

Background: NO is involved in the induction of mitochondrial biogenesis.
Results: Mitochondrial biogenesis is induced only when neuronal NO synthase (nNOS) is recruited to nuclei, an event that is mediated by α-Syntrophin.
Conclusion: Nuclear NO production is crucial for the induction of mitochondrial biogenesis.
Significance: Impairment of nuclear nNOS localization could be the cause of myopathies associated with mitochondrial dysfunction.
Keywords: Mitochondria, Myogenesis, Nitric-oxide Synthase, S-Nitrosylation, Scaffold Proteins
Abstract
Neuronal nitric-oxide synthase (nNOS) has various splicing variants and different subcellular localizations. nNOS can be found also in the nucleus; however, its exact role in this compartment is still not completely defined. In this report, we demonstrate that the PDZ domain allows the recruitment of nNOS to nuclei, thus favoring local NO production, nuclear protein S-nitrosylation, and induction of mitochondrial biogenesis. In particular, overexpression of PDZ-containing nNOS (nNOSα) increases S-nitrosylated CREB with consequent augmented binding on cAMP response element consensus sequence on peroxisome proliferator-activated receptor γ co-activator (PGC)-1α promoter. The resulting PGC-1α induction is accompanied by the expression of mitochondrial genes (e.g., TFAM, MtCO1) and increased mitochondrial mass. Importantly, full active nNOS lacking PDZ domain (nNOSβ) does not localize in nuclei and fails in inducing the expression of PGC-1α. Moreover, we substantiate that the mitochondrial biogenesis normally accompanying myogenesis is associated with nuclear translocation of nNOS. We demonstrate that α-Syntrophin, which resides in nuclei of myocytes, functions as the upstream mediator of nuclear nNOS translocation and nNOS-dependent mitochondrial biogenesis. Overall, our results indicate that altered nNOS splicing and nuclear localization could be contributing factors in human muscular diseases associated with mitochondrial impairment.
Introduction
Nitric oxide is a lipophilic and diffusible gaseous radical molecule, which is physiologically produced by a class of enzymes called NO synthases (NOSs).3 NO has been implicated as signaling molecule in a plethora of processes such as vasodilatation, neurotransmission, and immune response (1, 2). Due to its chemical nature, NO is highly reactive and is generally synthesized on demand by NOSs in precise subcellular compartments (3). In particular, the principal mechanism by which the specificity of NO signaling is conferred is the differential targeting of NOSs enzymes with close apposition to effector protein targets, thus facilitating local NO signaling (4, 5). One example is represented by the plasma membrane anchoring of neuronal NOS (nNOS) through specific interaction with scaffold proteins including PSD-95 in neuronal cells and α-Syntrophin in myocytes, which assures the coupling of nNOS activity to calcium influx during glutamatergic transmission and skeletal muscle contraction, respectively (6, 7).
The regulatory mechanisms controlling the expression, localization, and activity of nNOS are very complex and multifactorial. In addition to protein-protein interactions (e.g., with Hsp90, PIN, Ca2+-calmodulin) and post-translational covalent modifications (e.g., S-nitrosylation, phosphorylation), alternate mRNA splicing is also included (4, 8–11). nNOS is transcribed from the nos1 gene in at least four splicing variants that are mainly expressed in neurons and skeletal muscle cells: (i) nNOSα or full-length nNOS (fl-nNOS) that is predominantly found in neuronal cells and contains the N-terminal PDZ domain, which allows the interaction with cell membrane proteins (6, 12); (ii) cytosolic nNOSβ that lacks the PDZ domain (ΔnNOS) and maintains full NO synthetizing activity (13, 14); (iii) nNOSγ that lacks both PDZ and oxygenase domain and therefore has limited capacity to produce NO (11, 14); and (iv) nNOSμ that is expressed in skeletal muscle and contains an additional internal sequence with respect to nNOSα (15, 16).
NO is also involved in different signaling pathways in mitochondria, including respiration, apoptosis, and more recently mitochondrial biogenesis (17, 18), having the capacity to influence the activity and the expression of its crucial regulator peroxisome proliferator activated receptor γ co-activator 1α (PGC-1α) (19). Mitochondrial biogenesis is an intricate process consisting in the growth and division of pre-existing mitochondria that requires the replication of the mtDNA and the synthesis and import of proteins and lipids to the existing mitochondria (20). Mitochondrial biogenesis is substantially driven by the nuclear genome. Upon conditions that cause cell energetic stress (i.e., physical exercise, cold, fasting), PGC-1α is induced and impinges the co-activation of transcription factors regulating the expression of mitochondrial proteins including those controlling replication and transcription of mtDNA as well as oxidative phosphorylation genes (21). Notably, NO can either stimulate PGC-1α phosphoactivation by AMPK or increase its expression via CREB-dependent transcription (19, 22, 23). The modulation of mitochondrial respiratory chain is another way by which NO impacts on mitochondrial function. The main site of inhibition is at complex IV, because NO reacts with heme iron competing with oxygen (24). NO can also inhibit the electron transport chain at complex I and III reacting with iron-sulfur clusters (24).
Several human diseases including neurodegenerative disorders and myopathies are characterized by substantial alteration of mitochondrial content and function (20, 22, 25). To date, different therapeutic approaches have been suggested with the aim of implementing mitochondrial content to compensate mitochondrial dysfunction and to increase energy supply. Increased nNOS activity and expression were observed in myocytes undergoing mitochondrial proliferation, suggesting the direct involvement of such an enzyme in mitochondrial biogenesis (26). Similarly, several studies suggest that NO-donating drugs, NO precursors, or analogs are effective in restoring mitochondrial homeostasis via the induction of mitochondrial biogenesis and could be promising to treat patients with neurodegenerative diseases or myopathies (27–29). Although the induction of mitochondrial biogenesis by NO has been definitively demonstrated in different cell lines and tissues, in myocytes and neuronal cells (18, 30), it remains unclear whether NO involved in mitochondrial biogenesis originates from a specific nNOS isoform.
nNOSβ is abundantly expressed both in neuronal cells and myocytes and has generally a cytosolic localization (31). Although both nNOSα and nNOSμ are found associated with plasma membrane through their PDZ domain, they can be present also in a plasma membrane unbound form (6, 32–34). However, the exact function of cytoplasmic nNOSα and nNOSμ as well as nNOSβ remains still unclear. We previously demonstrated that the PDZ domain of nNOS is fundamental to recruit nNOS to the nucleus, allowing the interaction with Sp1 with consequent inhibition of its transcriptional activity on sod1 gene promoter (13). Thus, this finding gives fresh insight into how modulation of gene transcription may be achieved by nNOS redistribution to nucleus. The aim of this study was at analyzing the impact of nNOSα and nNOSβ on mitochondrial function. Our findings demonstrate that only nNOSα is able to induce PGC-1α and its downstream oxidative phosphorylation genes, implying that generation of NO in the nucleus is a mandatory event for the onset of the mitochondrial biogenesis pathway.
EXPERIMENTAL PROCEDURES
Materials
Protease inhibitor mixture, monoclonal anti-β-tubulin, ascorbate, S-nitrosoglutathione (GSNO), Triton X-100, soluble guanylate cyclase (sGC) inhibitor 1H-[1,2,4] oxadiazolo [4,3-a]quinoxalin-1-one (ODQ), protein A/G-agarose, salmon sperm, and primers for RT-qPCR were from Sigma-Aldrich. Polyclonal and monoclonal anti-Sp1, anti-nNOS (N terminus), anti-TFAM, anti-PGC-1α, anti-LDH, anti-CREB, and anti-Syntrophin were from Santa Cruz Biotechnology (Santa Cruz, CA). IgG (H + L)-HRP conjugate anti-mouse and anti-rabbit secondary antibodies were from Bio-Rad. l-NG-Nitroarginine methyl ester (l-NAME), and sGC inhibitor LY83583 were from Merck. Mouse monoclonal anti-nNOS (C terminus) was from Transduction Laboratories (Lexington, KY). MitoTracker Red was from Invitrogen. Nylon membrane was from Amersham Biosciences. ChemiGlow chemiluminescence substrate was from Alpha Innotech Corporation (San Leandro, CA). Streptavidin HRP-conjugated was from Calbiochem-Novabiochem Corporation (La Jolla, CA). All other chemicals were obtained from Merck.
