

Background: Latrophilins are large adhesion-type GPCRs that may mediate cell adhesion via heterophilic interactions.
Results: Latrophilin-1 binding to teneurins exhibits nanomolar affinity, is regulated by alternative splicing, and mediates intercellular adhesion.
Conclusion: Latrophilins are cell-adhesion molecules with multiple trans-synaptic ligands.
Significance: Our data support a role for latrophilin in trans-neuronal interactions by binding to multiple heterophilic ligands.
Keywords: Alternative Splicing, Cell Adhesion, G Protein-coupled Receptors (GPCR), Ligand-binding Protein, Neurons, FLRT3, Neurexin, α-Latrotoxin, Synapse Formation, Teneurin
Abstract
Latrophilin-1, -2, and -3 are adhesion-type G protein-coupled receptors that are auxiliary α-latrotoxin receptors, suggesting that they may have a synaptic function. Using pulldowns, we here identify teneurins, type II transmembrane proteins that are also candidate synaptic cell-adhesion molecules, as interactors for the lectin-like domain of latrophilins. We show that teneurin binds to latrophilins with nanomolar affinity and that this binding mediates cell adhesion, consistent with a role of teneurin binding to latrophilins in trans-synaptic interactions. All latrophilins are subject to alternative splicing at an N-terminal site; in latrophilin-1, this alternative splicing modulates teneurin binding but has no effect on binding of latrophilin-1 to another ligand, FLRT3. Addition to cultured neurons of soluble teneurin-binding fragments of latrophilin-1 decreased synapse density, suggesting that latrophilin binding to teneurin may directly or indirectly influence synapse formation and/or maintenance. These observations are potentially intriguing in view of the proposed role for Drosophila teneurins in determining synapse specificity. However, teneurins in Drosophila were suggested to act as homophilic cell-adhesion molecules, whereas our findings suggest a heterophilic interaction mechanism. Thus, we tested whether mammalian teneurins also are homophilic cell-adhesion molecules, in addition to binding to latrophilins as heterophilic cell-adhesion molecules. Strikingly, we find that although teneurins bind to each other in solution, homophilic teneurin-teneurin binding is unable to support stable cell adhesion, different from heterophilic teneurin-latrophilin binding. Thus, mammalian teneurins act as heterophilic cell-adhesion molecules that may be involved in trans-neuronal interaction processes such as synapse formation or maintenance.
Introduction
In the brain, neurons form complex overlapping networks in which each neuron communicates with other neurons at synapses, which are specialized intercellular junctions dedicated to trans-neuronal information transfer. Synapses are thought to be established and maintained by synaptic cell-adhesion molecules (1, 2). Moreover, different synapses exhibit distinct properties depending on their position in a neural network, and these synaptic properties are also likely determined by synaptic cell-adhesion molecules. Many synaptic cell-adhesion molecules have been intensely studied, such as neurexins, neuroligins, and cadherins, but the biochemical properties of synaptic cell-adhesion molecules are incompletely understood. Of particular interest here is the role of cell-adhesion molecules that are conserved in invertebrates as they might provide insights on the universal events leading to synapse formation and/or maintenance. Latrophilins (also referred to as CIRLs for “calcium-independent receptors for α-latrotoxin” (3, 4)), teneurins (also called “Odz” (5, 6)), and fibronectin leucine-rich repeat transmembrane proteins (FLRTs)3 are evolutionarily conserved cell-adhesion molecules that have been linked to synapses and that bind to each other (7–10), but their relative interaction properties have not been characterized.
Latrophilins are adhesion-type GPCRs that include a large extracellular sequence (∼1000 residues) composed of an N-terminal lectin domain, central olfactomedin-like, serine/threonine-rich, and hormone-binding domains, and a C-terminal GAIN domain containing a GPS motif that represents an auto-cleavage domain (11, 12). Latrophilins were identified as α-latrotoxin-binding proteins that together with neurexins may act as co-receptors for this potent neurotoxin (3, 4, 11). Three latrophilin proteins (latrophilin-1, -2, and -3, abbreviated as Lphn1, Lphn2, and Lphn3 after their gene symbols) are expressed in brain; of these, Lphn2 is also widely expressed in non-neuronal tissues (11).
As GPCRs, latrophilins are likely present on plasma membranes, but it is unclear which cells in brain and outside of brain express them and where they are localized in these cells. Knock-out of Lphn1 produces an impairment in α-latrotoxin responsiveness but no other major phenotype (13), suggesting that latrophilins may be functionally redundant in vertebrates.
Latrophilins are the only adhesion-type GPCRs besides flamingo-like CESLR proteins that are conserved in invertebrates. The Caenorhabditis elegans genome encodes two latrophilin genes (lat-1 and lat-2). Lat-1 is required for the alignment of cell division planes to the anterior-posterior axis during development (14). lat-1 function in C. elegans requires its GAIN domain but not its actual GPS sequence (which is the small sequence motif in the overall GAIN domain that contains the cleavage site for GAIN domain-mediated autoproteolysis; see Ref. 12). This result suggested that Lat-1 acts via extracellular interactions but does not need to be cleaved at the GPS motif (15).
Two endogenous ligands for latrophilins have been described, teneurin-2 (7) and FLRT3 (10). Teneurins are large type II transmembrane proteins (∼2800 residues) that are composed of a relatively short N-terminal cytoplasmic sequence, a single transmembrane region (TMR), and a long extracellular sequence containing multiple EGF-like repeats (6). Teneurins form constitutive disulfide-bonded homodimers and are highly glycosylated. Vertebrates express four teneurin genes, whereas invertebrates have usually one or two related genes.
Conflicting data were presented about teneurin function. Teneurins were discovered as pair-rule genes in Drosophila (5, 16). However, later studies suggested that the original phenotype of the teneurin mutation was an inadvertent result of gene manipulations, and was not due to a loss of teneurin expression (17). In mice, deletions of teneurin-3 result in impairments of axonal guidance in the visual system and cause mistargeting of axons from the retina to the geniculate nucleus and the superior colliculus (18, 19). In contrast, deletions of teneurin-4 impair oligodendrocyte differentiation and myelination (20). In Drosophila, conversely, teneurin mutations produce a defect in motor axon guidance, although overexpression studies suggested a role for teneurins in target selection during synaptogenesis (8, 9). Notably, reciprocal pre- and postsynaptic expression experiments in Drosophila combined with binding studies suggested that these phenotypes are caused by loss of a homophilic cell-adhesion function of teneurins.
FLRTs are also a family of cell-surface molecules encoded by three genes (FLRT1, FLRT2, and FLRT3). FLRTs are widely expressed in all tissues (21, 22) and are composed of 10 extracellular leucine-rich repeats, a fibronectin type III domain, a TMR, and a cytoplasmic tail. FLRTs were implicated in FGF signaling (22) and cell adhesion during development (23). Knock-out experiments established that both FLRT2 and FLRT3 are essential for embryonic development and likely required for normal development of the heart (23, 24). In neurons, FLRT3 mRNA is up-regulated after nerve injury (25–27). FLRT bind to the axonal guidance receptors Unc5b and Unc5d (28, 29), and FLRT ectodomains have been proposed to be shed from neurons to act as repellents for migrating neurons (29). Finally, FLRTs have been shown to bind to latrophilins and may act in synapse development (10).
In this study, we have examined potential ligands for latrophilins and characterized the properties of their interactions. Our data show that all teneurins tested and FLRT3 bind to all latrophilins tested with high affinity and that teneurin binding is regulated by alternative splicing of latrophilins, whereas FLRT3 binding to latrophilins is not. We also found that mammalian teneurins are capable of homophilic binding in a cis-configuration but do not stably interact homophilically in a trans-configuration, suggesting that they are not homophilic cell-adhesion molecules. Furthermore, we observed that addition of excess latrophilin fragments containing the teneurin- and FLRT3-binding sites to the medium of cultured neurons decreases synapse numbers. Our data suggest that latrophilins are potential synaptic cell-adhesion molecules that interact trans-synaptically with teneurins and FLRTs.
EXPERIMENTAL PROCEDURES
Materials
Antibodies were obtained from the following sources: mouse monoclonal HA antibody, Covance (clone 16B12); mouse monoclonal Myc antibody (clone 9E10), the Developmental Studies Hybridoma Bank; rabbit polyclonal FLAG antibody, Sigma; monoclonal GFP antibody (clone 3E6), Invitrogen; guinea pig polyclonal vGlut1 antibody, Millipore; rabbit polyclonal vGAT antibody, Synaptic Systems; and mouse monoclonal MAP2 antibody (clone HM-2), Sigma. Secondary antibodies against human IgG coupled to HRP were purchased from MP Biomedical, and secondary fluorescent antibodies coupled to AlexaFluor-488, -546, or -633 were from Invitrogen. Agarose beads coated with anti-FLAG M2 antibody were from Sigma, and protein A-Sepharose beads from GE Healthcare. 3,3′,5,5′-Tetramethylbenzidine peroxidase enzyme immunoassay solution was from Sigma. All oligonucleotides and probes were purchased from IDT except for ready-made qRT-PCR assays for mouse β-actin (4352933E) and GAPDH (4352933E) that were purchased from Applied Biosystems.
Construction of Expression Vectors
1) For full-length Lphn1 vectors, pCMV-Lp1-1 encodes full-length rat Lphn1 lacking inserts in the 5′ (SSA) and 3′ sites of alternative splicing, whereas pCMV-Lp1-5 encodes full-length rat Lphn1 containing an insert in the 5′ site of alternative splicing (SSA) (11). 2) PDGF receptor TMR fusion proteins were cloned into the pDisplay vector (Invitrogen) and contain an Igκ signal peptide and an N-terminal HA epitope before and a c-Myc epitope and the PDGF receptor TMR after the insert. Various pDisplay-Lp1 expression plasmids (30) were constructed by subcloning the respective PCR-amplified fragments into the SacII-SalI sites of the pDisplay vector and encode the following rat Lphn1 residues fused to the PDGF receptor TMR: pDisplay-Lp1ECD, Ser26–Glu856; pDisplay-Lp1LEC, Ser26–Tyr131; pDisplay-Lp1L/O/S), Ser26–Ser427 lacking an insert in splice site A; pDisplay-Lp1L/O/S+, Ser26–Ser427 with inclusion of an insert in splice site A; pDisplay-Lp1Olf/STR, Val137–Ser475; pDisplay-Lp1H/G, Leu472–Glu856. The pDisplay-FLRT3 vector was generated by subcloning the BglII-SalI fragment into the same sites of pDisplay vector and encoding the following rat FLRT3 residues fused to the PDGF receptor TMR, Cys31–Leu529. 3) pCMV-IgLp1 plasmids were described previously (30) and generated by subcloning the EcoRI-SalI fragments from the various Lphn1 pDisplay vectors into the same sites of pCMVIg9 (31), resulting in constructs encoding the Igκ signal peptide and HA epitope from the pDisplay vector as well as the different Lphn1 sequences fused to the human IgG-Fc domain. Only pCMV-IgLp1LEC that encodes Lphn1 residues Met1–Tyr131 fused to human IgG Fc fragment was cloned by direct insertion of a PCR fragment into the EcoRI-SalI site of pCMVIg vector. 4) For full-length teneurin constructs, pCMV-Ten4-HA (Ten4-HA) encodes full-length mouse teneurin-4 containing an HA epitope at the very C terminus. pcDNA3-Ten2-myc (Ten2-myc) was a gift from Dr. Ushkaryov (7). 5) For soluble teneurin constructs, pCMV-Ten1ECD and pCMV-Ten4ECD were generated by deleting the N terminus as well as the TMR and inserting a FLAG epitope at Gly359 of mouse teneurin-1 and Ser564 of mouse teneurin-4. pCDNA3-Ten2ECD (LassoB) was a gift from Dr. Ushkaryov.
