD4-β or δ loops were fixed, permeabilized, and double-stained with anti-CD4 antibody and an anti-giantin antibody to stain the Golgi complex. CD4 exhibited a diffuse intracellular and cell surface distribution, with little overlap with the giantin staining. In contrast, both CD4-β and δ loops exhibited extensive co-localization with the Golgi marker (see merged images). B, transfected myotubes were immunostained with anti-CD4 and an anti-protein disulfide isomerase (PDI) antibody to stain the endoplasmic reticulum. CD4-β or δ loops showed minimal co-localization with the ER marker. All confocal images are centered on a pair of nuclei within the multinucleate myotubes.
Another process that could contribute to the low surface expression of CD4-β and δ loops is selective endocytosis from the plasma membrane. To test this, we transfected C2 muscle cells in duplicate with each CD4-subunit loop chimera, and after 3 days for expression, we incubated the live cells with anti-CD4 antibody at 4 °C. After a brief wash to remove unbound antibody, one set of cultures was fixed immediately with 2% paraformaldehyde, blocked, and incubated with Alexa Fluor 594-secondary antibody to label surface CD4 chimeras. The second set was incubated for 15 min at 37 °C to allow endocytosis to occur before fixing, permeabilizing with 0.5% Triton X-100, and labeling with secondary antibody; this labeled both surface and internalized chimeras. For CD4 or CD4-α, γ, or ϵ loops, we observed similar patterns of staining in both conditions (Fig. 3A), indicating that most CD4 chimeras remained on the cell surface and were not internalized within the 15-min period. In contrast, we found prominent endocytosis of CD4-β and δ loops during the 15 min at 37 °C, with most labeling in permeabilized cultures being associated with punctate vesicular structures (Fig. 3A). The contrasting immunostaining patterns for CD4-β and δ compared with the other chimeras were observed regardless of the expression levels in individual myotubes. Moreover, the CD4-β and δ loop puncta were intracellular, as they were only observed after permeabilization in sequential labeling experiments (see “Experimental Procedures”). Indeed, >90% of myotubes expressing CD4-β or δ loops showed prominent endocytosis compared with 20% of myotubes expressing CD4-α loop and <1% of myotubes expressing CD4 or CD4-γ or ϵ loops (Fig. 3B; p < 0.01; n = 3–4; ANOVA with Tukey's post hoc test). Similarly, the number of CD4-labeled puncta/unit area was significantly higher in myotubes expressing CD4-β and δ loop, compared with CD4 or CD4-γ or ϵ loop (Fig. 3B).
FIGURE 3.
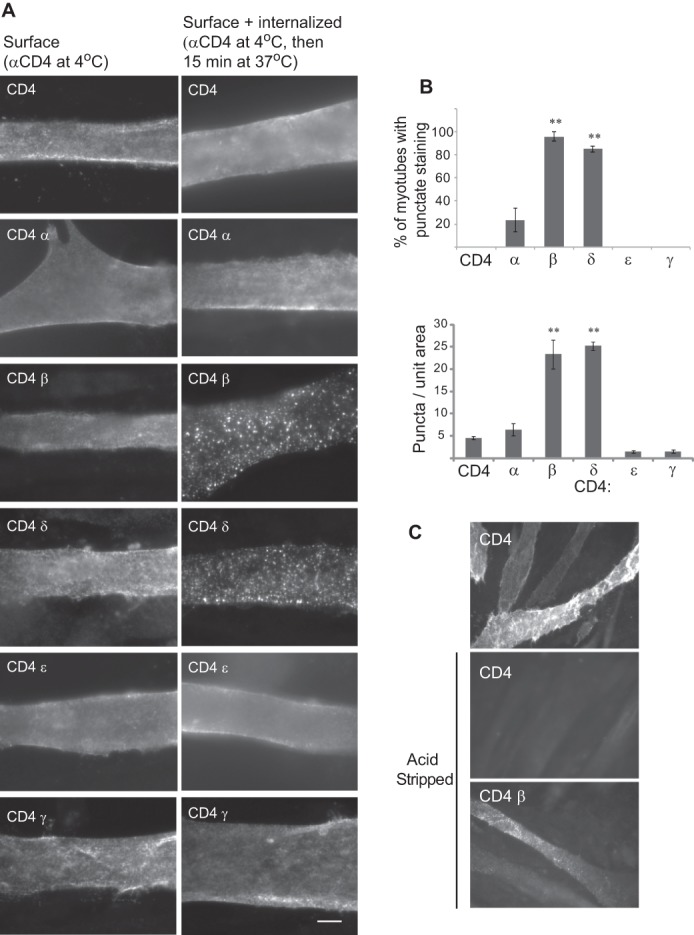
CD4-β and δ cytoplasmic loops are rapidly internalized from the plasma membrane. A, C2 myotubes were transfected in duplicate with CD4-subunit loop chimeras and surface-labeled with anti-CD4 antibody, live at 4 °C. One set was fixed immediately and labeled with secondary antibody (Surface). This showed diffuse surface labeling for all CD4 chimeras. The second set was incubated at 37 °C for 15 min prior to fixation, permeabilization, and labeling with a secondary antibody (Surface + internalized). This showed diffuse surface staining for CD4 and CD4-α, ϵ, and γ loops but punctate, vesicular-like