Cell Culture and Treatments
The murine skeletal muscle C2C12 cells and human HeLa cervix carcinoma cells were obtained from the European Collection of Cell Cultures (Salisbury, UK). C2C12 myoblasts and HeLa cells were cultured in growth medium composed of DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin/streptomycin, and 2 mm glutamine (Lonza Sales, Basel, Switzerland) and maintained at 37 °C in an atmosphere of 5% CO2 in air. Prior differentiation, C2C12 cells were plated at 80% of confluence and cultured in growth medium for 24 h. To induce differentiation, C2C12 cells were washed in PBS, and growth medium was replaced with differentiation medium, which contains 2% heat-inactivated horse serum and antibiotics. l-NAME, LY83583, and ODQ were used at concentrations of 100, 2, and 10 μm, respectively.
Transfections
Twenty-four hours after plating, 80% confluent cells were trypsinized and transfected by electroporation with pcDNA3.1 empty vector (Life Technologies) or pcDNA3.1 vector containing cDNA coding for full-length rat nNOS (nNOSα, fl-nNOS) or rat nNOS lacking the PDZ domain (nNOSβ, ΔnNOS) (13). Cells were used 24 h after transfection, because this time was sufficient to significantly increase the expression and the activity of nNOS (13). Transfection efficiency was estimated by co-transfecting nNOS cDNA containing vector with p-Tracer GFP vector (Lonza Sales) and by analyzing GFP fluorescence by microscopy.
C2C12 cells were transfected with a siRNA duplex directed against the mouse α-Syntrophin (synt−) and PGC-1α (PGC-1α−) target sequences (SASI_Mm02_00315675;SASI_Mm01_00157757). Transfection with a scramble siRNA duplex (scr), with no homology to other mouse mRNAs, was used as control. Cells were transfected by electroporation as described previously (13), and transfection efficiency was evaluated by co-transfecting siRNAs with nonspecific rhodamine-conjugated oligonucleotides. Only experiments that gave transfection efficiency >80% were considered.
Measurement of NOx
Nitrites and nitrates (NOx) released in culture medium were measured by the Griess reaction as described previously (35). Briefly, after nitrate reduction, the concentration of nitrites was determined by a standard curve obtained with a known amount of sodium nitrite and expressed as μmol/mg protein.
Isolation of Nuclei
Cell pellets were resuspended in nucleus lysis buffer (NLB) containing 50 mm Tris-HCl, pH 8.1, 10 mm EDTA, 1% SDS, 10 mm sodium butyrate, protease inhibitors and incubated 1 h at 4 °C. Nuclei were collected by centrifugation at 600 × g for 5 min at 4 °C, and pellets were resuspended in 1 ml of NLB. Subsequently, nuclei were purified on NLB containing 30% sucrose (w/v) and centrifuged at 700 × g for 10 min. The pellets were resuspended in NLB to remove nuclear debris and finally used for Western blot, oligonucleotide pulldown, or ChIP assays.
Western Blot Analysis
Cell pellets were resuspended in radioimmunoprecipitation buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 12 mm deoxycholic acid, 0.5% Nonidet P-40, and protease inhibitors) or in lysis buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA, 50 mm NaCl, 0.5% Igepal CA-630, and protease inhibitors). Protein samples were used for SDS-PAGE followed by Western blotting. Nitrocellulose membrane were stained with primary antibodies against β-tubulin (1:1,000), PGC-1α (1:500), TFAM (1:1,000), CREB (1:500), LDH (1:5,000), α-Syntrophin (1:500), and Sp1 (1:500). To detect nNOSα and nNOSβ contemporaneously, we probed nitrocellulose membrane with nNOS antibody directed against its C-terminal region (1:500). To detect nNOS during myocyte differentiation, we used nNOS antibody directed against its N-terminal region (1:500). Afterward, the membranes were incubated with the appropriate HRP conjugate secondary antibody, and immunoreactive bands were detected by a Fluorchem imaging system upon staining with ChemiGlow chemiluminescence substrate. Immunoblots reported in the figures are representative of at least four experiments that gave similar results. β-Tubulin or Sp1 were used as loading control of total and nuclear extracts, respectively. To exclude the cytoplasm contaminants, the nitrocellulose was probed with LDH antibody.
Determination of Protein Oxidation
Carbonylated proteins were detected using the OxyblotTM protein oxidation detection kit (Millipore-Merck) as described previously (36). Briefly, 20 μg of proteins were reacted with 2,4-dinitrophenylhydrazine for 15 min at 25 °C. Samples were resolved on 12% SDS-polyacrylamide gels, and 2,4-dinitrophenylhydrazine-derivatized proteins were identified by immunoblot using an anti-2,4-dinitrophenylhydrazine antibody.
RT-qPCR Analysis
Total RNA was extracted using TRI® reagent (Sigma-Aldrich). Three μg of RNA was used for retrotranscription with M-MLV (Promega, Madison, WI). qPCR was performed in triplicates by using validated qPCR primers (BLAST), Ex TAq qPCR Premix, and the real time PCR LightCycler II (Roche Diagnostics) as described previously (37). mRNA levels were normalized to ribosomal protein large subunit (RPL) mRNA, and the relative mRNA levels were determined by using the 2−ΔΔCt method. The primer sequences are listed in supplemental Table S1.
DNA Extraction and Determination of Mitochondrial Mass
DNA was isolated by Wizard SV Genomic DNA purification kit (Promega). qPCR was performed in triplicate by using validated qPCR primers (Blast), Ex TAq qPCR Premix (Lonza Sales), and the Roche real time PCR LightCycler II (Roche Applied Science) as described previously (38). mtDNA content was assayed by analyzing D-loop levels (noncoding mtDNA region). D-loop was normalized to genomic RPL, and the relative levels were determined by using the 2−ΔΔCt method. The primers sequences are listed in supplemental Table S1. Alternatively, mitochondrial mass was detected by cytofluorimetric analysis after incubation of cells with 100 nm Mito Tracker Red for 30 min.
Chromatin Immunoprecipitation Assay
ChIP was carried out as described previously (39). Briefly, samples were precleared with preadsorbed salmon sperm protein A/G-agarose beads (1 h, 4 °C), and immunoprecipitated overnight using anti-CREB or control IgG antibody. After de-cross-linking (1% SDS at 65 °C for 3 h), qPCR was used to quantify the promoter binding with 30 cycles total (95 °C for 1 s, 60 °C for 30 s, and 72 °C for 60 s). The results are expressed as fold enrichment with respect to IgG control or as percentages of input (1%) values. The primers used are reported in supplemental Table S1.
Oligonucleotide Pulldown Assay
GSNO was added to purified nuclei at a concentration of 5 mm at 4 °C for 30 min in NLB. After treatment, oligonucleotide pulldown assay was carried out as described previously (40) by using the cAMP response element sequence on the mouse PGC-1α gene (ppargc1a) promoter (supplemental Table S1). Oligonucleotide pulldown specificity was demonstrated with mutant oligonucleotides used as negative controls (data not shown).
Biotin Switch Assay
Biotin switch assay was performed as described previously (41). Briefly, proteins were subjected to S-NO derivatization by incubation in the presence of ascorbate, which reduces S-NO groups. The same sample incubated in the presence of biotin without ascorbate was used as negative control. After protein separation by nonreducing SDS-PAGE and Western blot, biotinylated proteins were detected by incubation of nitrocellulose membrane with HRP-conjugated streptavidin (1:1,000). Protein concentration was determined by the method of Lowry et al. (42).
Statistical Analysis
The results are presented as means ± S.D. Statistical evaluation was conducted by analysis of variance, followed by the post-Student-Newmann-Keul's test. Differences were considered to be significant at p < 0.05.
