

Background: PGC-1α regulates mitochondrial biogenesis, and may participate in antioxidant gene regulation.
Results: PGC-1α-deficient mice in sepsis demonstrated increased hepatocellular mitochondrial oxidative stress and impaired antioxidant enzyme induction, reflecting PGC-1α interaction with the ARE-dependent Nfe2l2 transcription factor and Sod2 activation.
Conclusion: PGC-1α is critical to mitochondrial SOD-2 induction during hepatic inflammation.
Significance: This novel pathway offers unique opportunities to mitigate oxidative mitochondrial damage.
Keywords: Antioxidants, Mitochondrial Metabolism, Oxidative Stress, Sepsis, Superoxide Dismutase (SOD), Co-immunoprecipitation, Peroxisome Proliferator-activated Receptor γ, Co-activator 1 α;
Abstract
A key transcriptional regulator of cell metabolism, the peroxisome proliferator-activated receptor γ co-activator 1-α (PPARGC-1-α or PGC-1α), also regulates mitochondrial biogenesis, but its role in antioxidant gene regulation is not well understood. Here, we asked whether genetic heterozygosity of PGC-1α modulates gene expression for the mitochondrial antioxidant enzyme SOD-2 during hepatic inflammatory stress. Using Staphylococcus aureus peritonitis in mice, we found significant Sod2 gene induction in WT mice, whereas PGC-1α heterozygotes (PGC-1α+/−) failed to augment Sod2 mRNA and protein levels. Impaired Sod2 regulation in PGC-1α+/− mice was accompanied by oxidative stress shown by elevated mitochondrial GSSG/GSH and protein carbonyls. In silico analysis of the mouse proximal Sod2 promoter region revealed consensus binding sites for the Nfe2l2 (Nrf2) transcription factor. Chromatin immunoprecipitation demonstrated diminished Nfe2l2 protein binding to the antioxidant response element promoter site proximal to the Sod2 start site in PGC-1α heterozygous mice, implicating PGC-1α in facilitation of Nfe2l2 DNA binding. Nuclear protein co-immunoprecipitation demonstrated an interaction between hepatic Nfe2l2 and PGC-1α in WT mice that was greatly reduced in PGC-1α+/− mice. The data indicate that PGC-1α promotes mitochondrial antioxidant enzyme expression through Nfe2l2-mediated SOD-2 expression in sepsis. The presence of this new PGC-1α-dependent signaling axis indicates that PGC-1α opposes mitochondrial oxidative stress by means of selective induction of one or more antioxidant response element-driven genes. By implication, exploitation of this axis could lead to new pharmacological interventions to improve the antioxidant defenses during oxidative stress-induced mitochondrial damage.
Introduction
Aerobic energy conservation within living cells is dependent on the functional density of mitochondria, which in turn is regulated by energy requirements (1). Indeed, the physiological demands of thermogenesis (2), exercise (3), and postnatal growth (4), all states of up-regulated metabolism, induce mitochondrial biogenesis. This induction is responsive to external cues, indicating the need for increased ATP production, and is mediated by a variety of transcription factors, co-activators, and co-repressors.
The PPARg co-activator 1 (PGC-1)2 family of co-activators is centrally important to the regulation of mitochondrial biogenesis. PGC-1α, one of three known co-activators of the family, partners with multiple transcription factors including nuclear respiratory factor-1 (NRF-1), nuclear respiratory factor-2 (NRF-2; GABPA), MEF-2, estrogen-related receptor α (ERRα), and the peroxisome proliferator-activated receptors, all of which regulate respiratory gene expression (5). PGC-1α is often designated a master regulator of the metabolic responses to cold exposure, fasting, exercise, and mitochondrial stress (6–12).
As the rate of oxidative metabolism increases, so does the rate of mitochondrial reactive oxygen species (ROS) production (13). An imbalance between this higher mitochondrial ROS production and ROS scavenging leads to the induction of ROS-detoxifying enzymes such as superoxide dismutase (SOD-2) and others. Normally, functional nuclear PGC-1α is integral to the expression of pathways that enhance oxidative metabolism, but the protein has also been implicated in the up-regulation of ROS-detoxifying enzymes (14–17).
Given the importance of PGC-1α to the maintenance of metabolism and energy production for a broad range of conditions, it is not surprising that PGC-1α knock-out mice display a number of maladaptive phenotypes including miniature stature, exercise and cold intolerance, fasting hypoglycemia, hepatic steatosis, decreased cardiac reserve and heart failure, hyperactivity, and in the brain, evidence of axonal degeneration (18–20). However, PGC-1α heterozygous knock-out (PGC-1α+/−) mice are viable and healthy, and they are capable of maintaining basal metabolism and mitochondrial biogenesis.
During severe inflammatory stress, such as bacterial sepsis, mitochondrial damage can be extensive. Such damage leads to increased synthesis and activity of PGC-1α and its transcription factor partners, with the induction of mitochondrial biogenesis and restoration of respiratory function (21, 22). Here, using an established mouse model of Staphylococcus aureus peritonitis and sepsis, we compared the impact of variable PGC-1α induction on the regulation of the mitochondrial antioxidant defenses in wild type (WT) and PGC-1α heterozygote knock-out (PGC-1α+/−) mice. We hypothesized that a single Ppargc1α (PGC-1α) allele would be insufficient for the transcriptional co-activation and maintenance of high expression levels of critical target antioxidant genes in a sentinel organ, the liver, under stress. This could lead to depressed enzyme function and decreased defenses against increased hepatic ROS production in sepsis. After a series of exploratory studies, we further evaluated the role of PGC-1α in the antioxidant response element (ARE)-dependent expression of the key mitochondrial antioxidant enzyme SOD-2.
EXPERIMENTAL PROCEDURES
Mouse Studies
The mouse studies were conducted on a protocol approved by the Duke University (Durham, NC) Institutional Animal Care and Use Committee. Young adult C57Bl6/J mice purchased from The Jackson Laboratory (Bar Harbor, ME) served as WT control animals. Heterozygote PGC-1α null mice (PGC-1α+/−) on a C57Bl6/J background were obtained from The Jackson Laboratory and bred at our institution. Healthy mice underwent sterile midline laparotomy after induction of anesthesia with a single injection of ketamine and xylazine. Using an established sepsis model (21, 22), the peritoneal cavities were implanted with 500-μl fibrin clots containing 1 × 106 CFU/ml S. aureus. Closure was achieved using proline sutures. Mice were killed at 6 and 24 h by aortic transection under isoflurane anesthesia. The livers were harvested immediately for fresh use or flash-frozen for later use. Control livers were obtained from WT and PGC-1a heterozygote knock-out mice without clot implantation.
Preparation of Total, Nuclear, and Mitochondrial Protein Samples
Total protein extracts were prepared from flash-frozen liver using mechanical homogenization followed by sonication in radioimmunoprecipitation assay buffer. High speed centrifugation (13,200 rpm for 18 min) was used to eliminate remaining heavy cellular components. Fresh liver nuclei were isolated immediately after organ harvest using Dounce homogenization (glass/glass) and low speed centrifugation (3000 rpm for 10 min) followed by resuspension and a 15-min incubation in cell lysis buffer (5 mm PIPES, 85 mm KCl, and 0.5% Nonidet P-40 with protease inhibitors/PMSF). After low speed centrifugation (5000 rpm for 5 min), the pellet was stored at −80 °C. Before freezing, nuclear samples for chromatin immunoprecipitation (ChIP) assays were treated with 1% formaldehyde followed by quenching with 2.5 m glycine for DNA-protein cross-linking.
