

Background: The suppression of selenoprotein P production may be a novel therapeutic target for reducing insulin resistance.
Results: Selenoprotein P expression was suppressed by metformin treatment, but co-administration of AMPK inhibitor or FoxO3a siRNA cancelled this suppression.
Conclusion: Metformin suppresses selenoprotein P expression via the AMPK/FoxO3a pathway.
Significance: The AMPK/FoxO3a pathway in the liver may be a therapeutic target for type 2 diabetes.
Keywords: AMP-activated Kinase (AMPK), Gene Expression, Hepatocyte, Insulin Resistance, Transcription Promoter, FoxO3a, Hepatokine, Metformin, Selenoprotein P
Abstract
Selenoprotein P (SeP; encoded by SEPP1 in humans) is a liver-derived secretory protein that induces insulin resistance in type 2 diabetes. Suppression of SeP might provide a novel therapeutic approach to treating type 2 diabetes, but few drugs that inhibit SEPP1 expression in hepatocytes have been identified to date. The present findings demonstrate that metformin suppresses SEPP1 expression by activating AMP-activated kinase (AMPK) and subsequently inactivating FoxO3a in H4IIEC3 hepatocytes. Treatment with metformin reduced SEPP1 promoter activity in a concentration- and time-dependent manner; this effect was cancelled by co-administration of an AMPK inhibitor. Metformin also suppressed Sepp1 gene expression in the liver of mice. Computational analysis of transcription factor binding sites conserved among the species resulted in identification of the FoxO-binding site in the metformin-response element of the SEPP1 promoter. A luciferase reporter assay showed that metformin suppresses Forkhead-response element activity, and a ChIP assay revealed that metformin decreases binding of FoxO3a, a direct target of AMPK, to the SEPP1 promoter. Transfection with siRNAs for Foxo3a, but not for Foxo1, cancelled metformin-induced luciferase activity suppression of the metformin-response element of the SEPP1 promoter. The overexpression of FoxO3a stimulated SEPP1 promoter activity and rescued the suppressive effect of metformin. Metformin did not affect FoxO3a expression, but it increased its phosphorylation and decreased its nuclear localization. These data provide a novel mechanism of action for metformin involving improvement of systemic insulin sensitivity through the regulation of SeP production and suggest an additional approach to the development of anti-diabetic drugs.
Introduction
Selenoprotein P (SeP2; encoded by SEPP1 in humans) is a secretory protein produced mainly by the liver (1, 2). SeP contains 10 selenocysteine residues and is known to transport the essential trace element selenium from the liver to the rest of the body (3, 4). Our laboratory reported recently that SeP functions as a hepatokine that contributes to insulin resistance in type 2 diabetes (5). Using comprehensive gene expression analyses in humans, hepatic gene expression levels of SEPP1 were found to be positively correlated with the severity of insulin resistance in patients with type 2 diabetes. Moreover, treatment with purified SeP protein impairs insulin signal transduction in both cell culture and animal models. Importantly, the RNA interference-mediated knockdown of SeP improves insulin resistance and hyperglycemia in a mouse model of type 2 diabetes, suggesting that the suppression of SeP production in the liver may be a novel therapeutic target for reducing insulin resistance in type 2 diabetes (5). However, few drugs that inhibit the production of SeP by hepatocytes have been identified to date.
Metformin is widely used as an anti-diabetic drug globally. The primary target of metformin action is the liver, which abundantly expresses organic cation transporter (Oct)-1, a transporter for metformin (6, 7). Adenosine monophosphate-activated protein kinase (AMPK) mediates primarily the glucose-lowering actions of metformin, including the suppression of hepatic gluconeogenesis (8). In contrast, several reports indicate that the oral administration of metformin in humans increases insulin sensitivity in skeletal muscle, increases serum adiponectin, and improves aortic arteriosclerosis (9–11). These reports suggest that orally administered metformin also exerts beneficial actions on tissues other than the liver, in which expression levels of Octs are lower. To date, however, the molecular mechanisms underlying the systemic actions of metformin are not fully understood.
Forkhead box protein O3a (FoxO3a), which belongs to the Forkhead transcription factors of the FoxO subfamily (FoxOs), is reported to be involved in cell cycle arrest (12), apoptosis (13), and the oxidative stress response (14, 15). Recently, Greer et al. (16) showed that FoxO3a, but not FoxO1, is directly phosphorylated and activated by AMPK in vitro. FoxO3a is reported to positively regulate mitochondria-related genes, such as uncoupling proteins, in mouse embryonic fibroblasts, suggesting that the direct regulation of FoxO3a by AMPK plays a crucial role in the control of the cellular energy balance. The phosphorylation of FoxO3a by AMPK was also identified in C2C12 myotubes (17), aortic vascular endothelial cells (18), and A549 lung cancer cells (19). However, the role of the AMPK/FoxO3a pathway in hepatocytes, an important target of metformin, remains unknown.
We demonstrate here that metformin suppresses SEPP1 gene expression by activating AMPK and subsequently inactivating FoxO3a in H4IIEC3 hepatocytes. These results suggest a novel mechanism underlying the glucose-lowering action of metformin.
EXPERIMENTAL PROCEDURES
Materials
The antibodies against AMPKα, phospho-AMPKα, FoxO1, FoxO3a, acetylated Lys, and Lamin A/C were purchased from Cell Signaling Technology (Beverly, MA). Antibody against phospho-Ser/Thr/Tyr was purchased from AnaSpec (San Jose, CA). Antibody against GAPDH was purchased from Santa Cruz Biotechnology, Inc. 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) and 6-[4-(2-Piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo[1,5-a]-pyrimidine (compound C) were purchased from Sigma-Aldrich. FoxO3a expression vector was provided from Ajinomoto Pharma (Tokyo, Japan) described before (20). Selenious acid was purchased from WaKo Pure Chemical Industries, Ltd. (Osaka, Japan). Metformin was provided by Dainippon Sumitomo Pharma (Osaka, Japan).
Generation of Plasmid Constructs
The human SEPP1 promoter region has been described previously (21). Fragments of ∼1800 bp from the human SEPP1 promoter region and the deletion promoter region were amplified by PCR using normal human genomic DNA as a template and the primer pairs shown in Table 1. The PCR product was subcloned into the luciferase reporter gene plasmid pGL3-basic (Promega, Madison, WI) and termed “SEPP1-Promoter-Luc,” “Mut-A,” “Mut-B,” “Mut-C,” “Mut-D,” “Mut-E,” “Mut-DΔ1,” “Mut-DΔ2,” and “Mut-DΔ3.” Putative FoxO binding site-deficient vector were generated using QuikChange Lightning site-directed mutagenesis kits (Agilent Technologies, Santa Clara, CA), according to the manufacturer's instructions. All inserts were confirmed by DNA sequencing.
