

Background: P-glycoprotein is an ATP-dependent drug pump.
Results: Mutations or cross-linking of intracellular loops (ICL) 2 and 3 inhibit folding and/or activity.
Conclusion: Hydrophobic residues in ICL2 and ICL3 and the second nucleotide-binding domain form a hydrophobic transmission network for folding and activity.
Significance: We identify a dual-purpose transmission interface required for folding and activity.
Keywords: ABC Transporter, Drug Resistance, Membrane Proteins, Protein Folding, Protein Misfolding, P-glycoprotein
Abstract
The P-glycoprotein (P-gp) drug pump (ABCB1) has two transmembrane domains and two nucleotide-binding domains (NBDs). Coupling of the drug-binding sites in the transmembrane domains to the NBDs occurs through interaction of the intracellular helices (IHs) with residues in the NBDs (IH1/IH4/NBD1 and IH2/IH3/NBD2). We showed previously that cross-linking of cysteines in IH3 and IH1 with a short cross-linker mimicked drug binding as it activated P-gp ATPase activity. Here we show that residue A259C(IH2) could be directly cross-linked to W803C(IH3). Cross-linking was inhibited by the presence of ATP and adenosine 5′-(β,γ-imino)triphosphate but not by ADP. Cross-linking of mutant A259C/W803C inhibited its verapamil-stimulated ATPase activity mutant, but activity was restored after addition of dithiothreitol. Because these residues are close to the ball-and-socket joint A266C(IH2)/Phe1086(NBD2), we mutated the adjacent Tyr1087(NBD2) close to IH3. Mutants Y1087A and Y1087L, but not Y1087F, were misprocessed, and all inhibited ATPase activity. Mutation of hydrophobic residues (F793A, L797A, L814A, and L818A) flanking IH3 also inhibited maturation. The results suggest that these residues, together with Trp803 and Phe804, form a large hydrophobic pocket. The results show that there is an important hydrophobic network at the IH2/IH3/NBD2 transmission interface that is critical for folding and activity of P-gp.
Introduction
The P-glycoprotein (P-gp,2 ABCB1) drug pump was the first human ABC protein to be discovered during efforts to understand how cancer cells developed multidrug resistance (1). P-gp was found to mediate the ATP-dependent efflux of a wide range of hydrophobic compounds (such as anticancer drugs, hydrophobic drugs, steroids, peptides, detergents, and lipids) (2, 3). It is expressed in the epithelium of the liver, kidney, and gastrointestinal tract and at the blood-brain or blood-testes barrier where it functions to protect us from cytotoxic compounds. It is one of the major causes of multidrug resistance in diseases such as cancer and AIDS (2).
Human P-gp has 1280 amino acids (4) that are organized as two tandem repeats of 610 amino acids that are joined by a linker region. Each repeat consists of an N-terminal transmembrane domain (TMD) containing six TM segments followed by a nucleotide-binding domain (NBD). Drug substrates appear to bind at multiple drug-binding sites located within a cavity located at the interface between the TMDs (5–7), likely through an induced-fit mechanism (8). ATP binds at the interface between the NBDs, and ATP hydrolysis occurs by an alternating site mechanism (9–13).
It has been proposed that a key feature of the P-gp mechanism is that it can exist in at least two major conformations during the catalytic cycle: an inward-facing conformation with separated NBDs and drug-binding sites exposed to the cytoplasm (open conformation) and an outward-facing conformation with close association of the NBDs and drug-binding sites exposed to the extracellular surface (closed conformation) (14).
There is a considerable degree of cross-talk between the TMDs and NBDs during the reaction cycle as drug binding activates ATPase activity by promoting formation of the closed conformation. ATP hydrolysis then leads to drug efflux from the TMDs. It appears that TMD/NBD cross-talk is mediated by four intracellular loops (ICLs) in the TMDs. We showed recently that it is possible to mimic the effects of drug binding to activate ATPase activity by cross-linking ICL1(TMD1) in close proximity to ICL3(TMD2) (15, 16).
Although there is no high-resolution crystal structure of human P-glycoprotein, the Caenorhabditis elegans P-gp crystal structure (17) appeared to be a good model for human P-gp in an open conformation because the structure was compatible with biochemical studies on the human P-gp TMDs (7, 18–23) and ICL2 (24). The structure of the C. elegans drug pump showed that TMDs were connected to the NBDs by four intracellular helices (IHs) within the ICLs that were proposed to act as “ball-and-socket” joints.
Models of human P-gp in a closed conformation have been built (25, 26) using the Sav1866 crystal structure (27) as a template. Sav1866 is a homodimeric bacterial ABC drug pump that is predicted to have a similar architecture to human P-gp. The predicted structure was also compatible with biochemical studies of human P-gp (7, 18, 20–23). It was predicted that the IHs of Sav1866 acted as interfaces to transmit conformational changes associated with ATP binding and hydrolysis at the NBDs to the TMDs (27).
In the open conformation, IH1 and IH2 connect to NBD1 and NBD2, respectively, whereas IH3 and IH4 connect to NBD2 and NBD1, respectively. In general, IH1 and IH3 are only found in ABC exporters but not in ABC importers (28) such as the maltose transporter (29).
In addition to acting as transmission interfaces, it is likely that the IHs play important roles in TMD/NBD interactions required for folding of P-gp because a truncation mutant lacking the NBDs will not mature in the absence of drug substrates (30). It appears that NBD interactions with the TMDs are important for packing of the 12 TM segments during synthesis (21). In the absence of the NBDs, the TMDs appear to accumulate as partially folded protease-sensitive proteins. The TMD1 + 2 truncation mutant can be rescued by carrying out expression in the presence of drug substrates (30).
P-gp differs from its sister protein, the CFTR chloride channel, because deletion of NBD2 in P-gp but not CFTR (31) inhibits maturation (32). Although P-gp and CFTR perform different functions, they are predicted to have similar structures (33). In this study, we tested our prediction that P-gp requires NBD2 because IH3-NBD2 interactions (predicted to involve Tyr1087 in NBD2 (34)) are critical for maturation of P-gp during synthesis. In addition, we tested whether cross-linking of IH3 to IH2 would have the opposite effect of cross-linking IH3 to IH1 (16) and cause inhibition, rather than stimulation, of ATPase activity.
EXPERIMENTAL PROCEDURES
Expression and Maturation of Mutants
Mutants were constructed to contain a C-terminal A52 epitope tag (35) for use in whole cell immunoblot assays or a 10-histidine tag for isolation of protein for measurement of activity (36). The presence of the epitope tag distinguished the mutant proteins from any endogenous P-gp.