Mitochondria were isolated from fresh tissue by glass homogenization in isolation buffer (0.25 m sucrose, 10 mm Tris base, 0.5 mm EDTA, and 0.5% BSA at pH 7.4) (23) followed by low speed centrifugation (4000 rpm for 4 min) to eliminate heavy aggregates. The supernatant was centrifuged at high speed (10,200 rpm for 8 min) to recover the mitochondrial fraction.
PAGE and Western Blot Analysis
After protein assay, individual proteins were resolved by electrophoresis on Novex Tris-glycine polyacrylamide gels (Invitrogen) followed by transfer to polyvinylidene difluoride membranes (PVDF, Millipore, Billerica, MA). Proteins were probed with primary antibodies to polyclonal rabbit anti-PGC-1α (Cayman Chemical, Ann Arbor, MI), rabbit anti-SOD-2 (Abcam, Cambridge, MA), rabbit anti-nuclear factor (erythroid-derived 2)-like 2 (Nfe2l2 H300, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-programmed cell death-1 (mPD-1, AnaSpec, Fremont, CA), rabbit anti-mitofusin-2 (Mfn-2, Epitomics, Burlingame, CA), and mouse anti-NAD(P)H dehydrogenase (quinone) 1 (QR1, Santa Cruz Biotechnology). Antibodies to NRF-1, NRF-2 (GABPA), and mitochondrial transcription factor A (mtTFA) developed in rabbits were characterized in our laboratory (24). After application of primary antibodies and washing, membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Membranes were developed with chemiluminescence reagent (Calbiochem RapidStep, EMD Chemicals; or luminol, Santa Cruz Biotechnology), and proteins were quantified on digitized images using Quantity One (Bio-Rad). Densitometry measurements were normalized to β-actin or tubulin in the same samples.
Extraction and Quantification of mtDNA and mRNA
Genomic DNA was purified from flash-frozen tissue after tissue digestion, cell lysis, protein denaturation (proteinase K), and RNA degradation (RNase) using a silica-based membrane (GenElute mammalian genomic DNA miniprep kit, Sigma-Aldrich). The mtDNA copy number was determined using real-time PCR (quantitative PCR) (25).
Total RNA was extracted from liver tissue using TRIzol reagent (Invitrogen). RNA purity was confirmed on 1.2% agarose, and the RNA was converted to cDNA using oligo(dT) (ImProm-II reverse transcription system; A3800). Real-time RT-PCR was performed on an ABI Prism 7000 using gene expression assays (Applied Biosystems). A ΔCt method was used to quantify mRNA levels for Ppargc1α, Nrf1, mitochondrial transcription factor A (Tfam), Sod2, Hmox1, mitochondrial thioredoxin (Txn2), tumor necrosis factor (Tnf), C-C motif chemokine 2 (Ccl2), and interleukin-10 (IL10). Amplification efficiency was checked with an internal 18 S rRNA (AB PN 4332078) over a 0.9–90-ng range of RNA, and gene expression was quantified using ABI Prism 7000 SDS and MS Excel software. Each sample was assayed in triplicate.
TUNEL Assay
Paraffin-embedded liver thin sections were used for the TUNEL assay. Samples from healthy control and 24-h post-sepsis livers were heated to 60 °C for 45 min and deparaffinized by two changes of xylene. Sections were rehydrated through a series of 5-min soaks in graded ethanol solutions. After washing for 10 min, sections were permeabilized for 30 min at 37 °C with proteinase K solution. A TUNEL reaction mixture (enzyme and label, Roche Applied Science, Mannheim, Germany) was then applied to the sections for 1 h at 37 °C. Negative and positive control sections were prepared with label solution only or by preinduction of strand breakage with recombinant DNase I. After incubation, samples were analyzed for the proportion of apoptotic cells using fluorescence microscopy.
GSSG/GSH and Glutathionylation Assays
Mitochondria (1 μg of protein/μl) were centrifuged at 700 × g for 10 min. Supernatant was removed, and protein was precipitated from the pelleted mitochondria via sonication in 5% metaphosphoric acid followed by centrifugation at 1000 × g for 10 min at 4 °C. The supernatant, containing mitochondrial GSH, was rapidly removed and stored at −80 °C until GSH assay. The total glutathione (GSH) and glutathione disulfide (GSSG) content of the mitochondria was determined by an enzymatic recycling assay based on glutathione reductase (26). GSSG levels were determined after derivatization of GSH by 2-vinylpyridine.
Mitochondrial protein S-glutathionylation levels were measured using a bacterial glutaredoxin 1 (GRX1) reduction assay with S-biotinylation and chemiluminescence detection. Mitochondrial samples were treated with 20 mm N-ethylmaleimide at room temperature for 30 min to alkylate free thiols. After repeated acetone precipitation, samples were resuspended in Tris buffer (pH 7.5), and S-glutathionylated proteins were reduced to free thiols by incubation with Escherichia coli GRX1 (Cayman Chemical), GSSG reductase (Sigma), 1 mm GSH, 1 mm NADPH, and 1 mm EDTA for 15 min at 37 °C. Reduced sulfhydryl groups were labeled by application of 2 mm biotin N-[6-(Biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide (Biotin-HPDP) for 1 h at room temperature. Negative controls were treated identically but without GRX1, whereas positive controls were treated with 2 mm DTT. Samples and controls were applied to an HSI vacuum slot blotter containing a PVDF membrane (Hoefer Scientific, San Francisco, CA). Streptavidin HRP (Thermo Fisher Scientific) was applied, and the membranes were developed using chemiluminescence detection (Santa Cruz Biotechnology) and quantified on digitized images using Image J (National Institutes of Health, Bethesda, MD).
DNPH-derivatized Protein Carbonyl Quantification
Flash-frozen tissue was homogenized (glass/glass) on ice in Tris/HCl containing 1 mm butylated hydroxytoluene, desferoxamine mesylate, and commercial protease and phosphatase inhibitor mixture. Aliquots of 1.5 mg of protein were reacted with 10 mm DNPH in 2.5 m HCl. After mixing with cold TCA, the samples were centrifuged at high speed, and the pellets were again washed with TCA and then repeatedly washed (×6) with 1:1 ethanol/ethyl acetate to remove free DNPH. The final three washes were accompanied by sonication. The pellets were allowed to dry, dissolved in 6 m guanidine HCl, and diluted ×0.1 with sodium phosphate. After high speed centrifugation to clarify the supernatant, the protein content was adjusted to a standard concentration (5 μg/100 μl). Samples, along with a negative control (treated with NaBH4), were applied to an HSI vacuum slot blotter containing a PVDF membrane (Hoefer Scientific). An immunoassay was performed using a primary antibody (rabbit anti-dinitrophenol, 1:150 dilution, Millipore) followed by a secondary antibody (goat anti-rabbit, 1:300 dilution, Millipore). The membranes were developed using chemiluminescence detection, and protein carbonyl concentration was quantified on digitized images using Quantity One (Bio-Rad).
Myeloperoxidase Assay
A myeloperoxidase activity assay was used as described to estimate the neutrophil content of liver tissue (27). Frozen liver tissue was mixed with 1% hexadecyltrimethylammonium bromide solution, homogenized, and sonicated. After centrifugation, the supernatant was mixed with o-dianisidine and 0.3% H2O2. Spectrophotometric measurement of change in optical density (406 nm) per minute per gram of liver was reported.
Bioinformatics
In silico analysis of the mouse Sod2 promoter (Ensembl Gene ID ENSMUSG00000006818) was performed for consensus sequences for transcription factors NRF-1, GABPA, and Nfe2l2 binding sites using DNASIS (Hitachi Software; Alameda, CA) and confirmed with MatInspector (Genomatix Software; München, Germany).
