

Background: Plasmodium falciparum merozoites invade erythrocytes via interaction of a pathogen protein PfRh4 with a host membrane receptor.
Results: The PfRh4-binding site on the human erythrocyte host receptor was mapped by mutagenesis and structural methodology.
Conclusion: PfRh4 binds at the receptor terminus in a region overlapping a regulatory functional site.
Significance: Understanding the molecular basis of erythrocyte invasion will aid in design of therapeutics.
Keywords: Cell Surface Receptor, Complement System, Convertases, Malaria, NMR, Plasmodium, Protein Structure, Surface Plasmon Resonance (SPR), Reticulocyte-binding Homologue Proteins
Abstract
To survive and replicate within the human host, malaria parasites must invade erythrocytes. Invasion can be mediated by the P. falciparum reticulocyte-binding homologue protein 4 (PfRh4) on the merozoite surface interacting with complement receptor type 1 (CR1, CD35) on the erythrocyte membrane. The PfRh4 attachment site lies within the three N-terminal complement control protein modules (CCPs 1–3) of CR1, which intriguingly also accommodate binding and regulatory sites for the key complement activation-specific proteolytic products, C3b and C4b. One of these regulatory activities is decay-accelerating activity. Although PfRh4 does not impact C3b/C4b binding, it does inhibit this convertase disassociating capability. Here, we have employed ELISA, co-immunoprecipitation, and surface plasmon resonance to demonstrate that CCP 1 contains all the critical residues for PfRh4 interaction. We fine mapped by homologous substitution mutagenesis the PfRh4-binding site on CCP 1 and visualized it with a solution structure of CCPs 1–3 derived by NMR and small angle x-ray scattering. We cross-validated these results by creating an artificial PfRh4-binding site through substitution of putative PfRh4-interacting residues from CCP 1 into their homologous positions within CCP 8; strikingly, this engineered binding site had an ∼30-fold higher affinity for PfRh4 than the native one in CCP 1. These experiments define a candidate site on CR1 by which P. falciparum merozoites gain access to human erythrocytes in a non-sialic acid-dependent pathway of merozoite invasion.
Introduction
Malaria continues to be a cause of significant disease burden worldwide, with over 700,000 deaths annually (1). Most cases of severe malaria are caused by Plasmodium falciparum. Sporozoites released after the bite of a female Anopheles mosquito travel via the bloodstream to the liver where they asexually reproduce into thousands of merozoites. The latter subsequently target and infect erythrocytes, where they asexually reproduce until the cell bursts to begin another merozoite infective cycle. Fatalities are common, especially in children, as a result of anemia or cerebral malaria, both of which occur during the erythrocytic phase of the parasite's life cycle (2). This is also a stage susceptible to vaccine-based prophylaxis. Although substantial efforts have been made to develop a vaccine, results have been largely disappointing (3, 4). A better understanding at the molecular level of the interaction between the P. falciparum merozoite and the host erythrocyte is important for development of potential therapeutics (5).
The process whereby merozoites enter erythrocytes takes only ∼30 s and was visualized nearly four decades ago for Plasmodium knowlesi and more recently for P. falciparum (6, 7), but the mechanisms involved are still not fully understood. In this multistep invasion process, parasite ligands first interact with erythrocyte membrane proteins (8). Subsequently, a tight junction forms between a merozoite and the erythrocyte membrane, which is triggered by and dependent on the initial receptor-parasite ligand interaction (5, 9).
Chief among these parasite ligands are members of the erythrocyte-binding-like antigens (5, 10, 11) and the reticulocyte-binding homologue proteins (PfRh) (5, 12, 13). Several host ligand protein pairs have been identified: glycophorin A:EBA-175 (14, 15), glycophorin B:EBL-1 (16), glycophorin C:EBA-140 (17), Basigin:PfRh5 (18), and complement receptor type 1 (CR1,9 CD35):PfRh4 (19, 20). The CR1:PfRh4 pathway is the major sialic acid-independent alternative to glycophorin-mediated invasion (19, 20). PfRh4 engages the large ectodomain of this single-pass membrane protein (21) to mediate a functional invasion pathway by P. falciparum parasites (20). CR1 is an ∼250-kDa type 1 membrane glycoprotein that is also known as the C3b/C4b receptor or immune adherence receptor (22–27). CR1 is displayed on primate peripheral blood cells including erythrocytes but is not produced by platelets or most T cells. Erythrocyte CR1 binds particles opsonized with C3b and/or C4b and transports them to the liver and spleen for destruction and to initiate an adaptive immune response. Population groups and rare individuals with low (<100) CR1 copy number expression on erythrocytes have been identified, but no individual with a complete deficiency has been reported (28, 29). CR1 also regulates the complement cascade by multiple mechanisms. It accelerates the dissociation or decay of C3 and C5 convertases that assemble after triggering of the alternative, classical, or lectin pathways of complement activation. The convertases cleave C3 and C5 to yield C3a and C5a (potent proinflammatory anaphylatoxins), C3b (an opsonin and initiator of a positive feedback amplification loop), and C5b (triggers formation of the membrane attack complex). Furthermore, CR1 serves as a cofactor for the factor I-mediated limited cleavage of C4b and C3b molecules that have become covalently attached to a target, producing smaller fragments that are no longer able to participate in complement activation cascades. Subsequently, the covalently attached C3 membrane fragments resulting from cofactor activity, iC3b and C3dg, can serve as ligands for other complement receptors including CR2, CR3, and CR4.
The CR1 gene lies within the regulators of complement activation cluster at 1q32 (30–33). Like other regulators of complement activation family members (34), it is predominantly composed of multiple compact β-sheet rich domains named complement control protein modules (CCPs) (also called sushi domains or short consensus repeats) (35). Thirty such modules comprise the entire ectodomain of the most common size (∼250 kDa) variant of CR1 (see Fig. 1). The CCPs, each consisting of 59–72 residues, mediate the interactions between CR1 and its ligands. The N-terminal 28 CCPs fall into four groups of seven sequential CCPs, called long homologous repeats (LHRs A–D), based on a high degree of internal sequence similarity (36, 37). These modules are linked like beads on a string by between four and eight amino acid residues. Within LHRs A, B, and C, the first three CCPs are required to mediate interactions with C3b or C4b and are also required for convertase decay accelerating and factor I cofactor activity (36–44). We previously observed that CCPs 1–3 (called functional site 1) are necessary and sufficient for binding to PfRh4 (21). Although binding of PfRh4 did not block C3b or C4b binding, it severely curtailed the ability of CCPs 1–3 to accelerate the decay of the AP and CP C3 convertases (21).
FIGURE 1.
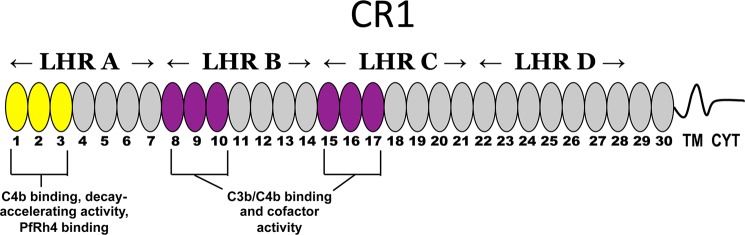
Schematic representation of CR1. The most common allelic form of CR1 contains 30 extracellular CCPs followed by a transmembrane domain (TM) of 28 amino acid residues and a cytoplasmic tail (CYT) of 43 residues. Based on homology, the first 28 CCPs are further grouped into LHRs A, B, C, and D. LHR A (CCPs 1–7) contains a C4b-binding site and possesses decay-accelerating activity (site 1, CCPs 1–3 in yellow), whereas LHRs B (CCPs 8–14) and C (CCPs 15–21) each contain site 2 (duplicated functional units CCPs 8–10 and 15–17 in purple) that are 99% identical, bind C3b and C4b, and have factor I cofactor activity. The binding site for PfRh4 was previously mapped to CCPs 1–3 in LHR A (yellow ovals) (21). LHR D+ refers to LHR D plus CCPs 29 and 30.
Employing the strategy of homologous substitution mutagenesis, we previously identified amino acids in CCPs 1–3 and CCPs 8–10 (duplicated in CCPs 15–17) necessary for C4b and C3b binding and for the complement regulatory activities (38–44). Further, we could convert a predominantly C4b-binding site to a C3b-binding site (42) and modulate primate forms of CR1 relative to their binding specificities and regulatory activities for these two opsonins (41).
In the current studies, we have mapped by mutagenesis the PfRh4 binding site to the most N-terminal and membrane-distal CCP of the CR1 ectodomain. We further identified key PfRh4-binding residues of CCP 1 using mutagenesis and placed them in a structural context by solving the three-dimensional structures of CCPs 1–3. These findings were further verified by the design and creation of a soluble engineered version of a CR1 fragment with a 30-fold enhanced affinity for PfRh4 that is a potential inhibitor of invasion. These studies will assist in the development of new therapeutic agents as well as strategies to prevent malaria.