Mutations were introduced into the human P-gp cDNA as described previously (33). Mutants were expressed in HEK 293 cells for 18 h in the presence or absence of 5 μm cyclosporine A. Expression in the presence of cyclosporine A promotes maturation of processing mutants (30, 37). Whole cell extracts were subjected to immunoblot analysis using monoclonal antibody A52 for A52-tagged proteins or with rabbit polyclonal antibody (38) against P-gp for histidine-tagged proteins, and the bands were visualized by enhanced chemiluminescence. The gel lanes were scanned, and the amount of mature 170-kDa product relative to total P-gp (immature 150-kDa plus mature 170-kDa protein) was analyzed using the NIH Image program and an Apple computer.
Purification of P-gp and Measurement of ATPase Activity
Histidine-tagged P-gps were expressed in HEK 293 cells and then isolated by nickel-chelate chromatography as described previously (36). Recovery of P-gp was monitored by immunoblot analysis with rabbit anti-P-gp polyclonal antibody (38). A sample of the isolated histidine-tagged P-gp was mixed with an equal volume of 10 mg/ml sheep brain phosphatidylethanolamine (type II-S, Sigma) that had been washed and suspended in TBS (pH 7.4). Samples of the P-gp/lipid mixture were assayed for ATPase activity by addition of an equal volume of 2× ATPase buffer (100 mm Tris-HCl (pH 7.5), 100 mm NaCl, 20 mm MgCl2, and 10 mm ATP) containing 1.2 mm verapamil. The samples were incubated for 30 min at 37 °C, and the amount of inorganic phosphate released was determined using the method of Chifflet et al. (39).
IH2-IH3 Cross-linking
Cysteine residues were introduced at each position within residues Leu258 to Ile-261 in IH2 and residues Trp803 and Phe804 in IH3 to generate eight different double cysteine mutants in a Cys-less background (40). The mutants were transiently expressed in HEK 293 cells in the presence of 5 μm cyclosporine A. Membranes were prepared, suspended in TBS (pH 7.4), and treated with 0.5 mm CuCl2 in the presence or absence of 5 mm MgATP, 5 mm MgADP, or 5 mm 5′-(β,γ-imino)triphosphate for 5 min at 0 °C. The reactions were performed using a protein concentration of 0.4 mg/ml. The reactions were stopped by addition of SDS sample buffer (125 mm Tris-HCl (pH 6.8), 20% (v/v) glycerol, and 4% (w/v) SDS) containing 50 mm EDTA and no thiol reducing agent. Samples of the reaction mixtures (1 μg of protein) were then subjected to SDS-PAGE (6.5% (w/v) polyacrylamide gels, 1.5-mm 15-slot minigels) and immunoblot analysis with a rabbit polyclonal antibody against P-gp. Intramolecular disulfide cross-linking between P-gp domains can be detected because the cross-linked product migrates with a slower mobility on SDS-PAGE gels (41).
RESULTS
Mutations to Hydrophobic Residues in IH3 and Flanking Regions Inhibit Maturation
According to the human P-gp model on the basis of the C. elegans structure, IH3 (joins TM segments 8 and 9, see Fig. 1A) consists of residues Asp800 to Asp806 in ICL3 and is predicted to connect TMD2 to NBD2 (Fig. 1A) (17). TMD2-NBD2 interactions may be particularly important for folding of P-gp into a native structure because an NBD2 truncation mutant missing amino acids 1024–1280 does not mature (32). By contrast, deletion of NBD2 (residues 1197–1480) from the structurally similar CFTR protein (42) yielded a mature product (32) with a similar stability as its full-length counterpart (31).
FIGURE 1.
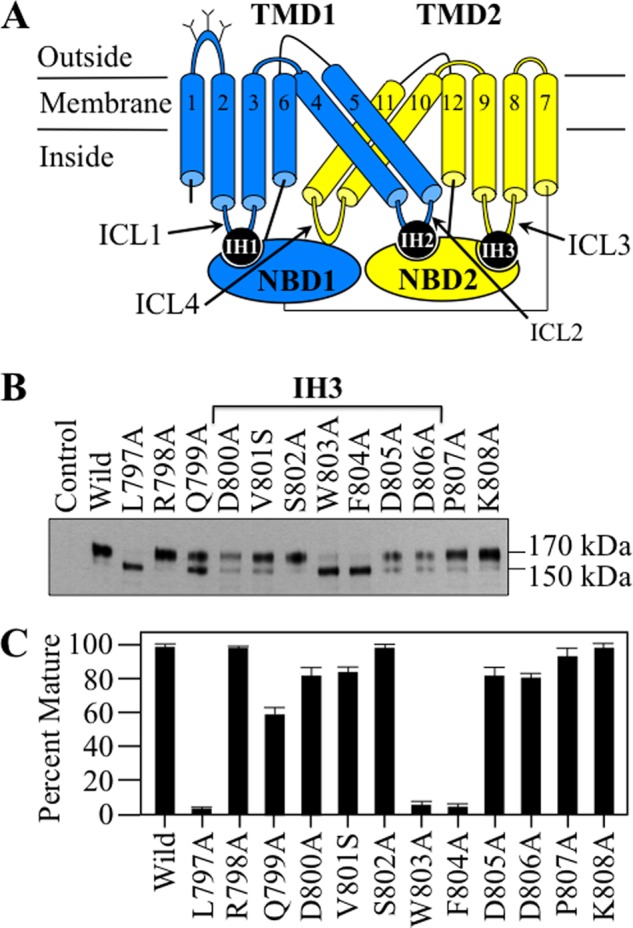
Point mutations to IH3 inhibit P-gp maturation. A, secondary structure of P-gp showing the location of coupling helix IH3 between TMD2 and the NBD2. The branched lines represent the glycosylation sites, and the numbered cylinders represent the TM segments. B, A52-tagged wild-type (Wild) P-gp and mutants containing mutations to IH3 and flanking regions were expressed in HEK 293 cells, and whole cell SDS extracts were subjected to immunoblot analysis. The positions of mature (170-kDa) and immature (150-kDa) forms of P-gp are shown. C, the amount of mature protein relative to total P-gp obtained from three different transfections + S.D. for each mutant.
To test whether IH3 was important for maturation, point mutations were made to residues in IH3 and some flanking residues (Phe793 to Leu818). The A52-tagged mutants were expressed in HEK 293 cells, and whole cell SDS extracts were subjected to immunoblot analysis. Maturation of P-gp can be monitored because it contains three N-linked glycosylation sites in the extracellular loop that connects TM segments 1 and 2 (Fig. 1A). Mutations that inhibit folding will trap P-gp as a 150-kDa core-glycosylated immature protein in the endoplasmic reticulum. Mutants containing mutations that do not grossly affect folding will undergo further processing in the Golgi to yield a 170-kDa mature protein. A representative immunoblot of whole cell extracts of cells expressing mutants with changes from Leu797 to Lys808 is shown in Fig. 1B. It was found that two mutations in IH3 (W803A and F804A) inhibited maturation of P-gp (Fig. 1, B and C) so that immature 150-kDa P-gp was the major product. By contrast, the D800A, V801S, S802A, D805A, and D806A mutations in IH3 appeared to have little or no effect on folding as they yielded mature 170-kDa P-gp as their major product. Point mutations to the residues flanking IH3 (L797A, Fig. 1B) along with F793A, L814A, and L818A (data not shown) also inhibited maturation so that the 150-kDa protein was the major product.