staining for CD4-β and δ loops. All images were acquired with automatic exposure times, which were longer for surface CD4-β and δ loops due to their low expression. Scale bar, 10 μm. B, quantification of the percentage of transfected myotubes with punctate immunostaining (top) and the number of puncta per unit area of myotube (bottom). Both measures demonstrate striking endocytosis of CD4-β and δ loops compared with CD4 or CD4-α, ϵ, and γ loops (**, p < 0.01, ANOVA with Tukey's post hoc test). Error bars, S.E. C, C2 myotubes expressing CD4 or CD4-β loop chimeras labeled with anti-CD4 antibody at 4 °C and incubated at 37 °C for 15 min. Surface-bound antibodies were then removed by acid stripping prior to cell fixation, permeabilization, and labeling with secondary antibody. Acid stripping efficiently removed surface labeling for CD4, but not the punctate labeling for CD4-β loop, confirming that it was internalized from the cell surface.
To further confirm internalization of CD4-β or δ loop chimeras we performed acid stripping experiments. Transfected myotubes were labeled with anti-CD4 antibody at 4 °C, incubated at 37 °C for 15 min to allow internalization, and then chilled on ice and incubated with 0.5 m NaCl/0.2 m acetic acid for 4 min. This procedure selectively strips bound antibody from the cell surface while leaving intracellular antibody-receptor complexes intact (20, 21). The cells were then fixed and permeabilized prior to incubating with Alexa Fluor 594-secondary antibody to detect internalized CD4-β loop. Acid stripping effectively removed anti-CD4 antibodies from the surface of CD4-expressing myotubes, confirming the efficiency of this procedure (Fig. 3C). CD4-β chimera, however, was readily detected after acid stripping and subsequent fixation and permeabilization, confirming that it was endocytosed (Fig. 3C). Taken together, these findings demonstrate that CD4-β and δ loop chimeras that reach the cell surface are rapidly internalized, whereas CD4 and CD4-α, γ, and ϵ loops are much more stably expressed on the plasma membrane.
Our findings suggest that β and δ subunit loops are selectively retained in the Golgi and recovered from the plasma membrane, indicating that both processes may be mediated by the same molecular determinants. To define these determinants, we first used deletion constructs to map the internalization signals in the β and δ loops. As shown in Fig. 4, we found prominent endocytosis of the proximal region of β and δ loops, but not of more C-terminal loop fragments. Progressive deletions then localized the internalization signal to β loop amino acids 333–360, and δ loop amino acids 337–359, with further truncations abolishing endocytosis (Fig. 4). In contrast, mutation of conserved residues in the membrane proximal region of the δ sequence (δP341A/S342A/T343A) had no significant effect on internalization. This identifies minimal 25–28 amino acid sequences that are sufficient for endocytosis and suggests that the C-terminal residues in each are critical determinants in the motif. Consistent with this, we found that transfer of β loop 353–369 to the analogous position in ϵ loop conferred endocytosis to CD4-ϵ, which otherwise was stably expressed on the plasma membrane (Fig. 3A).
FIGURE 4.