EXPERIMENTAL PROCEDURES
Recombinant Proteins for Functional Studies: Synthesis, Purification, and Quantification
The full-length ectodomain of CR1 (sCR1), produced in CHO cells, was a gift from Henry Marsh (Celldex Therapeutics, Needham, MA). LHR A, B, C, D, and D+ were prepared as previously described, employing transfected human embryonic kidney 293T (HEK 293T) cells (see Fig. 1) (39–43). LHR B and LHR C share >98% sequence identity and were therefore utilized interchangeably (36–42). In the same transfection system, we also prepared each individual LHR protein with a His6 tag on the C terminus to facilitate purification using nickel-nitrilotriacetic acid-agarose beads (Qiagen) following the manufacturer's instructions. These purified His-tagged samples were dialyzed against PBS and concentrated. Additional mutants of LHR A, in which we deleted either CCP 1, CCP 2, or CCP 3, were prepared as previously described (39, 42).
Mutations in which one or several amino acids were interchanged on a LHR A or LHR B template were prepared with either the QuikChange II or QuikChange lightning multi site-directed mutagenesis kit (Agilent Technologies). HEK 293T kidney epithelial cells were routinely transfected using LipoD293 (SignaGen) in serum-free media as per the manufacturer's directions. Supernatants were collected after 48 h.
Recombinant CR1 proteins were quantified by sandwich ELISA. In this procedure, 96-well flat-bottomed plates (Nunc) were coated overnight at 4 °C with 100 μl of rabbit polyclonal anti-CR1 antibody (45) diluted to 4.2 μg/ml in PBS. The plates were blocked for 2 h at 37 °C with 1% (w/v) BSA and 0.1% (v/v) Tween 20 in PBS. CR1 samples and a calibrated standard (CCPs 15–25) (46) were added to the wells and incubated therein for 1 h at 37 °C. Then the plates were washed with PBS containing 0.05% (v/v) Tween 20 (PBS-T). Next, the detection antibody diluted in PBS-T was added and incubated for 1 h at 37 °C followed by washing with PBS-T. The murine detection antibodies to CR1 were: 7G9, provided by R. Taylor from University of Virginia School of Medicine; 3D9, gifted by E. Brown at Genentech Inc., now a member of the Roche group; J3B11, provided by J. Cohen from Hôpital Robert Debré; or E11, purchased from Abcam. The HRP-conjugated anti-mouse IgG (1:5,000 dilution; GE Healthcare) diluted in PBS-T was added and incubated for 1 h at 37 °C followed by washing with PBS-T. ortho-Phenylenediamine (100 μl) was used to detect HRP activity. After 30 min, the reaction was stopped with 100 μl of 2 n sulfuric acid, and optical density was measured at 490 nm. Quantification of proteins was also performed using immobilized J3B11 or the rabbit polyclonal Ab to CR1 in surface plasmon resonance and in Western blotting. The production and purification of recombinant PfRh4 (Rh4.9), an 88-kDa protein comprised of amino acid residues 28–766, which behaved similarly to full-length PfRh4 (205 kDa) in its ability to bind to erythrocytes, were performed as previously described (20, 47).
ELISA to Detect PfRh4 Interaction with CR1 Proteins
Aliquots of PfRh4 (100 μl of 5 μg/ml in PBS) were coated on 96-well flat-bottomed plates (Nunc). After an overnight incubation at 4 °C, the plates were blocked as described above. Serum-free supernatants from transfected cells or purified recombinant His-tagged CR1 proteins (100 μl each) diluted in PBS-T containing 1% BSA and 1% Nonidet P-40 at a range of concentrations (between 1 and 20 pm) were added to the wells and incubated for 1 h at room temperature, and then the plates were washed with PBS-T. Polyclonal anti-human CR1 Ab (100 μl of 210 ng/ml in PBS) was added and incubated for 1 h at room temperature followed by washing with PBS-T. Next, HRP-conjugated donkey anti-rabbit IgG (100 μl of a 1:5,000 dilution) (GE Healthcare) was added and incubated for 1 h at room temperature followed by washing. Detection was with ortho-phenylenediamine as above. The means ± S.E. are shown for all samples, which were tested in duplicate in at least three independent experiments.
Immunoprecipitation and Western Blotting
Serum-free supernatants containing 1 μg (in 200–500 μl) of each CR1 construct were precleared, using 20 μl of a 50% (w/v) slurry of protein A/G-agarose (Thermo Scientific) and 0.5 μg of mouse IgG in PBS-T for 1 h at 4 °C. PfRh4 (2 μg) was added to the precleared supernatants of each CR1 sample and incubated overnight at 4 °C. In separate tubes either 4 μg of an anti-CR1 monoclonal Ab, J3B11, or 4 μg of an anti-Rh4 monoclonal Ab, 10C9, were added to 20 μl of protein A/G-agarose and preincubated for 1 h at room temperature. The CR1:PfRh4 mixture from above was then added to the antibody-bound beads and incubated for an additional 2 h at room temperature. Beads were washed five times with PBS-T, and proteins were eluted by incubating with SDS nonreducing sample buffer for 5 min at 95 °C. Eluants were analyzed using 10% SDS-PAGE (Invitrogen) and transferred to a nitrocellulose membrane. Western blotting for CR1 proteins was performed as follows: membrane was blocked overnight at 4 °C with 5% (w/v) nonfat dry milk in PBS-T. The blot was probed with rabbit anti-CR1 IgG (210 ng/ml diluted in blocking buffer) for 1 h at room temperature followed by washing in PBS-T. A 1:5,000 dilution of HRP-conjugated donkey anti-rabbit IgG (GE Healthcare) was added and incubated for 1 h at room temperature followed by washing in PBS-T. The signals were visualized by utilizing enhanced chemiluminescent substrate (Supersignal West Pico, Thermo Scientific). Western blotting for PfRh4 was performed similarly using rabbit anti-Rh4 polyclonal antibody (5 μg/ml in PBS-T).
Surface Plasmon Resonance
Experiments were performed at 25 °C on a Biacore 2000 instrument with Biacore CM5 sensor chips (GE Healthcare). Biosensor flow paths were activated with a fresh 1:1 mixture of 0.05 m N-hydroxysuccinimide and 0.2 m ethyl-dimethylaminopropyl carbodiimide injected at 5 μl/min for 7 min. Proteins were attached to the activated surface in 10 mm citrate buffer, pH 4.8. The flow path was then “blocked” by treatment with 1 m ethanolamine, pH 8.5, at 5 μl/min for 10 min. Analytes were introduced at 20 μl/min in HEPES running buffer (10 mm HEPES, 3 mm EDTA, and 0.005% Tween 20 at 150 mm NaCl) except as noted. Analyses of interactions with immobilized recombinant CR1 samples employed a reference flow path produced as above but without protein in the citrate buffer. Analyses of interactions with immobilized PfRh4 employed a reference flow path bearing immobilized mouse IgG (Millipore). Data were analyzed using BIAevaluation software version 4.1 (GE Healthcare). Dissociation constants were calculated from Req values derived from reference-subtracted curves and were fitted to a Langmuir 1:1 interaction model. Nonlinear regression analyses and KD calculations were made using GraphPad Prism software version 5.0.
NMR/SAXS-based Structural Determination of CR1 1,2, CR1 2,3, and CR1 1–3
A previously reported strategy (24, 46, 48–50) was deployed in which solution structures of overlapping CCP pairs (CR1 1,2 and CR1 2,3) were solved by NMR and then concatenated with the help of SAXS data (on CR1 1–3) to generate a structure of the C3b/C4b-binding and PfRh4-binding triple module N-terminal region of CR1 (amino acids 1–192).
Double-labeled (13C,15N) samples of CR1 1,2 (0.56 mm) and CR1 2,3 (0.80 mm) (produced from Pichia pastoris, essentially as described previously (24)) were prepared in 20 mm sodium acetate (deuterated) buffer at pH 4.0 (CR1 1,2) or 10 mm sodium phosphate buffer at pH 6.0 (CR1 2,3) with 10% (v/v) D2O. Data were collected at 25 °C (CR1 1,2) or 37 °C (CR1 2,3) on either Avance 600 or Avance 800 MHz NMR spectrometers (Bruker) (the lower temperature and pH used in the case of CR1 1,2 were necessary to avoid proteolytic degradation). The standard suite of two- and three-dimensional NMR spectra were collected and processed using the software package AZARA. The assignment of CR1 2,3 has already been described (51). The assignment of CR1 1,2 was accomplished using similar techniques. The assignment completeness of CR1 1,2 and CR1 2,3 is reported in Table 1. All Pro residues were found in the trans form.
TABLE 1.