Mutating Aromatic Residues Trp803 and Phe804 in IH3 Inhibits Maturation but Not Activity
We showed previously that hydrophobic residues at the IH2-NBD2 interface were particularly important for maturation and activity of P-gp (16). Because W803A and F804A mutations in IH3 yielded the immature 150-kDa immature P-gp as the major product, we tested whether the mutants could be rescued with cyclosporine A. It has been shown previously that substrates and modulators of P-gp could act as pharmacological chaperones to rescue misprocessed mutants (23, 37). Accordingly, the A52-tagged mutants W803A and F804A were expressed in HEK 293 cells in the absence or presence of 5 μm cyclosporine A, and whole cell SDS extracts were subjected to immunoblot analysis. Fig. 2A shows that expression of either W803A or F804A in the presence of cyclosporine A yielded the mature 170-kDa P-gp as the major product.
FIGURE 2.
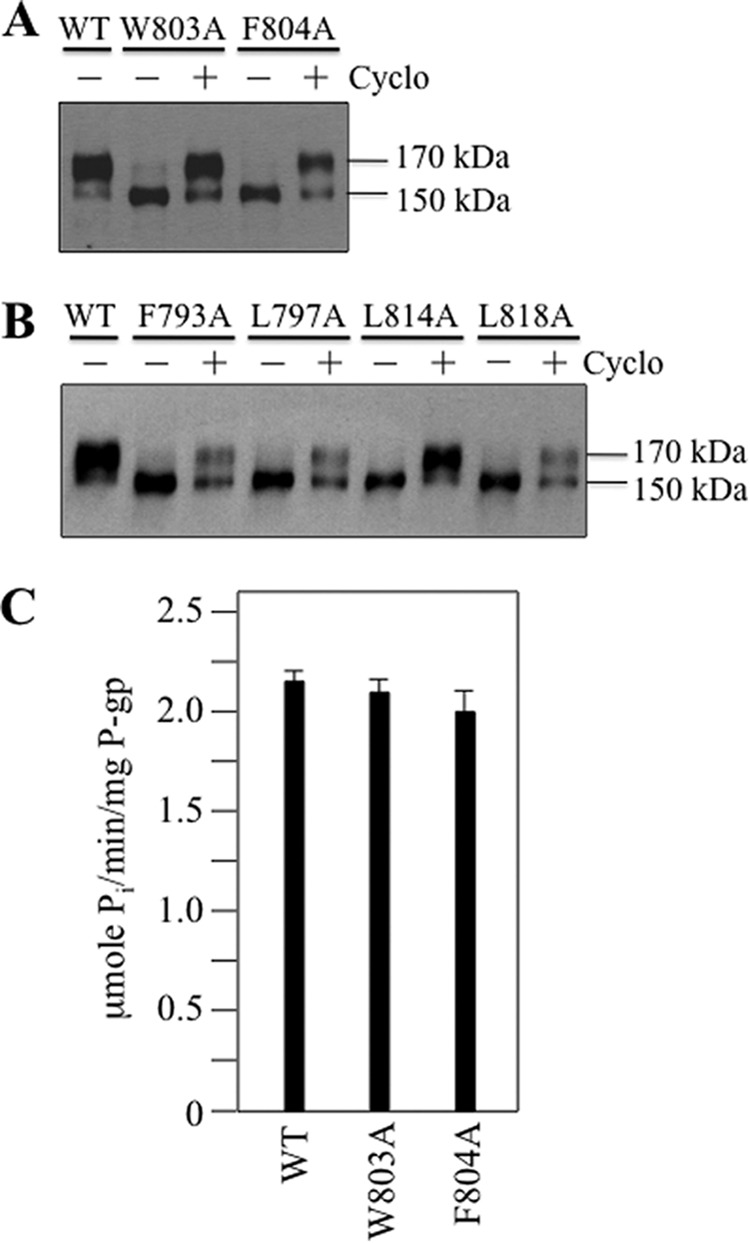
Drug rescue of processing mutants and activity of IH3 mutants W803A and F804A. A52-tagged WT P-gp, mutants W803A and F804A (A), or IH3 flanking mutants (F793A, L797A, L814A, and L818A) (B) were expressed in HEK 293 cells in the absence (−) or presence (+) of 5 μm cyclosporine A (Cyclo) for 18 h, and whole cell extracts were subjected to immunoblot analysis. The positions of mature (170-kDa) and immature (150-kDa) forms of P-gp are shown. C, verapamil-stimulated ATPase activities of histidine-tagged wild-type P-gp and W803A and F804A mutants. The results are derived from three different transfections + S.D.
Because the mutations F793A, L797A, L814A, and L818A in the flanking regions of IH3 also inhibited maturation, these mutants were also expressed in the presence of cyclosporine A to test for rescue. Expression of mutant L814A in the presence of cyclosporine A yielded the mature 170-kDa P-gp as the major product (Fig. 2B). Mutants F793A, L797A, and L818A were less efficiently rescued as they yielded about equivalent levels of mature 170-kDa and immature 150-kDa P-gp in the presence of cyclosporine A (Fig. 2B).
To determine whether the W803A and F804A mutants rescued with cyclosporine A were active, the histidine-tagged mutants W803A and F804A were expressed in HEK 293 cells in the presence of cyclosporine A, and P-gp was isolated by nickel-chelate chromatography. After reconstitution with lipid, they were assayed for verapamil-stimulated ATPase activity. Verapamil was used because it is transported by P-gp (43), and it highly stimulates the ATPase activity of human P-gp (over 10-fold) (44). It was found that the activity of both mutants was similar to wild-type P-gp (Fig. 2C). The results show that Trp803 or Phe804 were important for maturation but not essential for coupling drug binding to activation of ATPase activity.