RESULTS
nNOS PDZ Domain Is Essential for Inducing Mitochondrial Biogenesis
NOSs are master regulators of mitochondrial biogenesis (22, 43); however, the exact role of the splicing isoforms of nNOS in such a process remains to be elucidated. To this end, we transfected C2C12 myoblasts with either fl-nNOS (or nNOSα) or nNOS lacking the PDZ domain (ΔnNOS, or nNOSβ) (Fig. 1A). NO production was similarly increased with respect to control cells (mock), as assessed by measuring NO oxidation products (nitrites and nitrates, NOx) in culture medium (Fig. 1B) and the extent of S-nitrosylated proteins by biotin switch assay (Fig. 1C). These results confirmed our previous evidence that the lack of PDZ domain does not influence nNOS activity (13). We then analyzed the expression of genes implicated in mitochondrial biogenesis. mRNA of PGC-1α was increased (Fig. 2A) as well as mRNA of its downstream mitochondrial genes (i.e., TFAM, TFBM1, MtCO1) (Fig. 2, B–D). Coherently, an enhanced mitochondrial mass was observed only in fl-nNOS cells, as assessed by the qPCR analysis of mtDNA content (Fig. 2E) and by cytofluorimetric analysis using the mitochondrial specific probe MitoTracker Red (Fig. 2F). The NOS inhibitor l-NAME efficiently restrained fl-nNOS-mediated mitochondrial biogenesis. Indeed, l-NAME was able to inhibit the increase of mRNA of PGC-1α (Fig. 2A) and its downstream targets (Fig. 2, B–D) as well as mitochondrial mass (Fig. 2, E and F). NO can induce mitochondrial biogenesis via a NO/cGMP-dependent pathway (18). The use of sGC inhibitors such as LY83583 and ODQ was not able to reduce the fl-nNOS-mediated increase of mitochondrial biogenesis (Fig. 2, A–E). The same results were obtained by analyzing the PGC-1α and TFAM protein levels (Fig. 2G and supplemental Fig. S1, A and B). To demonstrate that PGC-1α is the crucial mediator of the expression of mitochondrial proteins in fl-nNOS cells, we co-transfected fl-nNOS cells with a siRNA against PGC-1α (PGC-1α−). As reported in supplemental Fig. S2A, TFAM level was not enhanced in fl-nNOS cells in which PGC-1α was down-regulated (fl-nNOS/PGC-1α−), confirming its role in fl-nNOS-mediated mitochondrial biogenesis.
FIGURE 1.

Effect of fl-nNOS overexpression in C2C12 cells. C2C12 cells were transiently transfected with pcDNA3.1 vector containing cDNA coding for full-length rat nNOS (fl-nNOS or nNOSα) or rat nNOS lacking the PDZ domain (ΔnNOS or nNOSβ) or with empty vector (mock). A, after 24 h from transfection, 20 μg of total protein extracts were loaded for detection of nNOS by Western blot using an antibody directed against the nNOS C terminus. Tubulin was used as loading control. B, the activity of nNOS was determined by measuring the total amount of nitrites plus nitrates (NOx) released in the culture medium. The data are reported as μmol/mg protein and expressed as means ± S.D. (n = 5; *, p < 0.001 versus mock). C, total proteins extracts (500 μg) were subjected to S-NO derivatization with biotin. After Western blot, biotin adducts were identified by incubating nitrocellulose membrane with HRP conjugate streptavidin. Proteins incubated in labeling buffer without ascorbate were used as negative control (− Ascorbate). Tubulin was used as loading control. Immunoblots reported are representative of at least four experiments that gave similar results.
FIGURE 2.

fl-nNOS overexpression elicits mitochondrial biogenesis in C2C12 cells. C2C12 cells were transiently transfected as reported in the legend to Fig. 1. After transfection, the cells were treated with l-NAME (100 μm), LY83583 (LY, 2 μm), or ODQ (10 μm) for 24 h. A–D, total RNA was isolated and relative mRNA levels of PGC-1α (A), TFAM (B), mitochondrial transcription factor B (TFBMI, C), and MtCO1 (D) were analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 7; *, p < 0.05 versus mock cells; °, p < 0.05 versus untreated fl-nNOS cells). E, DNA was extracted, and relative mitochondrial content was assayed by analyzing the D-loop noncoding mtDNA region through qPCR. The D-loop value was normalized to RPL. The data are expressed as means ± S.D. (n = 6; *, p < 0.001 versus mock; °, p < 0.01 versus untreated fl-nNOS). F, cells were incubated with MitoTracker Red for 30 min, and mitochondrial content was assayed by cytofluorimetric analysis. The data are expressed as percentages of MitoTracker Red-positive cells (n = 4; *, p < 0.05 versus mock cells; °, p < 0.05 versus untreated fl-nNOS cells). G, 20 μg of total protein extracts were loaded for detection of PGC-1α and TFAM by Western blot. Tubulin was used as loading control. Immunoblots reported are representative of at least four experiments that gave similar results.
To test whether fl-nNOS effects could depend on cell type or be of more general application, we transfected fl-nNOS or ΔnNOS in HeLa cells (Fig. 3A). Even though the NO production was equally increased in fl-nNOS and ΔnNOS with respect to mock cells (Fig. 3B), we found that only fl-nNOS was able to induce PGC-1α and its downstream mitochondrial genes. Indeed, mRNA expression of PGC-1α, TFAM, and TFBM1 was found to be increased, and, coherently, l-NAME, but not sGC inhibition, efficiently abrogated this event (Fig. 3, C–E).
FIGURE 3.

fl-nNOS overexpression induces mitochondrial biogenesis in HeLa cells. HeLa cells were transiently transfected with pcDNA3.1 vector containing cDNA coding for full-length rat nNOS (fl-nNOS or nNOSα) or rat nNOS lacking the PDZ domain (ΔnNOS or nNOSβ) or with empty vector (mock). A, 20 μg of total protein extracts were loaded for detection of nNOS by Western blot using an antibody directed against the nNOS C terminus. Tubulin was used as loading control. Immunoblots reported are representative of at least four experiments that gave similar results. B, the activity of nNOS was determined by measuring the total amount of nitrites plus nitrates (NOx) released in the culture medium. The data are reported as μmol/mg protein and expressed as means ± S.D. (n = 5; *, p < 0.001 versus mock). C–E, after transfection, cells were treated with l-NAME (100 μm) or ODQ (10 μm) for 24 h. Total RNA was isolated, and relative mRNA levels of PGC-1α (C), TFAM (D), and TFBM1 (E) were analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 7; *, p < 0.05 versus mock; °, p < 0.05 versus untreated fl-nNOS). TFBMI, mitochondrial transcription factor B.
Nuclear Recruitment of nNOS Promotes PGC-1α Expression via S-Nitrosylation of CREB in C2C12 Cells
The results obtained indicated that the induction of mitochondrial biogenesis could proceed via a transduction pathway involving protein S-nitrosylation. We previously demonstrated that the presence of PDZ domain is mandatory for nNOS localization at the nuclei of neuronal cells (13). Western blot analysis of nNOS carried out on nuclear fraction showed that only fl-nNOS was able to localize in the nuclei of C2C12 cells (Fig. 4A). fl-nNOS cells exhibited a higher extent of protein S-nitrosylation in nuclei with respect to ΔnNOS cells, which show S-nitrosylation level comparable to that of mock cells (Fig. 4B).
FIGURE 4.

fl-nNOS nuclear localization promotes CREB S-nitrosylation and its increased binding to PGC-1α promoter in C2C12 cells. C2C12 cells were transiently transfected as reported in Fig. 1. A, after 24 h from transfection, 20 μg of nuclear protein extracts were loaded for detection of nNOS by Western blot using an antibody directed against nNOS C terminus. Sp1 was used as loading control. The possible presence of cytoplasmic contaminants was tested by incubating nitrocellulose with rabbit anti-LDH. B, after 24 h from transfection, the nuclear proteins (500 μg) were subjected to S-NO derivatization with biotin. After Western blot, biotin adducts were identified by incubating nitrocellulose membrane with HRP conjugate streptavidin. Proteins incubated in labeling buffer without ascorbate were used as negative control (− Ascorbate). Sp1 was used as loading control. C, after transfection, cells were treated with l-NAME (100 μm), LY83583 (2 μm), or ODQ (10 μm) for 24 h. 20 μg of total protein extracts were loaded for detection of CREB by Western blot. Tubulin was used as loading control. D, after 24 h from transfection, the nuclear proteins (500 μg) were subjected to S-NO derivatization with biotin. After Western blot, the nitrocellulose was incubated with CREB antibody for detection of CREB-SNO. 20 μg of nuclear extracts were used also for detection of CREB by Western blot. Sp1 was used as loading control. The possible presence of nuclear contaminants was tested by incubating nitrocellulose with rabbit anti-LDH. The level of S-nitrosylated CREB (CREB-SNO) was quantified by densitometric analysis (right panel). The data are expressed as CREB-SNO/CREB (n = 3). E, ChIP assay was carried out on cross-linked nuclei from mock, fl-nNOS, and ΔnNOS cells, using CREB antibody followed by qPCR analysis of cAMP response element. The data are expressed as means ± S.D. (n = 3; *, p < 0.001 versus mock; °, p < 0.05 versus untreated fl-nNOS). F, intact nuclei of C2C12 cells were incubated with 5 mm GSNO at 4 °C for 30 min. Nuclear protein extracts (500 μg) were subjected to oligonucleotide pulldown by using the biotinylated oligonucleotide representing the cAMP response element on the PGC-1α promoter, and bound CREB was detected by Western blot. 20 μg of nuclear proteins (input) was used for Western blot analysis of Sp1. Immunoblots reported are representative of at least four experiments that gave similar results.