Summary of statistics gathered for ensembles of CR1 1,2, CR1 2,3, and CR1 1–3
The root mean square deviation values were calculated using segments between the first and fourth consensus Cys residues in a given module. The NOE categories were: sequential, residue i − residue i + 1; medium range, residue i − residue (i + (2–4)); long range, residue i − residue (i + (>4)). Tilt, twist, and skew angles were calculated using a described method (48). PROCHECK-NMR (66) software was used to derive the Ramachandran statistics. The areas of buried surfaces were calculated using the “get area” function in PyMOL (67).
| CR1 1,2 | CR1 2,3 | CR1 1–3a | |||
|---|---|---|---|---|---|
| Completeness of assignment | |||||
| 15N (excluding Pro) | 100% | 94% | |||
| 1H(N) | 100% | 94% | |||
| 13C( = O) | 95% | 94% | |||
| 13C(α) | 98% | 95% | |||
| 13C(β) | 99% | 95% | |||
| 1H(α) | 98% | 95% | |||
| 1H(β) | 88% | 95% | |||
| NOE statistics | |||||
| Intraresidue/sequential | 507/724 | 468/620 | 708/1,012b | ||
| Medium/long range | 348/999 | 203/950 | 427/1,458b | ||
| Total | 2578 | 2241 | 3,605b | ||
| Intermodular | 3 | 10 | |||
| CCP 1 to linker | 44 | ||||
| CCP 2 to linker | 85 | 89 | |||
| CCP 3 to linker | 51 | ||||
| RMSD values (Å) (superposition using heavy backbone atoms; mean ± S.D.) | |||||
| CCP 1 | 0.72 ± 0.12 | 0.73 ± 0.13 | |||
| CCP 2 | 0.75 ± 0.14 | 0.75 ± 0.14 | 0.88 ± 0.19 | ||
| CCP 3 | 1.21 ± 0.28 | 0.93 ± 0.15 | |||
| CCP 1 + CCP 2 | 1.05 ± 0.17 | 1.04 ± 0.18 | |||
| CCP 2 + CCP 3 | 1.31 ± 0.25 | 1.10 ± 0.21 | |||
| CCP 1 + CCP 2 + CCP 3 | 1.15 ± 0.19 | ||||
| CCP 1-CCP 2 | CCP 2-CCP 3 | ||||
| Intermodular angles (°) and buried surface area (Å2; mean ± S.D.) | |||||
| Tilt | 50 ± 4 | 64 ± 3 | 50 ± 2 | 60 ± 3 | |
| Skew | 92 ± 9 | −59 ± 9 | 90 ± 7 | −44 ± 8 | |
| Twist | 107 ± 4 | 85 ± 3 | 106 ± 3 | 76 ± 2 | |
| Buried surface area | 163 ± 51 | 515 ± 72 | 151 ± 48 | 469 ± 52 | |
| Ramachandran assessment (%) | |||||
| Most favored | 81.1 | 81.4 | 79.1 | ||
| Additionally allowed | 14.7 | 13.0 | 14.4 | ||
| Generously allowed | 2.0 | 3.0 | 4.5 | ||
| Disallowed | 2.2 | 2.6 | 2.0 | ||
a Calculated by concatenating NMR-derived data for CR1 1,2 and CR1 2,3 along with SAXS-derived data for CR1 1–3; the overall χ values calculated for the fittings across the 20 refined structures of CR1 1–3 were 1.45 ± 0.04.
b Derived from the NOE lists for CR1 1,2 and CR1 2,3 as described in text (i.e., not from NMR spectra collected on CR1 1–3).
To generate distance restraints, three-dimensional 15N-edited NOESY and three-dimensional 13C-edited NOESY spectra were collected and processed using AZARA in the CcpNmr suite of programs (52). The NOEs and assignments of CR1 1,2 and CR1 2,3 were transferred from CcpNmr Analysis to CYANA v. 2.1 (53) using the Format Converter software. Preliminary structural calculations were carried out in CYANA for both structures, until the published convergence. The final NOEs were converted to the Xplor-NIH format using the Format Converter.
Further structural calculations of CR1 1,2 and CR1 2,3 were carried out in Xplor-NIH version 2.30 (54). In the first stage of a simulated annealing protocol, the temperature was lowered from an initial 2,000 K to 600 K in 50-K intervals, with 300 steps of molecular dynamics at each temperature increment; within this temperature interval, the energy terms (including the NOE term and Ramachandran term) were progressively introduced by multiplicative ramping. In the second stage, the structures were cooled from 600 K to 100 K, in increments of 25 K, with 300 steps of molecular dynamics at each temperature; potential terms (including the NOE term) were statically set to the top values used in the first stage of cooling. One hundred structures were calculated for each molecule.
A concatenated CR1 1–3 NOE list was created by merging the CR1 1,2 and CR1 2,3 NOE lists. Only the NOEs from the CR1 1–2 list that involve residues in (and between) CCP 1, the CCP 1,2 linker, and the N-terminal half of the three-dimensional structure of CCP 2 were used; the only NOEs extracted for use from the CR1 2,3 list were those in (and between) the C-terminal half of the three-dimensional structure of CCP 2, the CCP 2,3 linker, and CCP 3. This procedure, similar to that described previously (24, 49), was designed to remove potential duplications and conflict involving the common CCP 2.
Structure calculations, incorporating the SAXS data for CR1 1–3 and the concatenated NOE list, were carried out in Xplor-NIH version 2.30. A naive model of CR1 1–3, prepared using MODELLER 9v10 (55) by overlaying closest to mean structures of CR1 1,2 and CR1 2,3 on the common CCP 2, was used as a starting structure. An identical protocol as for CR1 1,2 and CR1 2,3 was followed to calculate a refined structure of CR1 1–3, with the exception that at all times an additional SAXS-derived potential was used. In this calculation, 100 data points evenly distributed in a cubic spline constructed using the SAXS data were fitted against data back-calculated for the resulting structures using 500 solid angles.
Twenty structures (chosen based on the lowest overall energy and structural convergence) of CR1 1,2, CR1 2,3, and CR1 1–3 were submitted for water refinements. We included all experimental and statistical potentials employed at the previous stage of structural calculations, using a standard Xplor-NIH protocol. The resulting structures are summarized in Figs. 10 and 11 and in Table 1.
FIGURE 10.

Summary of NMR/SAXS-derived structures. Ensembles (for the 20 lowest energy structures) are shown as overlaid backbone traces; the root mean square deviation (heavy backbone atoms) value is shown for each overlay. Closest to mean structures are drawn as cartoons (PyMOL) based on β-strands determined by STRIDE (65), and cysteine sulfur atoms are drawn as spheres. A, ensemble for CR1 1–2. Upper left, overlay on module 1; upper right, overlay on module 2; lower left, overlay on both modules; lower right, closest to mean structure. B, ensemble for CR1 2–3. Upper left, overlay on module 2; upper right, overlay on module 3; lower left, overlay on both modules; lower right, closest to mean structure. C, ensemble for CR1 1–3. Top, from left to right, overlays on modules 1, 2, and 3; bottom left, overlay on all modules; bottom right, closest to mean structure.
FIGURE 11.

Identification of the PfRh4-binding site in CCP 1 of CR1. In the top left are two views of the surface of the (lowest energy) NMR-derived solution structure of CR1 1–3. Residues of CCP 1 that are conserved in CCP 8 are in white; nonconserved residues that may be substituted by equivalent CCP 8 residues without any measurable effect on PfRh4 binding are colored pink; residues substituted in the m6–9 mutant of LHR A (cyan) and in the m18,19 and m20,21 mutants (yellow) are also highlighted. Gly35 and other residues previously identified as important for C4b binding by CR1 1–3 are colored orange. Residues 6–9 and 18–21 are also shown by stick representation in the expansion (right), with carbons colored cyan and yellow (respectively), oxygens in red, and nitrogens in blue. Asp18 and Phe20 (blue labels) are essential for PfRh4 binding; substitution of Glu6 (red label) by Asp increases binding 3-fold. In the bottom right are shown (as sticks) the residues of CCP 8 (modeled on structure of CCP 15 that is nearly identical) equivalent to CCP 1 residues 6–9 and 18–21. Replacement of these (in LHR B) by the equivalent CCP 1 residues creates a novel, strong PfRh4-binding site in LHR B. These studies led to the proposal of the binding site shown in the bottom left. The surface here is cut away to reveal atom representations of Trp7 as well as Phe20 and the surrounding negatively charged side chains of Glu6, Asp18, Glu19, and Glu21 (colored as in the stick representation).
RESULTS
CCP 1 Is Required for PfRh4 Binding to CR1
We had previously mapped a PfRh4-binding site to CCPs 1–3 (functional Site 1) at the N terminus of CR1 (Fig. 1) and demonstrated that PfRh4 binding suppresses decay-accelerating activity of Site 1, but not binding to C4b (20, 21). In the current study, we sought to confirm that no other regions of CR1 are involved in binding to PfRh4, and we asked whether all three of the N-terminal CCPs are required for PfRh4 binding. To address these issues, we expressed a variety of CR1 truncation constructs in 293T cells and assessed the ability of the resultant proteins to bind PfRh4 in an ELISA-based assay. We first prepared LHRs A, B, C, and D+ (CR1 1–7, CR1 8–14, CR1 15–21, and CR1 22–30, respectively) and then assessed their binding to PfRh4 (Fig. 2, A and B). As expected (21), only LHR A bound to PfRh4 in this assay. We then deleted CCP 1, 2, or 3 from LHR A (LHR A Δ1, LHR A Δ2, and LHR A Δ3) and observed that the protein lacking CCP 1 did not bind detectably to PfRh4 (Fig. 2B), whereas proteins lacking CCP 2 or 3 bound normally. Thus, PfRh4 requires CCP 1 for binding to CR1; this is probably the most membrane-distal of the 30 CCPs in the extracellular domain of CR1.
FIGURE 2.
CCP 1 in LHR A is essential for PfRh4 binding. Microtiter plates were coated with PfRh4 (0.5 μg/well). The interaction between CR1 protein constructs in the supernatants of transfected HEK 293T cells and PfRh4 was detected by a rabbit anti-CR1 polyclonal Ab. A, only LHR A shows binding. B, in LHR A, deletion of CCP 1 abrogates PfRh4 binding, whereas deletion of CCP 2 or 3 has no effect. Displayed values are the means ± S.E. of at least three independent experiments in this figure and in Figs. 3 and 4.