We then tested whether mutating Trp803 to Phe (hydrophobic), Leu (hydrophobic), Ser (hydrophilic), Tyr (aromatic), or Asp (charged) and Phe804 to Leu, Ser, Tyr, Asp, or Trp affected maturation of the protein. The A52-tagged mutants were expressed in HEK 293 cells in the absence or presence of cyclosporine A, and whole cell SDS extracts were subjected to immunoblot analysis. Fig. 3, A and B, shows that mature 170-kDa P-gp was the major product if Trp803 was replaced with the aromatic residues Phe or Tyr. Replacement of Trp803 with Leu, Ser, or Asp inhibited maturation, although all three mutants could still be rescued with cyclosporine A (Fig. 3, A and B). Similar results were observed when Phe804 was mutated. Mature 170-kDa P-gp was the major product in mutants F804Y and F804W (Fig. 3, C and D), although the amount of mature protein in the absence of cyclosporine A in mutant F804W was lower than in F804Y (65% versus 85%, respectively). Mutating Phe804 to Leu, Ser, or Asp inhibited maturation, and only the F804A or F804S mutants could be rescued with cyclosporine A to yield mature 170-kDa P-gp as the major product (Fig. 3, C and D). These results show that Phe804 is more sensitive to changes than Trp803. The effects of mutating Trp803 and Phe804 at the IH3-NBD2 interface are also different from those observed by mutating Phe1086 (16) located at the IH2-NBD2 interface. P-gp maturation was not inhibited if Phe1086 was replaced with hydrophobic residues such as leucine (16), whereas maturation of both the Trp803 and Phe804 mutants was severely decreased unless replaced with an aromatic residue. The results suggest that both bulk and hydrophobicity at positions 803 and 804 are important for IH3 to adopt a structure that will mature in the absence of drug substrates.
FIGURE 3.
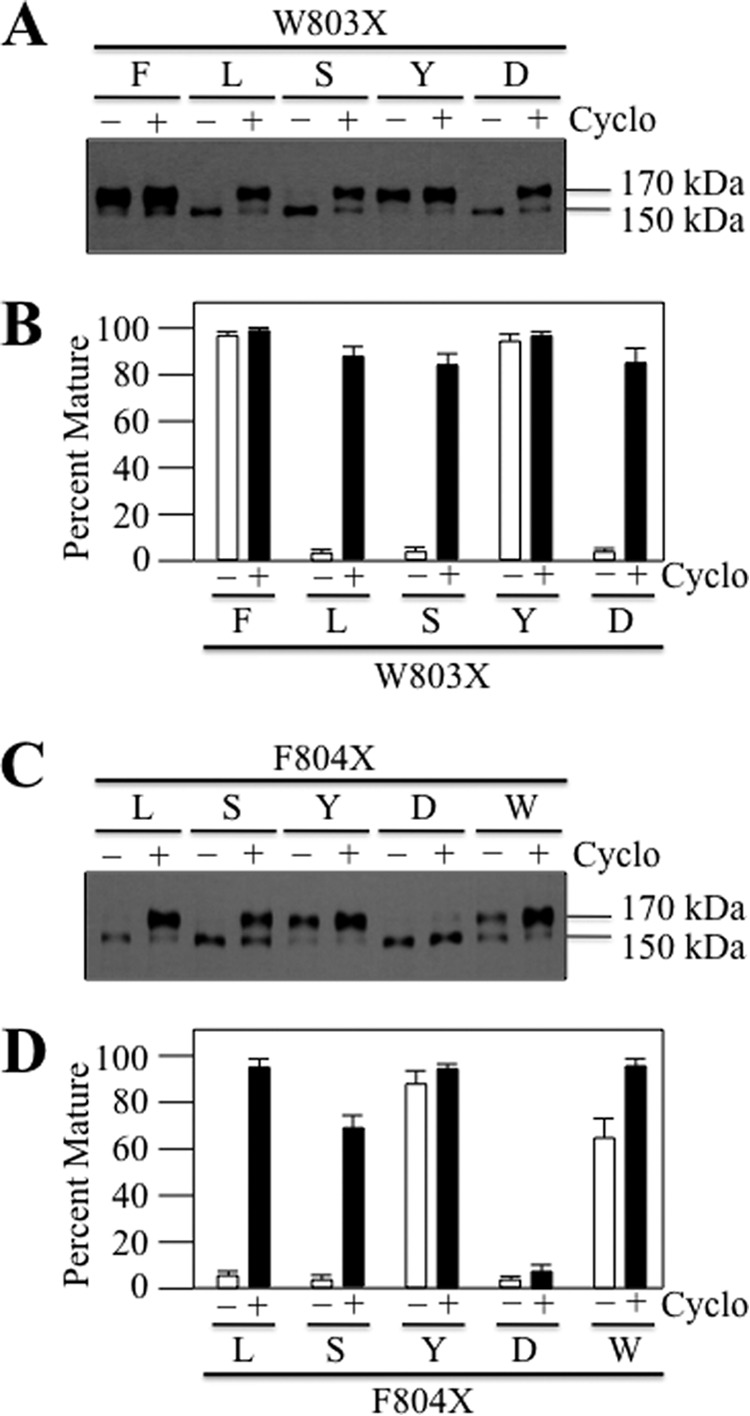
Only aromatic replacements of Trp803 or Phe804 yield mature P-gp as the major product. A52-tagged mutants containing changes to Trp803 (A and B) (W803F (F), W803L (L), W803S (S), W803Y (Y), or W803D (D)) or Phe804 (C and D) (F804L (L), F804S (S), F804Y (Y), F804D (D), or F804W (W)) were expressed in HEK 293 cells in the absence (−) or presence (+) of 5 μm cyclosporine A (Cyclo) for 18 h, and whole cell extracts were subjected to immunoblot analysis. The positions of mature (170-kDa) and immature (150-kDa) forms of P-gp are shown (A and C). B and D show the amount of mature protein relative to total P-gp (mature plus immature) obtained from three different transfections + S.D.
Cross-linking of IH2 and IH3 Inhibits Activity
In a previous cysteine mutagenesis and cross-linking study, we showed that clamping ICL3 (N820C) in close proximity to ICL1 (D175C or D177C) by cross-linking cysteines with short cross-linkers activated P-gp ATPase activity (15, 33). Because P-gp appears to be flexible so that the protein can adopt conformations in which the NBDs are close together (closed conformation) or far apart (open conformation), we postulated that cross-linking to bring IH1/IH3 in close proximity (in different halves of the protein) activated P-gp ATPase activity by trapping the protein in the closed conformation (Fig. 1A). We would not expect P-gp ATPase activity to be activated if IH3 was clamped (cross-linking of introduced cysteines) in close proximity to IH2 because human P-gp models in the open and closed conformation predict them to be close together, although they are in different halves of the molecule (17, 26). Molecular dynamic studies, however, predict that IH2/IH3 clamping might inhibit drug-stimulated ATPase activity because conformational changes in IH2 and IH3 during the reaction cycle would alter the relative positions of residues at the IH2/IH3 interface (34, 45).