Among the transcription factors that are implicated in PGC-1α induction and in mitochondrial biogenesis, CREB is included (44). Moreover, the DNA binding activity of CREB can be regulated by S-nitrosylation (45). Fig. 4C shows that fl-nNOS but not ΔnNOS was able to impinge CREB up-regulation, and l-NAME significantly abrogated this event. The sGC inhibitors did not prevent CREB up-regulation in fl-nNOS cells, confirming that NO/cGMP was not involved (Fig. 4C). Fig. 4D (left panel) shows that fl-nNOS cells have also increased levels of S-nitrosylated CREB (CREB-SNO). Conversely, ΔnNOS cells did not display increased levels of CREB-SNO. However, the CREB-SNO/CREB ratio was not enhanced in fl-nNOS cells with respect to ΔnNOS and mock cells (Fig. 4D, right panel), suggesting that the increase in CREB-SNO is due to the rise of total CREB. To investigate whether the up-regulation of CREB was associated with its increased binding activity on PGC-1α promoter, ChIP assay was carried out on nuclei isolated from fl-nNOS and ΔnNOS cells. We observed that in fl-nNOS cells the occupancy of CREB on consensus sequence located on PGC-1α promoter was significantly higher than ΔnNOS and mock cells (Fig. 4E). Consistent with the positive role of NO in regulating PGC-1α expression, the binding activity of CREB was efficiently reduced by l-NAME treatment (Fig. 4E). To test whether S-nitrosylation could be functional in enhancing CREB binding activity, we treated isolated nuclei with the NO donor GSNO, and we performed an oligonucleotide pulldown assay by using biotinylated oligonucleotides corresponding to CREB consensus sequence on PGC-1α promoter. Fig. 4F shows that the amount of CREB able to bind to DNA was markedly higher in GSNO-treated nuclei, implying that CREB S-nitrosylation is involved in impinging PGC-1α expression.
α-Syntrophin Is Implicated in nNOS Recruitment to Nuclei
α-Syntrophin, which is a known interactor of nNOS at the sarcolemma (6), was found in the nuclear compartment of HeLa cells, and at this level it is part of the nuclear Dystrophin complex together with nNOS (46). Interestingly, α-Syntrophin was also localized in nuclei of C2C12 cells (55). With all this in mind, we asked whether α-Syntrophin was really present in nuclei of C2C12 cells and involved in nNOS nuclear engagement. To this end, we analyzed nNOS localization in fl-nNOS cells after α-Syntrophin down-regulation through RNAi (synt− cells). α-Syntrophin was significantly down-regulated because its content was decreased both in total (Fig. 5A) and nuclear extracts (Fig. 5B). Under this condition, although no differences in nNOS expression were observed (supplemental Fig. S2B), fl-nNOS recruitment to nuclei was less efficient (Fig. 5B), and this event caused a significant reduction of the expression of PGC-1α, TFAM, MtCo1, and TFBM1 (Fig. 5, C–F), as well as of mtDNA content (Fig. 5G), indicating that α-Syntrophin plays a critical role in the induction of NO-mediated mitochondrial biogenesis. The same results were obtained by analyzing the protein level of PGC-1α and TFAM (Fig. 5H and supplemental Fig. S2B).
FIGURE 5.

α-Syntrophin is responsible for nNOS nuclear recruitment and induction of mitochondrial biogenesis. Mock and fl-nNOS C2C12 cells were transiently transfected with α-Syntrophin siRNA (synt−) or with a scramble siRNA (scr). A, 20 μg of total protein extracts were loaded for detection of α-Syntrophin by Western blot. B, 20 μg of total protein extracts were loaded for detection of α-Syntrophin and nNOS by Western blot (an antibody directed against nNOS N terminus was used). Sp1 was used as loading control. C and D, total RNA was isolated, and relative mRNA levels of PGC-1α (C) and TFAM (D) were analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 9; *, p < 0.001 versus mock/scr; °, p < 0.05 versus fl-nNOS). E and F, MtCO1 (E) and TFBM1 (F) were analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 4; *, p < 0.001 versus mock/scr; °, p < 0.05 versus fl-nNOS). G, DNA was extracted, and relative mitochondrial content was assayed by analyzing the D-loop noncoding mtDNA region through qPCR. D-loop value was normalized to RPL. The data are expressed as means ± S.D. (n = 4; *, p < 0.001 versus mock/scr; °, p < 0.05 versus fl-nNOS). H, 20 μg of total protein extracts were loaded for detection of nNOS, PGC-1α, and TFAM by Western blot. Immunoblots reported are representative of at least four experiments that gave similar results. TFBMI, mitochondrial transcription factor B.
α-Syntrophin-mediated nNOS Recruitment to Nuclei Is Mandatory for the Induction of Mitochondrial Biogenesis during Myocytes Differentiation
Cell differentiation including myogenesis is accompanied by mitochondrial biogenesis (47). Thus, we asked whether nNOS could undergo nuclear redistribution during myocytes differentiation. To this end, myogenesis was induced in C2C12 cells, and mitochondrial biogenesis, as well as nNOS expression and localization, was analyzed at different times of differentiation. Mitochondrial biogenesis was effective, because an accumulation of PGC-1α and TFAM protein (Fig. 6, A and B), together with an enhancement of mitochondrial mass (Fig. 6B), was detected in differentiating myocytes, with an extent comparable to that reported in the literature (48). Interestingly, nNOS content did not change in total extracts (Fig. 6C) and in the cytoplasm, whereas it significantly increased in nuclear compartment (Fig. 6C), suggesting its intracellular redistribution during differentiation. Differentiation was associated with a modest increase of S-nitrosylated proteins in total extracts (Fig. 6D), whereas S-nitrosylated proteins more clearly increased in nuclei (Fig. 6E).
FIGURE 6.

nNOS and α-Syntrophin are recruited to nuclei during C2C12 differentiation. C2C12 cells were differentiated for 0, 2, 4, and 6 days. A, 20 μg of total protein extracts were loaded for detection of PGC-1α and TFAM by Western blot. Tubulin was used as loading control. B, open symbols, DNA was extracted, and relative mtDNA content was assayed by analyzing D-loop level through qPCR. Filled symbols, the increase of PGC-1α (♦) and TFAM (■) was quantified by densitometric analysis of the immunoreactive bands. The data are reported as D-loop/RPL or PGC-1α/Tubulin and TFAM/Tubulin. The data are expressed as means ± S.D. (n = 4; *, p < 0.001 versus day 0). C, 20 μg of nuclear, cytoplasmic, and total protein extracts were loaded for detection of nNOS (using N terminus antibody) and α-Syntrophin by Western blot. Sp1 and LDH were used for assaying the purity of fractions and/or as loading controls. D and E, 500 μg of total (D) and nuclear (E) protein extracts were subjected to S-NO derivatization with biotin. After Western blot, biotin adducts were identified by incubating nitrocellulose membrane with HRP conjugate streptavidin. β-Tubulin and Sp1 were used as loading controls. Immunoblots reported are representative of at least four experiments that gave similar results.