Two Amino Acids in CCP 1 Are Essential for PfRh4 Binding
CCP1 has 55% sequence identity with both CCPs 8 and 15, which are 100% identical with one another (36, 37). Because CCP 1 is necessary for binding to PfRh4, whereas CCPs 8 and 15 in LHRs B and C, respectively, do not contain a PfRh4-binding site, we hypothesized that it might be possible to use homologous substitution mutagenesis to map more precisely the PfRh4 interaction. A similar exercise had previously achieved the delineation of C4b-binding and C3b-binding sites (39–44). Using the 27 amino acid residues that differ between the sequences of CCP 1 and CCP 8 (Fig. 3A) as an initial template for mutations, we substituted individual or clusters of amino acid residues from CCP 8 (Fig. 3B) into their homologous position(s) in CCP 1. These mutations (m) were named after their positions in CCP 1. For instance, the double mutant Q1H,N3Q was named m1,3.
FIGURE 3.

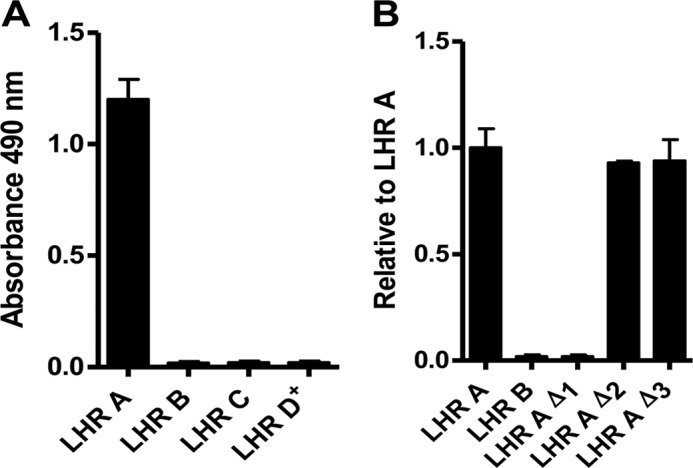
Identification of amino acid residues in CCP 1 important for PfRh4 binding. A, the 27 amino acid residues that differ between CCP 1 and CCP 8 are marked with braces. Because LHR B does not bind to PfRh4, amino acid residues in CCP 1 were substituted with their equivalents in CCP 8. Numbers following the “m” (for mutant) denote the position of substituted amino acids; i.e., m1,3 has both the Q1H and N3Q mutations in LHR A. B, m18–19 and m20–21 show negligible binding to PfRh4, whereas m6–9 and m35 display a >50% reduction in binding. All others are not significantly different from LHR A. C, amino acid residues Asp18 and Phe20 are essential for PfRh4 binding. Single amino acid substitutions were prepared in the LHR A template, and the proteins were tested for alterations in binding to PfRh4. Experimental design as in Fig. 2 including the use of supernatants of transfected HEK 293T cells containing the CR1 proteins.
The mutants were assessed for their ability to bind to PfRh4. For these experiments, LHR A was employed as a positive control, and LHR B was employed as a negative control. Of the 13 LHR A mutants tested (Fig. 3B), m18,19 and m20,21 showed the most striking decrease in binding. The mutants m6–9 and m35 also exhibited a significant (>60%) loss of affinity for PfRh4 compared with native LHR A. To further identify the exact residues that are required for binding, we mutated each individual amino acid residue in m6–9, m18,19, and m20,21 (Fig. 3C). We found that m18 and m20 abrogated PfRh4 binding, whereas m19 and m21 showed substantially reduced binding. Within the other region investigated, i.e., m6–9, the results of single-residue replacements were not as clear; in this case, m7, m8, and m9 individually had a more modest effect on binding compared with the large effect of the simultaneous replacement of all four residues. On the other hand, a mutant of LHR A, m6, carrying the single amino acid substitution E6D from LHR B bound to PfRh4 severalfold better than the native protein.
Conversion of LHR B to a PfRh4 Binding Site
By using reverse homologous substitution mutagenesis (i.e., substituting PfRh4-interacting amino acids residues from CCP 1 into CCP 8), we sought to create a PfRh4-binding site in LHR B (Fig. 4). The resultant LHR B mutants were named m18,20r or m18–21r, etc., to reflect the sequence numbers of the CCP 1 residues that replaced residues in homologous positions within LHR B. The double substitution of Asp18 and Phe20 into their appropriate positions in LHR B (replacing Asn468 and Ser470, respectively, in the context of m18,20r or m18–21r) was not sufficient to confer a PfRh4 binding capability on LHR B. Likewise, substitution of four residues to create m7–9,35r did not alone result in any measurable affinity for the parasite protein. However, if the six LHR A (CCP 1) amino acids (namely 7–9 and 18–20) were simultaneously substituted into LHR B, the resultant mutant (m7–9,18–20r) bound PfRh4 16-fold better than LHR A (Fig. 4). The additional substitution of a Gly with Glu at position 35 (as in m7–9,18–20,35r) reduced PfRh4 binding, but it was still severalfold better than LHR A. Taken together, these data provide strong evidence for the presence of a binding site on CCP 1, in functional Site 1 of CR1, primarily comprised of residues 7–9 and 18–20.
FIGURE 4.
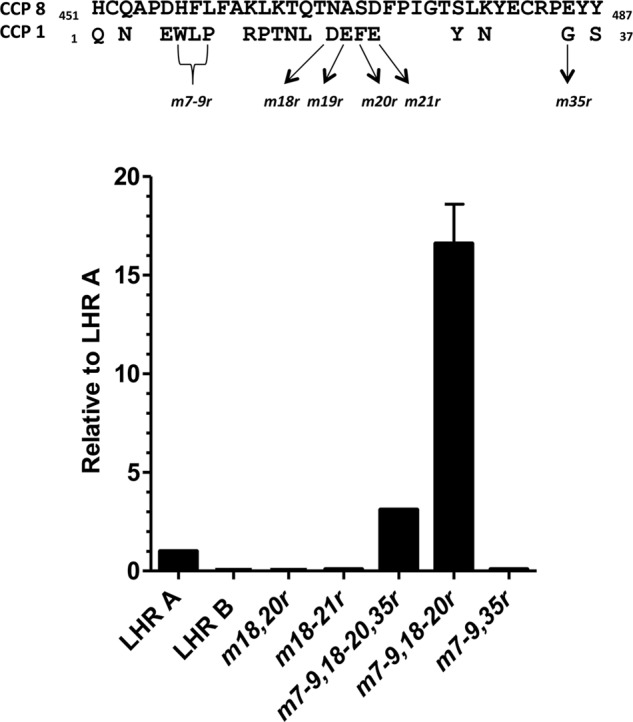
A PfRh4 binding capability can be conferred upon LHR B. The two amino acid residues in CCP 1 whose replacement had been shown to abrogate binding were first substituted into LHR B at their homologous positions (m18,20r) but were not sufficient to confer PfRh4 binding. Other mutations that decreased binding of LHR A were also substituted into m18,20r. m7–9,18–20,35r bound 3-fold better than LHR A. m7–9,18–20r bound ∼16-fold better than LHR A. On the other hand, m7–9,35r did not bind. The experimental design for this figure was similar to that in Figs. 2 and 3, except that the CR1 proteins were His-tagged and purified from the supernatants.
Immunoprecipitation Experiments Confirm Mutational Analyses
The recombinant proteins LHR A, LHR B, and LHR AΔ1 (i.e., CCPs 2–7) and the mutant m7–9,18–20r were individually incubated with PfRh4 and then immunoprecipitated using either a monoclonal Ab to CR1 (J3B11) or a monoclonal Ab to PfRh4 (10C9) (Fig. 5). Western blots of the eluants were evaluated for CR1 using a polyclonal anti-CR1 and for PfRh4 using a polyclonal anti-PfRh4 antibody. We observed that LHR A and m7–9,18–20r co-immunoprecipitated with PfRh4, whereas LHR B and LHR AΔ1 did not (Fig. 5A). The results also showed that, as expected, m7–9,18–20r was more efficiently pulled down with PfRh4 compared with LHR A. In our reciprocal immunoprecipitation experiments using anti-CR1 mAb J3B11, we observed that PfRh4 interacted with LHR A and m7–9,18–20r, but not with LHR B or LHR AΔ1, consistent with the results above (Fig. 5B). Furthermore, more PfRh4 co-precipitated with m7–9,18–20r than with LHR A itself. These results are in agreement with the ELISA data.
FIGURE 5.