TABLE 1.
Primers used in cloning
| Primer | Description | Sequence |
|---|---|---|
| SEPP1-Promoter-Luc | ||
| Forward | hSeP-promoter-F-BglII | ACTAGATCTACAAACCTTTCAGACACTGAGTTG |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-A | ||
| Forward | hSeP-promoter-Del-F1-BglII | ACTAGATCTGGGCTGCCTGTCTTTGATTTCACAT |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-B | ||
| Forward | hSeP-promoter-Del-F2-BglII | ACTAGATCTTTGTAGTTCCTGCACCTTGTACAAC |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-C | ||
| Forward | hSeP-promoter-Del-F3-BglII | ACTAGATCTGCATAGGTCTTCCAGGAAGTACGAC |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-D | ||
| Forward | hSeP-promoter-Del-F4-BglII | ACTAGATCTCAAATGTTTTTCCCTGTTATAGTTT |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-E | ||
| Forward | hSeP-promoter-F-BglII | ACTAGATCTACAAACCTTTCAGACACTGAGTTG |
| Reverse | hSeP-promoter-Del-R1-NocI | ACTCCATGGCTGAGCCAGCGAATTATGCTGCTGC |
| Mut-DΔ1 | ||
| Forward | hSeP-promoter-Del-F14-BglII | ACTAGATCTGATTCTAGGGTGACTGAAAAGGATA |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-DΔ2 | ||
| Forward | hSeP-promoter-Del-F15-BglII | ACTAGATCTATAACAATCAGCTCAGGGGTTTGCT |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-DΔ3 | ||
| Forward | hSeP-promoter-Del-F16-BglII | ACTAGATCTATAAATATCAGAGTGTGCTGCTGTG |
| Reverse | hSeP-promoter-R-NcoI | ACTCCATGGACAACCACTTCCAACGGGCCTGCTT |
| Mut-DΔ2-ΔFoxo A | ||
| Forward | del86–95 | GACTATACCTGAGGGGTGAGGGACTATAAATATCAGAGTG |
| Reverse | del86–95-antisense | CACTCTGATATTTATAGTCCCTCACCCCTCAGGTATAGTC |
| Mut-DΔ2-ΔFoxo B | ||
| Forward | hSeP-del-Foxo 3-F | GAGGTAAACAACAGGACTAAGAGTGTGCTGCTGTGG |
| Reverse | hSeP-del-Foxo 3-R | CCACAGCAGCACACTCTTAGTCCTGTTGTTTACCTC |
Cell Culture
Studies were performed using the rat hepatoma cell line H4IIEC3 (American Type Culture Collection, Manassas, VA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) and supplemented with 10% fetal bovine serum (Invitrogen), 2 mmol/liter l-glutamine (WaKo Pure Chemical Industries, Ltd.), 100 units/ml penicillin, and 0.1 mg/ml streptomycin (WaKo Pure Chemical Industries, Ltd.). The cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
Measurement of Glutathione Peroxidase Activity
To measure cellular glutathione peroxidase (cGPx) activity, a coupled enzyme assay, which was performed by following the oxidation of NADPH, was used as described previously (22). In brief, cells were cultured with 1) DMEM plus 10% FBS, 2) DMEM plus 10% FBS and 100 nm selenious acid, or 3) DMEM plus 10% FBS and 1000 nm selenious acid at 72 h. Then cells were fractured with homogenate buffer containing 0.25 m sucrose, 50 mm Tris-HCl (pH 7.4), 0.1 mm EDTA, 0.1 mm 2-mercaptoethanol. The assay conditions were as follows for the cGPx assay: 0.1 m phosphate buffer, pH 7.4, 0.2 mm NADPH, 0.5 mm EDTA, 1 mm NaN3, 2 mm GSH, 1 unit/ml GSH reductase, and 30 μm hydrogen peroxide. The oxidation of NADPH was followed at 340 nm at 37 °C, and units of the enzyme activity were expressed as μmol of NADPH oxidized/min.
Transfection and Luciferase Reporter Gene Assay
H4IIEC3 cells were grown in 24-well plates and transfected with 0.4 μg of plasmid DNA/well together with 1.2 μl of FuGENE6 (Promega). For the luciferase reporter gene assays, 0.4 μg of firefly luciferase promoter construct was co-transfected with 0.01 μg of Renilla luciferase control plasmid (pRL-SV40; Promega) and/or 0.05–0.4 μg of plasmids expressing FoxO3a or empty control plasmids, resulting in a total DNA amount of 0.41–0.81 μg/well. 24 h later, cells were treated with the indicated reagents, such as metformin, in DMEM plus 10% FBS for the indicated times. After 48 h, luciferase activities were measured using the Dual Luciferase assay system (Promega), as described previously (20).
siRNA Transfection in H4IIEC3 Hepatocytes
H4IIEC3 hepatocytes were grown in 24-well plates and transiently transfected with 10 nm small interfering RNA (siRNA) duplex oligonucleotides using 1 μl of LipofectamineTM RNAiMAX (Invitrogen) by the reverse transfection method according to the manufacturer's instructions. Foxo1- and Foxo3a-specific siRNAs with the following sequences were synthesized (Thermo Scientific): Foxo1 A, 5′-GACAGCAAAUCAAGUUAUG-3′ (sense); Foxo1 B, 5′-UUUGAUAACUGGAGUACAU-3′ (sense); Foxo3a A, 5′-GAACGUUGUUGGUUUGAAC-3′ (sense); and Foxo3a B, 5′-CGUCAUGGGUCACGACAAG-3′ (sense). Negative control siRNA was also utilized (Thermo Scientific). 24 h after transfection, the cells were treated with metformin for 24 h, followed by the extraction of total RNA.
Adenovirus-mediated Gene Transfer in H4IIEC3 Hepatocytes
Cells were transfected with adenoviruses as described previously (5). Briefly, H4IIEC3 hepatocytes were grown to 90% confluence in 24-well multiplates and transfected with adenoviruses encoding dominant negative (DN) α1 and α2 AMPK, constitutive active (CA) AMPK, or LacZ for 4 h. The cells were incubated with DMEM for 24 h after removing the adenoviruses; total RNA was then extracted.
Quantitative RT-PCR
Total RNA was extracted from cultured H4IIEC3 hepatocytes using a Genelute mammalian total RNA miniprep kit (Sigma). The reverse transcription of 100 ng of total RNA was performed using a high capacity cDNA reverse transcription kit (Invitrogen), according to the manufacturer's instructions. Quantitative RT-PCR was performed using TaqMan probes (ACTB, 4352340E; Foxo1, Rn01494868_m1; Foxo3, Rn01441087_m1; G6pc, Rn00565347_m1; Pck1, Rn01529014_m1; Sepp1, Rn00569905_m1) and the 7900HT fast real-time PCR system (Invitrogen), as described previously (23).