Chromatin Immunoprecipitation (ChIP)
Nuclei for ChIP assays were sonicated in shearing buffer, and shearing effectiveness was confirmed by electrophoresis on ethidium bromide-stained agarose gels. Samples were then processed for immunoprecipitation using a kit (ChIP IT assay kit, Active Motif, Carlsbad, CA) and antibodies to NRF-1 (Cayman), GABPA (Cayman), and Nfe2l2 according to the manufacturer's instructions. After precipitation, cross-linking was reversed, and PCR was carried out using 1 μl of each sample (input DNA dilution 1:10, immunoprecipitated fractions undiluted) in PCR buffer (Qiagen, Valencia, CA) containing dNTP (Invitrogen) and Taq DNA polymerase (Qiagen) with the primers 900CGCTTCGCTGTGTCCTTGC882 and 776TAATGTTGTGTCGGGCGGC794 to amplify NRF-1 site, 922CCGTCCTCCCCTCCGCTGAT941, and 774ACGCCGCCCGACACAACATT793 to amplify Nfe2l2 sites, and 1383GCTGCACCCGGAGTCCGCAA1364 and 1134TGGCTGTGAGTCGCAAAGCTTCC1156 to amplify GABPA sites for the Sod2 promoter. For the Hmox1 promoter, we used primers 1350CCTGAAGGGCTACTCCCGTCTTCC1327 and 1051TTGCAACATCCAGCCCGGAGGC1072. DNA concentration was measured and standardized across samples prior to PCR. PCR products were analyzed by agarose gel electrophoresis.
Protein Co-immunoprecipitation (Co-IP)
Nuclear protein (500 μg) in Co-IP buffer (120 mm sodium chloride, 50 mm Hepes [pH 7.5], anti-protease, and anti-phosphatase inhibitors) with 0.25% digitonin was incubated for 3 h with the primary antibody (10 μl of rabbit polyclonal PGC-1 primary antibody (Cayman Chemical)) for 3 h at 4 °C. A suspension of protein G-Sepharose beads (Sigma E3403 EZview Red protein G affinity gel) was added for 30 min to select for proteins bound to PGC-1α. The captured immune complexes were layered on a 1-ml 1 m sucrose cushion (in Co-IP buffer with 0.5% w/w digitonin) and centrifuged (10,000 × g for 5 min) followed by washing at least three times with Co-IP buffer (containing 0.25% w/w digitonin). The beaded complex was washed once with Co-IP buffer lacking digitonin. After washing, 1× sample buffer (Bio-Rad) with DTT was added to the samples, which were denatured by heating to 95 °C for 10 min. Separate 20-μg nuclear samples were diluted 1:1 with 2× sample buffer and heated as input controls. Co-IP samples were magnetized after heating, and supernatant aliquots were analyzed for Nfe2l2 (66 kDa; Santa Cruz Biotechnology) by Western blot analysis using bead and input control samples.
Statistical Analysis
All grouped data are presented as means ± S.D. Statistical significance was determined using two way analysis of variance and Student's t test for post hoc analysis with JMP software (version 11, SAS Institute Inc., Cary, NC). p ≤ 0.05 was considered significant.
RESULTS
Ppargc1α mRNA and Protein Expression in Heterozygous PGC-1α Mice
Differences in hepatic Ppargc1α mRNA levels were detectable by real-time RT-PCR in WT and in PGC-1α+/− mice following S. aureus clot implantation. There were no differences between the groups in base-line Ppargc1α transcript levels, but after peritoneal inoculation, the mRNA levels increased by 6 h and returned toward base line by 24 h (Fig. 1A). As expected, the magnitude of the increase in Ppargc1α mRNA was significantly less in the PGC-1α+/− mice than in WT mice. Western blot analysis of total liver PGC-1α protein showed a significant increase from base line in WT mice at 24 h but no change in the PGC-1α+/− animals (Fig. 1, B and C). Thus, PGC-1α heterozygosity significantly blunts hepatic Ppargc1α mRNA and protein induction in response to S. aureus sepsis.
FIGURE 1.
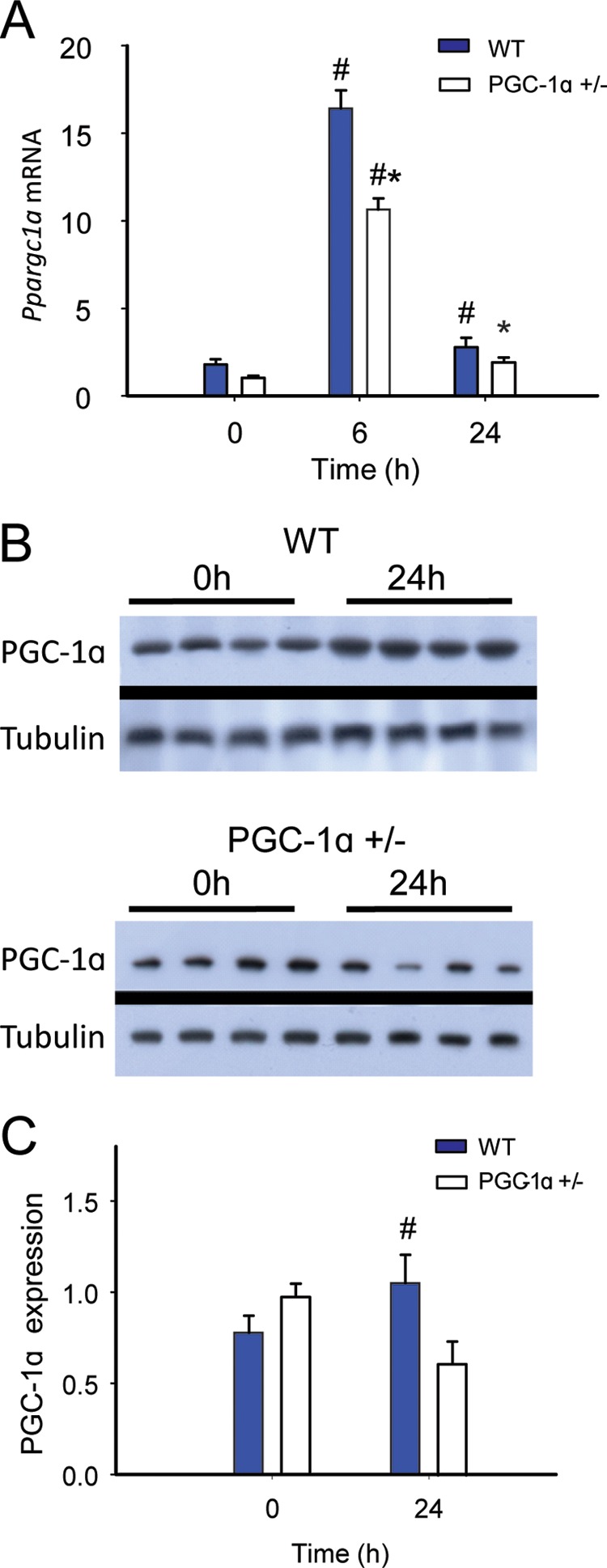
PGC-1α responsiveness in WT and PGC-1α heterozygote (+/−) mice during S. aureus sepsis. A, Ppargc1a mRNA expression levels at 0, 6, and 24 h after clot implantation (n = 4). B, PGC-1α Western blots before and 24 h after clot implantation. C, protein densitometry (n = 4). #, significant difference from 0 h; *, significant inter-strain difference; p < 0.05.