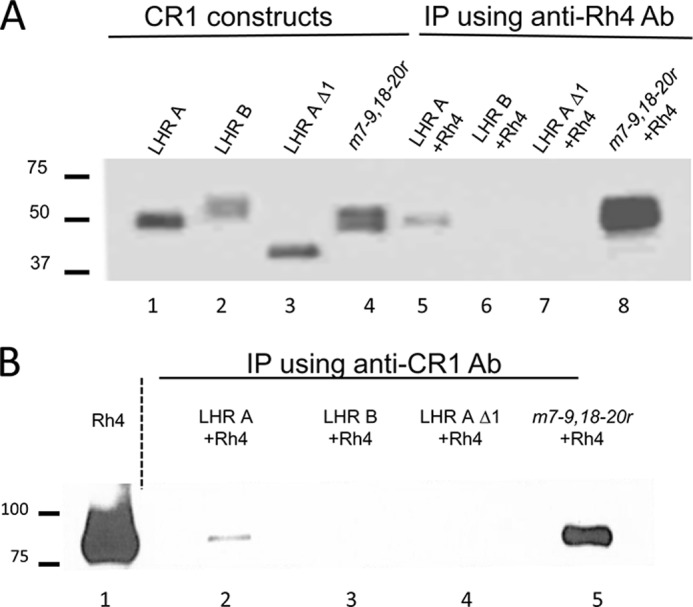
LHR A and m7–9,18–20r co-immunoprecipitate with PfRh4. Co-IP experiments were performed by mixing a CR1 variant in the supernatant of HEK 293T transfectants with PfRh4 and then incubating with protein A/G-agarose beads bearing either anti-CR1 monoclonal Ab J3B11 or anti-PfRh4 monoclonal Ab 10C9. A, Western blot analysis of eluants from IP with 10C9 using polyclonal anti-CR1 Ab. The first four lanes (controls) show LHR A, LHR B, LHR A Δ1, and m7–9,18–20r, whereas the last four lanes contain the eluants of IP with 10C9. LHR A (lane 5) and m7–9,18–20r (lane 8) co-IP with PfRh4, whereas LHR B (lane 6) and LHR A Δ1 (lane 7) do not. B, Western blot analysis of eluants from IP blotted with J3B11 shows that PfRh4 co-IP with LHR A (lane 2) and m7–9,18–20r (lane 5) but not with LHR B (lane 3) or LHR A Δ1 (lane 4).
Surface Plasmon Resonance Characterization of the PfRh4:CR1 Interaction
We first examined the binding of PfRh4 to immobilized LHR A, LHR B, and LHR D (Fig. 6). As anticipated, PfRh4 bound in a dose-dependent fashion to LHR A, whereas there was no detectable binding to LHR B or LHR D. The KD of PfRh4 binding to immobilized LHR A was 490 ± 60 nm (Fig. 6B is a representative example). We next assessed binding of LHR A and LHR B and selected mutants, this time employing immobilized PfRh4 (Fig. 7). The LHR B mutant carrying the six CCP 1 amino acid substitutions, m7–9,18–20r, bound many-fold better than LHR A. On the other hand, LHR A lacking CCP 1 did not bind detectably, and neither did the mutants m18 and m20. These observations are in accord with the ELISA and IP results.
FIGURE 6.
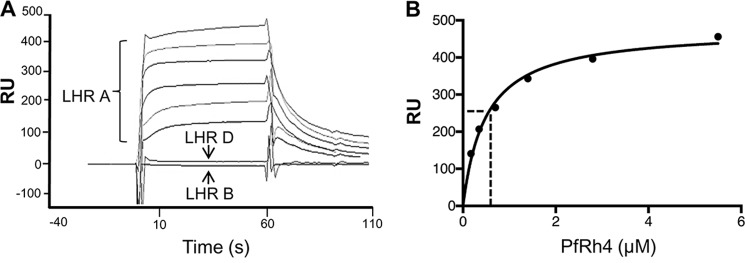
PfRh4 binds to immobilized CR1 LHR A. A, PfRh4 was injected at 5.5, 2.8, 1.4, 0.7, 0.35, and 0.18 μm over parallel flow cells of nickel-purified, surface plasmon resonance chip immobilized LHR A (3,000 RU), LHR B (3,600 RU), and LHR D (3,800 RU). The reference-subtracted sensorgrams demonstrate concentration-dependent binding to LHR A. No binding of PfRh4 to LHR B or LHR D was detected. B, the PfRh4:LHR A binding data conform to a 1:1 Langmuir interaction model. A nonlinear regression of response units versus PfRh4 concentrations demonstrate a KD (indicated by a dotted line) of 490 ± 60 nm.
FIGURE 7.
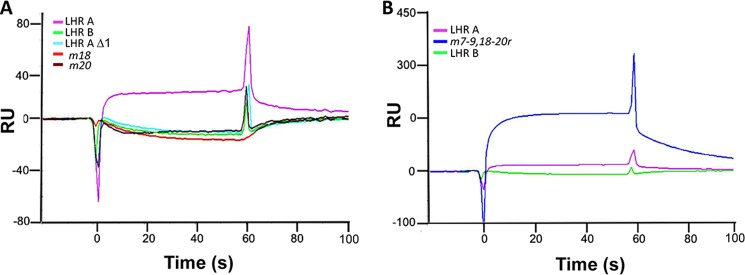
Mutations in CCP 1 of CR1 abrogate PfRh4 binding. A, supernatants containing His-tagged CR1 recombinant constructs were injected at 220 nm over immobilized PfRh4 (3,300 RU). Reference subtracted sensorgrams demonstrate that LHR A Δ1, m18, and m20 behave like LHR B and do not bind to immobilized PfRh4. A parallel flow cell bearing 4,000 RU immobilized mouse IgG was employed as the reference. B, duplicate injections of supernatants containing His-tagged constructs passed over immobilized PfRh4 demonstrate that m7–9,18–20r carrying six single amino acid substitutions from LHR A confers a PfRh4-binding site if placed in homologous positions of LHR B. Consistent with ELISA and IP results, m7–9,18–20 protein has a many-fold greater affinity for PfRh4 than LHR A.
In further experiments, LHR A, sCR1, and m7–9,18–20r were passed over immobilized PfRh4 to obtain quantitative affinities (Fig. 8). Dissociation constants were calculated from Req values derived from the resulting reference-subtracted curves and fitted to a Langmuir 1:1 interaction model. The KD of sCR1 binding to PfRh4 was estimated to be 8 ± 1 μm, whereas that of LHR A binding to PfRh4 was 2 ± 1 μm, which is somewhat weaker than when this experiment was performed in reverse (i.e., when LHR A was immobilized and PfRh4 passed over the chip; KD = 490 nm) (Fig. 6). Strikingly, the estimated KD for m7–9,18–20r passed over immobilized PfRh4 was 61 ± 9 nm. This is an ∼30-fold higher affinity than LHR A and is consistent with the ELISA and IP results.
FIGURE 8.

PfRh4 interactions with sCR1, LHR A, and m7–9,18–20r. CR1 constructs were injected at multiple concentrations over surface plasmon resonance chip immobilized PfRh4 (850 RU). A parallel flow path bearing 630 RU of immobilized mouse IgG was used as a reference. Dissociation constants were calculated from Req values derived from reference-subtracted curves and fitted to a Langmuir 1:1 interaction model. A, sCR1 was injected at 10, 6.7, 3.3, 2.2, 0.7, 0.2, and 0.08 μm. The KD (indicated by a dotted line) was calculated to be 8 ± 1 μm. B, supernatants from transfected 293T containing LHR A were injected at 5, 1.7, 0.6, 0.2, and 0.1 μm. KD = 2 ± 1 μm. C, supernatants from transfected 293T cells containing the m7–9,18–20r protein were injected in duplicate at 250, 128, 64, 32, and 17 nm. KD = 61 ± 9 nm, ∼30-fold greater than that observed for PfRh4:LHR A.
In prior studies of CR1 employing affinity chromatography with C3b or C4b Sepharose beads, we more efficiently isolated CR1 in solubilized cell extracts if a low salt (50 or 75 mm) buffer was used (56). To further investigate this ionic strength dependence, we passed sCR1, LHR A and m7–9,18–20r over immobilized PfRh4 at 75, 150, and 300 mm salt (Fig. 9). Consistent with the previous data, the affinities of all three analytes were many-fold higher at 75 mm NaCl compared with 150 mm NaCl, whereas there was no detectable binding at 300 mm. These data support the hypothesis that the PfRh4 interaction with CR1 also has a major electrostatic component.
FIGURE 9.

CR1 interaction with PfRh4 is ionic strength-dependent. Supernatants containing sCR1, LHR A, and m7–9,18–20r were injected over immobilized PfRh4 in 75, 150, or 300 mm NaCl buffer. A–C, representative reference-subtracted sensorgrams are presented for each protein: sCR1 (A), LHR A (B), and m7–9,18–20r (C).
Visualizing the PfRh4 Binding Site of CR1 1–3 in Three-dimensional Solution Structure
The three-dimensional solution structure of CR1 1–3 was solved using a combination of NMR and SAXS, thereby allowing visualization of the spatial positions of the mutated residues (Figs. 10 and 11). As expected from a previous study of CR1 15–17 (74% sequence identity with CR1 1–3) (24, 33), all three CCPs adopt a beta-sandwich-type fold and have an ovoid appearance. Two disulfide bridges within each CCP form the boundaries of a hydrophobic core containing conserved lipophilic side chains, whereas loops and turns contain conserved glycine and proline residues. In each CCP, the N and C termini lie at opposite ends of the long axis, and the three CCPs form an extended structure with small interfaces (and a few NOEs; Table 1) between modules (Fig. 10). This highly elongated shape is consistent both with intermodular flexibility and with the possibility of CCP 1 being projected well clear of the glycocalyx and accessible for binding by PfRh4.