Internalization is mediated by signals in the proximal β and δ loops. A, C2 myotubes transfected with CD4-β loop deletion constructs were surface-labeled with anti-CD4 antibody and incubated at 37 °C for 15 min. Proximal β loop fragments containing amino acids 333–360 were rapidly endocytosed, whereas more C-terminal segments of the loop were not. Further truncations (β(333–356)) blocked endocytosis, and substitution of β(353–369) into the analogous position in the ϵ loop conferred internalization. Scale bar, 10 μm. B, quantification of the percentage of transfected myotubes with punctate immunostaining (top) and the number of puncta per unit area of myotube (bottom) (**, p < 0.01, compared with CD4 control or CD4-β(370–406), β(407–469), and β(333–356); ANOVA with Tukey's post hoc test, n = 3–5 experiments for each construct). Error bars, S.E. These experiments identify β loop amino acids 333–360 as the minimal sequence sufficient for endocytosis. C, schematic of β loop and summary of the experiments mapping the internalization signal. D, analogous experiments with CD4-δ loop. Fragments containing amino acids 337–359 were rapidly internalized, whereas more C-terminal segments of the loop were not. Further truncation to δ(337–353) abolished endocytosis. E and F, quantification (E) and summary (F) identifying δ loop amino acids 337–359 as the minimal sequence sufficient for internalization from the plasma membrane.
The β and δ loop regions that mediate internalization are well conserved across species and share some sequence homology but no common endocytic motifs. Although the β sequence contains a putative YXXL motif for clathrin-mediated endocytosis (26), mutating these residues did not inhibit internalization (βY357A/L360A; Fig. 5, C and D), and the δ sequence does not contain either tyrosine- or dileucine-based endocytic signals. Consequently, we carried out a mutational analysis to define the critical determinants for endocytosis. Mutation of several conserved residues in β and δ failed to inhibit internalization (e.g. βP355A, βR362A/P363A, δF354A, δL358A/P359A). However, portions of both β and δ sequences are predicted to form α-helices, with similar, partially amphipathic structures (Jpred, psipred, and Heliquest secondary structure prediction programs; Fig. 5E). Therefore, we also tested mutations designed to alter the alignment of charged and hydrophobic residues, without disrupting the α-helical secondary structure. Notably, we found that K353L and L354K mutations dramatically reduced CD4-β endocytosis, as did single amino acid insertions or deletions at these positions (Fig. 5, A, C, and D). Similarly, the K351L mutation eliminated CD4-δ internalization (Fig. 5, B–D). These findings suggest that internalization of the β and δ subunit loops is mediated by a novel motif, with a possible α-helical secondary structure (Fig. 5E).
FIGURE 5.

Internalization is mediated by novel motifs that include lysine residues. A series of mutations was introduced in the proximal β and δ loop regions, and their internalization was assayed. A, C, and D, endocytosis of CD4-β(333–369) was decreased significantly by K353L and L354K mutations or by insertions (ins) or deletions (del) at these positions (**, p < 0.01 compared with β(333–369) wild type (WT), R347L, and P355A; ANOVA with Tukey's post hoc test). Error bars, S. E. In contrast, mutation of the putative YXXL motif (βY357A and L360A) did not inhibit internalization. B–D, endocytosis of CD4-δ(337–370) was inhibited by K351L and K354ins mutations. C, scale bar, 10 μm. E, sequence alignment of the proximal regions of β and δ loops is shown, with the minimal retention signals highlighted in gray, and asterisks marking the position of mutations that blocked internalization. For both subunits the critical determinants include lysine residues in predicted α-helixes (black box) with similar arrangements of hydrophobic and charged residues (helical wheel projection).
To test whether the same motif mediates intracellular retention in the Golgi complex we expressed CD4-β(333–369) and δ(337–370) in C2 muscle cells and assayed their cellular localization by immunostaining with anti-CD4 antibodies before and after permeabilization. As observed for the full β and δ loops, we found minimal surface expression of CD4-β(333–369) and δ(337–370) (Fig. 6, A and B). In both cases, they were retained largely intracellularly and exhibited a characteristic Golgi-like distribution. In addition, we found that βK353L and δK351L mutations resulted in significantly higher surface expression and increased the ratio of surface to intracellular labeling by >6-fold (Fig. 6, A and B). Similarly, in on-cell Western blot assays, we found that βK353L and δK351L mutations increased surface expression of CD4 chimeras containing the complete β and δ loops by >6-fold, as well as the proximal fragments (β(333–369) and δ(337–370)) by 2.4–3-fold (Fig. 6C). In all cases we observed higher surface expression for the proximal β and δ loops compared with the complete loops, suggesting that additional retention signals may exist in the C-terminal portion of these loops. To test whether the molecular machinery that recognizes the β and δ motifs is ubiquitous or specific to muscle cells, we repeated these experiments in HEK cells. As in muscle cells, we observed selective intracellular retention of CD4-β and δ loops, which was reduced substantially by βK351L and δK353L mutations (Fig. 6D). Surface expression increased with prolonged expression, however, suggesting that the retention mechanism is saturable in nonmuscle cells (data not shown). Next, we confirmed that the Golgi retention was due to arrest of forward trafficking in the Golgi complex rather than to internalization from the plasma membrane and retrograde trafficking to the Golgi. For this we expressed CD4-β(333–369) and δ(337–370) in C2 myotubes, labeled the surface chimeras live with anti-CD4 antibody, and then chased for variable periods (15 min to 2 h). Even with extended chase times (2 h), we found that internalized CD4-β(333–369) and δ(337–370) were localized in small punctate vesicular structures that had minimal overlap with the Golgi marker giantin (Fig. 6E), indicating that they are not trafficked back to the Golgi complex. Together, these results indicate that the β and δ subunits contain signals in their proximal cytoplasmic loops that mediate both Golgi retention and internalization from the plasma membrane. Moreover, retention and recovery are mediated by similar or identical sequence determinants that are recognized in both muscle and heterologous cells.