Western Blotting
Treated cells were collected and lysed as described previously (20). Protein samples (10 μg/lane) were subjected to SDS-PAGE and transferred to PVDF membranes using the iBlot Gel Transfer system (Invitrogen). The membranes were blocked, incubated with primary antibody, washed, and incubated with the secondary HRP-labeled antibody. Bands were visualized with the ECL Prime Western blotting Detection System (GE Healthcare) and LAS-3000 (Fujifilm, Tokyo, Japan). A densitometric analysis of blotted membranes was performed using ImageJ software.
Immunoprecipitation
Immunoprecipitation of serine/threonine/tyrosine-phosphorylated proteins or lysine-acetylated proteins was carried out using the Dynabeads protein G immunoprecipitation kit (Invitrogen) according to the manufacturer's instructions. The nuclear and cytoplasmic fractions were extracted using an NE-PER nuclear and cytoplasmic extraction reagent kit (Pierce).
Detection of the Conserved Transcription Factor Binding Sites Using Multiple-genome Alignments
The Ensembl 12-way Enredo-Pecan-Ortheus (EPO) eutherian multiple alignments (12-way EPO alignments) (24, 25) were downloaded. The 12-way EPO was excised to obtain the alignment block corresponding to the human genome coordinates from 10 kb upstream of the coding sequence of SEPP1, including the start codon. To predict the conserved transcription factor binding sites (TFBSs), the 10-kb upstream genome sequence for each of the 12 species was searched using the TRANSFAC (26) and the MATCHTM program (27) (version 6.1) with varied thresholds. Then the predicted TFBSs were mapped on the alignments, and the conserved TFBSs for SEPP1 were identified.
Chromatin Immunoprecipitation (ChIP) Assay
A ChIP assay was carried out using the ChIP IT Express enzymatic kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. In brief, HepG2 cells were treated with metformin 6 h before being fixed and homogenized. Following centrifugation, the supernatant was used for chromatin samples. Chromatin samples were incubated with protein G-coated magnetic beads and ChIP grade FoxO1 or FoxO3a antibodies (Abcam, Cambridge, MA) overnight at 4 °C. Following washing and elution, a reaction solution was used as the template for PCR. PCR primers were set for amplification of the Mut-DΔ2 region of the SEPP1 promoter, as follows: forward, 5′-GCACTTGCTACTTTCTTTTAAGTTG-3′; reverse, 5′-CACAGCAGCACACTCTGATATTTAT-3′.
Animals
12-week-old C57BL/6J female mice were obtained from CLEA Japan, Inc. (Tokyo, Japan). All animals were housed in a 12-h light/dark cycle and allowed free access to food and water. Following the fasting for 4 h, mice were administrated 300 mg/kg metformin. 4 h later, mice were anesthetized and sacrificed to allow isolation of liver tissue.
Statistical Analysis
Results are expressed as means ± S.D. Significance was tested by one-way analysis of variance with the Bonferroni method, and differences were considered statistically significant at a p value of less than 0.05.
RESULTS
Metformin Suppresses SEPP1 Expression at the Promoter Level
The effects of metformin on Sepp1 expression in H4IIEC3 hepatocytes were examined. Metformin suppressed Sepp1 mRNA expression in a concentration- and time-dependent manner, similarly to G6pc and Pck1, which encode representative gluconeogenic enzymes glucose-6-phosphatase and phosphoenolpyruvate carboxykinase 1, respectively (Fig. 1, A and B). These results are consistent with a previous report using rat primary hepatocytes (28). Next, the effects of metformin on SEPP1 promoter activity were examined. The human SEPP1 promoter region was cloned to a luciferase reporter vector as reported previously (21). The present sequence completely corresponded to the reference sequence of the National Center for Biotechnology Information, but it missed one thymidine against the sequence of the previous report (accession number Y12262) (supplemental Fig. S1). Similar to the mRNA results, metformin suppressed SEPP1 promoter activity in a concentration- and time-dependent manner (Fig. 1, C and D), suggesting that it directly decreases SEPP1 transcriptional activity in H4IIEC3 hepatocytes.
FIGURE 1.

Metformin suppressed Sepp1 gene expression in H4IIEC3 hepatocytes and livers of C57BL/6J mice. A and B, metformin suppressed Sepp1 mRNA expression in a concentration- and time-dependent manner. H4IIEC3 cells were treated with the indicated concentrations of metformin for the indicated times. Expression values were normalized to Actb mRNA. Data represent means ± S.D. (error bars) (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle-treated cells or 0 h. C and D, SEPP1 promoter activity was suppressed in a concentration- and time-dependent manner. H4IIEC3 cells were co-transfected with the SEPP1 promoter reporter vector and control reporter vector. 24 h later, the cells were treated with the indicated concentrations of metformin for the indicated times. Values were normalized to the activity of the control luciferase vector. Data represent means ± S.D. (n = 4). **, p < 0.01; ***, p < 0.001 versus vehicle-treated cells or 0 h. E and F, metformin suppressed Sepp1 mRNA expression in livers of C57BL/6J mice. Following fasting for 4 h, 12-week-old female C57BL/6J mice were administrated 300 mg/kg metformin. 4 h after metformin administration, mice were sacrificed, and liver mRNA expression was examined. Expression values were normalized to Actb mRNA. Data represent means ± S.D. (n = 7). *, p < 0.05 versus PBS-injected mice.
To confirm whether the present experimental condition (DMEM plus 10% FBS) supplied selenium sufficiently to synthesize selenoproteins for cultured cells, cGPx activity was measured with or without additional selenium supplement. Supplemental Fig. S2 indicates that supplementation of 100 or 1000 nm selenious acid to DMEM plus 10% FBS increased cGPx activity more than 3 times, suggesting that our experimental condition was insufficient to maximize selenoprotein synthesis. However, the current activity of cGPx in the cells cultured at DMEM plus 10% FBS (233 units/g) corresponded to the levels reported previously in the normal rat liver tissue (120–1800 units/g) (29, 30). Because these results suggest that the culture condition of DMEM plus 10% FBS was physiological, we used this condition in the following cellular experiments.
The action of metformin on Sepp1 was also examined in mice. Following fasting for 4 h, 12-week-old female C57BL/6J mice were administrated 300 mg/kg metformin. Metformin decreased blood glucose levels by 30% (Fig. 1E) and tended to down-regulate gene expression for G6pc and Pck1 after 4 h. Gene expression of Sepp1 was significantly decreased by metformin (Fig. 1F). These results indicate that metformin suppresses gene expression for Sepp1 in the liver of mice as well as in the cultured hepatocytes.