Production and Purification of Recombinant Proteins
For expression of recombinant Lphn1 Ig fusion proteins, HEK293T cells were cultured in 10-cm dishes until they reached 80–90% confluence. The medium was changed to fresh DMEM containing 25 mm chloroquine, incubated for 3 h, and transfected using calcium phosphate with 20 μg of cDNA corresponding to the various Ig fusion proteins. Media containing the soluble Ig fusion proteins were harvested 4 days post-transfection and cleared by centrifugation at 1000 × g. The supernatant was then adjusted to 10 mm HEPES-NaOH, pH 7.4, 1 mm EDTA with protease inhibitors (Roche Diagnostics) and incubated overnight with protein A-Sepharose (GE Healthcare) to bind the human IgG Fc domain. Beads were washed to remove unbound proteins. For recombinant FLAG-tagged teneurins, the same procedure was followed except that anti-FLAG M2-agarose beads (Sigma) were used to immobilize the secreted proteins. When needed, Lphn1 Ig fusion proteins were eluted from the beads using 3.5 m MgCl2 and diluted in dialysis buffer (50 mm HEPES-NaOH, pH 7.4, 150 mm NaCl); FLAG-tagged teneurins were eluted using 0.1 m glycine, pH 2.2, equilibrated with 100 mm HEPES-NaOH, pH 7.4, and diluted in dialysis buffer.
Brain Protein Affinity Chromatography
Brain affinity chromatography was performed as described previously with slight modifications (32). Nine frozen rat brains were homogenized with 6–8 strokes in 30 ml of 20 mm HEPES-NaOH, pH 7.4, 0.1 mm EGTA, 1 mm PMSF, 1 μg/ml pepstatin, 1 μg/ml leupeptin, and 2 μg/ml aprotinin, and 120 ml of the same buffer containing 150 mm NaCl and 1% Triton X-100 final concentration was added. Proteins from the homogenate were extracted for 2 h at 4 °C; insoluble material was removed by centrifugation (1 h at 100,000 × g), and 2 mm MgCl2 and 2 mm CaCl2 were added. For latrophilin pulldowns, protein A-Sepharose containing IgLp1ECD, IgLp1LEC, or Ig-Control (∼200, 500, and 500 μg of protein, respectively, in 0.2 ml) was equilibrated in buffer A (20 mm HEPES-NaOH, pH 7.4, 0.1 mm EDTA, 150 mm NaCl, 2 mm CaCl2, 2 mm MgCl2, 1% Triton X-100), incubated overnight at 4 °C with 50 ml of the brain extract, centrifuged (800 × g for 5 min), and washed five times with 15 ml of buffer A. Washed beads were then packed into polypropylene columns (Pierce), washed again with buffer A (2 ml), and sequentially eluted with 1 ml of buffer A containing 0.5 m NaCl and 1.0 m NaCl. Eluted proteins were precipitated with 30% TCA overnight at 4 °C, washed twice with acetone, and resuspended in 20 μl of sample buffer, and 5 μl were analyzed by SDS-PAGE and silver staining. The broad bands migrating at >200 kDa that were affinity-purified following the 1.0 m NaCl elution on the IgLp1ECD and IgLp1LEC columns were cut out of the gel and analyzed by mass spectrometry (Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University), which identified teneurin-1, -3, and -4 for IgLp1ECD and all four teneurins for IgLp1LEC.
Pulldown
HEK293T cells were transfected with constructs expressing FLAG-tagged extracellular domains of teneurin-1, -2, and -4 (Ten1ECD, Ten2ECD, and Ten4ECD), and the media were collected 4 days later. The proteins were immobilized on anti-FLAG M2-agarose beads washed and incubated in the presence of 200 nm IgLp1 proteins in binding buffer (20 mm HEPES-NaOH, pH 7.4, 0.1 mm EDTA, 2 mm CaCl2, 2 mm MgCl2, 0.1% Triton X-100, and 150 mm NaCl) for a period of 16 h at 4 °C with a gentle agitation. Agarose beads were washed three times with binding buffer, solubilized in SDS sample buffer, analyzed on SDS-polyacrylamide gel, and immunoblotted.
Cell-surface Labeling Assays
Cell surface labeling assays were performed essentially as described (30). For analysis of latrophilin Ig fusion protein binding to teneurins as receptors, HEK293T cells transfected with the indicated full-length teneurin expression vectors were incubated in DMEM containing 20 mm HEPES-NaOH, pH 7.4, 0.1% BSA, and 0.15 μm of Ig fusion protein for 16 h at 4 °C with a gentle agitation. Cells were washed three times with cold DMEM to remove excess Ig proteins and fixed with 4% paraformaldehyde for 10 min on ice. Cells were washed again three times with cold PBS and incubated at room temperature for 15 min in a blocking solution containing PBS and 3% BSA. Rabbit anti-human IgG antibody along with mouse anti-Myc (for Ten2) or anti-HA (for Ten4) were then added (1:500 ratio), and the incubation was prolonged for another 2 h. Cells were washed three times with blocking solution and incubated with anti-rabbit Alexa fluorescent antibody (AlexaFluor-488, green emission) to label bound Ig-latrophilins and anti-mouse Alexa fluorescent antibody (AlexaFluor-633, red emission) to label membrane-expressed teneurins, in the blocking solution for 1 h at room temperature. Cells were finally washed again three times with blocking solution and once with water before mounting on slides using media containing DAPI for nuclear staining. For analysis of soluble teneurin protein binding to latrophilins as receptors, the same procedure was followed except that soluble FLAG-tagged teneurins were used for incubation with cells expressing full-length latrophilins or latrophilin fragments fused to the PDGF receptor TMR. Bound teneurins were detected by immunofluorescence using rabbit anti-FLAG antibody (AlexaFluor-488, green emission), whereas latrophilins expressed as PDGF receptor fusion proteins were detected using mouse anti-HA antibody (AlexaFluor-633, red emission). For analysis of homophilic teneurin binding, soluble FLAG-tagged teneurins were incubated in the presence of cells expressing full-length Myc-tagged teneurin-2. Cells were incubated with rabbit anti-FLAG antibody to detect bound teneurins and mouse anti-Myc antibody to detect membrane expressed teneurin-2. For all samples, slides were then analyzed by confocal microscopy. Images were acquired using a Leica TCS2 confocal microscope with same confocal acquisition settings for all samples of an experiment. Collected z-section images were analyzed blindly using Leica confocal software.
Affinity Measurements
Affinity measurements were performed essentially as described (30). Briefly, transfected HEK293T cells were incubated for 16 h at 4 °C with gentle agitation in DMEM containing 50 mm HEPES-NaOH, pH 7.4, 2 mm CaCl2, 2 mm MgCl2, 0.1% BSA, and the indicated concentrations of Ig fusion proteins (added using serial dilutions). Cells were washed three times with cold DMEM to remove excess Ig proteins and fixed with 4% paraformaldehyde for 10 min on ice. Cells were washed again with cold PBS and incubated at room temperature for 15 min in blocking solution containing PBS and 3% BSA. Rabbit anti-human IgG antibody coupled to HRP was added (1:80,000 ratio in blocking solution), and the incubation was continued for an additional hour. Cells were washed three times with blocking solution and once with PBS before performing the colorimetric assay according to the manufacturer's instructions. Briefly, an HRP substrate solution, 3,3′,5,5′-tetramethylbenzidine peroxidase enzyme immunoassay (Bio-Rad), was added to each well and incubated at room temperature for 10 min under vigorous agitation until a blue coloration appeared. The reaction was stopped by adding an equal volume of 1 n sulfuric acid, producing a yellow coloration, and absorbance at 450 nm was measured in 96-well plates using an Apollo-8 LB912 plate reader (Berthold Technologies). The ligand concentration was plotted against the difference in absorbance measured between plasmid-transfected and mock-transfected cells. An apparent Kd value was calculated from the data using Scatchard analysis and SigmaPlot software.
Cell-adhesion Assays
Cell-adhesion assays were performed with HEK293T cells as described with slight modifications (30). HEK293T cells were individually transfected with the expression vectors as indicated in the figures. After 48 h, the cells were detached using 1 mm EGTA in PBS, mixed, and incubated under gentle agitation at room temperature in DMEM containing 10% FBS, 50 mm HEPES-NaOH, pH 7.4, 10 mm CaCl2, and 10 mm MgCl2. The extent of cell aggregation was assessed at 90 min by removing aliquots, spotting them onto culture slides (Falcon), and imaging by epifluorescence microscopy. The resulting images were then analyzed by counting the number and area of particles in the field using WCIF ImageJ. An arbitrary value for particle area was then set as a threshold (3000 pixels2), based on negative control values. The aggregation index was calculated by expressing the sum of the area of particles exceeding this threshold as a percentage of the total area occupied by all particles in the individual fields.
Acute Recombinant Protein Treatment on Hippocampal Neurons
This treatment was performed as described with slight modifications (33). Hippocampal neurons were dissected, cultured, and plated from DIV0 pups. Recombinant proteins (IgC, IgLp1L/O/S, and IgLp1LEC) were added to DIV10 neurons at a final concentration of 500 nm. At DIV17, neurons were washed with PBS, fixed with 4% paraformaldehyde in PBS for 10 min on ice, and blocked with PBS containing 3% BSA and 0.1% Triton X-100 for 30 min at room temperature. Mouse anti-MAP2, rabbit anti-vGAT, and guinea pig anti-vGLUT1 antibodies were then added (1:500 ratio), and the incubation was prolonged for another 2 h. Cells were washed three times with blocking solution and incubated for 1 h at room temperature with goat anti-mouse AlexaFluor-546, goat anti-rabbit AlexaFluor-488, and goat anti-guinea pig AlexaFluor-633 fluorescent antibodies to label dendrites as well as inhibitory and excitatory presynaptic sites. Cells were finally washed again three times with blocking solution and once with water before mounting on slides using media containing DAPI for nuclear staining. Slides were then analyzed by confocal microscopy. Images were acquired using the confocal microscope Leica TCS2. The same confocal acquisition settings were applied to all samples of the experiment. Collected z-section images were analyzed blindly using Leica confocal software. For each independent experiment, the first ∼50 μm of pyramidal neuron primary dendrites, defined as dendrites emanating directly from the soma, were imaged, and synapse densities/size were averaged. Individual puncta size and density were quantified using the imaging software MetaMorph (Molecular Devices). Dendritic branching numbers (arborization) and neuronal cell body areas were analyzed using MetaMorph software as described previously (34).