Induction of Mitochondrial Biogenesis
The redox-dependent induction of mitochondrial biogenesis is a key survival factor in sepsis (28); hence we evaluated mRNA and protein levels in our strains of mice for three key transcription factors involved in mitochondrial biogenesis: NRF-1, GABPA, and mtTFA. NRF-1 and GABPA are necessary for the up-regulation of mtTFA and other nucleus-encoded, mitochondria-targeted proteins involved in mtDNA transcription and replication. We found a progressive increase in Nrf1 and Tfam mRNA levels by real-time RT-PCR at 6 and 24 h in the WT mice, but nonsignificant changes in these mRNA levels in PGC-1α+/− animals (Fig. 2, A and B). Liver protein levels assessed at 0 and 24 h by Western blot revealed an increase in NRF-1, GABPA, and mtTFA levels by 24 h in WT and PGC-1α+/− mice, but equal induction of only GABPA in the PGC-1α+/− mice (Fig. 2, C–F). The mtDNA copy number was stable at 6 h, but we found an increase in both strains at 24 h that was significantly greater in the WT mice (Fig. 2G). These results support an adverse effect of impaired PGC-1α protein synthesis in the PGC-1α+/− mice on the induction of mitochondrial biogenesis and the bioenergetic response to sepsis. Moreover, hepatic nuclear protein levels for PGC-1α and NRF-1 were impressively elevated in WT mice when compared with PGC-1α+/− mice (Fig. 2, B and D). In addition, there were significant between strain differences in the nuclear levels of two other key redox-sensitive proteins involved in the regulation of mitochondrial biogenesis, Nfe2l2 and SIRT2 (Fig. 3, C and E).
FIGURE 2.

Mitochondrial biogenesis transcription factor expression in sepsis. A and B, changes in mRNA levels at 0, 6, and 24 h after clot implantation for Nrf1 (A) and Tfam (n = 4) (B). C, representative Western blot analysis. D–F, protein densitometry for NRF-1 (D); GABPA (E); and mtTFA (n = 4) (F). G, mtDNA copy number in WT and PGC-1α+/− mice at 0 h (controls) and 24h (n = 2). #, significant difference from 0 h; *, significant inter-strain difference; p < 0.05.
FIGURE 3.
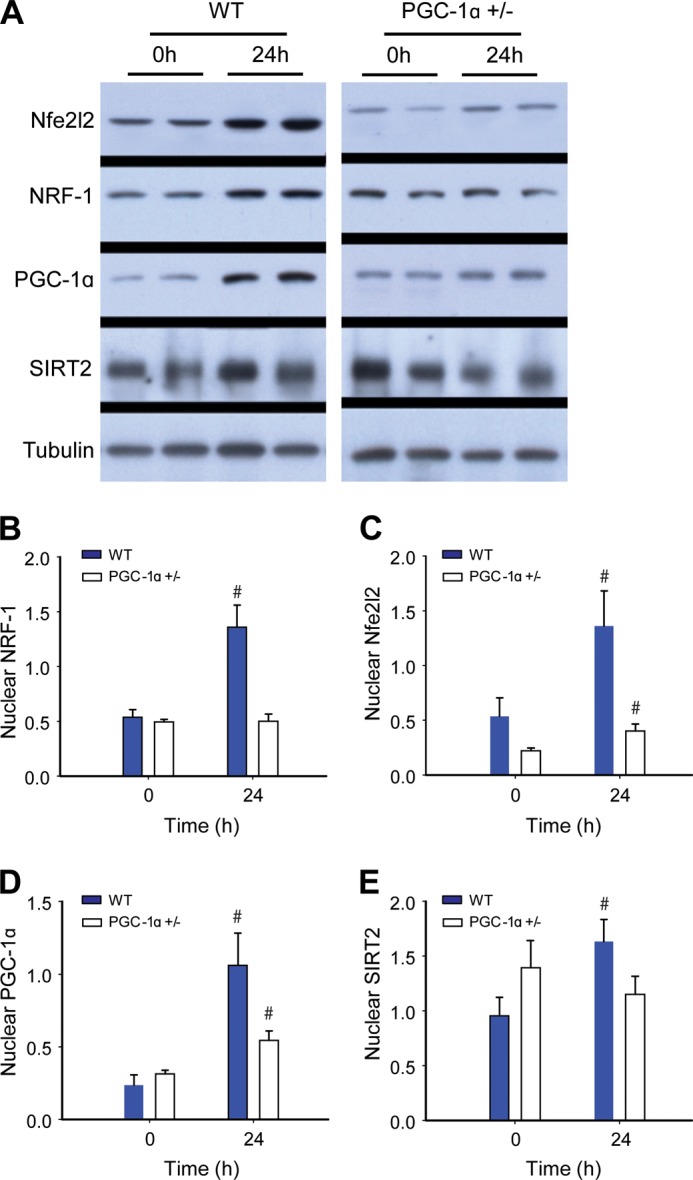
Western analysis for hepatic nuclear proteins. A, immunoblots for nuclear Nfe2l2 and NRF-1 transcription factors, PGC-1α, and Sir2 proteins. Tubulin is a reference protein. B–E, protein densitometry of nuclear Nfe2l2 (B); NRF-1 (C); PGC-1α (D); and SIRT2 (E) in WT and PGC-1α+/− mice at 0 (control) and 24 h after clot implantation (n = 4). #, significant difference from 0 h.
Oxidative Stress Assays
We evaluated overall hepatic oxidative stress by assaying mitochondrial glutathione levels. Base-line GSH levels were significantly lower in PGC-1α+/− mice than in the WT mice (Fig. 4A). After inoculation with S. aureus, GSH levels increased in both WT and PGC-1α+/− mice (10.6 ± 1.5 to 18.7 ± 1.4 μmol/mg of protein for WT; 6.6 ± 1.2 to 12.1 ± 1.2 μmol/mg of protein for PGC-1α+/−). Similarly, GSSG/GSH (%GSSG) increased after the induction of peritonitis (14.6 ± 2.5 to 62.7 ± 6.4% for WT; 19.6 ± 2.8 to 82 ± 9.9% for PGC-1α+/−). Also, %GSSG was increased in the PGC-1α+/− mice at base line, and relative to the WT, the onset of peritonitis led to a significant increase in %GSSG these mice (Fig. 4, A and B). There was no apparent difference in protein glutathionylation across time points for either strain (Fig. 4C).
FIGURE 4.

Mitochondrial oxidative stress responses during S. aureus sepsis in WT and PGC-1α+/− mice. A, mitochondrial GSH content. B, percentage of GSSG. C, mitochondrial protein carbonyls. D, mitochondrial protein S-glutathionylation (n = 4). x-axis shows 0 (controls), 6, and 24 h after clot implantation. #, significant difference from 0 h; *, significant inter-strain difference; p < 0.05.
Differences in oxidative stress levels between WT and PGC-1α+/− mice were confirmed by quantification of mitochondrial protein oxidation using a standardized protein carbonyl assay. Levels of oxidized mitochondrial protein increased in the livers of mice of both strains after induction of peritonitis (Fig. 4D). The PGC-1α+/− mice demonstrated higher base-line protein carbonyl levels than their WT counterparts, and this difference persisted at 6 h after peritonitis. By 24 h after inoculation, the levels of oxidized mitochondrial protein had decreased toward base-line level, particularly in WT mice, perhaps reflecting activation of GSH synthesis, more rapid disposal of oxidized protein, and/or an increase in other antioxidant defenses and an ability to limit protein carbonylation (29).