Metformin Suppresses SEPP1 Promoter Activity via AMPK Activation
Metformin is known to exert anti-diabetic effects by activating AMPK pathways (31). Hence, to determine whether AMPK pathways are involved in the metformin-induced suppression of SEPP1 promoter activity, cells were treated with compound C, a representative AMPK inhibitor. Findings confirmed that the metformin-induced phosphorylation of AMPK and acetyl-CoA carboxylase was cancelled by the co-administration of compound C in H4IIEC3 hepatocytes (Fig. 2A). Co-administration of compound C partly rescued the cells from the inhibitory effects of metformin on the SEPP1 promoter (Fig. 2B) and increased SEPP1 promoter activity in the absence of metformin (Fig. 2B). In contrast, treatment with AICAR, a known activator of AMPK, decreased SEPP1 promoter activity similarly to metformin (Fig. 2C). To determine whether AMPK pathways were involved in SEPP1 promoter activity, H4IIEC3 hepatocytes were infected with an adenovirus encoding CA- or DN-AMPK. Transfection with CA-AMPK suppressed Sepp1 and G6pc mRNA expression (Fig. 2D), whereas transfection with DN-AMPK enhanced Sepp1 and G6pc mRNA expression (Fig. 2E). These results suggest that metformin decreases SEPP1 promoter activity, at least partly, by activating AMPK.
FIGURE 2.

Metformin suppressed SEPP1 promoter activity via AMPK pathway in H4IIEC3 hepatocytes. A, metformin-induced AMPK phosphorylation in the absence or presence of compound C. H4IIEC3 cells were treated with the indicated concentrations of metformin and compound C for 24 h. AMPK phosphorylation was examined by Western blotting. B, compound C treatment recovered metformin-induced suppression of the SEPP1 promoter. H4IIEC3 cells were co-transfected with the SEPP1 promoter reporter vector and control reporter vector at 24 h and then treated with the indicated concentrations of metformin and compound C for 48 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (error bars) (n = 4). ***, p < 0.001. C, AICAR suppressed SEPP1 promoter activity. H4IIEC3 cells were co-transfected with the SEPP1 promoter reporter vector and control reporter vector at 24 h and then treated with 0.4 mm AICAR for 24 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (n = 4). ***, p < 0.001. D and E, influence of adenoviruses carrying constitutive active (CA) or dominant negative (DN) AMPK. H4IIEC3 cells were infected with adenoviruses encoding CA-AMPK, DN-AMPK, or LacZ. Expression values were normalized to Actb mRNA. Data represent means ± S.D. (n = 4). **, p < 0.01; ***, p < 0.001.
Metformin-response Element in the SEPP1 Promoter Includes the FoxO Binding Site
To determine the nature of the metformin-response element in the SEPP1 promoter region, several deletion mutants of the SEPP1 promoter were constructed (Fig. 3A). Promoter activity of Mut-A to Mut-D, but not Mut-E, was suppressed by metformin treatment (Fig. 3A), indicating that the metformin-response element of the SEPP1 promoter exists in Mut-D. Additional deletion mutants of Mut-D were constructed and named Mut-DΔ1 to DΔ3. Mut-DΔ1 and -DΔ2, but not Mut-DΔ3, were suppressed by metformin (Fig. 3B), indicating that the metformin-response element in the SEPP1 promoter is localized in the Mut-DΔ2 sequence. Using computational analysis to identify conserved TFBSs among the species (see “Experimental Procedures”), several putative TFBSs were identified in the Mut-DΔ2 sequence (supplemental Fig. S3). Because early reports indicate that AMPK directly phosphorylates FoxO3a and regulates its transcriptional activity (16), this investigation focused on the two putative FoxO binding sites (Fig. 4A).
FIGURE 3.
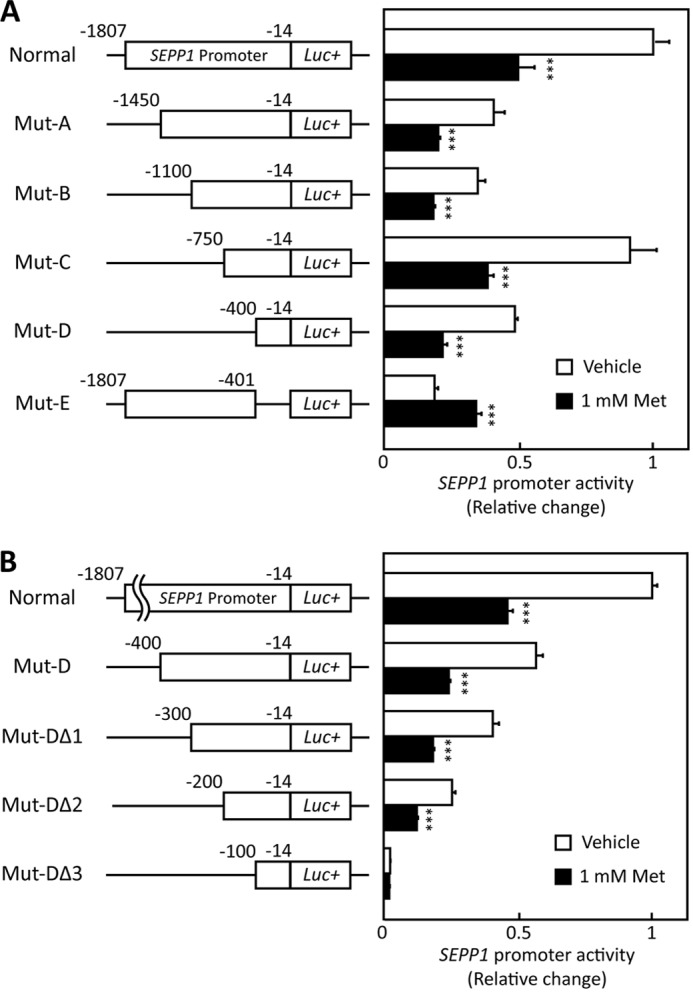
SEPP1 promoter activity of deletion mutants. A and B, structure and luciferase activity of promoter-deletion mutants. The sequences deleted within the constructs are shown as thin lines. The remaining parts of the SEPP1 promoter were fused to a luciferase reporter gene. H4IIEC3 cells were co-transfected with each reporter vector and control reporter vector at 24 h and then treated with the indicated concentrations of metformin for 48 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (error bars) (n = 4). ***, p < 0.001 versus vehicle-treated cells.
FIGURE 4.