Residues Leu258 to Ile261 are predicted to be in IH2, whereas Trp803 and Phe804 are predicted to be within IH3 (17). Jin et al. (17) proposed that IH2 consists of human P-gp residues Glu256 to Glu273 rather than residues Ile261 to Phe267, which make up the short α-helix connecting TM segments 8 and 9. To determine whether the residues Leu258 to Ile261 are in close proximity to Trp803 or Phe804 in IH3, we constructed eight double cysteine mutants, each of which contained one cysteine in the Leu258 to Ile261 segment and another at either Trp803 or Phe804 in a Cys-less background (40). The mutants were expressed in HEK 293 cells in the presence of cyclosporine A, and then membranes prepared for cross-linking with 0.5 mm CuCl2 for 5 min at 0 °C to reduce molecular motion. Samples were then subjected to immunoblot analysis. It was found that the yield of all the F804C double mutants was significantly lower than the W803C double cysteine mutants (data not shown). This would be consistent with Phe804 being more sensitive to mutation than Trp803 (Fig. 3). Therefore, the W803C double mutants were tested for cross-linking with cupric chloride oxidant. Cupric chloride, rather than copper phenanthroline, was used because it is a gentler oxidant than copper phenanthroline. When cross-linking was done with copper phenanthroline, the reaction was complete by 30 s, even when the reaction was done in an ice bath (data not shown). Cross-linked product was observed in all mutants (data not shown), with the greatest amount present in mutant A259C/W803C (about 75%, Fig. 4A).
FIGURE 4.
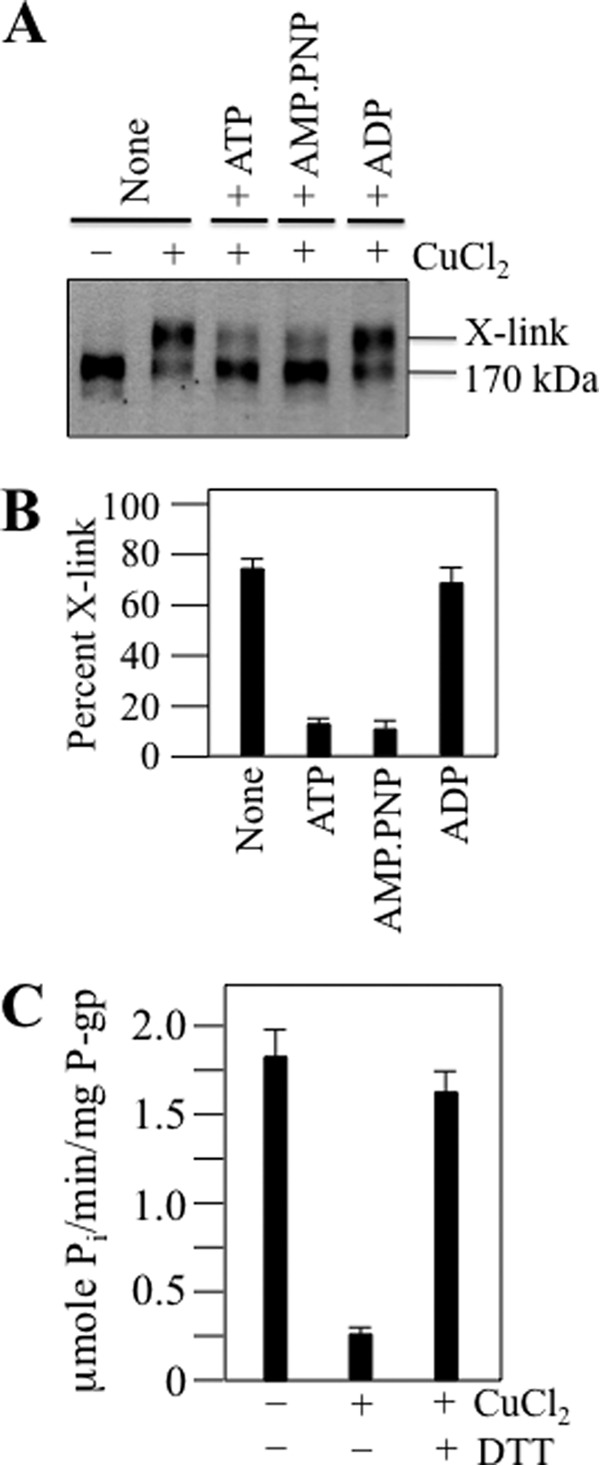
Cross-linking of A259C (IH2) to W803C (IH3) inhibits activity. A, membranes prepared from cells expressing histidine-tagged mutant A259C/W803C were treated without (−) or with (+) 0.5 mm CuCl2 for 10 min at 0 °C in the absence (None) or presence of 5 mm ATP, adenosine 5′-(β,γ-imino)triphosphate (AMP.PNP), or ADP. The reactions were stopped by addition of SDS sample buffer containing no reducing agent, and samples were then subjected to immunoblot analysis. The positions of the cross-linked (X-link) and mature (170-kDa) P-gps are indicated. B, the amount of cross-linked protein relative to total P-gp (170-kDa plus 150-kDa P-gp) was quantitated from three different transfections + S.D. C, the histidine-tagged mutant A259C/W803C was isolated by nickel-chelate chromatography, reconstituted with lipid, and treated without (−) or with (+) 0.2 mm CuCl2 for 10 min at 20 °C. The reaction was stopped with 1 mm EDTA. One sample was treated with 10 mm dithiothreitol (+DTT) after incubation with CuCl2. Samples were then assayed for verapamil-stimulated ATPase activity. Each value is the mean ± S.D. (n = 3).
We then tested whether cross-linking of mutant A259C/W803C could be inhibited by nucleotides. Fig. 4, A and B, shows that the presence of ATP or adenosine 5′-(β,γ-imino)triphosphate inhibited cross-linking. By contrast, the presence of ADP did not affect cross-linking (Fig. 4, A and B). These results suggest that ATP binding mediates conformational changes at the IH2/IH3 interface.
To test the effect of cross-linking on activity, histidine-tagged mutant A259C/W803C was expressed in HEK 293 cells in the presence of cyclosporine A and P-gp isolated by nickel-chelate chromatography. The isolated protein was reconstituted with lipid and treated with or without 0.2 mm CuCl2 oxidant at room temperature for 10 min. The reaction was performed at room temperature to promote cross-linking using reduced levels of CuCl2 that could be quenched with low levels of EDTA. The reaction was stopped by addition of 1 mm EDTA. Samples were then assayed for verapamil stimulation of ATPase activity. In the absence of oxidant, the mutant exhibited about 90% of wild-type P-gp activity (Fig. 4C). Cross-linking inhibited activity by about 85%. Inhibition by cross-linking could be reversed by treatment of the sample with 10 mm DTT (Fig. 4C). The results show that cross-linking IH2/IH3 in close proximity inhibits ATPase activity. It is possible that IH2/IH3 cross-linking blocks conformational changes associated with binding of ATP to the NBDs.