Measurements of mRNA Levels by Quantitative RT-PCR
All qRT-PCR analyses have been performed using total RNA samples of brains from P1, P8, P15, and adult C57BL/6 mice as well as selected adult tissues (n = 4). RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol and quantified using an ND-1000 spectrophotometer (ThermoScientific, Wilmington, DE). To correlate levels of transcripts with or without SSA directly, we combined reverse transcription with subsequent RNase H2-dependent PCR using blocked cleavable primers by modifying a strategy by Dobosy et al. (35) for qPCR and now for qRT-PCR. Briefly, 100 ng/μl of total RNA were mixed with 2× VeriQuest Probe one-step qRT-PCR master mix with ROX (Affymetrix Inc., Cleveland, OH) and 1 μl of transcript-specific qPCR assay and RNase-free H2O to a total volume of 15 μl. All assays contained 10 μm forward and reverse primers as well as 5 μm 6-carboxyfluorescein dye-labeled probe. The initial reverse transcription reaction was performed at 50 °C for 16 min using a T100 thermocycler (Bio-Rad). To allow cleavage of blocked primers, RNase H2 was added in a total volume of 5 μl of 1× VeriQuest MasterMix, supplemented with RNase H2 cleavage buffer and RNase H2 (both from IDT, Coralville, IA). Prior to the actual experiment, optimal RNase H2 concentrations have been determined for each assay. Assays detecting different transcripts of Lphn1 used final concentrations of 8 milliunits, Lphn2 20 milliunits, and Lphn3 4 milliunits of RNase H2. Every sample was loaded as a triplicate (now a final volume of 20 μl per well) onto an ABI7900 fast RT-PCR machine (Applied Biosystems, Foster City, CA). Each sample run consisted of an initial reverse transcriptase inactivation and DNA polymerase activation step at 95 °C for 10 min followed by 45 two-step amplification cycles consisting of the following: (a) denaturation at 95 °C for 15 s, and (b) annealing and extension at 60 °C for 30 s. The amplification curve was collected, and the relative transcript level of the target mRNA in each sample was calculated by normalization of Ct values to the reference mRNA (β-actin or GAPDH) using the following equation: V = 2CT[reference]/2CT[target]. V is the relative value of target gene normalized to the reference. The Ct value is the cycle number required for reaching the signal threshold on the quantitative RT-PCR machine. Two different sets of forward primers have been designed to determine the presence or absence of SSA for each latrophilin isoform. To determine all transcript types, forward primers have been used that fully compose sequence homology to the preceding exon of SSA resembling the “cleaved” state of the cleavable blocked primers that when used are hybridizing to a specific transcript isoform (with or without SSA). The reverse primer was always unblocked to allow initial cDNA synthesis and hybridized to the exon following SSA and so does the latrophilin isoform-specific 5′-6-carboxyfluorescein dye-coupled probe. The following oligonucleotides and probes have been used for quantification (forward cleaved, forward blocked, reverse primer, and probe; uppercase DNA base, lowercase RNA base, 3SpC3 capping residue): Lphn1, CAGTACGACTGTGTCCCTTACA, CAGTACGACTGTGTCCCTTACAaAGTG/3SpC3 (+SSA), CAGTACGACTGTGTCCCTTACAuCTTC/3SpC3 (−SSA), GCACCATGCACCAGACTG, TGCTGGAGCCCACCTCTACACAT; Lphn2, GTTCAATATGAATGTGTCCCTTACA, GTTCAATATGAATGTGTCCCTTACAuGGAG/3SpC3 (+SSA2), GTTCAATATGAATGTGTCCCTTACAaAGTG/3SpC3 (+SSA1), GTTCAATATGAATGTGTCCCTTACAuCTTT/3SpC3 (−SSA), GCACCAAGCCCCTGACTT, TGAAAGCAATTGTGGACTCACCATGTAT; Lphn3, GTGCAGTATGAATGTGTCCCTTATA; GTGCAGTATGAATGTGTCCCTTATAaAGTG/3SpC3 (+SSA), GTGCAGTATGAATGTGTCCCTTATAuTTTT/3SpC3 (−SSA), GCACCATGCCCCAGATTG, AGGAGTGTACCAGAGTGAACACTTGTT.
To determine differences in transcript levels of selected genes in WT and CSPα knock-out mice (CSPα KO), we isolated total brain RNA from two pairs of WT and CSPα KO littermates at 35 and 60 days of age. Assays for mouse β-actin (ACTB) (4352933E) and glyceraldehyde dehydrogenase (GAPDH) (4352933E) were purchased from Applied Biosystems. The following assays were purchased from IDT: APP (amyloid precursor protein) (Mm.PT.56a.10488606); Cdm1 (SynCAM) (Mm.PT.56a.30172490); Cdh2 (N-cadherin) (Mm.PT.56a.12378183); DAB (dystroglycan) (Mm.PT.56a.31997391); FLRT2 (Mm.PT.56a.8519890); FLRT3 (Mm.PT.56a.8839004.g); LRRTM2 (Mm.PT.45.12386236); Nlgn1 (NL1) (forward TTGGGCAATAAACTCTCCTGG, reverse TTCCAAGGGCAATACAGTCTC, and probe CCAAGGTTTAACGGTTCGCCACT), Nlgn2 (NL2) (Mm.PT.56a.10816796), Nrxn1 (forward ACTACATCAGTAACTCAGCACAG, reverse ACAAGTGTCCGTTTCAAATCTTG, and probe CTTCTCCTTGACCACAGCCCCAT); myelin-associated glycoprotein (MAG) (Mm.PT.56a.13580215); SNAP25 (Mm.PT.56a.11132039); SNCA (α-synuclein) (Mm.PT.56a.8495900); Syt1 (Mm.PT.56a.7011015.g); Tenm1 (Ten1) (Mm.PT.56a.31903272); Tenm2 (Ten2) (Mm.PT.56a.9415753), and VAMP2 (synaptobrevin-2) (Mm.PT.56a.12188030).
Miscellaneous Procedures
SDS-PAGE and immunoblotting. Protein samples were solubilized in gel sample buffer and loaded onto 6 or 8% SDS-polyacrylamide gels. Gels were stained with either Coomassie Blue or Silver Stain depending on the assay or analyzed by immunoblotting. For immunoblotting, gels were transferred onto nitrocellulose membranes and processed using standard procedures. Bound IgLp1 proteins were detected using a horseradish peroxidase-coupled secondary antibody to human IgG, incubated with ECL reagents, and revealed on x-ray films. Cell culture and transfection were performed as described previously (30). Briefly, HEK293T cells were cultured in 6-well plates until they reached 70–80% confluency. The cells were then transfected using 4 μl of FuGENE 6® (Roche Diagnostics) and 2 μg of respective cDNA constructs. The cells were trypsinized 24 h post-transfection and split into either 12-well plates containing coverslips that have been previously coated with 0.5 g/liter poly-l-lysine in borate buffer for cell surface labeling assays or into 96-well plates similarly coated for affinity measurement assays. The cells were grown for an additional 24 h before further experiments were done.
Statistics
All data are expressed as means ± S.E. and represent the results of at least three independent experiments. Statistical significance was determined by one-way ANOVA using StatView software (SAS Institute Inc.).
RESULTS
mRNA Levels and Alternative Splicing of Latrophilins
We measured the tissue distribution and relative levels of Lphn1, Lphn2, and Lphn3 mRNAs in mice using quantitative RT-PCR. We found that consistent with earlier studies (11), Lphn1 and Lphn3 were primarily expressed in brain, although Lphn1 was also significantly present in kidney (Fig. 1A). Lphn2, in contrast, was ubiquitously expressed in all tissues investigated, although even for Lphn2, the mRNAs levels were highest in brain. In general, the mRNA levels of different latrophilins were comparable, with Lphn1 being expressed at slightly higher levels than Lphn2 or Lphn3.
FIGURE 1.
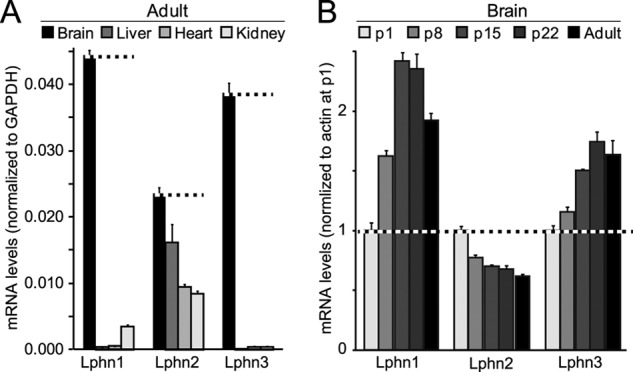
Latrophilin-1 and -3 (Lphn1 and Lphn3) are primarily expressed in brain in a developmentally regulated manner, whereas latrophilin-2 (Lphn2) is ubiquitously expressed. A, quantitative RT-PCR measurements of the latrophilin-1, -2, and -3 (Lphn1, Lphn2, and Lphn3) mRNAs in the brain, liver, heart, and kidney of adult mice. mRNA levels were normalized to those of GAPDH monitored in the same samples. B, quantitative RT-PCR measurements of the Lphn1, Lphn2, and Lphn3 mRNAs in brain at postnatal days 1 (p1), 8 (p8), 15 (p15), 22 (p22), and in adult brain. All mRNA levels were normalized to those of actin as an internal control and then to the mRNA levels of the respective latrophilin at p1. Data shown are means ± S.E. (n = 4).
We next studied the expression levels of latrophilins in mouse brain during postnatal development. Again, Lphn1 and Lphn3 exhibited a similar profile, with an ∼2.5- and 1.8-fold increase in expression during maturation of the brain, respectively (Fig. 1B). In contrast, Lphn2 displayed the opposite expression profile, with a decrease in mRNA levels during postnatal development.
In analyzing the sequences of mouse latrophilin mRNAs, we noted that Lphn1, Lphn2, and Lphn3 mRNAs contain a similar site of alternative splicing located between their lectin and olfactomedin-like domains (referred to as SSA for splice site A) (11). Analysis of genomic sequences revealed that all latrophilin genes contained a small alternatively spliced exon encoding 4 or 5 amino acids (Fig. 2A). This exon is preceded by noncanonical splice acceptor sites similar to the noncanonical acceptor sites preceding splice site 4 in neurexins (36). In the case of Lphn2, two different classes of transcripts were detected in which the differentially spliced exon was inserted into the mRNAs at two positions, creating shorter (4 residues) and longer variants (5 residues; Fig. 2A). Both transcripts were preceded by noncanonical splice acceptor sites, and sequence analyses of the differentially spliced exon suggested that Lphn1 and Lphn3 could potentially also contain both variants of this exon, although only the longer variant was observed in sequence analyses.
FIGURE 2.

All latrophilin genes contain an alternatively spliced 5′ exon. A, gene structures and sequences flanking the alternatively spliced 5′ exon of latrophilin genes. B, mRNA measurements of alternatively spliced variants of Lphn1, Lphn2, and Lphn3. C, tissue distribution of alternatively spliced Lphn2 variants.
We developed quantitative RT-PCR assays that selectively measure Lphn1 and Lphn3 mRNAs containing or lacking an insert in SSA, and Lphn2 mRNAs containing either the shorter or longer insert in SSA or lacking any insert (Fig. 2B; see under “Experimental Procedures”). Measurements of these mRNAs in mouse brain at different times of postnatal development demonstrated that the two SSA splice variants of Lphn1 are similarly abundant. For Lphn2, in contrast, the splice variant with the shorter insert predominated, and the splice variant with the longer insert was rare. For Lphn3, finally, the mRNA variant containing an insert in SSA was by far the most abundant (Fig. 2B). No major changes in alternative splicing during development were detected. Because Lphn2 is ubiquitously expressed, we also examined its alternative splicing in peripheral tissues in adult mice. Strikingly, peripheral tissues exclusively expressed the Lphn2 splice variants lacking an insert in SSA, demonstrating tissue-specific regulation of alternative splicing of Lphn2 at SSA (Fig. 1C).
Isolation of Teneurins on Immobilized Lphn1
Because Lphn1 is expressed at the highest levels among latrophilins (Fig. 1), we chose Lphn1 as the latrophilin isoform to search for potential ligands. We generated a series of Lphn1 expression vectors that produce various combinations of Lphn1 extracellular domains either as a secreted protein fused to the Fc domain of human immunoglobulin (31) or as a surface-displayed membrane protein fused to the TMR of the PDGF receptor (schematically shown in Fig. 3A). We then purified biochemical amounts of Ig fusion proteins containing either all of the extracellular domains of Lphn1 (IgLp1ECD) or only its N-terminal lectin domain (IgLp1LEC), as well as a control protein composed only of the human Fc domain (IgC). We immobilized these Ig fusion proteins on a protein A resin, and we performed affinity chromatography experiments using proteins solubilized from total rat brain (Fig. 3B). Analysis of the eluates of these columns by silver staining of SDS gels revealed only a single set of specific bands, a cluster of high molecular weight proteins that were found by mass spectrometry to include all four teneurin isoforms (Ten1–Ten4; Fig. 3B). Notably, in this initial identification of teneurin binding to Lphn1, which replicates a previous study reporting this interaction (7), we found that the Lphn1 lectin domain alone was sufficient for binding, although, as described below, it exhibits a lower affinity for teneurins than the full complement of Lphn1 extracellular domains.
FIGURE 3.