Western blot analysis of α-Syntrophin in cellular fractions showed that it was increased upon differentiation in both total and nuclei (Fig. 6C). According to its plasma and nuclear membrane localization, α-Syntrophin was undetectable in the cytoplasm (Fig. 6C). We then analyzed nNOS localization during myocytes differentiation after α-Syntrophin down-regulation through RNAi. α-Syntrophin was significantly down-regulated because its content was decreased both in total (Fig. 7A) and nuclear extracts (Fig. 7B). α-Syntrophin down-regulation did not affect the total content of nNOS (Fig. 7A); on the contrary, nNOS was no more able to be recruited to nuclei during differentiation (Fig. 7C), and this event was associated with an inhibition of S-nitrosylation of nuclear proteins (Fig. 7D). During differentiation, CREB binding of PGC-1α promoter was increased, and the down-regulation of α-Syntrophin efficiently restrained this event (Fig. 7E). Differentiating C2C12 cells lacking α-Syntrophin failed in inducing mitochondrial biogenesis. Indeed, PGC-1α as well as TFAM protein were not increased after the differentiation stimulus (Fig. 7, F–H).
FIGURE 7.

α-Syntrophin down-regulation impairs mitochondrial biogenesis during C2C12 differentiation. C2C12 cells were transiently transfected with α-Syntrophin siRNA (synt−) or with a scramble siRNA (scr). A, 20 μg of total protein extracts were loaded for detection of nNOS (an antibody directed against nNOS N terminus was used) and α-Syntrophin by Western blot. Tubulin was used as loading control. B, 20 μg of nuclear protein extracts were loaded for detection of α-Syntrophin by Western blot. Sp1 was used as loading control. C, 20 μg of nuclear protein extracts were loaded for detection of nNOS by Western blot using N terminus antibody. Sp1 was used as loading control. D, nuclear proteins (500 μg) were subjected to S-NO derivatization with biotin. After Western blot, biotin adducts were identified by incubating nitrocellulose membrane with HRP conjugate streptavidin. Sp1 was used as loading control. E, ChIP assay was carried out on cross-linked nuclei from scr and synt− cells, using CREB antibody followed by qPCR analysis of cAMP response element. The data are expressed as means ± S.D. (n = 5; *, p < 0.001 versus scr day 0; °, p < 0.001 versus scr day 4). F, 20 μg of total protein extracts were loaded for detection of PGC-1α and TFAM by Western blot. β-Tubulin was used as loading control. G and H, the increase of PGC-1α (G) and TFAM (H) was quantified by densitometric analysis. The data are expressed as PGC-1α/Tubulin and TFAM/Tubulin (n = 4; *, p < 0.001 versus scr day 0; °, p < 0.001 versus scr day 4). Immunoblots reported are representative of at least four experiments that gave similar results.
To assess the role of nNOS/α-Syntrophin-mediated mitochondrial biogenesis in myocyte differentiation, we checked the level of differentiation markers MyoD, myogenin, and muscle creatine kinase. Fig. 8 (A–C) shows that their mRNAs were increased in cells carrying α-Syntrophin; on the contrary, synt− cells failed to increase such markers, indicating defective myocytes differentiation. Indeed, synt− cells were not able to acquire the morphological features typical of myocytes undergoing differentiation but rather appeared nonfused and disordered (Fig. 8D). Next, by analyzing the level of atrophy markers atrogin 1 and Murf-1, we found a significant increase of their expression in both undifferentiated and differentiated synt− cells (Fig. 8, E and F), implying the occurrence of a degenerative process upon α-Syntrophin deficiency.
FIGURE 8.

α-Syntrophin down-regulation affects the expression of myogenic and atrophy genes in differentiating C2C12 cells. A–C, C2C12 cells were transiently transfected with α-Syntrophin siRNA (synt−) or with a scramble siRNA (scr). Total RNA was isolated, and relative mRNA levels of MyoD (A), myogenin (B), and muscle creatine kinase (MCK, C) were analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 6; *, p < 0.001 versus scr cells day 0; °, p < 0.05 versus scr day 2 or day 4). D, morphological analysis of differentiating cells (day 2) by optical microscopy. E and F, total RNA was isolated, and relative mRNA levels of Atrogin 1 (E) and Murf1 (F) were analyzed by RT-qPCR. The data are expressed as means ± S.D. (n = 6; *, p < 0.05 versus scr day 0; °, p < 0.001 versus scr day 4; #, p < 0.05 versus synt− day 0).
DISCUSSION
NO diffusion is limited by its interaction with different molecules within the cells, and therefore the subcellular location(s) of the NOS isoforms affect NO diffusion. Compelling evidence showed that NO can be produced at the nuclear level, because many factors involved in nNOS activity (e.g., subunits of the soluble guanylate cyclase, calmodulin, tetrahydrobiopterin synthetic enzymes) have been detected in nuclei (49–51). This evidence strongly suggests that direct nuclear production of NO could be implicated in transcriptional regulation. Notwithstanding, until now the role of nNOS in the proximity of the nucleus was still obscure. Here, we gave proof of the involvement of the PDZ in targeting the activity of nNOS directly in the nucleus, thus favoring the NO-dependent transcription of mitochondrial genes. Even though nNOS has been found localized in the nucleus of different cell lines also including neuroblastoma SH-SY5Y (13) and primary HUVEC endothelial cells (52), to our knowledge no data are available regarding the localization of nNOS in nuclei of myocytes. In this work we observed that nNOS resides in nuclei of C2C12 cells. Moreover, in line with what we previously observed in neuroblastoma cells (13), the nNOS isoform lacking the PDZ domain (nNOSβ) cannot be targeted to the nuclei of myocytes.
The S-nitrosylation of nuclear proteins, including NO-sensitive transcription factors, have been predominantly ascribed to the trans-S-nitrosylation activity of GAPDH (53). In this work, we demonstrated that NO can S-nitrosylate nuclear proteins only when it is produced in the nucleus. To do so, nNOS is recruited to the nuclear compartment thanks to its PDZ domain and therein orchestrates the signaling cascade culminating in the induction of mitochondrial biogenesis. In particular, our data suggest that NO derived from nuclear nNOS is the genuine mediator of post-translational modification of CREB, which in turn is implicated in the induction of mitochondrial biogenesis. Actually, CREB-SNO is higher in cells expressing nNOSα splicing variant, and such CREB modification is associated with its heightened binding capacity to PGC-1α promoter with consequent increased PGC-1α expression. Interestingly, here we give evidence of the functional role of S-nitrosylation in CREB activation that is frequently linked to NO/cGMP/PKA-dependent phosphorylation at the cytosolic level (54, 55). The fact that sGC inhibitors do not dampen fl-nNOS-mediated effects suggests that such a phosphorylation axis is not involved in CREB modulation. It is more likely that CREB activity is directly regulated by nuclear NO flux via its S-nitrosylation. Indeed, in-batch treatment of isolated nuclei with the NO donor GSNO significantly increases the binding capacity of CREB on its consensus sequence. A family of CREB co-activators has been identified, that is, TORCs (transducers of regulated CREB activity). Interestingly, TORCs induce PGC-1α transcription and mitochondrial biogenesis in muscle cells (56), thus our data highlight the possibility that also TORCs can be S-nitrosylated and contribute to activate CREB. This issue is currently under investigation in our laboratory.
α-Syntrophin has been predominantly found in the sarcolemma of skeletal muscle cells in association with Dystrophin. nNOS is a component of such a membrane complex and at the sarcolemma level is functional in regulating blood flow (32). However, α-Syntrophin and Dystrophin have been also observed in the inner nuclear membrane of cultured myocytes, and its level increases during myogenesis in nuclei (57, 58). Interestingly, nNOSα has been revealed in the nuclear membrane of HeLa cells in association with Dystrophin and α-Syntrophin (46). We have demonstrated that nNOS was no more able to be translocated in the nucleus when α-Syntrophin was down-regulated. Taking into account that PDZ of nNOS is the functional domain in the interaction with α-Syntrophin at the plasma membrane (32), overall our data corroborate the assumption that such a domain is pivotal for the interaction of nNOS with α-Syntrophin also at the nuclear envelope. Another interesting finding of our work is that during myogenesis, which is notably accompanied by mitochondrial biogenesis, the rate of nuclear nNOS protein and activity localized at the nucleus is increased. Similarly, also α-Syntrophin progressively accumulates in the nucleus, and its down-regulation dramatically affects nNOS nuclear recruitment as well as nuclear S-nitrosylation normally occurring during differentiation. On the basis of this evidence, it is likely that α-Syntrophin is the main carrier of nNOS toward nuclei and the upstream mediator of NO signaling cascade culminating in mitochondrial biogenesis (Fig. 9). Notably, Dystrophin complex, which includes α-Syntrophin, represents a crucial scaffold of signaling proteins at the plasma membrane of myocytes, among which nNOS is included (59, 60). Here we give evidence of the involvement of α-Syntrophin in driving the NO flux up to the nucleus favoring local S-nitrosylation and initiation of CREB/PGC-1α-dependent mitochondrial biogenesis also during myogenesis. It is worth mentioning that the nNOS variant we detected in the nuclei of untransfected C2C12 cells under basal condition or upon differentiation most likely is nNOSμ, because skeletal muscle cells do not express nNOSα (31). Moreover, an antibody directed against the N-terminal region of nNOS, where PDZ is located, was used; thus, we evaluated neither the behavior of nNOSβ during differentiation nor that after overexpression in C2C12 cells. Given the proven role of nNOSβ in skeletal muscle cells in regulating microtubule cytoskeleton integrity (31), studies are ongoing in our laboratory to investigate this issue.