FIGURE 6.

Golgi retention is mediated by the same sequence determinants that mediate internalization. A, C2 muscle cells transfected with CD4-β(333–369) were immunostained sequentially for surface and intracellular CD4 chimeras. CD4-β(333–369) wild type (WT) was mostly retained intracellularly in the Golgi complex, whereas CD4-β(333–369) K353L was robustly expressed on the cell surface. B, quantification shows that βK353L and δK351L mutations resulted in a significantly higher ratio of surface to intracellular staining compared with WT (**, p < 0.01, ANOVA with Tukey's post hoc test). Error bars, S.E. C, C2 muscle cells were transfected with CD4-β and δ loop chimeras, and the percentage of each protein that was expressed on the cell surface was determined in on-cell Western blot assays. Like CD4-β and δ full loops, CD4-β(333–369) and δ(337–370) were expressed on the cell surface at much lower levels than CD4 (#, p < 0.01, ANOVA with Tukey's post hoc test). Moreover, surface expression was increased significantly by βK353L and δK351 mutations (**, p < 0.01; *, p < 0.05, ANOVA with Tukey's post hoc test, n = 3–6 independent experiments). D, in analogous experiments in HEK cells, CD4-β and δ loops were expressed on the cell surface at significantly lower levels than CD4 (#, p < 0.01, ANOVA with Tukey's post hoc test), and their surface expression was increased significantly by K353L and K351L mutations, respectively (**, p < 0.01, ANOVA with Tukey's post hoc test). Thus, the retention/retrieval signals are also recognized in heterologous cells. E, C2 muscle cells transfected with CD4-β(333–369) or δ(337–370) were labeled with anti-CD4 antibody live for 10 min and then chased for 2 h. Co-staining for CD4 and giantin shows that the internalized CD4-β and δ chimeras remain in punctate-vesicular structures and do not traffic back to the Golgi complex. Together, these findings show that the β and δ subunit proximal loops mediate both Golgi retention and retrieval from the plasma membrane, via similar sequence determinants.
Finally, we tested the role of the β/δ subunit loop retention/recovery motifs in trafficking of assembled AChR. First, we expressed the AChR in HEK cells, either in wild-type form, or with βK353L and δK351L subunit mutations, and then measured surface levels of receptor by 125I-α-BuTX binding (Fig. 7A). Notably, AChR with combined mutations of the β and δ subunit motifs (βK353L and δK351L) was expressed at significantly higher levels on the cell surface than wild-type AChR (138% of WT levels, p = 0.02, t test, n = 4). Receptor with single β or δ subunit mutations was expressed at intermediate levels. These results indicate that the β and δ motifs are accessible and active in the assembled AChR and regulate its surface expression. To test whether this is due to increased trafficking to the plasma membrane, we sequentially isolated surface and intracellular AChR and then immunoblotted with anti-β subunit antibody (Fig. 7B). Compared with wild type, we found more βK353L/δK351L-AChR on the cell surface and less in the intracellular pool. Indeed, the percentage of AChR in the intracellular pool was only 15±5% for βK353L/δK351L-AChR, compared with 26±6% for WT-AChR (Fig. 7C; p = 0.02, t test, n = 4 independent experiments). In addition, we compared the turnover rate of wild-type and mutant AChR on the cell surface by labeling receptor with 125I-α-BuTx and following its degradation over time. Interestingly, we found no difference in the degradation rate of βK353L/δK351L-AChR compared with wild-type AChR (Fig. 7D), indicating that the β/δ motifs do not regulate basal turnover of surface receptor. Thus, mutation of the motifs increases surface levels of AChR by up-regulating receptor trafficking to the plasma membrane rather than down-regulating receptor turnover.