The three residues whose individual substitution had the largest effects on PfRh4 binding, Glu6 (3.5-fold increase when changed to Asp), Asp18 (no binding when changed to Asn), and Phe20 (no binding when changed to Ser) are all surface-exposed and lie close together on CCP 1 (Fig. 11). Likewise, residues 7–9 and 18–20, which upon incorporation into CCP 8 create a PfRh4-binding site, form a contiguous CCP 1 surface patch. Thus, the mutagenesis experiments identified a feasible and highly accessible PfRh4-binding site lying to the side of CCP 1 and well away from its interface with CCP 2; this includes all or part of Trp7 along with Phe20 and surrounding negative charges.
DISCUSSION
The binding of parasite ligand PfRh4 to CR1 mediates an important mode of entry for malaria parasites into human erythrocytes. By combining homology-guided mutagenesis with quantitative assays of protein-protein interactions and solution-structure determination, we mapped the PfRh4-interacting region on CR1. It lies entirely within the N-terminal CCP (CCP 1), which is potentially furthest from the membrane of the 30 tandem CCPs that comprise the highly extended extracellular domain of the most common size variant of CR1.
The functionally critical nature of a small number of individual residues with surface-exposed side chains on CCP 1 helped to delineate the PfRh4-binding site. The mutation W7H in LHR A (m7) halved the affinity of LHR A for the parasite ligand, whereas L8F and P9L had little effect, despite the drastic loss of activity when all three were mutated together (along with E6D) (i.e., in LHR A m6–9). Neither D18N (m18) nor F20S (m20) versions of LHR A retained any detectable affinity for PfRh4, whereas D6E LHR A (m6) exhibited a greatly enhanced affinity. Because both host and parasite are evolving, we would not expect the CR1:PfRh4 protein-protein interface to have been optimized by natural selection. That replacement of Glu6 by an Asp significantly improves binding is therefore not surprising; rather it reinforces our hypothesis that Glu6, along with Asp18 and Phe20, contributes to binding of PfRh4 and suggests that the carboxyl group of the shorter side chain is better placed to form a salt bridge with an electropositive group on PfRh4. Note that none of these homologous substitutions are likely to have disturbed the structure of CCP 1, given the high structural similarity evident between the relevant regions of CCP 1 (solved in this study) and CCP 8 (modeled on the structure of CCP 15 that is 100% identical in sequence to CCP 8).
Although G35E had decreased affinity for PfRh4, this probably does not reflect its direct contribution to a PfRh4-binding site. Gly occurs at equivalent positions in all of the CCPs of CR1 with the exception of CCPs 7 (and nearly identical CCP 14, CCP 21, and CCP 28) and CCP 8 (and nearly identical CCP 15). The same G35E mutation also disrupts C4b binding and decay-accelerating activity in the construct LHR A (39, 43). Gly35 occupies a loop of CCP 1 that contacts CCP 2, and its replacement by Glu in CCP 8 could help to explain the differences in intermodule angles that are apparent from a comparison of CR1 1–3 and CR1 15–17 structures. In sum, Gly35 is thought more likely to have an architectural role rather than being involved directly in binding.
The substitution of Asp18 and Phe20 (individually critical for PfRh4 binding in CCP 1 as discussed above) and the glutamates at positions 19 and 21 into their homologous positions in CCP 8 of LHR B (creating, m18–21r) was not sufficient to facilitate binding of the mutant LHR B to PfRh4 (Fig. 4). On the other hand, a PfRh4-binding mutant of LHR B was successfully created when three further CCP 1 amino acid residues (Trp7, Leu8, and Pro9) were additionally substituted into CCP 8 at their homologous positions (although in this case Asp471, equivalent to Glu21, was unchanged). Both Leu8 and Phe458 are buried in their respective modules, so this substitution is a conservative one, and we assume this does not affect function. As mentioned, W7H (in m7) reduced binding by only 50%, so it appears that the imidazole side chain of His can substitute partially for the indole ring of Trp provided Pro9 is present; in the context of P9L, however, the His side chain has swung out as may be observed in CCP 8 (Fig. 11), and it can no longer perform this surrogate role. The additional substitution of Glu458 with Gly35 in CCP 8 led to a reduction in binding (i.e., when m7–9,18-20 is compared with m7–9,18–20,35r), supporting the idea that Gly35 is not part of the primary binding site of PfRh4 on CR1. Unsurprisingly, m7–9,35r (lacking substitution by residues 18–20) did not bind to PfRh4.
All of these results were combined with knowledge gained from the newly determined structure of CR1 1–3 (Fig. 11) to infer the location and extent of the PfRh4-binding site. We propose that it includes the surface-exposed Phe20, four surrounding residues with electronegative side chains (Glu6, Asp18, Glu19, and Glu21), and all or part of Trp7. Thus, four of six negatively charged amino acid side chains of CCP 1 lie in the putative PfRh4-binding site, whereas the 88-kDa region of the recombinant PfRh4 protein used in these experiments has a very basic pI of 8.9. We showed that the CR1/PfRh4 interaction is sensitive to high ionic strength, supporting the notion that electrostatic forces including salt bridges are crucial to receptor-ligand engagement. Strikingly, if these putative binding site residues are collectively substituted into their equivalent positions within CCP 8, the product is a version of LHR B that has a high affinity for PfRh4; this convincingly confirms our identification of the PfRh4-binding site in CCP 1 of CR1. The residues thought to form the PfRh4-binding site on CCP 1 do not participate in the junction with CCP 2 and are also distant from other residues that were identified in a previous study as important for binding by CR1 1–3 to C4b (40). This is consistent with our previous demonstration of the formation of a C4b·PfRh4·CR1 1–3 ternary complex. Interestingly, one of the binding site residues, Trp7, was shown (43) to be required for decay-accelerating activity that probably involves contacts by CR1 functional site 1 with both components of the convertases, i.e., Bb as well as C3b in the alternative pathway or C2a as well as C4b in the classical pathway. The participation of this putative residue in the Bb/C2a and PfRh4-binding site could explain why PfRh4 blocks the decay-accelerating activity of CR1 1–3 (21).
The discovery that the LHR B mutant, m7–9,18-20r, binds nearly 30-fold better to PfRh4 than LHR A could potentially lead to an inhibitor of parasite invasion. Experiments to determine whether this construct blocks the PfRh4-CR1 merozoite invasion pathway of erythrocytes are in progress. It would also be of interest to determine whether insertion of these mutations into the CR1 protein enhances the ability of merozoites to invade erythrocytes (i.e., increases parasite virulence). However, it is possible that there is a level of PfRh4 affinity for CR1 that is sufficient for its role in invasion, and increased ability to bind may not increase its function.
The structure of PfRh4 is not known, and there are no reports of its successful crystallization. The formation of a complex of PfRh4 with the high affinity CR1 mutant m7–9,18–20r might facilitate crystal formation and subsequent structure determination. This could provide information that would be of use for the design of a highly efficacious component of a multicomponent vaccine that would include a combination of blood stage antigens to protect against disease and interrupt transmission of P. falciparum (57).
The evolution of parasite-host relationships is greatly intriguing. The malaria parasite uses erythrocyte invasion pathways that involving binding to glycophorins, which are likely to be ancient in origin because there has been significant negative pressure on human populations living in malaria endemic areas. For example, there are mutations in glycophorins B and C that have been associated with alterations in malarial severity or susceptibility (17, 58–62). Such mutations likely favored the use of other pathways in which a merozoite adhesin binds to different erythrocyte receptor, e.g., PfRh4:CR1. Both laboratory and field P. falciparum isolates with differing dependences on CR1 or glycophorins have been described, but notably, some P. falciparum lines can activate PfRh4 expression to switch receptor usage (63, 64). This is an example of phenotypic variation that provides a means for the malaria parasite population to vary its ligand receptor usage in the face of selective pressures from the host. Interestingly, SNPs in the putative PfRh4 binding site in CCP 1 of CR1 have not been reported in the 1,000-genome database, suggesting either that this is a more novel pathway or possibly that this binding site serves a crucial host CR1 function. Our results favor the latter possibility and are consistent with this being the decay-accelerating activity of functional Site 1 and specifically (because these residues are not needed for C3b/C4b binding) an important contact between CCP 1 and the C3 and C5 convertases. Mutations of at least some of these critical CR1 residues might thus be selected against, even in malaria-endemic populations.
In conclusion, our studies have identified a site in CR1 that the P. falciparum merozoite utilizes for erythrocyte invasion. These studies provide new understanding at the molecular level that may assist in development of future therapeutics.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AI041592, P30 AR48335, and T32 AR07279 (to J. P. A.) and R01 AI051436 (to D. H.). This work was also supported by Wellcome Trust Grants 081179 (to P. N. B., C. Q. S., and M. G.) and 085025 (to M. M.), National Health and Medical Research Council Grant 1026581 (to W.-H. T.), and an Australian Research Council Future Fellowship (to W.-H. T.).
The atomic coordinates and structure factors (codes 2MCZ and 2MCY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CR
- complement receptor
- CCP
- complement control protein
- LHR
- long homologous repeat
- SAXS
- small angle x-ray scattering
- IP
- immunoprecipitation
- RU
- response units
- PfRh4
- P. falciparum reticulocyte-binding homologue protein 4.