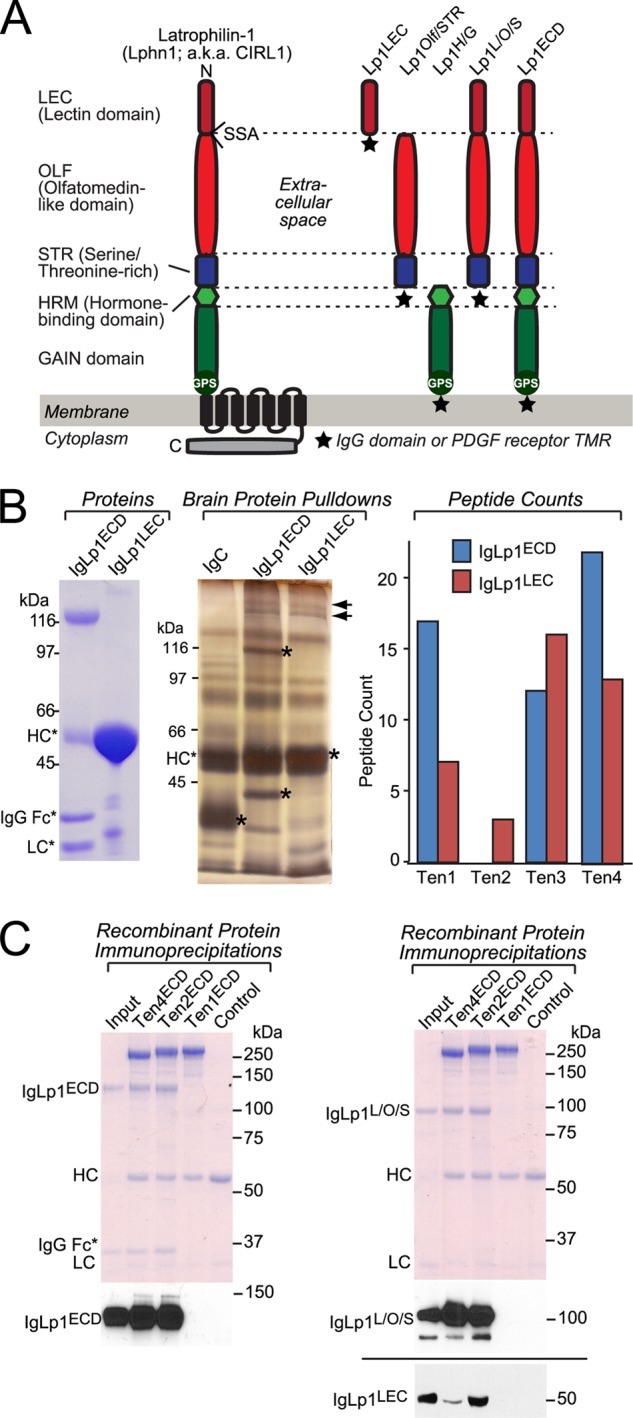
Affinity purification of teneurins with the lectin domain of latrophilin-1. A, domain structure of Lphn1 (left) and of Lphn1 constructs used in this study (right). SSA in Lphn1 contains or lacks a five-residue sequence (KVEQK, after Tyr131). The GAIN domain includes a C-terminal GPS sequence that determines the site of cleavage of the autocatalytic GAIN domain (12). The diagram on the right shows the structure of different Lphn1 fragments that were used for experiments either as purified secreted Ig domain fusion proteins or as surface-displayed fusion proteins with a C-terminal TMR from the PDGF receptor. B, purified recombinant Ig domain fusion proteins of the entire extracellular sequence (IgLp1ECD) or the lectin domain of Lphn1 (IgLp1LEC; see left panel for Coomassie-stained SDS gel of purified proteins) were used for affinity chromatography experiments with total solubilized rat brain proteins, using IgC as a negative control. Bound proteins were eluted with 1.0 m NaCl and analyzed by SDS-PAGE and silver staining (middle panel; asterisks mark eluted bait proteins.). Mass spectrometry of eluted proteins identified fragments of all four teneurin isoforms (right panel). C, pulldown of soluble recombinant Ig domain fusion proteins of the indicated Lphn1 fragments (see A) with immobilized teneurin fragments containing the entire extracellular sequences of teneurin-1 (Ten1ECD), teneurin-2 (Ten2ECD), and teneurin-4 (Ten4ECD). FLAG-tagged teneurins were immobilized on an anti-FLAG antibody column and incubated with the indicated Ig fusion proteins. Beads were washed with incubation buffer, eluted with sample buffer, run on an SDS-polyacrylamide gel, and either stained with Coomassie Blue or analyzed by immunoblotting using an anti-human IgG antibody (to visualize the Ig domain fusion protein) coupled to horseradish peroxidase. Data shown are representative images of experiments that were repeated at least three times.
To confirm the binding of teneurins to Lphn1, we performed pulldown experiments with purified recombinant proteins (Fig. 3C). We produced secreted HA-tagged proteins composed of the extracellular domains of Ten1, Ten2, and Ten4 in transfected HEK293T cells, purified and immobilized the proteins on an antibody column, and measured binding of purified Lphn1 Ig fusion proteins to the teneurin extracellular domains. Three Lphn1 Ig fusion proteins were examined as follows: proteins containing the entire extracellular sequence of Lphn1 (IgLp1ECD), its three N-terminal domains (the lectin, olfactomedin-like, and serine/threonine-rich domains; IgLp1L/O/S), or its lectin domain (IgLp1LEC). In this assay, we observed specific binding of Ten2 and Ten4 to all Lphn1 constructs but not of Ten1 (Fig. 3C). The lack of Ten1 binding in this assay could be due to a decreased affinity of this isoform or to a possible artifact in cDNA cloning even though sequencing did not identify any mutations in the construct used.
Binding of Teneurins to Latrophilins on the Cell Surface
To further characterize latrophilin binding to teneurins, we produced recombinant proteins of extracellular sequences of Lphn1, Ten2, and Ten4 (Figs. 3B and 4A). We then bound these proteins to HEK293T cells expressing Lphn1, Ten2, or Ten4 in a surface binding assay (30).
FIGURE 4.

Surface binding assays reveal domain-specific interactions between Lphn1 and Ten2 and Ten4. A, soluble IgC, Ig-Lp1ECD, IgLp1L/O/S, and IgLp1LEC were produced in transfected HEK293T cells and analyzed by SDS-PAGE and Coomassie Blue staining. B–D, cell surface labeling assays. Transfected HEK293T cells co-expressing membrane-anchored teneurin or latrophilin “receptor” with DsRed were incubated with control medium or medium containing soluble latrophilin Ig domain fusion proteins or soluble teneurin fragments as ligands. Receptors and bound ligands were visualized by immunocytochemistry except for C in which the Lphn1 receptor lacked a tag, and cells were visualized using co-expressed DsRed. B analyzes the binding of latrophilin-1 Ig fusion proteins containing the complete extracellular domains (IgLp1ECD) or only the lectin domain (IgLp1LEC) of Lphn1 to cells expressing DsRed, Myc-tagged Ten2, or HA-tagged Ten4. C examines the binding of soluble Myc-tagged Ten2 and HA-tagged Ten4 proteins containing their entire extracellular domains to cells expressing full-length Lphn1. D monitors the binding of recombinant teneurin proteins to cells expressing latrophilin-1 fusion proteins in which various extracellular latrophilin domains are fused C-terminally to the TMR of the PDGF receptor. The various Lphn1 proteins in D contained the following domains: Lp1ECD-PTMR, the entire extracellular domains; Lp1LEC-PTMR, the lectin domain only; Lp1O/S-PTMR, the olfactomedin-like and serine/threonine-rich sequence; Lp1L/O/S-PTMR, the N-terminal lectin-olfactomedin-like, and serine-rich sequences, and Lp1H/G-PTMR, the hormone binding and GAIN domains. Data shown are from a representative experiment that was independently repeated at least three times.
We first bound different Lphn1 Ig fusion proteins to HEK293T cells expressing Myc-tagged Ten2 or HA-tagged Ten4, and we observed strong binding of Lphn1 proteins containing all of the Lphn1 extracellular domains, or only its lectin domain, to cells containing either Ten2 or Ten4 but not to control cells (Fig. 4B). Conversely, when we bound soluble Ten2 and Ten4 proteins comprising their extracellular domains to HEK293T cells expressing Lphn1, we also observed strong binding (Fig. 4C). These findings confirm that Lphn1 binds to Ten2 and Ten4 on the cell surface.
We next examined in greater detail which Lphn1 domains suffice to bind Ten2. We expressed in HEK293T cells Lphn1 fusion proteins in which different Lphn1 domains are C-terminally fused to the TMR of the PDGF receptor, and we examined binding of soluble Ten2 to these cells (Fig. 4D). We observed strong binding of Ten2 to all Lphn1 proteins containing the lectin domain, whereas no binding was observed to Lphn1 proteins lacking the lectin domain. Thus, the Lphn1 lectin domain is both necessary and sufficient for teneurin binding in the cell surface binding assay, consistent with the pulldown results presented in Fig. 3.
Affinity of Lphn1 Binding to Teneurins and FLRT3
To assess the affinity of Lphn1 interactions with Ten2 and Ten4 and to compare it with the binding of another potential Lphn1 ligand, FLRT3 (10), we expressed these proteins on the surface of HEK293T cells, and we estimated the binding affinity of different Lphn1-Ig fusion proteins to the HEK293T cells using a standard assay (Fig. 5) (33). Specifically, in these experiments we expressed full-length teneurin-2 or -4 in HEK293T cells and incubated the cells in the presence of soluble Lphn1 Ig fusion proteins at concentrations ranging from 0.05 to 25 nm or 0.4 to 100 nm, respectively (Fig. 5, A and B). Mock-transfected cells were used as negative controls, and bound Ig proteins were detected using an HRP-conjugated secondary antibody measured with a colorimetric substrate. To calculate binding affinities, the net binding of Ig fusion proteins at any given concentration was plotted as a function of IgLp1 proteins concentrations on a logarithmic scale. The binding curves were then fitted to a Scatchard function, assuming a single independent binding site for Lphn1 in a teneurin molecule. Four different Lphn1-Ig fusion proteins were examined as follows: a protein containing the entire extracellular sequence of Lphn1 (IgLp1ECD); proteins containing its three N-terminal domains (the lectin, olfactomedin-like, and serine/threonine-rich domains) without (IgLp1L/O/S) or with an insert in SSA (IgLp1L/O/S+); or an Ig domain fusion protein containing only the lectin domain of Lphn1 (IgLp1LEC).
FIGURE 5.