FIGURE 7.

Mutations in the retention motif increase surface trafficking of assembled AChR. A, HEK cells were transfected with wild-type AChR or receptor with single or combined βK353L and δK351L mutations, and surface levels of receptor were measured by 125I-labeled α-BuTx binding. Receptor with combined βK353 and δK351 mutations was expressed on the cell surface at significantly higher levels than wild-type AChR (138% of WT levels, p = 0.02, t test, n = 4). Smaller increases were observed with individual mutations of the β or δ subunits. Error bars, S.E. B, surface and intracellular AChR were isolated sequentially from HEK cells transfected with wild-type (WT) or βK353L/δK351L (K*)-AChR and from control C2 muscle cells. The isolates were then immunoblotted with anti-β subunit antibody (mAb148) to compare receptor levels. Compared with WT-AChR, more βK353L/δK351L-AChR was present in the surface pool and less in the intracellular pool. C, quantification of the immunoblotting experiments shows the relative percentages of surface and intracellular AChR. A significantly higher percentage of total βK353L/δK351L-AChR was expressed on the cell surface compared with WT-AChR (p = 0.02; t test, n = 4), consistent with decreased Golgi retention. D, wild-type and βK353L/δK351L-AChR were surface-labeled with 125I-α-BuTx, and receptor degradation was measured over time as described under “Experimental Procedures.” The rate of receptor degradation is similar for wild-type and mutant AChR (n = 5). Thus, higher surface levels of βK353L/δK351L-AChR stem from increased trafficking to the cell surface rather than decreased turnover.
DISCUSSION
The correct assembly and trafficking of nAChRs are essential for their surface expression and synaptic function. Previous studies have defined several molecular determinants in the receptor that control ER export and quality control, but the determinants for later trafficking steps are unknown. Here, we define novel signals in the muscle β and δ subunit cytoplasmic loops that mediate Golgi retention and retrieval from the plasma membrane. We show that mutation of the motifs permits surface expression of unassembled subunit loops and also increases surface levels of assembled AChR. We propose that the β/δ retention signals regulate Golgi exit and surface trafficking of assembled AChR and may also contribute to Golgi- and plasma membrane-based quality control mechanisms that prevent surface expression of unassembled or incorrectly assembled subunits.
Molecular Determinants for AChR Trafficking
Trafficking of nAChRs through the secretory pathway is thought to be regulated by a series of compartment-specific retention and export signals. In the ER, unassembled muscle subunits are selectively retained by a conserved motif in the first transmembrane domain (10) and by dibasic motifs in α and possibly other subunit cytoplasmic loops (11). These motifs likely become buried upon subunit assembly, allowing export of pentameric receptor (10), whereas unassembled subunits are retained and targeted for degradation by ERAD (8). In addition, spaced pairs of hydrophobic residues in the α4 and β2 subunit cytoplasmic loops serve as export signals for assembled neuronal AChR (12). Together, these molecular signals provide critical quality control, ensuring that only assembled receptor is trafficked to later compartments in the secretory pathway.
Our findings now identify analogous molecular signals that regulate Golgi trafficking. In an unbiased screen we defined 25–28 amino acid sequences in the β and δ subunit cytoplasmic loops (Fig. 5E) that act as strong Golgi retention signals. These motifs largely prevent trafficking of β and δ loops to the plasma membrane, and the trace amounts of CD4-β and δ that reach the cell surface are rapidly internalized via the same motif. In contrast, other subunit loops are efficiently trafficked to and stably expressed on the muscle cell surface. Interestingly, the β and δ Golgi retention motifs are localized in the proximal, major cytoplasmic loop like two of the previously identified ER trafficking signals. However, Golgi retention does not correlate with the dibasic motifs present in the proximal loops of α, γ, and δ subunits (11), and the critical determinants for Golgi retention are distinct from the spaced pairs of hydrophobic residues present in all subunit proximal loops (12). Rather, our mutational analysis demonstrates that Golgi retention and recovery from the plasma membrane are dependent on an ordered array of basic and hydrophobic residues in the C-terminal portion of the motif with a predicted α-helical secondary structure. Most notably, this includes specific lysine residues and their immediate neighbors, which lie at slightly different positions in β and δ loops. Mutation of βK353 and δK351 dramatically decreased Golgi retention and plasma membrane recovery of CD4-subunit chimeras. Moreover, the same mutations increased surface levels of assembled AChR in heterologous cells by ∼38%, with an associated decrease in the intracellular pool of receptor. Together, these findings define novel motifs in the AChR β and δ subunits that regulate Golgi trafficking and surface expression of the AChR.