FIGURE 9.
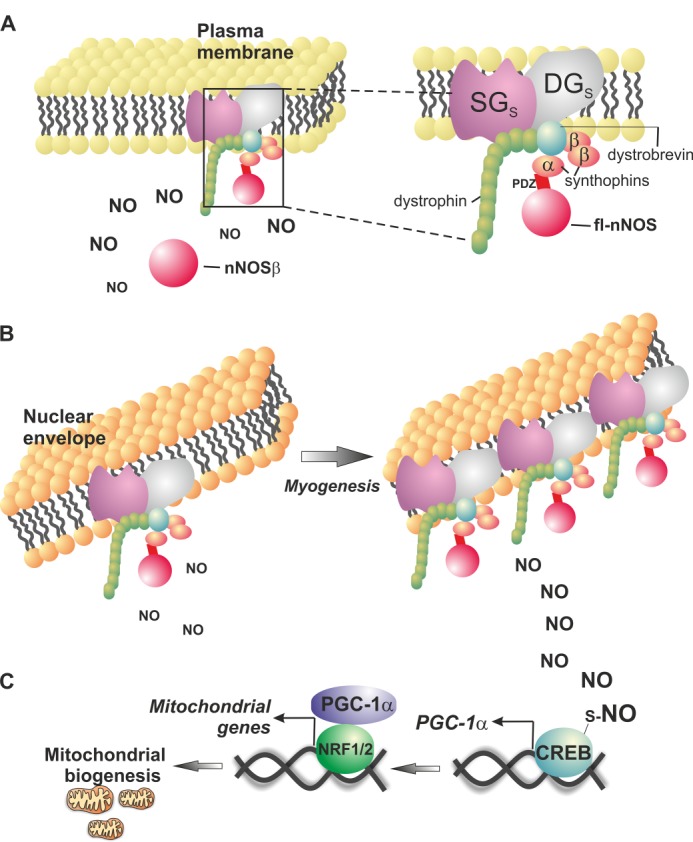
Schematic representation of the role of nuclear nNOS and α-Syntrophin in mitochondrial biogenesis. A, PDZ-containing nNOS (fl-nNOS) is located at the plasma membrane and is part of the Dystrophin complex via the anchoring to α-Syntrophin. PDZ-lacking nNOS (nNOSβ) maintains full NO synthetizing activity and localizes in the cytoplasm but not in the nucleus. B, the Dystrophin complex also localizes at the inner membrane of the nuclear envelope (58), and fl-nNOS is recruited into the nucleus via its interaction with α-Syntrophin. Under certain stimuli (e.g., induction of myogenesis), the PDZ domain is responsible for α-Syntrophin-mediated fl-nNOS recruitment to the nucleus and allows NO synthesis directly at the nuclear level. C, this process facilitates S-nitrosylation of nuclear proteins including CREB transcription factor, which binds to PGC-1α promoter and induces mitochondrial biogenesis. During myogenesis, nuclear fl-nNOS and α-Syntrophin content increases in the nucleus, and this triggers mitochondrial biogenesis. SGs, sarcoglycans; DGs, dystroglycans; NRF1/2, nuclear respiratory factor 1 or 2.
It is largely known that reduction of mitochondrial mass contributes to exercise intolerance, inflammation, and degeneration typical of skeletal muscle wasting and atrophy (61–63). Interestingly we have shown that the failure of mitochondrial biogenesis upon α-Syntrophin down-regulation is associated with increased marker of myotube degeneration. In contrast, α-Syntrophin knock-out mice do not show any effect of myopathy or skeletal muscle degeneration and have unchanged contractile function in vitro (64, 65). However, no information is available about the age of the knock-out mice used for these studies (64, 65). Conceivably, such mice could develop myopathy later in the adulthood. In addition, there are no data regarding muscle oxidative capacity and the effect of exercise on degeneration markers in such α-Syntrophin knock-out mice. Thus, it is possible to postulate that α-Syntrophin, by impinging myogenesis and mitochondrial biogenesis, could be implicated in skeletal muscle regeneration. Our findings could be helpful for the comprehension of the molecular mechanism underlying Duchenne's muscular dystrophy as well as other human myopathies that have been associated with the loss of sarcolemmal nNOS (66, 67) and defective mitochondrial biogenesis (68). It can be postulated that in addition to nNOS localization to sarcolemma, the lack of Dystrophin complex in Duchenne's muscular dystrophy may affect nNOS distribution to the nucleus, thus dramatically inhibiting PGC-1α expression and mitochondrial biogenesis and finally triggering the atrophy process. Accordingly, PGC-1α gene transfer efficiently recovers the lower mitochondrial biomass and oxidative capacity and ameliorates disease parameters, including muscle histology, running performance, and plasma creatine kinase levels in the mouse model of Duchenne's myopathy (69, 70). Similarly, treatments with NO donors favor mitochondrial biogenesis and myogenesis and restore oxidative capacity in affected myocytes (71, 72).
In conclusion, our results show that nNOS is anchored via the PDZ domain also at the Dystrophin-Syntrophin complex sited at the nuclear envelope of myocytes (58), and this event is mandatory for the NO/CREB/PGC-1α-driven mitochondrial biogenesis. On the basis of our results, it is conceivable that the lack of nNOS anchoring to the nucleus may contribute to myopathy because of impaired mitochondrial biogenesis. Thus, control of nNOS splicing in skeletal muscle could represent a novel avenue to prevent or treat myopathies. We hope that continuing research into the roles of nNOS splicing variants and NO in mitochondrial biogenesis will lead to a greater understanding of pathological mechanisms involved in myopathies that can ultimately be exploited therapeutically.
This work was partially supported by grants from MIUR PRIN 2009.

This article contains supplemental Tables S1 and Figs. S1 and S2.
- NOS
- nitric-oxide synthase
- CREB
- cAMP response element-binding protein
- GSNO
- S-nitrosoglutathione
- l-NAME
- l-NG-nitroarginine methyl ester
- MtCO1
- cytochrome c oxidase subunit I
- mtDNA
- mitochondrial DNA
- nNOS
- neuronal nitric-oxide synthase
- ODQ
- sGC inhibitor 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one
- PGC
- peroxisome proliferator-activated receptor γ co-activator
- RPL
- ribosomal protein large subunit
- sGC
- soluble guanylate cyclase
- TFAM
- mitochondrial transcription factor A
- fl-nNOS
- full-length nNOS
- qPCR
- quantitative PCR
- NLB
- nucleus lysis buffer.