REFERENCES
- 1.(2010) World malaria report 2010. in WHO Global Malaria Programme, World Health Organization [Google Scholar]
- 2. Olotu A., Fegan G., Wambua J., Nyangweso G., Awuondo K. O., Leach A., Lievens M., Leboulleux D., Njuguna P., Peshu N., Marsh K., Bejon P. (2013) Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. N. Engl. J. Med. 368, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agnandji S. T., Lell B., Fernandes J. F., Abossolo B. P., Methogo B. G., Kabwende A. L., Adegnika A. A., Mordmuller B., Issifou S., Kremsner P. G., Sacarlal J., Aide P., Lanaspa M., Aponte J. J., Machevo S., Acacio S., Bulo H., Sigauque B., Macete E., Alonso P., Abdulla S., Salim N., Minja R., Mpina M., Ahmed S., Ali A. M., Mtoro A. T., Hamad A. S., Mutani P., Tanner M., Tinto H., D'Alessandro U., Sorgho H., Valea I., Bihoun B., Guiraud I., Kaboré B., Sombié O., Guiguemdé R. T., Ouédraogo J. B., Hamel M. J., Kariuki S., Oneko M., Odero C., Otieno K., Awino N., McMorrow M., Muturi-Kioi V., Laserson K. F., Slutsker L., Otieno W., Otieno L., Otsyula N., Gondi S., Otieno A., Owira V., Oguk E., Odongo G., Woods J. B., Ogutu B., Njuguna P., Chilengi R., Akoo P., Kerubo C., Maingi C., Lang T., Olotu A., Bejon P., Marsh K., Mwambingu G., Owusu-Agyei S., Asante K. P., Osei-Kwakye K., Boahen O., Dosoo D., Asante I., Adjei G., Kwara E., Chandramohan D., Greenwood B., Lusingu J., Gesase S., Malabeja A., Abdul O., Mahende C., Liheluka E., Malle L., Lemnge M., Theander T. G., Drakeley C., Ansong D., Agbenyega T., Adjei S., Boateng H. O., Rettig T., Bawa J., Sylverken J., Sambian D., Sarfo A., Agyekum A., Martinson F., Hoffman I., Mvalo T., Kamthunzi P., Nkomo R., Tembo T., Tegha G., Tsidya M., Kilembe J., Chawinga C., Ballou W. R., Cohen J., Guerra Y., Jongert E., Lapierre D., Leach A., Lievens M., Ofori-Anyinam O., Olivier A., Vekemans J., Carter T., Kaslow D., Leboulleux D., Loucq C., Radford A., Savarese B., Schellenberg D., Sillman M., Vansadia P. (2012) A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N. Engl. J. Med. 367, 2284–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schofield L., Grau G. E. (2005) Immunological processes in malaria pathogenesis. Nat. Rev. Immunol. 5, 722–735 [DOI] [PubMed] [Google Scholar]
- 5. Cowman A. F., Crabb B. S. (2006) Invasion of red blood cells by malaria parasites. Cell 124, 755–766 [DOI] [PubMed] [Google Scholar]
- 6. Dvorak J. A., Miller L. H., Whitehouse W. C., Shiroishi T. (1975) Invasion of erythrocytes by malaria merozoites. Science 187, 748–750 [DOI] [PubMed] [Google Scholar]
- 7. Gilson P. R., Crabb B. S. (2009) Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int. J. Parasitol. 39, 91–96 [DOI] [PubMed] [Google Scholar]
- 8. Tham W. H., Healer J., Cowman A. F. (2012) Erythrocyte and reticulocyte binding-like proteins of Plasmodium falciparum. Trends Parasitol. 28, 23–30 [DOI] [PubMed] [Google Scholar]
- 9. Riglar D. T., Richard D., Wilson D. W., Boyle M. J., Dekiwadia C., Turnbull L., Angrisano F., Marapana D. S., Rogers K. L., Whitchurch C. B., Beeson J. G., Cowman A. F., Ralph S. A., Baum J. (2011) Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe 9, 9–20 [DOI] [PubMed] [Google Scholar]
- 10. Peterson D. S., Wellems T. E. (2000) EBL-1, a putative erythrocyte binding protein of Plasmodium falciparum, maps within a favored linkage group in two genetic crosses. Mol. Biochem. Parasitol. 105, 105–113 [DOI] [PubMed] [Google Scholar]
- 11. Adams J. H., Sim B. K., Dolan S. A., Fang X., Kaslow D. C., Miller L. H. (1992) A family of erythrocyte binding proteins of malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 89, 7085–7089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rayner J. C., Vargas-Serrato E., Huber C. S., Galinski M. R., Barnwell J. W. (2001) A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J. Exp. Med. 194, 1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Triglia T., Tham W. H., Hodder A., Cowman A. F. (2009) Reticulocyte binding protein homologues are key adhesins during erythrocyte invasion by Plasmodium falciparum. Cell. Microbiol. 11, 1671–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duraisingh M. T., Maier A. G., Triglia T., Cowman A. F. (2003) Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 4796–4801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sim B. K., Chitnis C. E., Wasniowska K., Hadley T. J., Miller L. H. (1994) Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science 264, 1941–1944 [DOI] [PubMed] [Google Scholar]
- 16. Mayer D. C., Cofie J., Jiang L., Hartl D. L., Tracy E., Kabat J., Mendoza L. H., Miller L. H. (2009) Glycophorin B is the erythrocyte receptor of Plasmodium falciparum erythrocyte-binding ligand, EBL-1. Proc. Natl. Acad. Sci. U.S.A. 106, 5348–5352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maier A. G., Duraisingh M. T., Reeder J. C., Patel S. S., Kazura J. W., Zimmerman P. A., Cowman A. F. (2003) Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbvich negativity in human populations. Nat. Med. 9, 87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crosnier C., Bustamante L. Y., Bartholdson S. J., Bei A. K., Theron M., Uchikawa M., Mboup S., Ndir O., Kwiatkowski D. P., Duraisingh M. T., Rayner J. C., Wright G. J. (2011) Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spadafora C., Awandare G. A., Kopydlowski K. M., Czege J., Moch J. K., Finberg R. W., Tsokos G. C., Stoute J. A. (2010) Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathog. 6, e1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tham W. H., Wilson D. W., Lopaticki S., Schmidt C. Q., Tetteh-Quarcoo P. B., Barlow P. N., Richard D., Corbin J. E., Beeson J. G., Cowman A. F. (2010) Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc. Natl. Acad. Sci. U.S.A. 107, 17327–17332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tham W. H., Schmidt C. Q., Hauhart R. E., Guariento M., Tetteh-Quarcoo P. B., Lopaticki S., Atkinson J. P., Barlow P. N., Cowman A. F. (2011) Plasmodium falciparum uses a key functional site in complement receptor type-1 for invasion of human erythrocytes. Blood 118, 1923–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fearon D. T. (1979) Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc. Natl. Acad. Sci. U.S.A. 76, 5867–5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krych-Goldberg M., Atkinson J. P. (2001) Structure-function relationships of complement receptor type 1. Immunol. Rev. 180, 112–122 [DOI] [PubMed] [Google Scholar]
- 24. Smith B. O., Mallin R. L., Krych-Goldberg M., Wang X., Hauhart R. E., Bromek K., Uhrin D., Atkinson J. P., Barlow P. N. (2002) Structure of the C3b binding site of CR1 (CD35), the immune adherence receptor. Cell 108, 769–780 [DOI] [PubMed] [Google Scholar]
- 25. Krych-Goldberg M., Moulds J. M., Atkinson J. P. (2002) Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends Mol. Med. 8, 531–537 [DOI] [PubMed] [Google Scholar]
- 26. Krych-Goldberg M., Barlow P. N., Mallin R. L., Atkinson J. P. (2005) C3b/C4b binding site of complement receptor type 1 (CR1, CD35), in Structural Biology of the Complement System (Lambris J. D., Morikis D., eds) pp. 179–212, Marcel Dekker, New York [Google Scholar]
- 27. Khera R., Das N. (2009) Complement receptor 1. Disease associations and therapeutic implications. Mol. Immunol. 46, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moulds J. M., Brai M., Cohen J., Cortelazzo A., Cuccia M., Lin M., Sadallah S., Schifferli J., Bala Subramanian V., Truedsson L., Wu G. W., Zhang F., Atkinson J. P. (1998) Reference typing report for complement receptor 1 (CR1). Exp. Clin. Immunogenet. 15, 291–294 [DOI] [PubMed] [Google Scholar]
- 29. Daniels G. L., Cartron J. P., Fletcher A., Garratty G., Henry S., Jørgensen J., Judd W. J., Levene C., Lin M., Lomas-Francis C., Moulds J. J., Moulds J. M., Overbeeke M., Reid M. E., Rouger P., Scott M., Sistonen P., Smart E., Tani Y., Wendel S., Zelinski T. (2003) International society of blood transfusion committee on terminology for red cell surface antigens. Vancouver Report. Vox Sang. 84, 244–247 [DOI] [PubMed] [Google Scholar]
- 30. Rodriguez de Cordoba S., Dykman T. R., Ginsberg-Fellner F., Ercilla G., Aqua M., Atkinson J. P., Rubinstein P. (1984) Evidence for linkage between the loci coding for the binding protein for the fourth component of human complement (C4BP) and for the C3b/C4b receptor. Proc. Natl. Acad. Sci. U.S.A. 81, 7890–7892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez de Cordoba S., Lublin D. M., Rubinstein P., Atkinson J. P. (1985) The human genes for three complement components that regulate the activation of C3 are tightly linked. J. Exp. Med. 161, 1189–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carroll M. C., Alicot E. M., Katzman P. J., Klickstein L. B., Smith J. A., Fearon D. T. (1988) Organization of the genes encoding complement receptors type 1 and 2, decay-accelerating factor, and C4-binding protein in the RCA locus on human chromosome 1. J. Exp. Med. 167, 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hourcade D., Garcia A. D., Post T. W., Taillon-Miller P., Holers V. M., Wagner L. M., Bora N. S., Atkinson J. P. (1992) Analysis of the regulators of complement activation (RCA) gene cluster with yeast artificial chromosomes (YACs). Genomics 12, 289–300 [DOI] [PubMed] [Google Scholar]