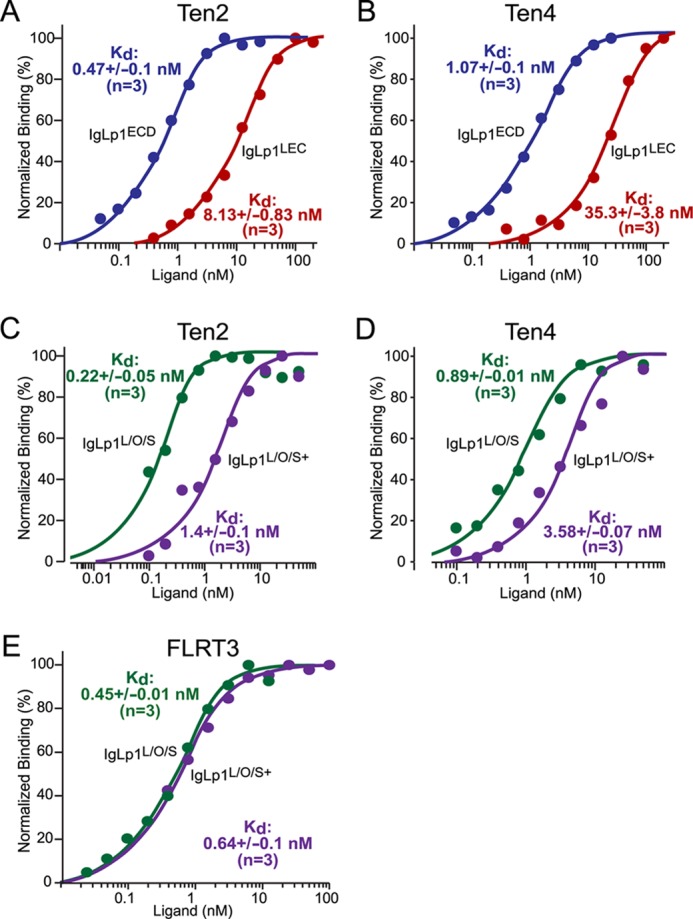
Binding affinity of Lphn1 to Ten2 and Ten4 and to FLRT3 and effect of alternative splicing of latrophilin-1 on such binding. HEK293T cells expressing Ten2, Ten4, or FLRT3 were used to measure the binding affinity of Ig fusion proteins containing the indicated extracellular Lphn1 domains. All data shown are binding after subtraction of the signal obtained with mock-transfected cells included in all assays. Binding data shown are representative graphs of experiments that were conducted at least three times. Data were fit to a Scatchard equation using SigmaPlot software and assuming uniform binding sites; Kd numbers displayed in the graphs show the averages determined in at least three independent experiments. A–E, binding data for the entire Lphn1 extracellular domains (IgLp1ECD; A and B), the N-terminal Lphn1 lectin domain only (IgLp1LEC; A and B), or the N-terminal three Lphn1 domains (i.e. its lectin, olfactomedin-like, and serine/threonine-rich domains); for the latter, splice variants lacking an insert in SSA (IgLp1L/O/S; C–E) or containing such insert (IgLp1L/O/S+; C–E) were analyzed.
We found that the Ig fusion protein of the entire Lphn1 extracellular region exhibited the highest affinity for Ten2 and Ten4, with subnanomolar binding constants. In contrast, the lectin domain alone exhibited a 30-fold lower affinity that was, however, still nanomolar (Fig. 5, A and B). The Ig fusion protein of the N-terminal three domains of Lphn1 also exhibited a very high affinity similar to the fusion proteins of the entire extracellular domains, but this affinity depended on the alternative splicing at SSA. For both Ten2 and Ten4, Lphn1 with an insert in SSA displayed an ∼5-fold lower affinity that Lphn1 lacking an insert in SSA (Fig. 5, C and D). We found that the Lphn1 Ig fusion protein of the three N-terminal Lphn1 domains exhibited the same overall affinity for FLRT3 and for Ten2 and Ten4. In contrast to teneurin binding, however, FLRT3 binding was independent of alternative splicing at SSA (Fig. 5E). Viewed together, these data not only demonstrate that Lphn1 binds with high affinities to both teneurins and FLRT3, but also that this binding is regulated by alternative splicing for teneurins but not for FLRT3, suggesting distinct binding mechanisms even though the same Lphn1 fragment binds to both ligands.
Heterophilic Teneurin Binding to Latrophilins Mediates Intercellular Adhesion
The binding of cell-surface proteins could represent a trans-interaction that mediates formation of intercellular junctions between cells as observed in synapses, or it could represent a cis-interaction that cannot operate in trans, i.e. cannot mediate the adhesion of cells to each other. To differentiate between these two possibilities, we performed cell aggregation assays, which measure the binding of cells to each other as a function of the surface expression of candidate cell-adhesion molecules.
When we simply expressed Lphn1, Lphn2, or Lphn3 in HEK293T cells, we observed no cell aggregates, suggesting that there is no homophilic cell adhesion (Fig. 6A). We also detected no cell aggregates when we mixed cells expressing these latrophilins with control-transfected HEK293T cells. However, when we mixed these latrophilin-expressing cells with cells expressing Ten2 or Ten4, we observed formation of large cell aggregates indicative of intercellular adhesion (Fig. 6A).
FIGURE 6.

Binding of latrophilins to teneurins mediates cell-cell adhesion. A, HEK293T cells expressing DsRed alone (Control) or together with Myc-tagged Ten2 or HA-tagged Ten4 were incubated for 90 min with cells expression EGFP alone (control) or together with Lphn1, Lphn2, or Lphn3 and imaged by fluorescence microscopy. Scale bar, 100 μm. B, summary graphs of the aggregation index determined in independent cell-adhesion assays as shown in A (means ± S.E.; n = 3). The aggregation index was calculated as the percentage of the total particle surface occupied by particles exceeding a threshold of 3000 pixels2 compared with the total particle surface. Statistical significance was assessed by one-way ANOVA comparing the test conditions to the control (***, p < 0.001). C and D, same as A and B, except that the effect of Lphn1 alternative splicing at SSA was analyzed.
Quantifications showed that for each combination of latrophilin and teneurin tested, highly significant cell aggregation occurred (Fig. 6B). We also examined the effect of Lphn1 alternative splicing at SSA on intercellular adhesion mediated by binding to either Ten2 or Ten4, but we found no difference (Fig. 6, C and D). This result is not surprising despite the fact that the SSA splice variants exhibit significantly different binding affinities to teneurins (Fig. 5), because even in the presence of an SSA insert in Lphn1 (the lower affinity variant), the binding affinity is still very high and sufficient to mediate intercellular adhesion.
Many cell-adhesion interactions, including those mediated by cadherins or neurexins/neuroligins (37), require binding of structural Ca2+ ions to one or both of the cell-adhesion molecules. Thus, we asked whether binding of latrophilins and teneurins and the intercellular adhesion mediated by them require Ca2+. However, we found that the Ca2+-chelator EGTA had no effect on either latrophilin binding to teneurin on a cell surface (Fig. 7A) or on cell adhesion mediated by Lphn1 interaction to Ten2 or Ten4 (Fig. 7, B and C). These data suggest that the teneurin-latrophilin interaction is Ca2+-independent, consistent with the role of latrophilins as Ca2+-independent α-latrotoxin receptors (3, 4).
FIGURE 7.
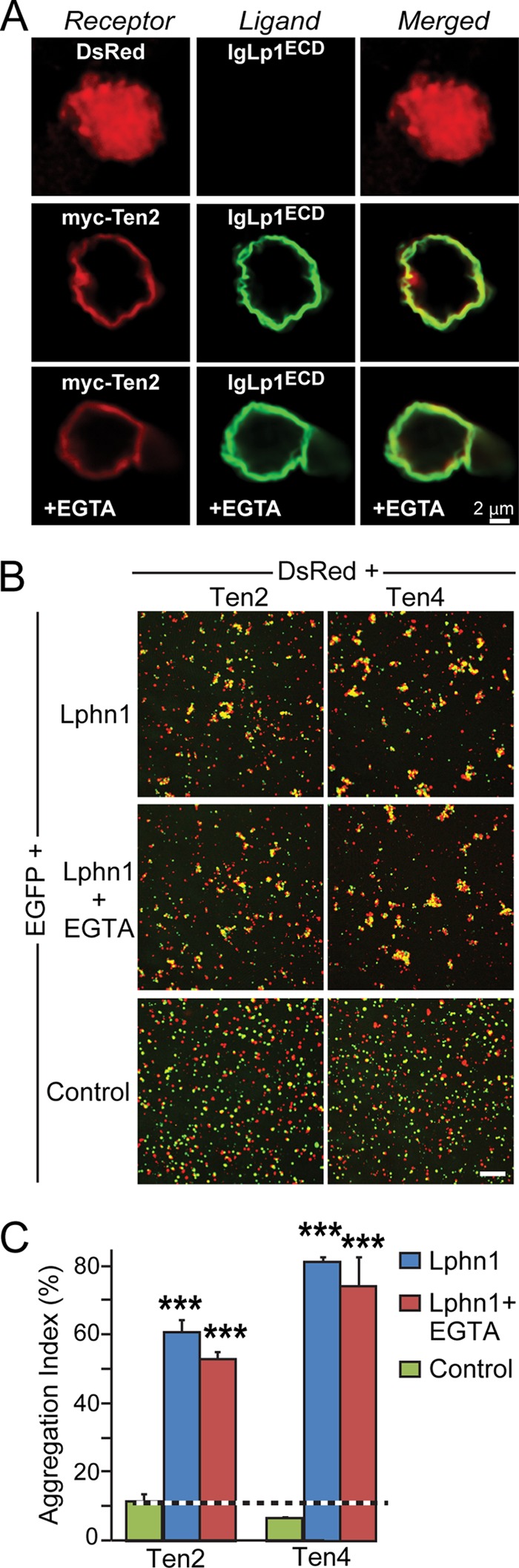
Interaction of Lphn1 with Ten2 and Ten4 is Ca2+-independent. A, cell surface binding assays. HEK293T cells expressing DsRed or Myc-tagged Ten2 were incubated with an Ig fusion protein of the extracellular Lphn1 domains (IgLp1ECD) with or without 5 mm EGTA as indicated. Lphn1 binding was assessed by immunofluorescence labeling using antibodies to IgG (AlexaFluor-488, green) or to the Myc epitope (AlexaFluor-633, red). B, cell aggregation assays. HEK cells expressing EGFP alone or together with Lphn1 (without an insert in splice site A) were mixed with cells expressing DsRed together with Ten2 or Ten4, incubated for 90 min in the absence or presence of 5 mm EGTA, and analyzed by fluorescence mircroscopy. Data shown are a representative experiment independently performed three times. Scale bar, 100 μm. C, summary graphs of the aggregation index determined in cell aggregation assays as described in B (n = 3). The aggregation index was calculated as the percentage of the total particle surface occupied by particles exceeding a threshold of 3000 pixels2. Data shown are means ± S.E. Statistical significance was assessed by comparing Lphn1- or teneurin-expressing cells with the control expressing EGFP alone using one-way ANOVA (***, p < 0.001).
Homophilic Binding of Teneurins Does Not Mediate Intercellular Adhesion
Teneurins are known to form homodimers (6, 38) and were proposed in Drosophila to mediate synapse specificity by trans-synaptic homophilic interactions (8, 9). When we tested, using the surface binding assay (Fig. 4), whether mammalian teneurins could bind to each other on a cell surface, we found that a soluble Myc-tagged Ten2 protein avidly and specifically bound to HEK293T cells expressing either Ten2 or Ten4, confirming a homophilic interaction (Fig. 8A).
FIGURE 8.