REFERENCES
- 1. Förstermann U., Sessa W. C. (2012) Nitric oxide synthases. Regulation and function. Eur. Heart J. 33, 829–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rotilio G., Aquilano K., Ciriolo M. R. (2003) Interplay of Cu,Zn superoxide dismutase and nitric oxide synthase in neurodegenerative processes. IUBMB Life 55, 629–634 [DOI] [PubMed] [Google Scholar]
- 3. Villanueva C., Giulivi C. (2010) Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol. Med. 49, 307–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou L., Zhu D. Y. (2009) Neuronal nitric oxide synthase. Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 20, 223–230 [DOI] [PubMed] [Google Scholar]
- 5. Sullivan J. C., Pollock J. S. (2003) NOS 3 subcellular localization in the regulation of nitric oxide production. Acta Physiol. Scand. 179, 115–122 [DOI] [PubMed] [Google Scholar]
- 6. Brenman J. E., Chao D. S., Gee S. H., McGee A. W., Craven S. E., Santillano D. R., Wu Z., Huang F., Xia H., Peters M. F., Froehner S. C., Bredt D. S. (1996) Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell 84, 757–767 [DOI] [PubMed] [Google Scholar]
- 7. Thomas G. D., Sander M., Lau K. S., Huang P. L., Stull J. T., Victor R. G. (1998) Impaired metabolic modulation of α-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 95, 15090–15095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qu Z. W., Miao W. Y., Hu S. Q., Li C., Zhuo X. L., Zong Y. Y., Wu Y. P., Zhang G. Y. (2012) N-Methyl-d-aspartate receptor-dependent denitrosylation of neuronal nitric oxide synthase increase the enzyme activity. PLoS One 7, e52788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saur D., Paehge H., Schusdziarra V., Allescher H. D. (2000) Distinct expression of splice variants of neuronal nitric oxide synthase in the human gastrointestinal tract. Gastroenterology 118, 849–858 [DOI] [PubMed] [Google Scholar]
- 10. Catania M. V., Aronica E., Yankaya B., Troost D. (2001) Increased expression of neuronal nitric oxide synthase spliced variants in reactive astrocytes of amyotrophic lateral sclerosis human spinal cord. J. Neurosci. 21, RC148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee M. A., Cai L., Hübner N., Lee Y. A., Lindpaintner K. (1997) Tissue- and development-specific expression of multiple alternatively spliced transcripts of rat neuronal nitric oxide synthase. J. Clin. Invest. 100, 1507–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aquilano K., Rotilio G., Ciriolo M. R. (2003) Proteasome activation and nNOS down-regulation in neuroblastoma cells expressing a Cu,Zn superoxide dismutase mutant involved in familial ALS. J. Neurochem. 85, 1324–1335 [DOI] [PubMed] [Google Scholar]
- 13. Baldelli S., Aquilano K., Rotilio G., Ciriolo M. R. (2011) Neuronal nitric oxide synthase interacts with Sp1 through the PDZ domain inhibiting Sp1-mediated copper-zinc superoxide dismutase expression. Int. J. Biochem. Cell Biol. 43, 163–169 [DOI] [PubMed] [Google Scholar]
- 14. Eliasson M. J., Blackshaw S., Schell M. J., Snyder S. H. (1997) Neuronal nitric oxide synthase alternatively spliced forms. Prominent functional localizations in the brain. Proc. Natl. Acad. Sci. U.S.A. 94, 3396–3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Larsson B., Phillips S. C. (1998) Isolation and characterization of a novel, human neuronal nitric oxide synthase cDNA. Biochem. Biophys. Res. Commun. 251, 898–902 [DOI] [PubMed] [Google Scholar]
- 16. Silvagno F., Xia H., Bredt D. S. (1996) Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. J. Biol. Chem. 271, 11204–11208 [DOI] [PubMed] [Google Scholar]
- 17. Piantadosi C. A., Suliman H. B. (2012) Redox regulation of mitochondrial biogenesis. Free Radic. Biol. Med. 53, 2043–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nisoli E., Falcone S., Tonello C., Cozzi V., Palomba L., Fiorani M., Pisconti A., Brunelli S., Cardile A., Francolini M., Cantoni O., Carruba M. O., Moncada S., Clementi E. (2004) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lira V. A., Brown D. L., Lira A. K., Kavazis A. N., Soltow Q. A., Zeanah E. H., Criswell D. S. (2010) Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J. Physiol. 588, 3551–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lettieri Barbato D., Baldelli S., Pagliei B., Aquilano K., Ciriolo M. R. (2012) Caloric restriction and the nutrient-sensing PGC-1α in mitochondrial homeostasis. New perspectives in neurodegeneration. Int. J. Cell Biol. 2012, 759583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baldelli S., Aquilano K., Ciriolo M. R. (2013) Punctum on two different transcription factors regulated by PGC-1α. Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 1830, 4137–4146 [DOI] [PubMed] [Google Scholar]
- 22. Tengan C. H., Rodrigues G. S., Godinho R. O. (2012) Nitric oxide in skeletal muscle. Role on mitochondrial biogenesis and function. Int. J. Mol. Sci. 13, 17160–17184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vercauteren K., Pasko R. A., Gleyzer N., Marino V. M., Scarpulla R. C. (2006) PGC-1-related coactivator. Immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol. Cell Biol. 26, 7409–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stamler J. S., Meissner G. (2001) Physiology of nitric oxide in skeletal muscle. Physiol. Rev. 81, 209–237 [DOI] [PubMed] [Google Scholar]
- 25. Aquilano K., Baldelli S., Pagliei B., Ciriolo M. R. (2013) Extranuclear localization of SIRT1 and PGC-1α. An insight into possible roles in diseases associated with mitochondrial dysfunction. Curr. Mol. Med. 13, 140–154 [PubMed] [Google Scholar]
- 26. Tengan C. H., Kiyomoto B. H., Godinho R. O., Gamba J., Neves A. C., Schmidt B., Oliveira A. S., Gabbai A. A. (2007) The role of nitric oxide in muscle fibers with oxidative phosphorylation defects. Biochem. Biophys. Res. Commun. 359, 771–777 [DOI] [PubMed] [Google Scholar]
- 27. El-Hattab A. W., Emrick L. T., Craigen W. J., Scaglia F. (2012) Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 107, 247–252 [DOI] [PubMed] [Google Scholar]
- 28. El-Hattab A. W., Hsu J. W., Emrick L. T., Wong L. J., Craigen W. J., Jahoor F., Scaglia F. (2012) Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol. Genet. Metab. 105, 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brunelli S., Sciorati C., D'Antona G., Innocenzi A., Covarello D., Galvez B. G., Perrotta C., Monopoli A., Sanvito F., Bottinelli R., Ongini E., Cossu G., Clementi E. (2007) Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. U.S.A. 104, 264–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nisoli E., Clementi E., Paolucci C., Cozzi V., Tonello C., Sciorati C., Bracale R., Valerio A., Francolini M., Moncada S., Carruba M. O. (2003) Mitochondrial biogenesis in mammals. The role of endogenous nitric oxide. Science 299, 896–899 [DOI] [PubMed] [Google Scholar]
- 31. Percival J. M., Anderson K. N., Huang P., Adams M. E., Froehner S. C. (2010) Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle. J. Clin. Invest. 120, 816–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas G. D., Shaul P. W., Yuhanna I. S., Froehner S. C., Adams M. E. (2003) Vasomodulation by skeletal muscle-derived nitric oxide requires α-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ. Res. 92, 554–560 [DOI] [PubMed] [Google Scholar]
- 33. Corso-Díaz X., Krukoff T. L. (2010) nNOSα and nNOSβ localization to aggresome-like inclusions is dependent on HSP90 activity. J. Neurochem. 114, 864–872 [DOI] [PubMed] [Google Scholar]
- 34. Frandsen U., Lopez-Figueroa M., Hellsten Y. (1996) Localization of nitric oxide synthase in human skeletal muscle. Biochem. Biophys. Res. Commun. 227, 88–93 [DOI] [PubMed] [Google Scholar]
- 35. Baldelli S., Aquilano K., Rotilio G., Ciriolo M. R. (2008) Glutathione and copper, zinc superoxide dismutase are modulated by overexpression of neuronal nitric oxide synthase. Int. J. Biochem. Cell Biol. 40, 2660–2670 [DOI] [PubMed] [Google Scholar]
- 36. Aquilano K., Vigilanza P., Rotilio G., Ciriolo M. R. (2006) Mitochondrial damage due to SOD1 deficiency in SH-SY5Y neuroblastoma cells. A rationale for the redundancy of SOD1. FASEB J. 20, 1683–1685 [DOI] [PubMed] [Google Scholar]