Inflammation and Hepatocellular Damage
To quantify the degree of hepatocellular damage induced by oxidative stress during S. aureus sepsis, we measured myeloperoxidase activity as a surrogate for acute inflammation, which typically reflects the early neutrophilic inflammatory response to sepsis. Myeloperoxidase levels were similarly elevated at 6 and 24 h in both strains of mice (Fig. 5A). However, the hepatic induction of early-phase proinflammatory cytokines and chemokines differed in WT and PGC-1α+/− mice before and after S. aureus inoculation (Fig. 5, B and C). The mRNA levels for two key potentially damaging mediators, Tnf and Ccl2, increased significantly more in PGC-1α+/− than in WT mice, whereas anti-inflammatory IL10 cytokine levels were lower in PGC-1α+/− mice than in WT mice (Fig. 5D). The two proinflammatory cytokine levels peaked at 6 h and returned to base line by 24 h in WT mice, but remained elevated in PGC-1α+/− mice. Quantification of hepatocellular cell death by TUNEL staining suggested a trend toward more base-line apoptosis in PGC-1α+/− mice, but a significant increase in the fraction of apoptotic cells at 24 h in PGC-1α+/− when compared with WT mice (0.01 ± 0.01 to 0.16 ± 0.06 for WT versus 0.08 ± 0.04 to 0.41 ± 0.02 for PGC-1α+/−) (Fig. 6, A and B). Of further interest, the extent of hepatic cell death indicated by programmed cell death protein 1 (mPD-1) increased from base line in both strains of mice at 24 h after S. aureus inoculation, but remained significantly higher in the PGC-1α+/− mice (Fig. 6, C and D).
FIGURE 5.

Inflammation during S. aureus sepsis in WT and PGC-1α+/− mice. A, myeloperoxidase activity (n = 4). B–D, changes in mRNA expression in WT and PGC-1α+/− mice at 0 (controls), 6, and 24 h after clot implantation: Ccl2 (B), Tnf (C), and IL10 (D) mRNA levels (n = 4). x-axis shows 0 (controls), 6, and 24 h after clot implantation. #, significant difference from 0 h; *, significant inter-strain difference; p < 0.05. OD, optical density.
FIGURE 6.
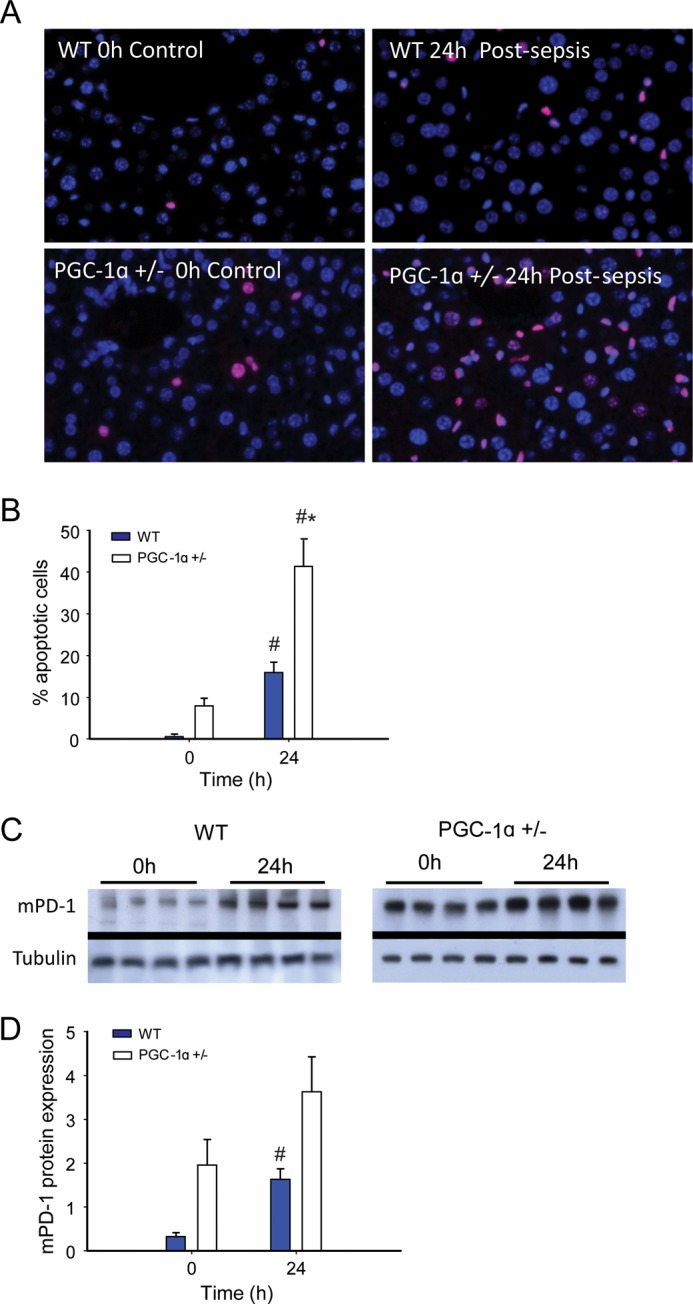
Cell death in sepsis. A and B, TUNEL staining showing pronounced increase in hepatic apoptosis in sepsis in PGC-1α+/− mice (A) and quantification of apoptotic liver cells in WT and PGC-1α+/− mice at 0 and 24h (n = 6 for WT; 4 for PGC-1α+/−) (B). C and D, Western blot for mPD-1 protein (C) and protein quantification by densitometry for WT and PGC-1α+/− mice (D) at 0 h (control) and 24 h after clot implantation (n = 4). #, significant difference from 0 h; *, significant inter-strain difference; p < 0.05.
Antioxidant Enzyme Induction in WT and PGC-1α+/− Mice in Sepsis
The significant oxidative stress in PGC-1α+/− during the infection relative to WT mice raised the possibility of differences in antioxidant enzyme induction between the two strains. Examination of hepatic mRNA expression for Sod2, Txn2, and Hmox1 mRNA in WT mice by real-time RT-PCR revealed sustained increases in Txn2 and Hmox1 and an increase in Sod2 mRNA level at 24 h after inoculation (Fig. 7, A–C). In the PGC-1α+/− mice, however, Sod2 and Hmox1 mRNA levels had increased only marginally at 24 h, whereas Txn2 increased marginally at 6 h and returned to base line at 24 h. Among the three antioxidant enzymes, we also noted pronounced differences in mRNA levels in the PGC-1α+/− mice in sepsis (0 versus 24 h), suggesting that PGC-1α contributes differentially to the regulation of these genes (Fig. 7D).
FIGURE 7.

Mitochondrial antioxidant enzyme responses during S. aureus sepsis in WT and PGC-1α+/− mice. A, Sod2 mRNA expression levels. B, Hmox1 mRNA expression. C, Txn2 mRNA expression. D, -fold increase in mRNA levels 0 versus 24 h sepsis, (n = 4). x-axis shows 0 (controls), 6, and 24 h after clot implantation. #, significant difference from 0 h (significant difference between antioxidant mRNA -fold change for panel D); **, significant inter-strain difference; p < 0.05.
PGC-1α and Regulation of Nfe2l2 and NRF-1 Downstream Genes
To evaluate PGC-1α as a transcriptional co-activator, we next measured protein content for three specific nucleus-encoded downstream genes: the mitochondrial antioxidant enzyme SOD-2, Mfn-2, regulated in part by NRF-1, and the NAD(P)H dehydrogenase QR1, regulated by Nfe2l2. The PGC-1α+/− mice displayed less Mfn-2 and QR1 protein induction during sepsis when compared with the WT (Fig. 8, A–C). In contrast to a robust increase in SOD-2 protein in WT mice, SOD-2 did not increase after inoculation in PGC-1α+/− mice (Fig. 8D). Thus, the capacity to express PGC-1α was important for the activation of genes downstream of both NRF-1 and Nfe2l2 during sepsis. This activation enhances steady-state expression of QR1, Mfn-2, and SOD-2. The regulatory role of PGC-1α in NRF-1-dependent genes is known, but the possibility of PGC-1α contributing to redox-regulated gene expression through an interaction with Nfe2l2 is novel. Therefore we evaluated the association of these two proteins at 0, 6, and 24 h in our strains of mice. A protein-protein interaction for PGC-1α-Nfe2l2 was readily discernible, as was evidence of significantly less PGC-1α binding to Nfe2l2 in PGC-1α+/− mouse liver nuclei at each time point (depicted in Fig. 9A, repeated twice with separate samples for verification).