Activation of the AMPK suppressed FoxO activity. A, putative FoxO3a binding sites of Mut-DΔ2 sequence. Detection of the conserved TFBSs was performed using multiple-genome alignments and the highlighted putative transcriptional factor binding sites. B, FoxO activity in the absence or presence of metformin and compound C. H4IIEC3 cells were co-transfected with the FHRE-Luc vector and control reporter vector at 24 h and then treated with the indicated concentrations of metformin and compound C for 48 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (error bars) (n = 4). **, p < 0.01; ***, p < 0.001. N.S., not significant. C, FoxO activity in the absence or presence of AICAR. H4IIEC3 cells were co-transfected with the FHRE-Luc vector and control reporter vector at 24 h and then treated with the indicated concentrations of AICAR for 24 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (n = 4). *, p < 0.05 versus vehicle-treated cells. D, deficiency of putative FoxO binding site cancelled metformin-induced suppression of SEPP1 promoter activity. H4IIEC3 cells were co-transfected with each reporter vector and control reporter vector at 24 h and then treated with the indicated concentrations of metformin for 24 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (n = 4). ***, p < 0.001 versus vehicle-treated cells. E, chromatin immunoprecipitation assay of HepG2 cells treated with metformin. HepG2 cells were treated with metformin for 6 h. Chromatin samples precipitated with anti-FoxO3a, anti-FoxO1, or normal IgG were amplified using primers for the Mut-DΔ2 region of the human SEPP1 promoter.
Metformin Suppresses FoxO Activity via AMPK Activation
To determine whether metformin treatment influences FoxO activity, a forkhead-response element (FHRE)-Luc vector that includes three tandems of FHREs ligated with a luciferase gene was utilized (32). This vector was used as a reporter of FoxO-responsive promoter activity (33). Metformin treatment suppressed FHRE activity, and concurrent treatment with compound C completely cancelled this suppression (Fig. 4B). In addition, treatment with compound C stimulated FHRE activity in the absence of metformin (Fig. 4B), whereas AICAR treatment suppressed FHRE activity (Fig. 4C). These results suggest that metformin suppresses FHRE activity via AMPK activation. To determine the critical FoxO binding site for metformin-induced SEPP1 suppression, we constructed luciferase vectors that deleted either of two putative FoxO binding sites and were named Mut-DΔ2-ΔFoxo A or B, respectively (supplemental Fig. S4). Luciferase assay using these vectors revealed that putative FoxO binding site B was essential for metformin-induced SEPP1 suppression (Fig. 4D). Because the assays using these vectors are not specific to FoxO3a activity, the interaction of FoxO proteins with DNA sequences in the SEPP1 promoter was examined using a ChIP assay. For the ChIP assay, HepG2 cells were utilized to evaluate the human SEPP1 promoter. Metformin suppressed SEPP1 expression in HepG2 cells as well as H4IIEC3 cells (data not shown). The ChIP assay indicates that treatment with metformin decreased the binding of FoxO3a to SEPP1 promoter, whereas it increased the binding of FoxO1 (Fig. 4E). These results suggest that FoxO3a, but not FoxO1, is associated with the metformin-induced suppression of SEPP1 expression.
Metformin Suppresses SEPP1 Expression via FoxO3a Inactivation
Next, we examined whether the specific knockdown of endogenous Foxo3a or Foxo1 affects Sepp1 expression in H4IIEC3 hepatocytes. Transfection with Foxo3a- or Foxo1-specific siRNA resulted in a ∼50% reduction in mRNA levels of Foxo3a or Foxo1 (Fig. 5A). Knockdown of both Foxo1 and Foxo3a resulted in a significant down-regulation of Sepp1 expression (Fig. 5A). Interestingly, mRNA levels of G6pc were decreased by Foxo3a knockdown (Fig. 5A), suggesting that not only FoxO1 but also FoxO3a positively regulates the expression of the gluconeogenesis-related genes in H4IIEC3 hepatocytes. Next, we assessed whether knockdown of Foxo3a selectively affects the inhibitory action of metformin on the SEPP1 promoter. Transfection with siRNAs for Foxo3a, but not for Foxo1, cancelled metformin-induced suppression of Mut-DΔ2 luciferase activity (Fig. 5B). These results suggest that the metformin-induced suppression of Sepp1 is dependent on FoxO3a but not on FoxO1.
FIGURE 5.

Metformin suppressed SeP expression via FoxO3a. A, efficiency of Foxo3a siRNA and Foxo1 siRNA. H4IIEC3 cells were transfected with Foxo3a siRNAs or Foxo1 siRNAs or a negative control (NC) siRNA at 48 h. Knockdown efficiency was assessed by real-time PCR. Expression values were normalized to Actb mRNA. Data represent means ± S.D. (error bars) (n = 4). **, p < 0.01; ***, p < 0.001 versus negative control siRNA-treated cells. B, luciferase activity of Mut-DΔ2 treated with Foxo3a or Foxo1 siRNA and metformin. H4IIEC3 cells were transfected with Foxo3a siRNAs or Foxo1 siRNAs or negative control siRNA at 24 h and then co-transfected with Mut-DΔ2 vector and control reporter vector. 24 h after transfection, cells were treated with the indicated concentrations of metformin for 24 h. Signals were normalized to the control reporter vector. Data represent means ± S.D. (n = 4). ***, p < 0.001 versus vehicle-treated cells. N.S., not significant. C, protein levels in the presence of the FoxO3a overexpression vector. H4IIEC3 cells were transfected with the pCMV-FoxO3a vector or pCMV empty vector at 24 h. FoxO3a protein levels were then assessed by Western blotting. D, SEPP1 promoter activity transfected with the FoxO3a overexpression vector. H4IIEC3 cells were co-transfected with the expression vectors for FoxO3a, SEPP1 promoter reporter and control reporter at 24 h and then treated with the indicated concentrations of metformin for 48 h. Data represent means ± S.D. (n = 3–4). ***, p < 0.001 versus control; ††, p < 0.01; †††, p < 0.001 versus vehicle-treated cells. E, percentage inhibition of SEPP1 promoter activity by metformin. Suppression ratios of SEPP1 promoter activity were calculated based on the data in E. Data represent means ± S.D. (n = 3–4). **, p < 0.01; ***, p < 0.001 versus control.
Whether FoxO3a overexpression influences the action of metformin on SEPP1 promoter activity was also investigated. The FoxO3a protein was overexpressed in a concentration-dependent manner in cells transfected with the pCMV6-FoxO3a expression vector (Fig. 5C). Overexpression of FoxO3a significantly enhanced SEPP1 promoter activity (Fig. 5D), and transfection with pCMV6-FoxO3a rescued the cells from the suppressive effect of metformin on SEPP1 promoter activity in a concentration-dependent manner (Fig. 5, D and E). These results indicate that metformin decreases SEPP1 promoter activity and gene expression via FoxO3a inactivation in H4IIEC3 hepatocytes.