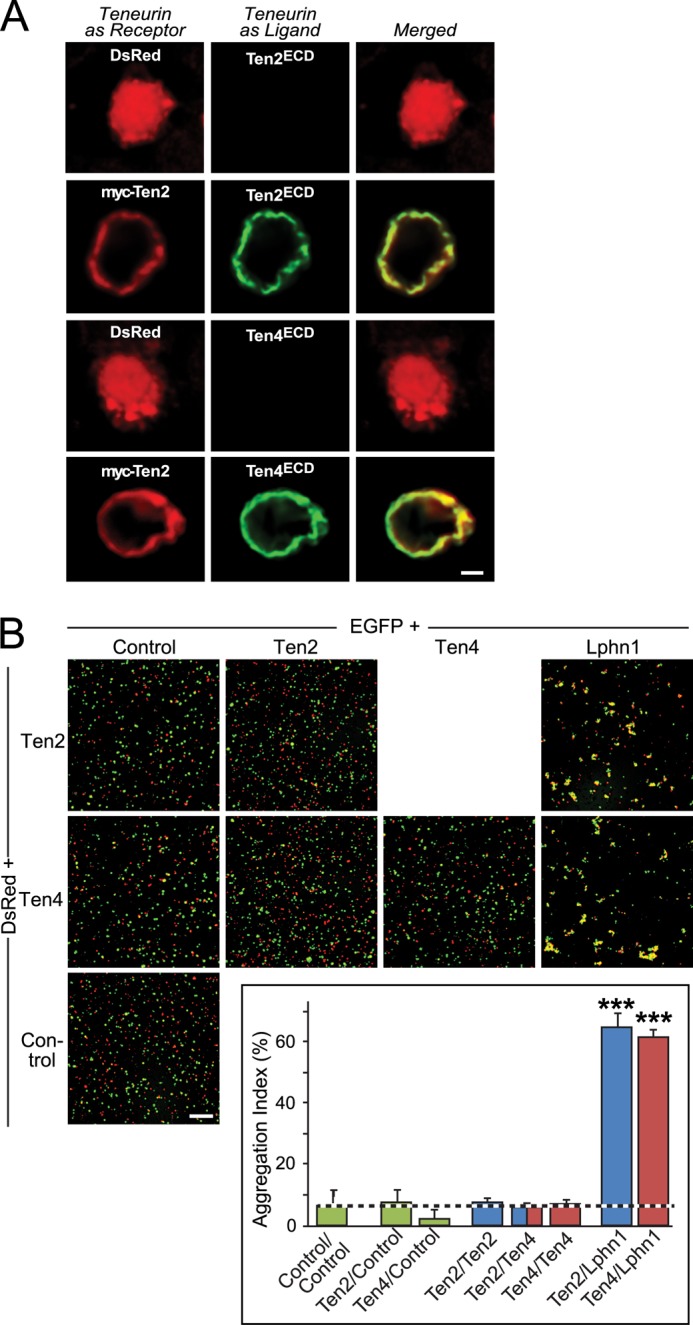
Teneurin-2 and -4 form homophilic complexes but do not mediate homophilic cell adhesion. A, surface binding assays in which HEK293T cells expressing DsRed alone or together with full-length, Myc-tagged Ten2 were incubated with soluble recombinant proteins of the FLAG-tagged Ten2 or Ten4 extracellular domains as indicated, washed, fixed, and analyzed by immunofluorescence labeling using antibodies to FLAG (AlexaFluor-488, green) and to the Myc epitope (AlexaFluor-633, red). Data shown are representative images of experiments that were repeated at least three times (scale bars, 2 μm; apply to all images). Note that teneurin fragments only bind to Ten2-expressing but not control cells. B, cell aggregation assays. HEK293T cells expressing EGFP alone or together with full-length Ten2, Ten4, or Lphn1 (without an insert in SSA, used as a positive control) were mixed with cells expressing DsRed alone or together with Ten2 or Ten4. After a 90-min incubation, cells were imaged by fluorescence microscopy. Scale bar, 100 μm. The aggregation index determined in three independent experiments was determined as the percentage of total particle surface that is present in particles exceeding a threshold of 3000 pixels2 and is depicted in the inserted bar diagram (means ± S.E.). Statistical significance was assessed using one-way ANOVA comparing test conditions to the control expressing EGFP alone (***, p < 0.001).
We next investigated whether homophilic teneurin binding could mediate intercellular cell adhesion, i.e. whether this binding is potentially a trans-interaction. Strikingly, no cell aggregation was observed between Ten2- or Ten4-expressing cells, either alone or in combination (Fig. 8B). As a positive control, when we added Lphn1-expressing cells to Ten2- or Ten4-expressing cells, cell aggregation became immediately apparent. Quantifications confirmed that Ten2 or Ten4 cells did not by themselves aggregate (Fig. 8B). These data suggest that although mammalian teneurins bind to each other, they are incapable of mediating intercellular cell adhesion and thus cannot function as homophilic synaptic cell-adhesion molecules.
Addition of Excess Soluble N-terminal Lphn1 Domain Recombinant Proteins Decreases Synapse Density
Previous reports suggested that Lphn1, FLRT3, and teneurins may functionally be related to synapses (7–10). As an initial in vitro test of such a role, we hypothesized that the presence of a large concentration of the Lphn1 N-terminal domain as a soluble ligand for FLRT3 and teneurins might block endogenous interactions and interfere with synapse formation and/or maintenance. Thus, we added purified recombinant Ig fusion proteins of either only the lectin domain of Lphn1 or of the N-terminal three Lphn1 domains (the lectin, olfactomedin-like, and serine/threonine-rich domains) to the medium of cultured hippocampal neurons at DIV10, and we analyzed the neurons by quantitative fluorescence microscopy at DIV17 (Fig. 8). As a control, we used equivalent concentrations of IgC.
We found that the recombinant Lphn1 proteins had no effect on the overall morphology of the neurons as assessed by quantifications of the size and the number of neurites of the neurons (Fig. 9, A and B). However, we observed that the recombinant N-terminal Lphn1 domains reduced the density of both excitatory (visualized by immunofluorescence staining with antibodies to the vesicular glutamate transporter vGlut1) and inhibitory synapses (visualized by staining with antibodies to the vesicular GABA transporter vGAT; Fig. 9C). Excitatory synapses were suppressed by ∼25% and inhibitory synapses by ∼33% (Fig. 9D, left panels). Moreover, the N-terminal Lphn1 domains decreased the staining intensity of synaptic puncta for vGlut1 and vGAT (Fig. 9D, right panels), suggesting that the synapses may have been rendered smaller. Thus, the recombinant Lphn1 proteins had a specific effect on synapses in cultured neurons.
FIGURE 9.
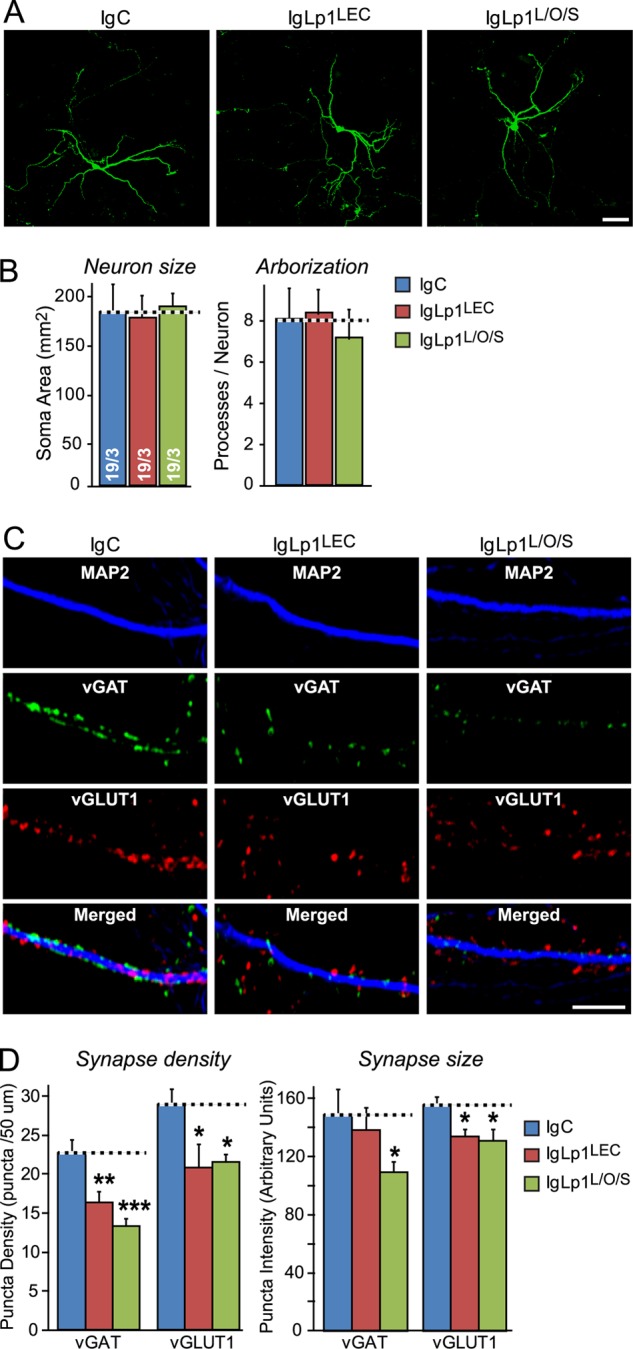
Addition of recombinant extracellular latrophilin-1 domains to cultured neurons decreases synapse density. A, hippocampal neurons were sparsely transfected with EGFP at DIV7 and incubated from DIV10 to DIV17 with 0.5 μm of a control Ig fusion protein (IgC), an Ig fusion protein of the latrophilin-1 lectin domain (IgLp1LEC), or an Ig fusion protein of the N-terminal lectin, olfactomedin-like and serine/threonine-rich domains of latrophilin-1 (IgLp1L/O/S). At DIV17, neurons were fixed, stained by immunofluorescence with antibodies to EGFP, and examined by confocal microscopy (scale bar, 200 μm). B, summary graphs of neuronal size (soma size) and arborization (processes/neuron) determined by MetaMorph image analysis of EGFP-stained neurons in the experiments described in A. C, same as A, except that neurons were not transfected and were examined by triple immunofluorescence labeling with antibodies to MAP2, vGlut1, and vGat (the excitatory and inhibitory vesicular amino acid transporters, respectively; scale bar, 20 μm). D, summary graphs of the synapse density (puncta density) and synapse size (puncta intensity) determined in experiments as shown in C. Data shown in B and D are means ± S.E. (n = 3 independent experiments for B, n = 5 for D). Statistical significance was assessed by one-way ANOVA comparing test to control conditions (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Lphn mRNA Levels Are Differentially Regulated in a Mouse Model of Neurodegeneration
Our results suggest that cell adhesion mediated in part by latrophilins and their ligands constitutes an important aspect of synapse stability and function. To probe a potential contribution of this particular cell-adhesion process to brain function, we sought to investigate if interfering with synapse stability in the first place would alter the gene expression profile of cell-adhesion molecules in brain. As a template for our investigation, we chose a mouse model of neurodegeneration in which the deletion of CSPα, a synaptic co-chaperone protein, leads to early defects in synapse stability and function (39–42). Neurodegeneration in CSPα KO mice can be observed as early as 2 weeks of age (39) and is characterized in part by decreased neuronal and synapse density in the brain (40). Hence, we collected brain samples from wild-type and mutant mice at 5–8 weeks of age to allow for complete synaptogenesis and for observable signs of neurodegeneration to appear. We postulated that the gene expression of adhesion molecules might represent a molecular marker of synapse stability in this synaptic model of neurodegeneration, and therefore reiterate their role as physiological synaptic stabilizers. Therefore, we performed quantitative RT-PCR assays to detect the mRNA levels of 20 genes encoding neuronal proteins comprising cell-adhesion molecules, SNARE proteins, and others. Cell-adhesion molecule targets were selected on the basis that they do not constitute the group of described CSPα clients, therefore excluding a direct contribution of CSPα-chaperoning function (43).
Consistent with a loss of neurons, mRNAs for SNARE protein SNAP25 was found to be reduced in CSPα KO mouse samples compared with WT littermates (Fig. 10A). Interestingly, mRNA levels for myelin-associated glycoprotein were drastically reduced, a change that is consistent with its role in maintaining the integrity of axon-glial interactions, and suggests that a loss in myelin proteins might accompany synaptic instability in this mouse model of neurodegeneration (Fig. 10A) (44, 45). From the Lphn gene family, only Lphn2 mRNA was found to be down-regulated along with its ligand FLRT2 and with neurexin-1. However, mRNAs for Lphn1 and its ligand Ten2 remained constant compared with WT littermates (Fig 10B). Quite surprisingly, mRNA levels for Lphn3 and its ligand FLRT3 were both up-regulated, denoting that elevated gene expression was induced for this pair of adhesion molecules following neurodegenerative signals, a mechanism reminiscent of neuronal response to nerve injury regarding FLRT3 mRNA (Fig. 10C) (25–27). Lphn3 and FLRT3 are part of a specific array of cell-adhesion molecule mRNAs that were found to be up-regulated, including neuroligin-1, SynCAM, dystroglycan, and N-cadherin, which might constitute a signature for CSPα KΟ-induced neurodegeneration. These data indicate that the gene expression of Lphns and their ligands is actively modulated in this mouse model of neurodegeneration.
FIGURE 10.