FIGURE 8.

Redox-regulated mitochondrial proteins during S. aureus sepsis in WT and PGC-1α+/− mice. A, Western blot analysis. Tubulin acts as a reference protein. B–D, protein densitometry for mitofusin-2 (B); QR1 (C); and SOD-2 (D) in WT and PGC-1α+/− mice at 0 h (control) and 24 h after clot implantation (n = 4). #, significant difference from 0 h.
FIGURE 9.
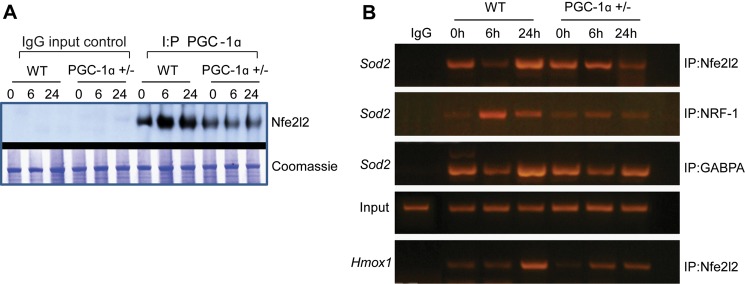
PGC-1α co-immunoprecipitation with Nfe2l2. A, representative Western blot for Nfe2l2 transcription factor after pulldown with anti-PGC-1α (n = 1). B, representative chromatin IP of the Sod2 promoter region demonstrating Nfe2l2, NRF-1, and GABPA transcription factor binding to promoter recognition sites (n = 1). Binding for all three transcription factors is more pronounced at 6 or 24 h of sepsis in WT mice than in PGC-1α+/− mice. The bottom lane shows greater induction of Nfe2l2 recruitment to the Hmox1 promoter in WT mice than in PGC-1α+/− mice in sepsis.
Active NRF-1, GABPA, and Nfe2l2 Binding Sites in the Sod2 Proximal Promoter Region
Because SOD-2 expression was so strongly induced in WT, but not in PGC-1α+/− mice, we specifically evaluated Sod2 gene promoter regulation in the mouse liver. By in silico analysis of mouse Sod2 5′-UTR (DNAsis and Genomatix), we identified potential NRF-1, GABPA, and Nfe2l2 binding sites within the conserved Sod2 promoter sequence. Putative core ARE (RTGAYnnnGC) canonical binding sites of 100% homology were identified at positions −1701 and −910. A canonical binding site (100% homology) for the NRF-1 consensus was found at position 843. We also found two putative GABPA binding sites of at least 85% homology at positions −1383 and −1100. We performed ChIP assays for Nfe2l2, NRF1, and GABPA occupancy of the Sod2 promoter at 0, 6, and 24 h after S. aureus inoculation. The data demonstrated significant occupancy of the known PGC-1α co-activator partners NRF-1 and GABPA on the Sod2 promoter (Fig. 9B). Concurrently, we found that the Nfe2l2 transcription factor, which regulates the antioxidant response, displayed higher Sod2 promoter occupancy in WT when compared with PGC-1α+/− mice. Notably, the difference in responses between the WT and PGC-1α+/− mice suggested a role for PGC-1α in hepatic Sod2 gene regulation during infectious inflammation. Moreover, occupancy of the Sod2 promoter by NRF-1 and Nfe2l2 was accompanied by recruitment of DNA polymerase II, indicative of initiation of transcription (data not shown). Also, because Nfe2l2 is a powerful regulator of Hmox1 gene expression, we confirmed Nfe2l2 binding to the murine Hmox1 promoter by ChIP assay. Nfe2l2 occupancy of the Hmox1 promoter was also suppressed in PGC-1α+/− when compared with WT mice, particularly at 24 h after inoculation (Fig. 9B), providing more support for the interaction of Nfe2l2 with PGC-1α.
DISCUSSION
Our rationale was to address our limited understanding of redox-based mechanisms for the resolution of mitochondrial damage in sepsis. Multiple organ failure in sepsis is a leading cause of death in the United States (30), and the resolution of oxidative mitochondrial damage (31) is an area of concentrated investigation (32). The failure to induce mitochondrial biogenesis negates an important prosurvival mechanism that simultaneously activates antioxidant and anti-inflammatory responses in the liver and other tissues (33, 34). Also, although the transcriptional network for mitochondrial biogenesis is regulated by the PGC-1α co-activator (35), a role for this co-activator in the antioxidant and counter-inflammatory defenses in sepsis is supported only indirectly. Hence, we tested the idea that the loss of even a single Ppargc1α allele would adversely affect the ability of the liver not only to induce mitochondrial biogenesis, but also to avoid excess cytokine elaboration, oxidative damage, and apoptosis.
Normally, the response to experimental S. aureus sepsis in mice includes PGC-1α up-regulation in connection with the induction of mitochondrial biogenesis (21). Here, we have not only confirmed an essential role for PGC-1α in regulating hepatic mitochondrial biogenesis in sepsis, but confirmed that PGC-1α induction protects against mitochondrial oxidative stress. The limitation in mitochondrial biogenesis is reflected in part by the less pronounced increases in mRNA and protein for mtTFA, NRF-1, GABPA, and in mtDNA copy number in PGC-1α heterozygous mice.
Sepsis also produces an increase in cellular ROS production rates, leading to oxidative stress driven both by the host inflammatory response and by mitochondrial damage (36, 37). In this mouse model, hepatic oxidative stress was demonstrated both by the increased GSSG/GSH ratio and by increased protein carbonyl content. Although hepatic GSH levels increased in both strains, confirming prior observations in WT mice, the level of oxidative stress was exaggerated in PGC-1α+/− mice. Similarly, the heavy catalytic subunit of hepatic glutamate cysteine ligase (GCLC), which is essential for GSH synthesis, is rapidly induced after LPS administration (24). LPS administration causes transient mitochondrial GSH depletion in the rodent heart (38), whereas here PGC-1α heterozygosity led to a higher mitochondrial GSSG/GSH ratio in sepsis as well as to greater increases in mitochondrial protein oxidation, suggesting that loss of the PGC-1α allele negatively impacted hepatic GSH recycling. The implications of mitochondrial oxidative stress in PGC-1α+/− mice after S. aureus inoculation are seen in the failure to increase mtDNA copy number, along with an increase in proapoptotic protein activation and greater hepatocyte apoptosis.
The normal cytoprotective response to sepsis-induced oxidative stress is the up-regulation in antioxidant defense mechanisms. In particular, SOD-2 up-regulation and increased superoxide scavenging modulate damage to mtDNA and dysfunction of the TCA cycle and the electron transport chain (39). PGC-1α+/− mice display diminished mitochondrial antioxidant enzyme induction despite clear evidence of oxidative stress and ongoing hepatic damage. For instance, in WT mice, we confirmed that this experimental model strongly induces hepatic SOD-2 (21), but in PGC-1α+/− mice, we found significant blunting of Sod2 mRNA up-regulation. Similarly, two other antioxidant enzymes normally induced in sepsis, mitochondrial thioredoxin (MRTX) and HO-1, show significant decreases in responsiveness in PGC-1α+/− mice. Given that SOD-2 and MTRX function together at sites of mitochondrial superoxide production, the association between the levels of the two enzymes may regulate the leak rate of H2O2 and impact redox signaling (40, 41). A change in sensitivity to redox state would implicate hepatic PGC-1α as an essential regulator of antioxidant enzyme induction during the host response to S. aureus infection.