Mutations to Tyr1087 in NBD2 Inhibit Maturation and Activity
In a molecular dynamics simulation study of human P-gp, Tyr1087 was predicted to mediate the key contact between IH3 and NBD2 (34). To test whether Tyr1087 was important for folding or mediating drug stimulation of ATPase activity, we tested the effect of Y1087A, Y1087L, and Y1087F mutations. The A52-tagged mutants were expressed in HEK 293 cells in the absence or presence of cyclosporine A, and samples of whole cell SDS extracts were subjected to immunoblot analysis. Fig. 5A shows that the mature 170-kDa protein was the major product in mutant Y1087F, whereas the 150-kDa immature protein was the major product in mutants Y1087A and Y1087L. Both mutants Y1087A and Y1087L, however, could be rescued when they were expressed in the presence of cyclosporine A (Fig. 5A). These results show that maturation of P-gp was more sensitive to changes to Tyr1087 compared with Phe1086 because the F1086L mutation did not inhibit maturation (16).
FIGURE 5.
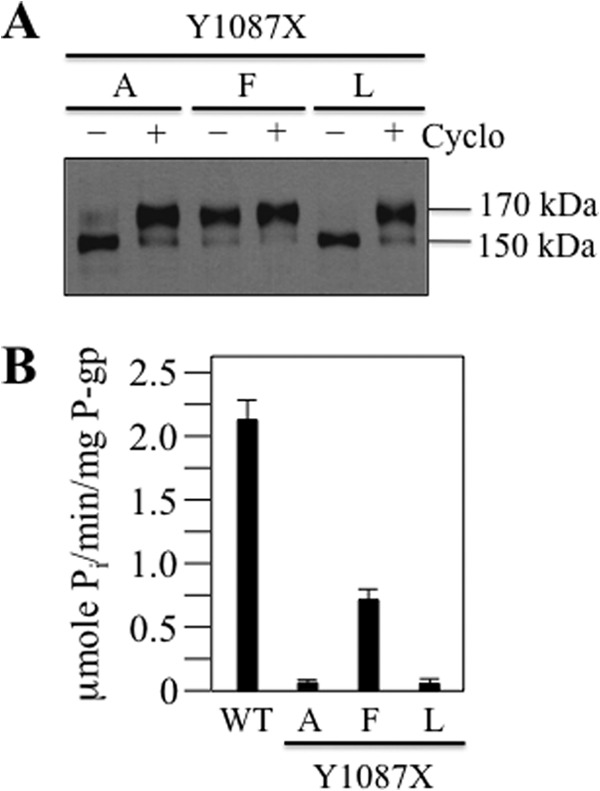
Tyr1087 mutations inhibit maturation and activity. A, A52-tagged Y1087A (A), Y1087F (F), and Y1087L (L) mutants were expressed in the absence (−) or presence (+) of 5 μm cyclosporine A (Cyclo). Samples of whole cell SDS extracts were subjected to immunoblot analysis. The positions of mature (170-kDa) and immature (150-kDa) P-gps are indicated. B, histidine-tagged WT, Y1087A (A), Y1087F (F), and Y1087L (L) P-gp were expressed in HEK 293 cells and isolated by nickel-chelate chromatography. Mutants Y1087A and Y1087L were expressed in the presence of cyclosporine A to promote maturation. After reconstitution with lipid, the ATPase activities were measured in the presence of verapamil. Each value is the mean ± S.D. (n = 3).
To test the effects of the mutations on activity, histidine-tagged mutants Tyr1087 mutants were expressed in the presence of cyclosporine A, isolated by nickel-chelate chromatography, and assayed for verapamil-stimulated ATPase activity. It was found that the Y1087A and Y1087L mutations reduced activity by over 90% (Fig. 5B). Mutant Y1087F showed about one-third of wild-type P-gp ATPase activity. These results show that Tyr1087 makes important contributions to both folding and activity to P-gp. Mutations of Tyr1087 to Ala or Leu severely reduced both maturation and activity of P-gp, and even the small change of replacing Tyr with Phe caused about a 70% reduction in activity.
DISCUSSION
The first IHs (IH1 and IH3) in each TMD (Fig. 1A) that link the TMDs to NBDs in the same half of the protein are generally only found in ABC exporters (46), whereas IH2 and IH4, which make the TMD1/NBD2 or TMD2/NBD1 connections, respectively (Fig. 1A), are found in all ABC transporters. The recent crystal structure of P-gp from C. elegans (17) showed that IH2 and IH4 make more extensive contacts with the NBDs compared with IH1 and IH3 (salt bridges, hydrogen bonds, or van der Waals interactions).
In TMD1, only three residues in IH1 were found to make contacts with NBD1, whereas 14 residues in IH2 make contact with NBD2. IH2 appeared to be more important for folding as point mutations to five of nine residues between Ile261 to Gly269 inhibited P-gp maturation, whereas no point mutations to IH1 or flanking amino acids (residues Gln158 to Gly169) inhibited maturation (16).
For TMD2, five residues in IH3 interacted with NBD2, whereas 11 residues in IH4 make contacts with NBD1 (17). In contrast to IH1, mutations to two of seven residues (Trp803 and Phe804) in IH3 inhibited P-gp maturation. The relatively high number of point mutations in IH2 and IH3 that inhibit maturation suggests that TMD/NBD2 interactions are particularly important for P-gp to fold into a native structure. P-gp is different from CFTR, its structurally similar sister protein (42), because deletion of NBD2 only inhibits P-gp maturation. It should be noted, however, that simulation studies suggest that IH1 and IH3 are predicted to make contacts with NBD2 and NBD1, respectively, when the protein adopts a closed conformation (45).
The effects of mutations to residues in IH2 and IH3 may be different. For IH2, four of five processing mutations were to residues predicted to form contacts with NBD2 (17). For IH3, only mutations to residues not predicted to interact with NBD2 (Trp803, Phe804) inhibited P-gp maturation. In the human P-gp model (17), Trp803 and Phe804 face away from NBD2 and appear to form part of a hydrophobic pocket network (with Phe793, Leu797, Leu814, and Leu818) between the cytoplasmic extensions of TM segments 8 and 9 (Fig. 6). In support of the prediction that Trp803 and Phe804 form a hydrophobic pocket with these residues, alanine-scanning mutagenesis of the 793–797 and 814–818 regions showed that only the F793A, L797A, L814A, and L818A mutations inhibited maturation (Fig. 2B). The bulkiness and hydrophobicity of Trp803 and Phe804 may be critical to fold into the native hairpin structure around IH3 because only replacement with aromatic residues yielded substantial levels of mature protein in the absence of drug substrates.
FIGURE 6.