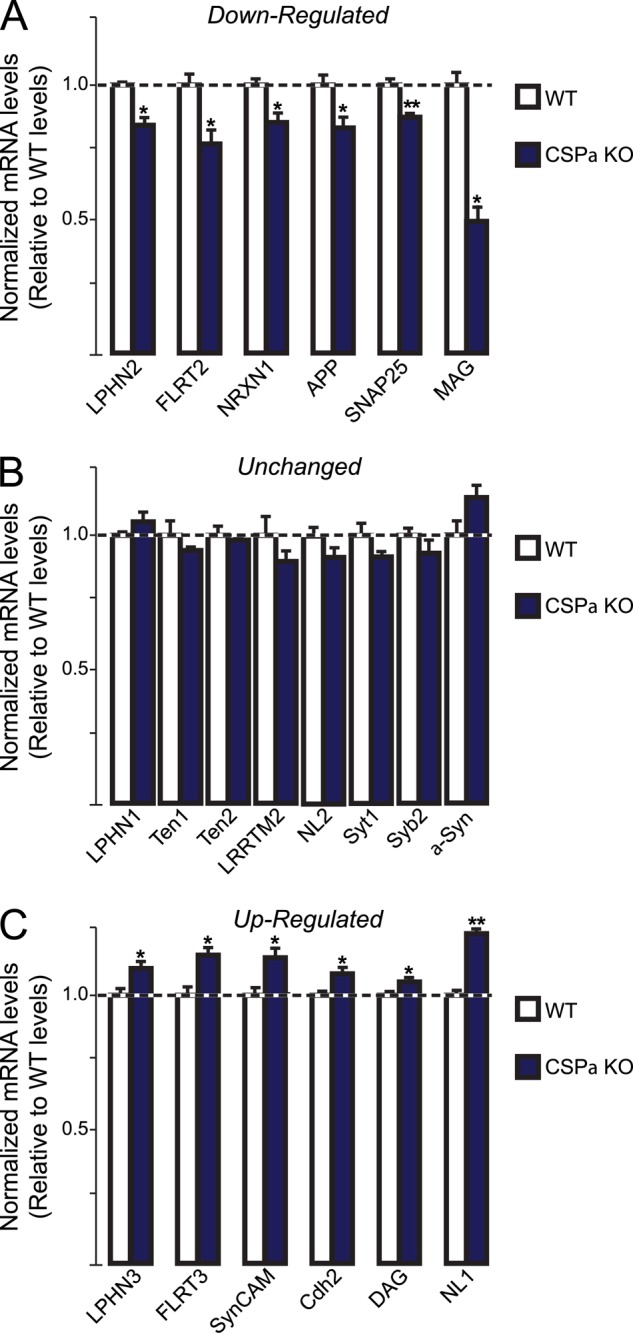
Latrophilins and their ligands are differentially regulated during neurodegeneration in CSPα KO mice. Quantitative RT-PCR measurements of selected proteins in brains of adult CSPα KO mice and WT littermates. Results are represented for mRNA samples as follows: A, were down-regulated; B, were unchanged, or C, were up-regulated in brain samples of CSPα KO mice compared with WT littermates. All mRNA levels were normalized to those of actin as an internal control and then to the mRNA levels of the respective proteins in WT littermates. Data shown are means ± S.E. (n = 4). Statistical significance was assessed by Student's t test comparing KO to WT conditions (*, p < 0.05; **, p < 0.01). α-Syn, α-synuclein; APP, amyloid precursor protein; Cdh2, N-cadherin; DAG, dystroglycan; LRRTM2, leucine-rich repeat transmembrane-2; MAG, myelin-associated glycoprotein; NL1, neuroligin-1; NL2, neuroligin-2; NRXN1, neurexin-1; SNAP25, synaptosomal associated protein-25; SynCAM, synaptic cell-adhesion molecule-1; Syb2, synaptobrevin-2; Syt1, synaptotagmin-1.
DISCUSSION
Our data confirm previous studies showing that teneurins and FLRT3 specifically bind to latrophilins (7, 10), and extend these data by making the following principal observations.
1) In quantifying the expression levels of latrophilins and their N-terminal splice variants, we demonstrate that all three latrophilin genes are expressed at similar levels in brain but that Lphn2 is the only latrophilin that is expressed at significant levels in peripheral tissues. We show that the alternative splicing of latrophilins at the N-terminal SSA site differs among latrophilins and that in particular most Lphn2 mRNAs in brain contain an insert in SSA, whereas most Lphn2 mRNAs in non-neural tissues lack an insert (Figs. 1 and 2).
2) We found that teneurins are the predominant binding partners for latrophilins in brain as analyzed by pulldowns (Fig. 3), and we mapped the minimal teneurin-binding site of latrophilins to their N-terminal lectin domain (Fig. 4). The lectin domain of Lphn1 by itself had a lower affinity for Ten2 and Ten4 than the combination of the lectin domain with the olfactomedin-like domain and the serine/threonine-rich sequence (Fig. 5), but the lectin domain still bound more tightly to Ten2 and Ten4 than most cell-adhesion interactions that we previously characterized. Thus, surprisingly, the lectin domain of latrophilins serves as a protein interaction module.
3) In measuring the relative affinities of different Lphn1 domains for Ten2, Ten4, and FLRT3, we estimated nanomolar binding affinities of all these interactions (Fig. 5). The same fragment of Lphn1 composed of the lectin and the olfactomedin-like domains and the serine/threonine-rich region bound to Ten2, Ten4, and FLRT3 with affinities of ∼0.1–1.0 nm, suggesting that teneurins and FLRT3 bind to the same site. Nevertheless, although we observed a significant regulation of the teneurin-binding affinity of Lphn1 by alternative splicing at SSA, the same alternative splicing had no effect on the affinity of Lphn1 for FLRT3, suggesting that their binding sites differ and that the observed effect of alternative splicing is specific.
4) Binding of latrophilins to teneurins is Ca2+-independent and can mediate cell-cell adhesion between cells expressing these molecules on their surface (Figs. 6 and 7). Thus, these molecules form a classical heterophilic cell-adhesion pair.
5) Confirming previous data (8, 9, 46), we find that teneurins can bind to each other in a homophilic interaction (Fig. 8A). Because teneurins are thought to be disulfide-bonded dimers (6, 38), this result indicates that teneurins may form higher order multimers of disulfide-bonded dimers.
6) Despite their homophilic interaction, however, we observed that teneurins were unable to form homophilic cell-adhesion complexes. Specifically, we observed no aggregation of teneurin-expressing cells with each other, despite their strong aggregation with cells expressing latrophilins (Fig. 8B). This assay indicates that teneurins are not strong homophilic cell-adhesion molecules, possibly because they exhibit a large elongated size that may only be able to multimerize in a parallel cis-configuration. Our results are at odds with the conclusion reached by Beckmann et al. (46), who observed adhesion between cells expressing teneurins. The primary difference between our experiments and those of Beckmann et al. (46) is not the detection method used for measuring cell adhesion (light microscopy versus atomic force microscopy), but the overall approach. We analyzed spontaneous aggregates formed between cells expressing cell-adhesion molecules, whereas Beckmann et al. (46) brought two HEK293T cells into contact for a predefined time (ranging from 1 to 120 s) and force (1 nN) to induce formation of adhesive contacts between cells. After this initial contact time, adhering cells were separated, and the force needed for separation was measured. These sophisticated experiments thus allow an unprecedented measurement of the force of homophilic binding of teneurins, but they do not actually measure the formation of adhesion complexes between cells expressing teneurins.
7) Addition of soluble, teneurin-, and FLRT3-binding fragments of Lphn1 to the medium of cultured neurons decreased the density of synapses in these cultures, suggesting that latrophilin directly or indirectly contributes to the formation and/or maintenance of synapses (Fig. 9).
8) Synaptic instability induced by the deletion of a co-chaperone, CSPα, leads to differential regulation of mRNAs encoding Lphns and their endogenous ligands, teneurins, and FLRT as well as neurexins (Fig. 10). This highly specific and differential pattern of gene expression in response to neuronal loss suggests that different sets of Lphns may be linked to different sets of ligands in brain to maintain synapse stability and function. In a broader scope, these data indicate that cell-adhesion genes can be specifically solicited in a response to counteract deteriorating synapses in brain.
Viewed together, our data describe high affinity interactions of latrophilins with two unrelated cell-adhesion molecules, teneurins and FLRT3. These interactions are mediated by the same domains of latrophilins and in the case of teneurins are capable of supporting the formation of an intercellular junction. The interactions of Lphn1 with Ten2, Ten4, and FLRT3 are differentially regulated by alternative splicing, suggesting that they are mediated by distinct binding surfaces. Our results establish that the interactions of Lphn1 with teneurin and FLRT3 exhibit the requisite specificity and affinity to be physiologically meaningful. It should be noted that although we describe latrophilins, teneurins, and FLRT3 here as heterophilic cell-adhesion molecules, this term might be misleading because it is quite likely that the primary function of these molecules is not the mechanical establishment of a tissue structure, such as a synapse. The term “cell adhesion” only refers to the measurable adhesion of cells that is conferred by the interaction of these molecules (but not, for example, by the interaction of teneurins with each other; see Fig. 8). Cell-adhesion assays detect the establishment of intercellular interaction, but such an interaction does not imply a specific function in intercellular adhesion as opposed to intercellular signaling. Indeed, we believe it is probable that a primary function of the interaction of latrophilins with various ligands consists of activating bidirectional signaling pathways, as beautifully demonstrated first for the Notch-Delta pathway (47). Identifying a possible signaling function of latrophilins will require the development of new tools and in vivo assays because classical methods for measuring GPCR activity have been largely unsuccessful.
Finally, despite all of the pre-existing and present data, a role for latrophilins, FLRT3, and teneurins at synapses cannot be considered as established. The binding of latrophilins to α-latrotoxin, which acts as a presynaptic toxin that causes neurotransmitter release, is strongly suggestive of a synaptic localization, as is the binding of Lphn1 to neurexins, which are presynaptic molecules (30). Moreover, functional experiments that we (Fig. 9) and others (7, 10) have performed point to a synaptic role. However, no specific localization of these proteins to synapses has been reported, and all functional assays described are indirect. All of these molecules are widely expressed outside of brain and thus must have general functions. Even more uncertain than the synaptic localization of these molecules is the assignment of a specific synaptic function to these molecules, e.g. as synaptic cell-adhesion molecules or as synapse-specification molecules. Even if latrophilins, teneurins, and FLRT3 perform an important function at neurons in addition to their broader non-neuronal function as cell-adhesion molecules, their major roles may occur in nonsynaptic processes in neurons, as suggested by the phenotype of mutations in the C. elegans latrophilin gene Lat-1 (14) and by the phenotype of teneurin mutations in mice (18–20). Elucidating these roles and the contribution of their binding to each other will pose a major opportunity and challenge in understanding animal development.
Acknowledgments
We thank Yuri Ushkaryov (Kent University, United Kingdom) and Liqun Luo (Stanford University) for providing plasmids; Manu Sharma for providing the CSPα KO mice; Özgün Gokce, Theodoris Tsetsenis, and Garret Anderson for advice.
This work was supported, in whole or in part, by National Institutes of Health Grants MH052804 and NS077906.
- FLRT
- fibronectin leucine-rich repeat transmembrane protein
- GPCR
- G protein-coupled receptor
- SSA
- splice site A
- TMR
- transmembrane region
- EGFP
- enhanced GFP
- ANOVA
- analysis of variance
- qRT
- quantitative RT
- DIV
- days in vitro
- CSPα
- cysteine string protein α.