Previously, PGC-1α expression has been associated with the suppression of cellular ROS production. For instance, in cultured neuronal cells, PGC-1α protein is rapidly up-regulated after exogenous oxidative stress (H2O2). Further investigations in PGC-1α null cells revealed decreased induction of specific ROS-detoxifying enzymes (SOD-1, SOD-2, catalase, and GPx1) during an oxidative stress (14). In vascular endothelial cells, PGC-1α overexpression led to the induction of the mitochondrial antioxidant defenses and a reduction in oxidative stress (42). In addition, PGC-1α−/− mice show low basal cardiac mitochondrial antioxidant enzyme levels that fail to respond to systolic heart overload (16).
The dampening of PGC-1α induction found in PGC-1α+/− mice in association with decreased antioxidant enzyme induction with the oxidative stress of sepsis also suggests a causative role for PGC-1α in the antioxidant response, but the molecular pathways involved could be diverse. The literature emphasizes PGC-1α regulation of mitochondrial biogenesis and metabolic pathways (43), but its role in the genetic regulation of mitochondrial antioxidant enzymes such as SOD-2 has been circumstantial. Although it is well known that the Nfe2l2 transcription factor interacts with ARE motifs in the promoter regions of many antioxidant genes, including Sod1, Hmox1, and enzymes of the glutathione cycle (44), PGC-1α has not been recognized as an Nfe2l2 binding partner. The diminished levels of NRF-1 and Nfe2l2 protein and the lack of SOD-2 induction in PGC-1α+/− mice in sepsis raised the prospect of one or more interactions between the PGC-1α co-activator and one or both genes for these transcription factors or with the nuclear proteins at the Sod2 gene promoter.
The in silico identification of putative ARE binding sites for Nfe2l2 on the Sod2 promoter led us to evaluate occupancy of those sites by ChIP assay. This demonstrated both NRF-1 and Nfe2l2 occupancy of binding sites on the Sod2 promoter, which led us to check the interaction of the PGC-1α co-activator with the Nfe2l2 hepatic nuclear protein by co-immunoprecipitation. This demonstrated the association of PGC-1α with Nfe2l2 protein (Fig. 9A) and demonstrated by subsequent ChIP assay that Nfe2l2 binding to murine Sod2 was decreased in PGC-1α+/− mice in sepsis when compared with WT mice. These data support PGC-1α co-activation of Nfe2l2 during Sod2 induction.
As a result, although mPD-1 levels increased in sepsis in both strains of mice, this response was exaggerated in the PGC-1α+/− mice. Activation of mPD-1 was initially reported in apoptotic lymphoid cell lines in mice (45), and the protein is well known to repress the immune response by inhibiting T cells and decreasing cytokine synthesis (46). More recently, the protein was implicated in the immune response to sepsis because the knock-out mice exhibit decreased lymphocyte apoptosis in sepsis (47) and show improved regional and systemic bacterial clearance after cecal ligation and puncture (48). In human patients, differences in mPD-1 levels in sepsis survivors versus nonsurvivors correlate with rates of secondary nosocomial infections (49). Here in mice, we observed greater mPD-1 induction in PGC-1α+/− mice corresponding to the observed increase in hepatocyte apoptosis, yet it remains unclear whether the mPD-1 up-regulation was the proximate cause or merely associated with the apoptosis, for example through possible modulation of the immune response. We did evaluate the acute neutrophilic response in the livers by myeloperoxidase assay and found no significant inter-strain differences in activity. Nonetheless the low PGC-1α expression in the heterozygous mice did vary directly with the early proinflammatory response and inversely with counter-inflammatory IL-10 production.
In conclusion, we provide new evidence that PGC-1α is required not only for the induction of hepatic mitochondrial biogenesis in sepsis, but also for mitochondrial antioxidant enzyme induction, in particular SOD-2. The partnership of PGC-1α with both NRF-1 and Nfe2l2 is detectable on mouse Sod2 promoter by ChIP. Further evidence for this antioxidant role of PGC-1α is seen in the dampening of induction of two other ARE-regulated genes, Txn2 and Hmox1, in sepsis in the PGC-1α+/− mice. The significant phenotypic manifestations in the PGC-1α+/− mice during the infections include greater mitochondrial oxidative stress, higher proinflammatory cytokine levels, and a higher proportion of apoptotic cells within the liver. Thus, the absence of even a single Ppargc1α allele impairs the murine host response to S. aureus peritonitis, resulting in increased levels of oxidative stress and hepatic cell death.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1-A1095424 and R01-HL108801 (to C. A. P.).
- PGC-1
- PPARg co-activator 1
- PPAR
- peroxisome proliferator-activated receptor
- NRF-1
- nuclear respiratory factor-1
- GABPA
- GA-binding protein α chain
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- mtTFA
- mitochondrial transcription factor A
- ARE
- antioxidant response element
- DNPH
- 2,4-dinitrophenylhydrazine
- Co-IP
- co-immunoprecipitation.
REFERENCES
- 1. Hoppeler H., Lüthi P., Claassen H., Weibel E. R., Howald H. (1973) The ultrastructure of the normal human skeletal muscle. A morphometric analysis on untrained men, women and well-trained orienteers. Pflugers Arch. 344, 217–232 [DOI] [PubMed] [Google Scholar]
- 2. Cannon B., Nedergaard J. (2004) Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 [DOI] [PubMed] [Google Scholar]
- 3. Holloszy J. O., Booth F. W. (1976) Biochemical adaptations to endurance exercise in muscle. Annu. Rev. Physiol. 38, 273–291 [DOI] [PubMed] [Google Scholar]
- 4. Valcarce C., Navarrete R. M., Encabo P., Loeches E., Satrústegui J., Cuezva J. M. (1988) Postnatal development of rat liver mitochondrial functions: the roles of protein synthesis and of adenine nucleotides. J. Biol. Chem. 263, 7767–7775 [PubMed] [Google Scholar]
- 5. Scarpulla R. C. (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638 [DOI] [PubMed] [Google Scholar]
- 6. Baar K., Wende A. R., Jones T. E., Marison M., Nolte L. A., Chen M., Kelly D. P., Holloszy J. O. (2002) Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 16, 1879–1886 [DOI] [PubMed] [Google Scholar]
- 7. Goto M., Terada S., Kato M., Katoh M., Yokozeki T., Tabata I., Shimokawa T. (2000) cDNA cloning and mRNA analysis of PGC-1 in epitrochlearis muscle in swimming-exercised rats. Biochem. Biophys. Res. Commun. 274, 350–354 [DOI] [PubMed] [Google Scholar]
- 8. Lin J., Wu H., Tarr P. T., Zhang C. Y., Wu Z., Boss O., Michael L. F., Puigserver P., Isotani E., Olson E. N., Lowell B. B., Bassel-Duby R., Spiegelman B. M. (2002) Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418, 797–801 [DOI] [PubMed] [Google Scholar]
- 9. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 10. Russell A. P., Hesselink M. K., Lo S. K., Schrauwen P. (2005) Regulation of metabolic transcriptional co-activators and transcription factors with acute exercise. FASEB J. 19, 986–988 [DOI] [PubMed] [Google Scholar]
- 11. Wu H., Kanatous S. B., Thurmond F. A., Gallardo T., Isotani E., Bassel-Duby R., Williams R. S. (2002) Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296, 349–352 [DOI] [PubMed] [Google Scholar]
- 12. Yoon J. C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C. R., Granner D. K., Newgard C. B., Spiegelman B. M. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 [DOI] [PubMed] [Google Scholar]