- 37. Lettieri Barbato D., Tatulli G., Aquilano K., Ciriolo M. R. (2013) FoxO1 controls lysosomal acid lipase in adipocytes. Implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 4, e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pagliei B., Aquilano K., Baldelli S., Ciriolo M. R. (2013) Garlic-derived diallyl disulfide modulates peroxisome proliferator activated receptor gamma co-activator 1α in neuroblastoma cells. Biochem. Pharmacol. 85, 335–344 [DOI] [PubMed] [Google Scholar]
- 39. Lettieri Barbato D., Aquilano K., Baldelli S., Cannata S. M., Bernardini S., Rotilio G., Ciriolo M. R. (2013) Proline oxidase-adipose triglyceride lipase pathway restrains adipose cell death and tissue inflammation. Cell Death Differ. 10.1038/cdd.2013.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aquilano K., Baldelli S., Pagliei B., Cannata S. M., Rotilio G., Ciriolo M. R. (2013) p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox. Signal. 18, 386–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aquilano K., Baldelli S., Cardaci S., Rotilio G., Ciriolo M. R. (2011) Nitric oxide is the primary mediator of cytotoxicity induced by GSH depletion in neuronal cells. J. Cell Sci. 124, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 42. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 43. Bossy-Wetzel E., Lipton S. A. (2003) Nitric oxide signaling regulates mitochondrial number and function. Cell Death Differ. 10, 757–760 [DOI] [PubMed] [Google Scholar]
- 44. Wu H., Kanatous S. B., Thurmond F. A., Gallardo T., Isotani E., Bassel-Duby R., Williams R. S. (2002) Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296, 349–352 [DOI] [PubMed] [Google Scholar]
- 45. Riccio A., Alvania R. S., Lonze B. E., Ramanan N., Kim T., Huang Y., Dawson T. M., Snyder S. H., Ginty D. D. (2006) A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol. Cell 21, 283–294 [DOI] [PubMed] [Google Scholar]
- 46. Fuentes-Mera L., Rodríguez-Muñoz R., González-Ramírez R., García-Sierra F., González E., Mornet D., Cisneros B. (2006) Characterization of a novel Dp71 dystrophin-associated protein complex (DAPC) present in the nucleus of HeLa cells. Members of the nuclear DAPC associate with the nuclear matrix. Exp. Cell Res. 312, 3023–3035 [DOI] [PubMed] [Google Scholar]
- 47. Kraft C. S., LeMoine C. M., Lyons C. N., Michaud D., Mueller C. R., Moyes C. D. (2006) Control of mitochondrial biogenesis during myogenesis. Am. J. Physiol. Cell Physiol. 290, C1119–C1127 [DOI] [PubMed] [Google Scholar]
- 48. Remels A. H., Langen R. C., Schrauwen P., Schaart G., Schols A. M., Gosker H. R. (2010) Regulation of mitochondrial biogenesis during myogenesis. Mol. Cell Endocrinol. 315, 113–120 [DOI] [PubMed] [Google Scholar]
- 49. Pifarré P., Baltrons M. A., Földi I., García A. (2009) NO-sensitive guanylyl cyclase beta1 subunit is peripherally associated to chromosomes during mitosis. Novel role in chromatin condensation and cell cycle progression. Int. J. Biochem. Cell Biol. 41, 1719–1730 [DOI] [PubMed] [Google Scholar]
- 50. Bachs O., Agell N., Carafoli E. (1992) Calcium and calmodulin function in the cell nucleus. Biochim. Biophys. Acta 1113, 259–270 [DOI] [PubMed] [Google Scholar]
- 51. Elzaouk L., Laufs S., Heerklotz D., Leimbacher W., Blau N., Résibois A., Thöny B. (2004) Nuclear localization of tetrahydrobiopterin biosynthetic enzymes. Biochim. Biophys. Acta 1670, 56–68 [DOI] [PubMed] [Google Scholar]
- 52. Chakrabarti S., Chan C. K., Jiang Y., Davidge S. T. (2012) Neuronal nitric oxide synthase regulates endothelial inflammation. J. Leukocyte Biol. 91, 947–956 [DOI] [PubMed] [Google Scholar]
- 53. Kornberg M. D., Sen N., Hara M. R., Juluri K. R., Nguyen J. V., Snowman A. M., Law L., Hester L. D., Snyder S. H. (2010) GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Delghandi M. P., Johannessen M., Moens U. (2005) The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 17, 1343–1351 [DOI] [PubMed] [Google Scholar]
- 55. Ciani E., Virgili M., Contestabile A. (2002) Akt pathway mediates a cGMP-dependent survival role of nitric oxide in cerebellar granule neurones. J. Neurochem. 81, 218–228 [DOI] [PubMed] [Google Scholar]
- 56. Wu Z., Huang X., Feng Y., Handschin C., Feng Y., Gullicksen P. S., Bare O., Labow M., Spiegelman B., Stevenson S. C. (2006) Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proc. Natl. Acad. Sci. U.S.A. 103, 14379–14384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim M. J., Hwang S. H., Lim J. A., Froehner S. C., Adams M. E., Kim H. S. (2010) α-Syntrophin modulates myogenin expression in differentiating myoblasts. PLoS One 5, e15355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. González-Ramírez R., Morales-Lázaro S. L., Tapia-Ramírez V., Mornet D., Cisneros B. (2008) Nuclear and nuclear envelope localization of dystrophin Dp71 and dystrophin-associated proteins (DAPs) in the C2C12 muscle cells. DAPs nuclear localization is modulated during myogenesis. J. Cell Biochem. 105, 735–745 [DOI] [PubMed] [Google Scholar]
- 59. Constantin B. (2013) Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 10.1016/j.bbamem.2013.08.023 [DOI] [PubMed] [Google Scholar]
- 60. Bhat H. F., Adams M. E., Khanday F. A. (2013) Syntrophin proteins as Santa Claus. Role(s) in cell signal transduction. Cell Mol. Life Sci. 70, 2533–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tarnopolsky M. A., Raha S. (2005) Mitochondrial myopathies. Diagnosis, exercise intolerance, and treatment options. Med. Sci. Sports Exerc. 37, 2086–2093 [DOI] [PubMed] [Google Scholar]
- 62. Hernández-Aguilera A., Rull A., Rodríguez-Gallego E., Riera-Borrull M., Luciano-Mateo F., Camps J., Menéndez J. A., Joven J. (2013) Mitochondrial dysfunction. A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013, 135698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marzetti E., Calvani R., Cesari M., Buford T. W., Lorenzi M., Behnke B. J., Leeuwenburgh C. (2013) Mitochondrial dysfunction and sarcopenia of aging. From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 45, 2288–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Adams M. E., Kramarcy N., Krall S. P., Rossi S. G., Rotundo R. L., Sealock R., Froehner S. C. (2000) Absence of α-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 150, 1385–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kameya S., Miyagoe Y., Nonaka I., Ikemoto T., Endo M., Hanaoka K., Nabeshima Y., Takeda S. (1999) α1-Syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J. Biol. Chem. 274, 2193–2200 [DOI] [PubMed] [Google Scholar]
- 66. Meinen S., Lin S., Rüegg M. A., Punga A. R. (2012) Fatigue and muscle atrophy in a mouse model of myasthenia gravis is paralleled by loss of sarcolemmal nNOS. PLoS One 7, e44148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Finanger Hedderick E. L., Simmers J. L., Soleimani A., Andres-Mateos E., Marx R., Files D. C., King L., Crawford T. O., Corse A. M., Cohn R. D. (2011) Loss of sarcolemmal nNOS is common in acquired and inherited neuromuscular disorders. Neurology 76, 960–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Percival J. M., Siegel M. P., Knowels G., Marcinek D. J. (2013) Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 22, 153–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Godin R., Daussin F., Matecki S., Li T., Petrof B. J., Burelle Y. (2012) Peroxisome proliferator-activated receptor gamma coactivator1- gene α transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol. 590, 5487–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Handschin C., Kobayashi Y. M., Chin S., Seale P., Campbell K. P., Spiegelman B. M. (2007) PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 21, 770–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Archer J. D., Vargas C. C., Anderson J. E. (2006) Persistent and improved functional gain in mdx dystrophic mice after treatment with l-arginine and deflazacort. FASEB J. 20, 738–740 [DOI] [PubMed] [Google Scholar]
- 72. Buono R., Vantaggiato C., Pisa V., Azzoni E., Bassi M. T., Brunelli S., Sciorati C., Clementi E. (2012) Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells 30, 197–209 [DOI] [PMC free article] [PubMed] [Google Scholar]