Metformin Decreases FoxO3a Protein in the Nuclear Components
To elucidate the mechanism by which metformin inactivates FoxO3a, phosphorylation and acetylation of FoxO3a were examined in hepatocytes treated with metformin. Metformin treatment altered neither mRNA levels of Foxo3a (Fig. 6A) nor protein levels of FoxO3a (Fig. 6B). However, immunoprecipitation experiments revealed that treatment with metformin phosphorylated FoxO3a but not FoxO1 in H4IIEC hepatocytes (Fig. 6, B and C, and supplemental Fig. S5). Because a previous report indicated that FoxO3a, as well as FoxO1, is deacetylated by sirtuin family proteins downstream of AMPK (34), we examined the deacetylation of FoxO3a and FoxO1. Acetylation of both FoxO1 and FoxO3a was unaffected by metformin administration (Fig. 6, B and C, and supplemental Fig. S5). To determine the intracellular localization of FoxO3a, the cytosolic and nuclear components of the FoxO3a protein were fractionated. FoxO3a and FoxO1 protein levels were decreased by metformin treatment in the nuclear fraction (Fig. 6D). These results suggest that metformin inactivates FoxO3a by decreasing FoxO3a protein levels in the nucleus and subsequently inhibiting the binding of FoxO3a to the SEPP1 promoter.
FIGURE 6.

Metformin treatment did not suppress FoxO3a expression but did suppress its activity. A, FoxO3a mRNA expression in H4IIEC3 hepatocytes treated with metformin for 6 h. Expression values were normalized to Actb mRNA. Data represent means ± S.D. (n = 5–6). B and C, modification of FoxOs proteins by metformin treatment. Proteins were extracted after 6 h of metformin treatment. Immunoblotting was performed using anti-FoxO3a antibody (B) or anti-FoxO1 antibody (C). Data represent means ± S.D. (n = 3). **, p < 0.01. N.S., not significant. IP, immunoprecipitation; IB, immunoblot. D, intracellular localization of FoxO3a and FoxO1 in H4IIEC3 hepatocytes upon treatment with metformin. Proteins were extracted after 6 h of metformin treatment. E, scheme of SeP suppression by metformin in the liver. FoxO3a positively regulates SEPP1 promoter activity. Metformin suppresses FoxO3a activity via AMPK activation, resulting in suppression of SeP expression. Thus, the hypoglycemic effects of metformin may be mediated at least in part by SeP suppression in the liver.
DISCUSSION
Our data demonstrate that metformin suppresses production of the insulin resistance-inducing hepatokine SeP by activating AMPK and subsequently inactivating FoxO3a in H4IIEC3 hepatocytes. During the course of this study, it was reported that metformin decreases mRNA levels of Sepp1 in rat primary hepatocytes (28); however, the molecular mechanisms by which metformin reduces the expression of Sepp1 were not fully understood. Our data demonstrate that the AMPK/FoxO3a pathway downstream of metformin action plays a major role in the regulation of SEPP1 expression in cultured hepatocytes. Our data suggest a previously unrecognized mechanism of action of metformin in combating the systemic insulin resistance in type 2 diabetes.
The finding that FoxO3a positively regulates Sepp1 and G6pc expression in H4IIEC3 hepatocytes supports the suggestion that FoxO3a plays an important role in glucose homeostasis. The ability of FoxO1 to increase the expression of gluconeogenic genes has been confirmed (35). To date, however, little information concerning the involvement of FoxO3a in glucose metabolism is available. Certainly, no defects in glucose metabolism have been described in FoxO3a-deficient mice (36), suggesting that the function of FoxO3a in glucose metabolism is compensated for by FoxO1. Indeed, Haeusler et al. (37) reported that triple liver-specific ablation of FoxO1, FoxO3a, and FoxO4 causes a more pronounced hypoglycemia and increased insulin sensitivity in mice compared with a single knockout of FoxO1. The present findings indicate that FoxO proteins, including FoxO3a, regulate hepatic glucose metabolism in a coordinated manner. These data reveal that FoxO3a, the downstream target of metformin/AMPK, positively regulates SEPP1 transcriptional activity in cultured hepatocytes independently of FoxO1 and suggest that FoxO3a participates in glucose homeostasis via regulation of the hepatic production of SeP, an insulin resistance-inducing hepatokine.
Knockdown of Foxo3a, but not Foxo1, rescued the cells from metformin-induced inactivation of the SEPP1 promoter, although knockdown of both Foxo3a and Foxo1 down-regulated Sepp1 in the absence of metformin (Fig. 5, A and B). These results are in harmony with early reports showing that FoxO1 positively regulates Sepp1 expression in cultured hepatocytes (38, 39). The current data suggest that both FoxO3a and FoxO1 positively regulate expression of SEPP1 in the basal conditions, but FoxO3a has a dominant role in the suppression of SEPP1 downstream of metformin/AMPK pathway in H4IIEC3 hepatocytes. Interestingly, metformin selectively phosphorylated and deacetylated FoxO3a but not FoxO1 (Fig. 6, B and C). This FoxO3a-selective phosphorylation by metformin is consistent with the previous report showing that AMPK-induced phosphorylation displays a strong preference toward FoxO3 compared with FoxO1 by using in vitro kinase assays (16).
The current study is the first to demonstrate the decreased nuclear localization and subsequent transcriptional inactivation of FoxO3a by AMPK downstream of metformin in the cultured hepatocytes. Greer et al. (16) identified FoxO3a as a direct phosphorylation target of AMPK using in vitro kinase assays. However, the authors reported that phosphorylation by AMPK increases FoxO3a transcriptional activity without affecting FOXO3A subcellular localization in mouse embryonic fibroblasts or 293T cells. A similar activation of FoxO3a by AMPK was reported in C2C12 myotubes (17). In this respect, our results suggest that the AMPK-induced inactivation of FoxO3a is hepatocyte-specific. When FoxO proteins are phosphorylated by Akt, the dissociation of nuclear co-factors from FoxO is thought to be required for nuclear exclusion of FoxO (40). Hence, the difference in nuclear co-activator/co-repressor recruitment between hepatocytes and other cells might explain differences in the action of AMPK on FoxO3a cellular localization and transcriptional activity. Notably, the siRNA-induced knockdown of Foxo3a decreased Sepp1 and G6pc mRNA levels in H4IIEC3 hepatocytes, suggesting that the AMPK/FoxO3a pathway in the liver regulates gluconeogenesis and the production of the hepatokine SeP. These findings shed light on a previously unrecognized role for the AMPK/FoxO3a pathway in the regulation of glucose metabolism in the liver.