REFERENCES
- 1. Benson D. L., Huntley G. W. (2012) Synapse adhesion: a dynamic equilibrium conferring stability and flexibility. Curr. Opin. Neurobiol. 22, 397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Missler M., Südhof T. C., Biederer T. (2012) Synaptic cell adhesion. Cold Spring Harbor Perspect. Biol. 4, a005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krasnoperov V. G., Bittner M. A., Beavis R., Kuang Y., Salnikow K. V., Chepurny O. G., Little A. R., Plotnikov A. N., Wu D., Holz R. W., Petrenko A. G. (1997) α-Latrotoxin stimulates exocytosis by the interaction with a neuronal G-protein-coupled receptor. Neuron 18, 925–937 [DOI] [PubMed] [Google Scholar]
- 4. Lelianova V. G., Davletov B. A., Sterling A., Rahman M. A., Grishin E. V., Totty N. F., Ushkaryov Y. A. (1997) α-Latrotoxin receptor, latrophilin, is a novel member of the secretin family of G protein-coupled receptors. J. Biol. Chem. 272, 21504–21508 [DOI] [PubMed] [Google Scholar]
- 5. Levine A., Bashan-Ahrend A., Budai-Hadrian O., Gartenberg D., Menasherow S., Wides R. (1994) Odd Oz: a novel Drosophila pair rule gene. Cell 77, 587–598 [DOI] [PubMed] [Google Scholar]
- 6. Oohashi T., Zhou X. H., Feng K., Richter B., Mörgelin M., Perez M. T., Su W. D., Chiquet-Ehrismann R., Rauch U., Fässler R. (1999) Mouse ten-m/Odz is a new family of dimeric type II transmembrane proteins expressed in many tissues. J. Cell Biol. 145, 563–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Silva J. P., Lelianova V. G., Ermolyuk Y. S., Vysokov N., Hitchen P. G., Berninghausen O., Rahman M. A., Zangrandi A., Fidalgo S., Tonevitsky A. G., Dell A., Volynski K. E., Ushkaryov Y. A. (2011) Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. U.S.A. 108, 12113–12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mosca T. J., Hong W., Dani V. S., Favaloro V., Luo L. (2012) Trans-synaptic Teneurin signalling in neuromuscular synapse organization and target choice. Nature 484, 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hong W., Mosca T. J., Luo L. (2012) Teneurins instruct synaptic partner matching in an olfactory map. Nature 484, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Sullivan M. L., de Wit J., Savas J. N., Comoletti D., Otto-Hitt S., Yates J. R., 3rd, Ghosh A. (2012) FLRT proteins are endogenous latrophilin ligands and regulate excitatory synapse development. Neuron 73, 903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugita S., Ichtchenko K., Khvotchev M., Südhof T. C. (1998) α-Latrotoxin receptor CIRL/latrophilin 1 (CL1) defines an unusual family of ubiquitous G-protein-linked receptors. G-protein coupling not required for triggering exocytosis. J. Biol. Chem. 273, 32715–32724 [DOI] [PubMed] [Google Scholar]
- 12. Araç D., Boucard A. A., Bolliger M. F., Nguyen J., Soltis S. M., Südhof T. C., Brunger A. T. (2012) A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31, 1364–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tobaben S., Südhof T. C., Stahl B. (2002) Genetic analysis of α-latrotoxin receptors reveals functional interdependence of CIRL/latrophilin 1 and neurexin 1α. J. Biol. Chem. 277, 6359–6365 [DOI] [PubMed] [Google Scholar]
- 14. Langenhan T., Prömel S., Mestek L., Esmaeili B., Waller-Evans H., Hennig C., Kohara Y., Avery L., Vakonakis I., Schnabel R., Russ A. P. (2009) Latrophilin signaling links anterior-posterior tissue polarity and oriented cell divisions in the C. elegans embryo. Dev. Cell 17, 494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prömel S., Frickenhaus M., Hughes S., Mestek L., Staunton D., Woollard A., Vakonakis I., Schöneberg T., Schnabel R., Russ A. P., Langenhan T. (2012) The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep. 2, 321–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minet A. D., Rubin B. P., Tucker R. P., Baumgartner S., Chiquet-Ehrismann R. (1999) Teneurin-1, a vertebrate homologue of the Drosophila pair-rule gene ten-m, is a neuronal protein with a novel type of heparin-binding domain. J. Cell Sci. 112, 2019–2032 [DOI] [PubMed] [Google Scholar]
- 17. Zheng L., Michelson Y., Freger V., Avraham Z., Venken K. J., Bellen H. J., Justice M. J., Wides R. (2011) Drosophila Ten-m and filamin affect motor neuron growth cone guidance. PLoS One 6, e22956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dharmaratne N., Glendining K. A., Young T. R., Tran H., Sawatari A., Leamey C. A. (2012) Ten-m3 is required for the development of topography in the ipsilateral retinocollicular pathway. PLoS One 7, e43083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Merlin S., Horng S., Marotte L. R., Sur M., Sawatari A., Leamey C. A. (2013) Deletion of Ten-m3 induces the formation of eye dominance domains in mouse visual cortex. Cereb. Cortex 23, 763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki N., Fukushi M., Kosaki K., Doyle A. D., de Vega S., Yoshizaki K., Akazawa C., Arikawa-Hirasawa E., Yamada Y. (2012) Teneurin-4 is a novel regulator of oligodendrocyte differentiation and myelination of small-diameter axons in the CNS. J. Neurosci. 32, 11586–11599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lacy S. E., Bönnemann C. G., Buzney E. A., Kunkel L. M. (1999) Identification of FLRT1, FLRT2, and FLRT3: a novel family of transmembrane leucine-rich repeat proteins. Genomics 62, 417–426 [DOI] [PubMed] [Google Scholar]
- 22. Böttcher R. T., Pollet N., Delius H., Niehrs C. (2004) The transmembrane protein XFLRT3 forms a complex with FGF receptors and promotes FGF signalling. Nat. Cell Biol. 6, 38–44 [DOI] [PubMed] [Google Scholar]
- 23. Maretto S., Müller P. S., Aricescu A. R., Cho K. W., Bikoff E. K., Robertson E. J. (2008) Ventral closure, headfold fusion and definitive endoderm migration defects in mouse embryos lacking the fibronectin leucine-rich transmembrane protein FLRT3. Dev. Biol. 318, 184–193 [DOI] [PubMed] [Google Scholar]
- 24. Müller P. S., Schulz R., Maretto S., Costello I., Srinivas S., Bikoff E., Robertson E. (2011) The fibronectin leucine-rich repeat transmembrane protein Flrt2 is required in the epicardium to promote heart morphogenesis. Development 138, 1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson M., Parsons Perez M. C., Tébar L., Palmer J., Patel A., Marks D., Sheasby A., De Felipe C., Coffin R., Livesey F. J., Hunt S. P. (2004) FLRT3 is expressed in sensory neurons after peripheral nerve injury and regulates neurite outgrowth. Mol. Cell. Neurosci. 27, 202–214 [DOI] [PubMed] [Google Scholar]
- 26. Tanabe K., Bonilla I., Winkles J. A., Strittmatter S. M. (2003) Fibroblast growth factor-inducible-14 is induced in axotomized neurons and promotes neurite outgrowth. J. Neurosci. 23, 9675–9686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsuji L., Yamashita T., Kubo T., Madura T., Tanaka H., Hosokawa K., Tohyama M. (2004) FLRT3, a cell surface molecule containing LRR repeats and a FNIII domain, promotes neurite outgrowth. Biochem. Biophys. Res. Commun. 313, 1086–1091 [DOI] [PubMed] [Google Scholar]
- 28. Söllner C., Wright G. J. (2009) A cell surface interaction network of neural leucine-rich repeat receptors. Genome Biol. 10, R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamagishi S., Hampel F., Hata K., Del Toro D., Schwark M., Kvachnina E., Bastmeyer M., Yamashita T., Tarabykin V., Klein R., Egea J. (2011) FLRT2 and FLRT3 act as repulsive guidance cues for Unc5-positive neurons. EMBO J. 30, 2920–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boucard A. A., Ko J., Südhof T. C. (2012) High affinity neurexin binding to cell adhesion G-protein-coupled receptor CIRL1/latrophilin-1 produces an intercellular adhesion complex. J. Biol. Chem. 287, 9399–9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ushkaryov Y. A., Hata Y., Ichtchenko K., Moomaw C., Afendis S., Slaughter C. A., Südhof T. C. (1994) Conserved domain structure of β-neurexins. Unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J. Biol. Chem. 269, 11987–11992 [PubMed] [Google Scholar]
- 32. Boucard A. A., Chubykin A. A., Comoletti D., Taylor P., Südhof T. C. (2005) A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to α- and β-neurexins. Neuron 48, 229–236 [DOI] [PubMed] [Google Scholar]
- 33. Bolliger M. F., Martinelli D. C., Südhof T. C. (2011) The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 2534–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ko J., Soler-Llavina G. J., Fuccillo M. V., Malenka R. C., Südhof T. C. (2011) Neuroligins/LRRTMs prevent activity- and Ca2+/calmodulin-dependent synapse elimination in cultured neurons. J. Cell Biol. 194, 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobosy J. R., Rose S. D., Beltz K. R., Rupp S. M., Powers K. M., Behlke M. A., Walder J. A. (2011) RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 11, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aoto J., Martinelli D. C., Malenka R. C., Tabuchi K., Südhof T. C. (2013) Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 154, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen T., Südhof T. C. (1997) Binding properties of neuroligin 1 and neurexin 1beta reveal function as heterophilic cell adhesion molecules. J. Biol. Chem. 272, 26032–26039 [DOI] [PubMed] [Google Scholar]
- 38. Feng K., Zhou X. H., Oohashi T., Mörgelin M., Lustig A., Hirakawa S., Ninomiya Y., Engel J., Rauch U., Fässler R. (2002) All four members of the Ten-m/Odz family of transmembrane proteins form dimers. J. Biol. Chem. 277, 26128–26135 [DOI] [PubMed] [Google Scholar]
- 39. Chandra S., Gallardo G., Fernández-Chacón R., Schlüter O. M., Südhof T. C. (2005) α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123, 383–396 [DOI] [PubMed] [Google Scholar]
- 40. Fernández-Chacón R., Wölfel M., Nishimune H., Tabares L., Schmitz F., Castellano-Muñoz M., Rosenmund C., Montesinos M. L., Sanes J. R., Schneggenburger R., Südhof T. C. (2004) The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42, 237–251 [DOI] [PubMed] [Google Scholar]
- 41. Schmitz F., Tabares L., Khimich D., Strenzke N., de la Villa-Polo P., Castellano-Muñoz M., Bulankina A., Moser T., Fernández-Chacón R., Südhof T. C. (2006) CSPα deficiency causes massive and rapid photoreceptor degeneration. Proc. Natl. Acad. Sci. U.S.A. 103, 2926–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sharma M., Burré J., Bronk P., Zhang Y., Xu W., Südhof T. C. (2012) CSPα knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 31, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y. Q., Henderson M. X., Colangelo C. M., Ginsberg S. D., Bruce C., Wu T., Chandra S. S. (2012) Identification of CSPα clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nguyen T., Mehta N. R., Conant K., Kim K. J., Jones M., Calabresi P. A., Melli G., Hoke A., Schnaar R. L., Ming G. L., Song H., Keswani S. C., Griffin J. W. (2009) Axonal protective effects of the myelin-associated glycoprotein. J. Neurosci. 29, 630–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yin X., Crawford T. O., Griffin J. W., Tu Ph, Lee V. M., Li C., Roder J., Trapp B. D. (1998) Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J. Neurosci. 18, 1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beckmann J., Schubert R., Chiquet-Ehrismann R., Müller D. J. (2013) Deciphering teneurin domains that facilitate cellular recognition, cell-cell adhesion, and neurite outgrowth using atomic force microscopy-based single-cell force spectroscopy. Nano Lett. 13, 2937–2946 [DOI] [PubMed] [Google Scholar]
- 47. Rebay I., Fleming R. J., Fehon R. G., Cherbas L., Cherbas P., Artavanis-Tsakonas S. (1991) Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell 67, 687–699 [DOI] [PubMed] [Google Scholar]