- 13. Nohl H., Hegner D. (1978) Do mitochondria produce oxygen radicals in vivo? Eur. J. Biochem. 82, 563–567 [DOI] [PubMed] [Google Scholar]
- 14. St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D. K., Bachoo R., Spiegelman B. M. (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 [DOI] [PubMed] [Google Scholar]
- 15. Valle I., Alvarez-Barrientos A., Arza E., Lamas S., Monsalve M. (2005) PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 66, 562–573 [DOI] [PubMed] [Google Scholar]
- 16. Lu Z., Xu X., Hu X., Fassett J., Zhu G., Tao Y., Li J., Huang Y., Zhang P., Zhao B., Chen Y. (2010) PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 13, 1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geng T., Li P., Yin X., Yan Z. (2011) PGC-1α promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am. J. Pathol. 178, 1738–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P. H., Rybkin I. I., Shelton J. M., Manieri M., Cinti S., Schoen F. J., Bassel-Duby R., Rosenzweig A., Ingwall J. S., Spiegelman B. M. (2005) Transcriptional coactivator PGC-1α controls the energy state and contractile function of cardiac muscle. Cell Metab 1, 259–271 [DOI] [PubMed] [Google Scholar]
- 19. Leone T. C., Lehman J. J., Finck B. N., Schaeffer P. J., Wende A. R., Boudina S., Courtois M., Wozniak D. F., Sambandam N., Bernal-Mizrachi C., Chen Z., Holloszy J. O., Medeiros D. M., Schmidt R. E., Saffitz J. E., Abel E. D., Semenkovich C. F., Kelly D. P. (2005) PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jäger S., Vianna C. R., Reznick R. M., Cui L., Manieri M., Donovan M. X., Wu Z., Cooper M. P., Fan M. C., Rohas L. M., Zavacki A. M., Cinti S., Shulman G. I., Lowell B. B., Krainc D., Spiegelman B. M. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135 [DOI] [PubMed] [Google Scholar]
- 21. Haden D. W., Suliman H. B., Carraway M. S., Welty-Wolf K. E., Ali A. S., Shitara H., Yonekawa H., Piantadosi C. A. (2007) Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 176, 768–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sweeney T. E., Suliman H. B., Hollingsworth J. W., Piantadosi C. A. (2010) Differential regulation of the PGC family of genes in a mouse model of Staphylococcus aureus sepsis. PLoS One 5, e11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clark J. B., Nicklas W. J. (1970) The metabolism of rat brain mitochondria. Preparation and characterization. J. Biol. Chem. 245, 4724–4731 [PubMed] [Google Scholar]
- 24. Suliman H. B., Carraway M. S., Welty-Wolf K. E., Whorton A. R., Piantadosi C. A. (2003) Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J. Biol. Chem. 278, 41510–41518 [DOI] [PubMed] [Google Scholar]
- 25. Suliman H. B., Welty-Wolf K. E., Carraway M. S., Schwartz D. A., Hollingsworth J. W., Piantadosi C. A. (2005) Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J. 19, 1531–1533 [DOI] [PubMed] [Google Scholar]
- 26. Baker M. A., Cerniglia G. J., Zaman A. (1990) Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal. Biochem. 190, 360–365 [DOI] [PubMed] [Google Scholar]
- 27. Clayton C. E., Carraway M. S., Suliman H. B., Thalmann E. D., Thalmann K. N., Schmechel D. E., Piantadosi C. A. (2001) Inhaled carbon monoxide and hyperoxic lung injury in rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L949–L957 [DOI] [PubMed] [Google Scholar]
- 28. Brealey D., Brand M., Hargreaves I., Heales S., Land J., Smolenski R., Davies N. A., Cooper C. E., Singer M. (2002) Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360, 219–223 [DOI] [PubMed] [Google Scholar]
- 29. Suliman H. B., Carraway M. S., Piantadosi C. A. (2003) Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am. J. Respir. Crit. Care Med. 167, 570–579 [DOI] [PubMed] [Google Scholar]
- 30. Guidet B., Aegerter P., Gauzit R., Meshaka P., Dreyfuss D. (2005) Incidence and impact of organ dysfunctions associated with sepsis. Chest 127, 942–951 [DOI] [PubMed] [Google Scholar]
- 31. Kantrow S. P., Taylor D. E., Carraway M. S., Piantadosi C. A. (1997) Oxidative metabolism in rat hepatocytes and mitochondria during sepsis. Arch. Biochem. Biophys. 345, 278–288 [DOI] [PubMed] [Google Scholar]
- 32. Carré J. E., Orban J. C., Re L., Felsmann K., Iffert W., Bauer M., Suliman H. B., Piantadosi C. A., Mayhew T. M., Breen P., Stotz M., Singer M. (2010) Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am. J. Respir. Crit. Care Med. 182, 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacGarvey N. C., Suliman H. B., Bartz R. R., Fu P., Withers C. M., Welty-Wolf K. E., Piantadosi C. A. (2012) Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 185, 851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piantadosi C. A., Withers C. M., Bartz R. R., MacGarvey N. C., Fu P., Sweeney T. E., Welty-Wolf K. E., Suliman H. B. (2011) Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 286, 16374–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., Spiegelman B. M. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 [DOI] [PubMed] [Google Scholar]
- 36. Schulze-Osthoff K., Bakker A. C., Vanhaesebroeck B., Beyaert R., Jacob W. A., Fiers W. (1992) Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions: evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 267, 5317–5323 [PubMed] [Google Scholar]
- 37. Taylor D. E., Ghio A. J., Piantadosi C. A. (1995) Reactive oxygen species produced by liver mitochondria of rats in sepsis. Arch. Biochem. Biophys. 316, 70–76 [DOI] [PubMed] [Google Scholar]
- 38. Suliman H. B., Welty-Wolf K. E., Carraway M., Tatro L., Piantadosi C. A. (2004) Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 64, 279–288 [DOI] [PubMed] [Google Scholar]
- 39. Holley A. K., Bakthavatchalu V., Velez-Roman J. M., St Clair D. K. (2011) Manganese superoxide dismutase: guardian of the powerhouse. Int. J. Mol. Sci. 12, 7114–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Das K. C., Lewis-Molock Y., White C. W. (1997) Elevation of manganese superoxide dismutase gene expression by thioredoxin. Am. J. Respir. Cell Mol. Biol. 17, 713–726 [DOI] [PubMed] [Google Scholar]
- 41. Lowes D. A., Galley H. F. (2011) Mitochondrial protection by the thioredoxin-2 and glutathione systems in an in vitro endothelial model of sepsis. Biochem. J. 436, 123–132 [DOI] [PubMed] [Google Scholar]
- 42. Zhang K., Lu J., Mori T., Smith-Powell L., Synold T. W., Chen S., Wen W. (2011) Baicalin increases VEGF expression and angiogenesis by activating the ERRα/PGC-1α pathway. Cardiovasc. Res. 89, 426–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scarpulla R. C. (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kensler T. W., Wakabayashi N., Biswal S. (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116 [DOI] [PubMed] [Google Scholar]
- 45. Ishida Y., Agata Y., Shibahara K., Honjo T. (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Francisco L. M., Sage P. T., Sharpe A. H. (2010) The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brahmamdam P., Inoue S., Unsinger J., Chang K. C., McDunn J. E., Hotchkiss R. S. (2010) Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J. Leukoc. Biol. 88, 233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang X., Venet F., Wang Y. L., Lepape A., Yuan Z., Chen Y., Swan R., Kherouf H., Monneret G., Chung C. S., Ayala A. (2009) PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. U.S.A. 106, 6303–6308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guignant C., Lepape A., Huang X., Kherouf H., Denis L., Poitevin F., Malcus C., Chéron A., Allaouchiche B., Gueyffier F., Ayala A., Monneret G., Venet F. (2011) Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit. Care 15, R99. [DOI] [PMC free article] [PubMed] [Google Scholar]