The cancellation of the metformin-induced suppression of SeP by compound C, a known inhibitor of the AMPK pathway, was only partial (Fig. 2B). Likewise, the overexpression of FoxO3a only partially cancelled the suppressive action of metformin on Sepp1 gene expression (Fig. 6, D and E). These results suggest that metformin decreases Sepp1 gene expression through both AMPK/FoxO3a-dependent and other independent pathways. Recently, Kalender et al. (41) reported that metformin acts to suppress mTORC1 signaling in an AMPK-independent manner. In addition, Guigas et al. (42) found that metformin inhibits glucose phosphorylation in primary cultured hepatocytes independently of AMPK activity. Additional studies are needed to elucidate the AMPK-independent actions of metformin on Sepp1 expression in H4IIEC3 hepatocytes.
The present sequence of SEPP1 promoter completely corresponds to the refseq of the National Center for Biotechnology Information, but it misses one thymidine against the sequence of a previous report (21). This site had been reported as an SNP site (reference SNP ID rs201851607). Because both allele origin and minor allele frequency of this SNP site are not available, it is difficult to prove which genome sequence is “correct.” At least this SNP site does not seem to affect basal SEPP1 promoter activity. In addition, the metformin-responsible element identified in the current paper locates in the other region of the SNP site. Thus, we consider that the effect of this SNP on the conclusion of this paper is negligible.
A limitation of the present study is that the effects of metformin on SeP expression were not investigated in human samples. The metformin concentrations used in this study (0.25–1 mm) were higher than the blood levels of metformin in patients treated with conventional doses of the drug (10–40 μm). However, it has been pointed out that concentrations of metformin in liver tissue are much higher than those in the blood because the liver receives portal vein blood, which may contain materially higher doses of metformin than plasma (43). An early report indicated that metformin concentrations in the liver were greater than 250 μmol/kg in an STZ diabetic mouse model treated with 50 mg/kg metformin (44). One previous study used 0.25–1 mm metformin in rat primary hepatocytes as a more physiological range of intrahepatic concentration (43). In addition, we show that administration of 300 mg/kg metformin was effective on hepatic expression for Sepp1 in C57BL/6J mice (Fig. 1F). Although clinical trials are necessary, we speculate here that treatment with metformin decreases blood levels of SeP in patients with diabetes. Additionally, the contribution of SeP suppression to the anti-diabetic actions of metformin should be confirmed by additional investigations using Sepp1-knock-out mice.
In summary, the present data provide a novel mechanism of action for metformin involving improvement of systemic insulin sensitivity via the regulation of SeP production (Fig. 6E) and suggest that AMPK/FoxO3a pathway in the liver may be a therapeutic target to the development of new anti-diabetic drugs.
Acknowledgments
We thank Dr. Atsushi Hirao (Kanazawa University) for providing a vector for FHRE-Luc and Maki Wakabayashi (Kanazawa University) for technical assistance. We thank Fabienne Foufelle (Université Pierre et Marie Curie) for providing adenovirus vector encoding DN-AMPK. We thank In-kyu Lee (Kyungpook National University) for providing adenovirus vector encoding CA-AMPK.
This work was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to H. M., T. T., and S. K.) and research grants from Dainippon Sumitomo Pharma (to S. K.) and Takeda Science Foundation (to H. M.).

This article contains supplemental Figs. S1–S5.
- SeP
- selenoprotein P
- AMPK
- AMP-activated kinase
- AICAR
- 5-aminoimidazole-4-carboxamide ribonucleotide
- compound C
- 6-[4-(2-piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo[1,5-a]-pyrimidine
- cGPx
- cellular glutathione peroxidase
- DN
- dominant negative
- CA
- constitutive active
- TFBS
- transcription factor binding site
- FHRE
- forkhead-response element.
REFERENCES
- 1. Burk R. F., Hill K. E. (2005) Selenoprotein P. An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr. 25, 215–235 [DOI] [PubMed] [Google Scholar]
- 2. Carlson B. A., Novoselov S. V., Kumaraswamy E., Lee B. J., Anver M. R., Gladyshev V. N., Hatfield D. L. (2004) Specific excision of the selenocysteine tRNA[Ser]Sec (Trsp) gene in mouse liver demonstrates an essential role of selenoproteins in liver function. J. Biol. Chem. 279, 8011–8017 [DOI] [PubMed] [Google Scholar]
- 3. Schomburg L., Schweizer U., Holtmann B., Flohé L., Sendtner M., Köhrle J. (2003) Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Atkins J. F., Gesteland R. F., Burk R. F. (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 [DOI] [PubMed] [Google Scholar]
- 5. Misu H., Takamura T., Takayama H., Hayashi H., Matsuzawa-Nagata N., Kurita S., Ishikura K., Ando H., Takeshita Y., Ota T., Sakurai M., Yamashita T., Mizukoshi E., Yamashita T., Honda M., Miyamoto K., Kubota T., Kubota N., Kadowaki T., Kim H.-J., Lee I., Minokoshi Y., Saito Y., Takahashi K., Yamada Y., Takakura N., Kaneko S. (2010) A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 12, 483–495 [DOI] [PubMed] [Google Scholar]
- 6. Wang D.-S., Jonker J. W., Kato Y., Kusuhara H., Schinkel A. H., Sugiyama Y. (2002) Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J. Pharmacol. Exp. Ther. 302, 510–515 [DOI] [PubMed] [Google Scholar]
- 7. Shu Y., Sheardown S. A., Brown C., Owen R. P., Zhang S., Castro R. A., Ianculescu A. G., Yue L., Lo J. C., Burchard E. G., Brett C. M., Giacomini K. M. (2007) Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest. 117, 1422–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyle J. G., Salt I. P., McKay G. A. (2010) Metformin action on AMP-activated protein kinase. A translational research approach to understanding a potential new therapeutic target. Diabet. Med. 27, 1097–1106 [DOI] [PubMed] [Google Scholar]
- 9. Malin S. K., Gerber R., Chipkin S. R., Braun B. (2012) Independent and combined effects of exercise training and metformin on insulin sensitivity in individuals with prediabetes. Diabetes Care 35, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh S., Akhtar N., Ahmad J. (2012) Plasma adiponectin levels in women with polycystic ovary syndrome. Impact of Metformin treatment in a case-control study. Diabetes Metab. Syndr. 6, 207–211 [DOI] [PubMed] [Google Scholar]
- 11. Shargorodsky M., Omelchenko E., Matas Z., Boaz M., Gavish D. (2012) Relation between augmentation index and adiponectin during one-year metformin treatment for nonalcoholic steatohepatosis. Effects beyond glucose lowering? Cardiovasc. Diabetol. 11, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Medema R. H., Kops G. J., Bos J. L., Burgering B. M. (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787 [DOI] [PubMed] [Google Scholar]