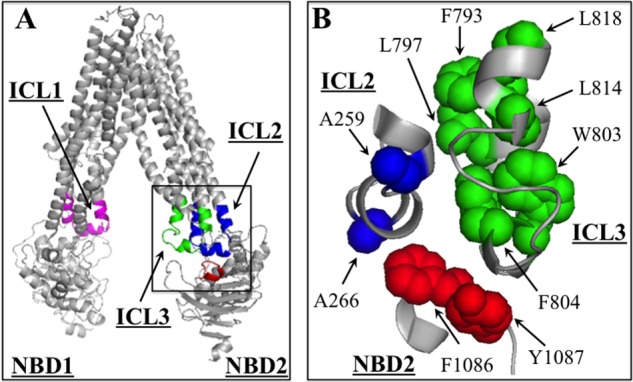
Model of the IH2/IH3-NBD2 interface critical for P-gp maturation and activity. A, model of human P-gp in the open conformation (17). The locations of ICL1, ICL2, and ICL3 are shown. Some regions within the highlighted inset are expanded in B. B, segments 256–269 (ICL2), 793–818 (ICL3), and 1081–1089 (NBD2) are shown. The location of residues Phe793, Leu797, Trp803, Phe804, Leu814, and Leu818 (green), predicted to form a hydrophobic pocket critical for maturation, is shown in green. Cross-linking of A259C (blue) in ICL2 to W803C (green) in ICL3 inhibited activity. Residue Phe1086 (red) has been shown previously to interact with A266C (red) (16) and, together with Tyr1087 (red) in NBD2, is predicted to form an important part of the IH2/IH3-NBD2 network important for folding and activity of P-gp.
The importance of the IH3/TM8/TM9 region for folding has been established previously with the observation that TMDs 8 and 9 were not inserted into the membrane in some processing mutants (47). It was observed that some processing mutants were core-glycosylated at Asn809 (Asn809-Thr810-Thr811 consensus glycosylation site), adjacent to IH3. This Asn809 site is normally not glycosylated because IH3 is located on the cytoplasmic side of the membrane. Remarkably, the defect can be corrected by carrying out expression in the presence of a drug substrate to yield an active molecule. It appears that drug substrates promote packing of the TM segments into a native structure. Drug substrates also promote maturation of P-gp mutants lacking NBD2 (30). Expression in the presence of drug substrates could also promote maturation of most Trp803- and Phe804-processing mutants (Fig. 3).
Molecular dynamic simulation studies of P-glycoprotein predicted that IH2/IH3-NBD2 interactions would be important in driving the necessary conformational changes to couple drug binding to binding and hydrolysis of ATP (34, 45). Studies of several crystal structures of the bacterial ABC maltose transporter have demonstrated that its reaction cycle is accompanied by rotation of IH segments within the cleft of the NBDs (29). In addition, mutations to the IH-NBD interfaces disrupted activity and structure of the protein (48).
A detailed molecular simulation study of the IH2, IH3, NBD2 interaction network in human P-gp was recently reported (34). The simulation predicted that rotations of IH2 relative to the adjacent IH3 might be a key feature of the coupling reaction and that a key contact between IH3 and NBD2 was Tyr1087. In support of these predictions, we found that clamping IH2 to IH3 by oxidative cross-linking of mutant A259C/W803C or mutations to Tyr1087 inhibited activity.
Binding of ATP appeared to influence conformational changes at the IH2/IH3 interface because ATP, but not ADP, inhibited cross-linking (Fig. 4). Although we showed previously that mutation of the adjacent Phe1086 to Ala also inhibited activity (16), the activity of P-gp was much more sensitive to changes to Tyr1087. For example, mutation of Phe1086 to Leu, Phe, or Trp yielded P-gps with activities similar to the wild-type protein. Mutation of Tyr1087 to Leu severely inhibited activity, whereas the conservative Y1087F change caused about a 70% reduction in activity. The molecular simulation study (34) suggested that Tyr1087 interacted with IH3 through a hydrogen bond with Asp805. In support of this prediction, it has been reported recently that a P-gp mutant containing the D805C mutation reduced verapamil-stimulated ATPase activity by about 70% but had no effect on the Km for ATP (49).
In summary, we have identified a key IH2/IH3/NBD2 hydrophobic network with dual functions. It forms an important TMD/NBD transmission interface to couple drug binding to ATPase activity, and the interface is also critical for folding of P-gp into a native structure.
Acknowledgment
We thank M. Claire Bartlett for technical contributions.
This work was supported by a grant from the Canadian Institutes of Health Research (to D. M. C.).
- P-gp
- P-glycoprotein
- ABC
- ATP-binding cassette
- TMD
- transmembrane domain
- NBD
- nucleotide-binding domain
- ICL
- intracellular loop
- IH
- intracellular helix.
REFERENCES
- 1. Juliano R. L., Ling V. (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162 [DOI] [PubMed] [Google Scholar]
- 2. Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., Gottesman M. M. (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398 [DOI] [PubMed] [Google Scholar]
- 3. Eckford P. D., Sharom F. J. (2009) ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109, 2989–3011 [DOI] [PubMed] [Google Scholar]
- 4. Chen C. J., Chin J. E., Ueda K., Clark D. P., Pastan I., Gottesman M. M., Roninson I. B. (1986) Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 47, 381–389 [DOI] [PubMed] [Google Scholar]
- 5. Dey S., Ramachandra M., Pastan I., Gottesman M. M., Ambudkar S. V. (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 10594–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lugo M. R., Sharom F. J. (2005) Interaction of LDS-751 with P-glycoprotein and mapping of the location of the R drug binding site. Biochemistry 44, 643–655 [DOI] [PubMed] [Google Scholar]
- 7. Loo T. W., Bartlett M. C., Clarke D. M. (2009) Identification of residues in the drug-translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J. Biol. Chem. 284, 24074–24087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 278, 13603–13606 [DOI] [PubMed] [Google Scholar]
- 9. Loo T. W., Clarke D. M. (1995) Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J. Biol. Chem. 270, 22957–22961 [DOI] [PubMed] [Google Scholar]
- 10. Sauna Z. E., Kim I. W., Nandigama K., Kopp S., Chiba P., Ambudkar S. V. (2007) Catalytic cycle of ATP hydrolysis by P-glycoprotein. Evidence for formation of the E.S reaction intermediate with ATP-γ-S, a nonhydrolyzable analogue of ATP. Biochemistry 46, 13787–13799 [DOI] [PubMed] [Google Scholar]
- 11. Siarheyeva A., Liu R., Sharom F. J. (2010) Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides. Implications for catalysis. J. Biol. Chem. 285, 7575–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delannoy S., Urbatsch I. L., Tombline G., Senior A. E., Vogel P. D. (2005) Nucleotide binding to the multidrug resistance P-glycoprotein as studied by ESR spectroscopy. Biochemistry 44, 14010–14019 [DOI] [PubMed] [Google Scholar]
- 13. Urbatsch I. L., Sankaran B., Weber J., Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 [DOI] [PubMed] [Google Scholar]
- 14. Gutmann D. A., Ward A., Urbatsch I. L., Chang G., van Veen H. W. (2010) Understanding polyspecificity of multidrug ABC transporters. Closing in on the gaps in ABCB1. Trends Biochem. Sci. 35, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loo T. W., Bartlett M. C., Clarke D. M. (2010) Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 395, 436–440 [DOI] [PubMed] [Google Scholar]