- 34. Kirkitadze M. D., Barlow P. N. (2001) Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunol. Rev. 180, 146–161 [DOI] [PubMed] [Google Scholar]
- 35. Soares D. C., Barlow P. N. (2005) Complement control protein modules in the regulators of complement activation, in Structural Biology of the Complement System (Morikis D., Lambris J.D., eds) pp. 19–62, CRC Press, Boca Raton, FL [Google Scholar]
- 36. Klickstein L. B., Wong W. W., Smith J. A., Weis J. H., Wilson J. G., Fearon D. T. (1987) Human C3b/C4b receptor (CR1). Demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristics of C3/C4 binding proteins. J. Exp. Med. 165, 1095–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hourcade D., Miesner D. R., Atkinson J. P., Holers V. M. (1988) Identification of an alternative polyadenylation site in the human C3b/C4b receptor (CR1) transcriptional unit and prediction of a secreted form of CR1. J. Exp. Med. 168, 1255–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hourcade D., Miesner D. R., Bee C., Zeldes W., Atkinson J. P. (1990) Duplication and divergence of the aminoterminal coding region of the complement receptor 1 (CR1) gene. J. Biol. Chem. 265, 974–980 [PubMed] [Google Scholar]
- 39. Krych M., Hourcade D., Atkinson J. P. (1991) Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. Proc. Natl. Acad. Sci. U.S.A. 88, 4353–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krych M., Clemenza L., Howdeshell D., Hauhart R., Hourcade D., Atkinson J. P. (1994) Analysis of the functional domains of complement receptor type 1 (C3b/C4b receptor; CD35) by substitution mutagenesis. J. Biol. Chem. 269, 13273–13278 [PubMed] [Google Scholar]
- 41. Subramanian V. B., Clemenza L., Krych M., Atkinson J. P. (1996) Substitution of two amino acids confers C3b binding to the C4b binding site of CR1 (CD35). Analysis based on ligand binding by chimpanzee erythrocyte complement receptor. J. Immunol. 157, 1242–1247 [PubMed] [Google Scholar]
- 42. Krych M., Hauhart R., Atkinson J. P. (1998) Structure-function analysis of the active sites of complement receptor type 1. J. Biol. Chem. 273, 8623–8629 [DOI] [PubMed] [Google Scholar]
- 43. Krych-Goldberg M., Hauhart R. E., Subramanian V. B., Yurcisin B. M., 2nd, Crimmins D. L., Hourcade D. E., Atkinson J. P. (1999) Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J. Biol. Chem. 274, 31160–31168 [DOI] [PubMed] [Google Scholar]
- 44. Krych-Goldberg M., Hauhart R. E., Porzukowiak T., Atkinson J. P. (2005) Synergy between two active sites of human complement receptor type 1 (CD35) in complement regulation. Implications for the structure of the classical pathway C3 convertase and generation of more potent inhibitors. J. Immunol. 175, 4528–4535 [DOI] [PubMed] [Google Scholar]
- 45. Makrides S. C., Scesney S. M., Ford P. J., Evans K. S., Carson G. R., Marsh H. C. (1992) Cell surface expression of the C3b/C4b receptor (CR1) protects Chinese hamster ovary cells from lysis by human complement. J. Biol. Chem. 267, 24754–24761 [PubMed] [Google Scholar]
- 46. Tetteh-Quarcoo P. B., Schmidt C. Q., Tham W. H., Hauhart R., Mertens H. D., Rowe A., Atkinson J. P., Cowman A. F., Rowe J. A., Barlow P. N. (2012) Lack of evidence from studies of soluble protein fragments that Knops blood group polymorphisms in complement receptor-type 1 are driven by malaria. PLoS One 7, e34820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tham W. H., Wilson D. W., Reiling L., Chen L., Beeson J. G., Cowman A. F. (2009) Antibodies to reticulocyte binding protein-like homologue 4 inhibit invasion of Plasmodium falciparum into human erythrocytes. Infect. Immun. 77, 2427–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barlow P. N., Steinkasserer A., Norman D. G., Kieffer B., Wiles A. P., Sim R. B., Campbell I. D. (1993) Solution structure of a pair of complement modules by nuclear magnetic resonance. J. Mol. Biol. 232, 268–284 [DOI] [PubMed] [Google Scholar]
- 49. Hocking H. G., Herbert A. P., Kavanagh D., Soares D. C., Ferreira V. P., Pangburn M. K., Uhrín D., Barlow P. N. (2008) Structure of the N-terminal region of complement factor H and conformational implications of disease-linked sequence variations. J. Biol. Chem. 283, 9475–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Makou E., Mertens H. D., Maciejewski M., Soares D. C., Matis I., Schmidt C. Q., Herbert A. P., Svergun D. I., Barlow P. N. (2012) Solution structure of CCP modules 10–12 illuminates functional architecture of the complement regulator, factor H. J. Mol. Biol. 424, 295–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Black G., Wenzler M., Wang X., Krych-Goldberg M., Atkinson J. P., Barlow P. N. (2004) 1H, 15N and 13C resonance assignments of complement control protein module pair 2–3 from the C4b-binding site of complement receptor type 1. J. Biomol. NMR 30, 227–228 [DOI] [PubMed] [Google Scholar]
- 52. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy. Development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 53. Schmidt C. Q., Herbert A. P., Mertens H. D., Guariento M., Soares D. C., Uhrin D., Rowe A. J., Svergun D. I., Barlow P. N. (2010) The central portion of factor H (modules 10–15) is compact and contains a structurally deviant CCP module. J. Mol. Biol. 395, 105–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 55. Fiser A., Sali A. (2003) Modeller. Generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 [DOI] [PubMed] [Google Scholar]
- 56. Cole J. L., Housley G. A., Jr., Dykman T. R., MacDermott R. P., Atkinson J. P. (1985) Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc. Natl. Acad. Sci. U.S.A. 82, 859–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alonso P. L., Brown G., Arevalo-Herrera M., Binka F., Chitnis C., Collins F., Doumbo O. K., Greenwood B., Hall B. F., Levine M. M., Mendis K., Newman R. D., Plowe C. V., Rodríguez M. H., Sinden R., Slutsker L., Tanner M. (2011) A research agenda to underpin malaria eradication. PLoS Med 8, e1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ko W. Y., Kaercher K. A., Giombini E., Marcatili P., Froment A., Ibrahim M., Lema G., Nyambo T. B., Omar S. A., Wambebe C., Ranciaro A., Hirbo J. B., Tishkoff S. A. (2011) Effects of natural selection and gene conversion on the evolution of human glycophorins coding for MNS blood polymorphisms in malaria-endemic African populations. Am. J. Hum. Genet. 88, 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zimmerman P. A., Patel S. S., Maier A. G., Bockarie M. J., Kazura J. W. (2003) Erythrocyte polymorphisms and malaria parasite invasion in Papua New Guinea. Trends Parasitol. 19, 250–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Patel S. S., King C. L., Mgone C. S., Kazura J. W., Zimmerman P. A. (2004) Glycophorin C (Gerbich antigen blood group) and band 3 polymorphisms in two malaria holoendemic regions of Papua New Guinea. Am. J. Hematol. 75, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kwiatkowski D. P. (2005) How malaria has affected the human genome and what human genetics can teach us about malaria. Am. J. Hum. Genet. 77, 171–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tarazona-Santos E., Castilho L., Amaral D. R., Costa D. C., Furlani N. G., Zuccherato L. W., Machado M., Reid M. E., Zalis M. G., Rossit A. R., Santos S. E., Machado R. L., Lustigman S. (2011) Population genetics of GYPB and association study between GYPB*S/s polymorphism and susceptibility to P. falciparum infection in the Brazilian Amazon. PLoS One 6, e16123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baum J., Maier A. G., Good R. T., Simpson K. M., Cowman A. F. (2005) Invasion by P. falciparum merozoites suggests a hierarchy of molecular interactions. PLoS Pathog. 1, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stubbs J., Simpson K. M., Triglia T., Plouffe D., Tonkin C. J., Duraisingh M. T., Maier A. G., Winzeler E. A., Cowman A. F. (2005) Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science 309, 1384–1387 [DOI] [PubMed] [Google Scholar]
- 65. Frishman D., Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23, 566–579 [DOI] [PubMed] [Google Scholar]
- 66. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 67. Schrodinger L. (ed) (2011) PyMOL Molecular Graphics System, version 1.4 [Google Scholar]