- 13. Luo X., Puig O., Hyun J., Bohmann D., Jasper H. (2007) Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J. 26, 380–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kops G. J., Dansen T. B., Polderman P. E., Saarloos I., Wirtz K. W., Coffer P. J., Huang T.-T., Bos J. L., Medema R. H., Burgering B. M. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 15. Olmos Y., Valle I., Borniquel S., Tierrez A., Soria E., Lamas S., Monsalve M. (2009) Mutual dependence of Foxo3a and PGC-1α in the induction of oxidative stress genes. J. Biol. Chem. 284, 14476–14484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greer E. L., Oskoui P. R., Banko M. R., Maniar J. M., Gygi M. P., Gygi S. P., Brunet A. (2007) The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 282, 30107–30119 [DOI] [PubMed] [Google Scholar]
- 17. Sanchez A. M., Csibi A., Raibon A., Cornille K., Gay S., Bernardi H., Candau R. (2012) AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell Biochem. 113, 695–710 [DOI] [PubMed] [Google Scholar]
- 18. Li X.-N., Song J., Zhang L., LeMaire S. A., Hou X., Zhang C., Coselli J. S., Chen L., Wang X. L., Zhang Y., Shen Y. H. (2009) Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 58, 2246–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lützner N., Kalbacher H., Krones-Herzig A., Rösl F. (2012) FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS One 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Honda M., Takehana K., Sakai A., Tagata Y., Shirasaki T., Nishitani S., Muramatsu T., Yamashita T., Nakamoto Y., Mizukoshi E., Sakai Y., Yamashita T., Nakamura M., Shimakami T., Yi M., Lemon S. M., Suzuki T., Wakita T., Kaneko S. (2011) Malnutrition impairs interferon signaling through mTOR and FoxO pathways in patients with chronic hepatitis C. Gastroenterology 141, 128–140, 140.e1–140.e2 [DOI] [PubMed] [Google Scholar]
- 21. Dreher I., Jakobs T. C., Köhrle J. (1997) Cloning and characterization of the human selenoprotein P promoter. Response of selenoprotein P expression to cytokines in liver cells. J. Biol. Chem. 272, 29364–29371 [DOI] [PubMed] [Google Scholar]
- 22. Takebe G., Yarimizu J., Saito Y., Hayashi T., Nakamura H., Yodoi J., Nagasawa S., Takahashi K. (2002) A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 277, 41254–41258 [DOI] [PubMed] [Google Scholar]
- 23. Nakamura S., Takamura T., Matsuzawa-Nagata N., Takayama H., Misu H., Noda H., Nabemoto S., Kurita S., Ota T., Ando H., Miyamoto K., Kaneko S. (2009) Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 284, 14809–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flicek P., Amode M. R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S., Gil L., Gordon L., Hendrix M., Hourlier T., Johnson N., Kähäri A. K., Keefe D., Keenan S., Kinsella R., Komorowska M., Koscielny G., Kulesha E., Larsson P., Longden I., McLaren W., Muffato M., Overduin B., Pignatelli M., Pritchard B., Riat H. S., Ritchie G. R., Ruffier M., Schuster M., Sobral D., Tang Y. A., Taylor K., Trevanion S., Vandrovcova J., White S., Wilson M., Wilder S. P., Aken B. L., Birney E., Cunningham F., Dunham I., Durbin R., Fernández-Suarez X. M., Harrow J., Herrero J., Hubbard T. J., Parker A., Proctor G., Spudich G., Vogel J., Yates A., Zadissa A., Searle S. M. (2012) Ensembl 2012. Nucleic Acids Res. 40, D84–D90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paten B., Herrero J., Beal K., Fitzgerald S., Birney E. (2008) Enredo and Pecan. Genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 18, 1814–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wingender E. (2008) The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 9, 326–332 [DOI] [PubMed] [Google Scholar]
- 27. Kel A. E., Gössling E., Reuter I., Cheremushkin E., Kel-Margoulis O. V., Wingender E. (2003) MATCH. A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31, 3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Speckmann B., Sies H., Steinbrenner H. (2009) Attenuation of hepatic expression and secretion of selenoprotein P by metformin. Biochem. Biophys. Res. Commun. 387, 158–163 [DOI] [PubMed] [Google Scholar]
- 29. Łukaszewicz-Hussain A., Moniuszko-Jakoniuk J. (2004) Liver catalase, glutathione peroxidase, and reductase activity, reduced glutathione and hydrogen peroxide levels in acute intoxication with chlorfenvinphos, an organophosphate insecticide. Pol. J. Environ. Stud. 13, 303–309 [Google Scholar]
- 30. Magwere T., Naik Y. S., Hasler J. A. (1997) Effects of chloroquine treatment on antioxidant enzymes in rat liver and kidney. Free Radic. Biol. Med. 22, 321–327 [DOI] [PubMed] [Google Scholar]
- 31. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 33. Eckers A., Sauerbier E., Anwar-Mohamed A., Hamann I., Esser C., Schroeder P., El-Kadi A. O., Klotz L.-O. (2011) Detection of a functional xenobiotic response element in a widely employed FoxO-responsive reporter construct. Arch. Biochem. Biophys. 516, 138–145 [DOI] [PubMed] [Google Scholar]
- 34. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 36. Hosaka T., Biggs W. H., 3rd, Tieu D., Boyer A. D., Varki N. M., Cavenee W. K., Arden K. C. (2004) Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl. Acad. Sci. U.S.A. 101, 2975–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haeusler R. A., Kaestner K. H., Accili D. (2010) FoxOs function synergistically to promote glucose production. J. Biol. Chem. 285, 35245–35248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Speckmann B., Walter P. L., Alili L., Reinehr R., Sies H., Klotz L.-O., Steinbrenner H. (2008) Selenoprotein P expression is controlled through interaction of the coactivator PGC-1α with FoxO1a and hepatocyte nuclear factor 4α transcription factors. Hepatology 48, 1998–2006 [DOI] [PubMed] [Google Scholar]
- 39. Walter P. L., Steinbrenner H., Barthel A., Klotz L.-O. (2008) Stimulation of selenoprotein P promoter activity in hepatoma cells by FoxO1a transcription factor. Biochem. Biophys. Res. Commun. 365, 316–321 [DOI] [PubMed] [Google Scholar]
- 40. Van Der Heide L. P., Hoekman M. F., Smidt M. P. (2004) The ins and outs of FoxO shuttling. Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 380, 297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kalender A., Selvaraj A., Kim S. Y., Gulati P., Brûlé S., Viollet B., Kemp B. E., Bardeesy N., Dennis P., Schlager J. J., Marette A., Kozma S. C., Thomas G. (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 11, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 43. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilcock C., Bailey C. J. (1994) Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 24, 49–57 [DOI] [PubMed] [Google Scholar]