- 16. Loo T. W., Bartlett M. C., Clarke D. M. (2013) Human P-glycoprotein contains a greasy ball-and-socket joint at the second transmission interface. J. Biol. Chem. 288, 20326–20333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin M. S., Oldham M. L., Zhang Q., Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loo T. W., Bartlett M. C., Clarke D. M. (2004) Val-133 and Cys-137 in transmembrane segment 2 are close to residues Arg-935 and Gly-939 in transmembrane segment 11 of human P-glycoprotein. J. Biol. Chem. 279, 18232–18238 [DOI] [PubMed] [Google Scholar]
- 19. Loo T. W., Bartlett M. C., Clarke D. M. (2004) Disulfide cross-linking analysis shows that transmembrane segments 5 and 8 of human P-glycoprotein are close together on the cytoplasmic side of the membrane. J. Biol. Chem. 279, 7692–7697 [DOI] [PubMed] [Google Scholar]
- 20. Loo T. W., Bartlett M. C., Clarke D. M. (2006) Transmembrane segment 1 of human P-glycoprotein contributes to the drug binding pocket. Biochem. J. 396, 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loo T. W., Bartlett M. C., Clarke D. M. (2007) Suppressor mutations in the transmembrane segments of P-glycoprotein promote maturation of processing mutants and disrupt a subset of drug-binding sites. J. Biol. Chem. 282, 32043–32052 [DOI] [PubMed] [Google Scholar]
- 22. Loo T. W., Bartlett M. C., Clarke D. M. (2008) Arginines in the first transmembrane segment promote maturation of a P-glycoprotein processing mutant by hydrogen bond interactions with tyrosines in transmembrane segment 11. J. Biol. Chem. 283, 24860–24870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loo T. W., Clarke D. M. (2013) Drug rescue distinguishes between different structural models of human P-glycoprotein. Biochemistry 52, 7167–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loo T. W., Clarke D. M. (2013) A salt bridge in intracellular loop 2 is essential for folding of human P-glycoprotein. Biochemistry 52, 3194–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ravna A. W., Sylte I., Sager G. (2007) Molecular model of the outward facing state of the human P-glycoprotein (ABCB1), and comparison to a model of the human MRP5 (ABCC5). Theor. Biol. Med. Model. 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Globisch C., Pajeva I. K., Wiese M. (2008) Identification of putative binding sites of P-glycoprotein based on its homology model. ChemMedChem. 3, 280–295 [DOI] [PubMed] [Google Scholar]
- 27. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 28. Rees D. C., Johnson E., Lewinson O. (2009) ABC transporters. The power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khare D., Oldham M. L., Orelle C., Davidson A. L., Chen J. (2009) Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell 33, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loo T. W., Clarke D. M. (1999) The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem. 274, 24759–24765 [DOI] [PubMed] [Google Scholar]
- 31. Cui L., Aleksandrov L., Chang X. B., Hou Y. X., He L., Hegedus T., Gentzsch M., Aleksandrov A., Balch W. E., Riordan J. R. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 365, 981–994 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y., Loo T. W., Bartlett M. C., Clarke D. M. (2007) Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol. Pharmacol. 71, 751–758 [DOI] [PubMed] [Google Scholar]
- 33. Loo T. W., Bartlett M. C., Detty M. R., Clarke D. M. (2012) The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem. 287, 26806–26816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pajeva I. K., Hanl M., Wiese M. (2013) Protein contacts and ligand binding in the inward-facing model of human P-glycoprotein. ChemMedChem. 8, 748–762 [DOI] [PubMed] [Google Scholar]
- 35. Loo T. W., Clarke D. M. (1994) Functional consequences of glycine mutations in the predicted cytoplasmic loops of P-glycoprotein. J. Biol. Chem. 269, 7243–7248 [PubMed] [Google Scholar]
- 36. Loo T. W., Clarke D. M. (1995) Rapid purification of human P-glycoprotein mutants expressed transiently in HEK 293 cells by nickel-chelate chromatography and characterization of their drug-stimulated ATPase activities. J. Biol. Chem. 270, 21449–21452 [DOI] [PubMed] [Google Scholar]
- 37. Loo T. W., Clarke D. M. (1997) Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem. 272, 709–712 [DOI] [PubMed] [Google Scholar]
- 38. Loo T. W., Clarke D. M. (1995) P-glycoprotein. Associations between domains and between domains and molecular chaperones. J. Biol. Chem. 270, 21839–21844 [DOI] [PubMed] [Google Scholar]
- 39. Chifflet S., Torriglia A., Chiesa R., Tolosa S. (1988) A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein. Application to lens ATPases. Anal. Biochem. 168, 1–4 [DOI] [PubMed] [Google Scholar]
- 40. Loo T. W., Clarke D. M. (1995) Membrane topology of a cysteine-less mutant of human P-glycoprotein. J. Biol. Chem. 270, 843–848 [DOI] [PubMed] [Google Scholar]
- 41. Loo T. W., Bartlett M. C., Clarke D. M. (2006) Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem. J. 399, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Loo T. W., Bartlett M. C., Shi L., Clarke D. M. (2012) Corrector-mediated rescue of misprocessed CFTR mutants can be reduced by the P-glycoprotein drug pump. Biochem. Pharmacol. 83, 345–354 [DOI] [PubMed] [Google Scholar]
- 43. Omote H., Al-Shawi M. K. (2002) A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J. Biol. Chem. 277, 45688–45694 [DOI] [PubMed] [Google Scholar]
- 44. Loo T. W., Bartlett M. C., Clarke D. M. (2003) Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J. Biol. Chem. 278, 50136–50141 [DOI] [PubMed] [Google Scholar]
- 45. Wise J. G. (2012) Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 51, 5125–5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oldham M. L., Davidson A. L., Chen J. (2008) Structural insights into ABC transporter mechanism. Curr. Opin. Struct. Biol. 18, 726–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Loo T. W., Clarke D. M. (1999) The glycosylation and orientation in the membrane of the third cytoplasmic loop of human P-glycoprotein is affected by mutations and substrates. Biochemistry 38, 5124–5129 [DOI] [PubMed] [Google Scholar]
- 48. Mourez M., Hofnung M., Dassa E. (1997) Subunit interactions in ABC transporters. A conserved sequence in hydrophobic membrane proteins of periplasmic permeases defines an important site of interaction with the ATPase subunits. EMBO J. 16, 3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kapoor K., Bhatnagar J., Chufan E. E., Ambudkar S. V. (2013) Mutations in intracellular loops 1 and 3 lead to misfolding of human P-glycoprotein (ABCB1) that can be rescued by cyclosporine A, which reduces its association with chaperone Hsp70. J. Biol. Chem. 288, 32622–32636 [DOI] [PMC free article] [PubMed] [Google Scholar]