Function of Golgi Retention Signals
Our findings suggest two possible functions for the β/δ Golgi retention signals in regulating AChR trafficking. First, the Golgi retention signals might help ensure that unassembled subunits or partially assembled intermediates that escape from the ER are not expressed on the cell surface (Fig. 8). This would be detrimental because partially assembled intermediates that include the α subunit could uncouple transmitter binding from channel function or form aggregates of misfolded protein. Support for such a quality control checkpoint at the Golgi complex comes from studies in yeast, where it has been demonstrated that some misfolded proteins that escape ERAD are recognized in the Golgi complex and trafficked to vacuoles for degradation (for review, see Ref. 27). In some cases, this involves monoubiquitination of misfolded proteins by Golgi-localized ubiquitin ligases, which leads to their recognition by Golgi-localized, γ-ear-containing ARF-binding proteins and vacuolar trafficking. Similar, ubiquitin-based endocytosis and sorting also contribute to quality maintenance at the plasma membrane in yeast (27). Intriguingly, such mechanisms are consistent with our finding that specific lysine residues are critical for β/δ subunit retention in the Golgi and retrieval from the plasma membrane. Thus, ER-, Golgi-, and plasma membrane-based quality control mechanisms may combine to ensure that only correctly assembled, functional AChR is expressed on the cell surface.
FIGURE 8.
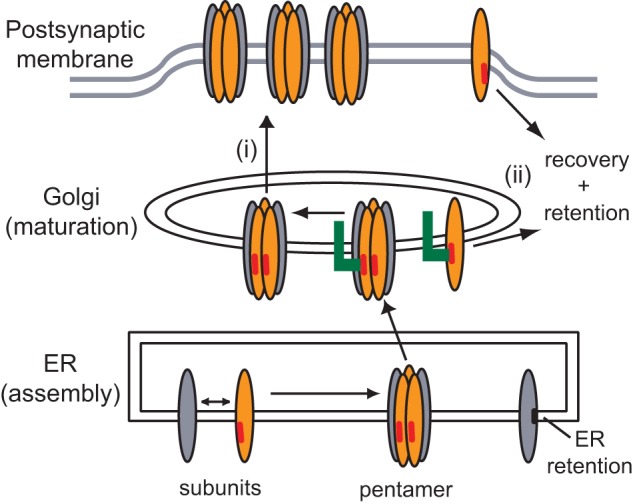
Model for the role of the β/δ subunit loop retention motifs in AChR trafficking. The biogenesis and trafficking of the AChR are regulated by a series of compartment-specific signals. First, in the ER, pentameric receptor is exported to the Golgi whereas most unassembled or partially assembled subunits are retained by transmembrane (black bar) and dibasic motifs and targeted for degradation. Second, in the Golgi complex, β and δ subunit cytoplasmic loop motifs (red bars) mediate retention by interacting with an as-yet unidentified protein (green). We propose that these motifs regulate the Golgi to surface trafficking of assembled AChR (i) and might also act in the Golgi and at the plasma membrane to retain and retrieve assembly intermediates that have escaped the ER (ii). This additional quality control mechanism could help ensure that only correctly assembled and fully functional AChR is expressed on the cell surface.
Second, the β/δ Golgi retention signals could regulate export of assembled AChR from the Golgi complex (Fig. 8). Importantly, mutation of the β/δ motifs increased surface levels of receptor by up-regulating forward trafficking to the cell surface. Thus, the Golgi retention signals are not masked upon subunit assembly, as shown for ER retention signals (10), and remain functional in intact AChR. Potential roles for the retention signals in assembled AChR could be controlling the size of the intracellular receptor pool, facilitating processing of N-linked oligosaccharides on the γ and δ subunits to complex forms (16, 28), monitoring AChR association with rapsyn (29, 30), or assessing the correct order or stoichiometry of subunits in the pentameric receptor.
Several previous studies support our conclusion that AChR export from the Golgi is regulated, and they identify possible components of the trafficking machinery. In the nematode C. elegans, genetic screens identified the Golgi resident protein unc-50 as being required for trafficking of levamisole-sensitive nAChRs to the neuromuscular junction (17). In unc-50 mutants, this subtype of AChRs is sorted to the lysosomal system and degraded, whereas other AChR types and GABA receptors are trafficked normally to the plasma membrane. This suggests that unc-50 is involved in the recognition and sorting of AChRs in the Golgi, likely by interacting with subtype-specific trafficking motifs. Similarly, in mammalian systems, the neuronal calcium sensor VILIP-1 binds the neuronal α4 subunit at a site in its proximal cytoplasmic loop (14) and up-regulates surface expression of α4β2-AChRs. Notably, VILIP-1 associates with α4β2 receptor in the trans-Golgi network in a calcium-regulated manner and appears to promote receptor trafficking to the cell surface (31). Given that unc-50 and VILIP-1 promote AChR export, however, it seems likely that other Golgi components will mediate AChR retention via interaction with the β/δ subunit motifs.
In summary, our findings identify novel molecular determinants in the muscle AChR β/δ subunit cytoplasmic loops that regulate Golgi to surface trafficking. An intriguing possibility is that these retention signals provide an additional quality control step that monitors AChR biogenesis and surface expression.
This work was supported by Muscular Dystrophy Association Grant 134412 (to M. J. F.) and a University of California Davis Physician Scientist Training Program award (to J. C. R.).
- nAChR
- nicotinic AChR
- AChR
- acetylcholine receptor
- ANOVA
- analysis of variance
- α-BuTx
- α-bungarotoxin
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation.
REFERENCES
- 1. Vincent A. (2002) Unravelling the pathogenesis of myasthenia gravis. Nat. Rev. Immunol. 2, 797–804 [DOI] [PubMed] [Google Scholar]
- 2. Engel A. G., Ohno K., Sine S. M. (2003) Congenital myasthenic syndromes: progress over the past decade. Muscle Nerve 27, 4–25 [DOI] [PubMed] [Google Scholar]
- 3. Albuquerque E. X., Pereira E. F., Alkondon M., Rogers S. W. (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dani J. A., Bertrand D. (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 47, 699–729 [DOI] [PubMed] [Google Scholar]
- 5. Miwa J. M., Freedman R., Lester H. A. (2011) Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron 70, 20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Millar N. S., Harkness P. C. (2008) Assembly and trafficking of nicotinic acetylcholine receptors. Mol. Membr. Biol. 25, 279–292 [DOI] [PubMed] [Google Scholar]
- 7. Merlie J. P., Lindstrom J. (1983) Assembly in vivo of mouse muscle acetylcholine receptor: identification of an α subunit species that may be an assembly intermediate. Cell 34, 747–757 [DOI] [PubMed] [Google Scholar]
- 8. Christianson J. C., Green W. N. (2004) Regulation of nicotinic receptor expression by the ubiquitin-proteasome system. EMBO J. 23, 4156–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Valkova C., Albrizio M., Röder I. V., Schwake M., Betto R., Rudolf R., Kaether C. (2011) Sorting receptor Rer1 controls surface expression of muscle acetylcholine receptors by ER retention of unassembled α-subunits. Proc. Natl. Acad. Sci. U.S.A. 108, 621–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang J. M., Zhang L., Yao Y., Viroonchatapan N., Rothe E., Wang Z. Z. (2002) A transmembrane motif governs the surface trafficking of nicotinic acetylcholine receptors. Nat. Neurosci. 5, 963–970 [DOI] [PubMed] [Google Scholar]
- 11. Keller S. H., Lindstrom J., Ellisman M., Taylor P. (2001) Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor α-subunit. J. Biol. Chem. 276, 18384–18391 [DOI] [PubMed] [Google Scholar]
- 12. Ren X. Q., Cheng S. B., Treuil M. W., Mukherjee J., Rao J., Braunewell K. H., Lindstrom J. M., Anand R. (2005) Structural determinants of α4β2 nicotinic acetylcholine receptor trafficking. J. Neurosci. 25, 6676–6686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Halevi S., McKay J., Palfreyman M., Yassin L., Eshel M., Jorgensen E., Treinin M. (2002) The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. 21, 1012–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin L., Jeanclos E. M., Treuil M., Braunewell K. H., Gundelfinger E. D., Anand R. (2002) The calcium sensor protein visinin-like protein-1 modulates the surface expression and agonist sensitivity of the α4β2 nicotinic acetylcholine receptor. J. Biol. Chem. 277, 41872–41878 [DOI] [PubMed] [Google Scholar]
- 15. Rezvani K., Teng Y., Pan Y., Dani J. A., Lindstrom J., García Gras E. A., McIntosh J. M., De Biasi M. (2009) UBXD4, a UBX-containing protein, regulates the cell surface number and stability of α3-containing nicotinic acetylcholine receptors. J. Neurosci. 29, 6883–6896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gu Y., Ralston E., Murphy-Erdosh C., Black R. A., Hall Z. W. (1989) Acetylcholine receptor in a C2 muscle cell variant is retained in the endoplasmic reticulum. J. Cell Biol. 109, 729–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eimer S., Gottschalk A., Hengartner M., Horvitz H. R., Richmond J., Schafer W. R., Bessereau J. L. (2007) Regulation of nicotinic receptor trafficking by the transmembrane Golgi protein UNC-50. EMBO J. 26, 4313–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacob M. H., Lindstrom J. M., Berg D. K. (1986) Surface and intracellular distribution of a putative neuronal nicotinic acetylcholine receptor. J. Cell Biol. 103, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borges L. S., Yechikhov S., Lee Y. I., Rudell J. B., Friese M. B., Burden S. J., Ferns M. J. (2008) Identification of a motif in the acetylcholine receptor β subunit whose phosphorylation regulates rapsyn association and postsynaptic receptor localization. J. Neurosci. 28, 11468–11476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou J., Valletta J. S., Grimes M. L., Mobley W. C. (1995) Multiple levels for regulation of TrkA in PC12 cells by nerve growth factor. J. Neurochem. 65, 1146–1156 [DOI] [PubMed] [Google Scholar]
- 21. Wilde A., Beattie E. C., Lem L., Riethof D. A., Liu S. H., Mobley W. C., Soriano P., Brodsky F. M. (1999) EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 96, 677–687 [DOI] [PubMed] [Google Scholar]
- 22. Salpeter M. M., Andreose J., O'Malley J. P., Xu R., Fumagalli G., Lomo T. (1993) Degradation of acetylcholine receptors at vertebrate neuromuscular junctions. Ann. N.Y. Acad. Sci. 681, 155–164 [DOI] [PubMed] [Google Scholar]
- 23. Phillips W. D., Vladeta D., Han H., Noakes P. G. (1997) Rapsyn and agrin slow the metabolic degradation of the acetylcholine receptor. Mol. Cell. Neurosci. 10, 16–26 [DOI] [PubMed] [Google Scholar]
- 24. Wang Z. Z., Mathias A., Gautam M., Hall Z. W. (1999) Metabolic stabilization of muscle nicotinic acetylcholine receptor by rapsyn. J. Neurosci. 19, 1998–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu Z., Joseph D., Bugnard E., Zaal K. J., Ralston E. (2001) Golgi complex reorganization during muscle differentiation: visualization in living cells and mechanism. Mol. Biol. Cell 12, 795–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 27. MacGurn J. A., Hsu P. C., Emr S. D. (2012) Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81, 231–259 [DOI] [PubMed] [Google Scholar]
- 28. Ramanathan V. K., Hall Z. W. (1999) Altered glycosylation sites of the δ subunit of the acetylcholine receptor (AChR) reduce αδ association and receptor assembly. J. Biol. Chem. 274, 20513–20520 [DOI] [PubMed] [Google Scholar]
- 29. Marchand S., Bignami F., Stetzkowski-Marden F., Cartaud J. (2000) The myristoylated protein rapsyn is cotargeted with the nicotinic acetylcholine receptor to the postsynaptic membrane via the exocytic pathway. J. Neurosci. 20, 521–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park J. Y., Ikeda H., Ikenaga T., Ono F. (2012) Acetylcholine receptors enable the transport of rapsyn from the Golgi complex to the plasma membrane. J. Neurosci. 32, 7356–7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao C. J., Noack C., Brackmann M., Gloveli T., Maelicke A., Heinemann U., Anand R., Braunewell K. H. (2009) Neuronal Ca2+ sensor VILIP-1 leads to the up-regulation of functional α4β2 nicotinic acetylcholine receptors in hippocampal neurons. Mol. Cell. Neurosci. 40, 280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]