

Background: NO-induced conformational changes of guanylate cyclase were analyzed.
Results: FRET experiments show a movement of two tryptophans toward a fluorescent substrate.
Conclusion: Activation is transmitted to the catalytic domain by direct interaction of the heme binding domain and through the coiled-coil helix.
Significance: The results help to advance understanding of the molecular events associated with activation of NOsGC.
Keywords: Cyclic GMP (cGMP), Fluorescence Resonance Energy Transfer (FRET), Guanylate Cyclase (Guanylyl Cyclase), Nitric Oxide, Signal Transduction, Ciguate
Abstract
The complete structure of the assembled domains of nitric oxide-sensitive guanylate cyclase (NOsGC) remains to be determined. It is also unknown how binding of NO to heme in guanylate cyclase is communicated to the catalytic domain. In the current study the conformational change of guanylate cyclase on activation by NO was studied using FRET. Endogenous tryptophan residues were used as donors, the substrate analog 2′-Mant-3′-dGTP as acceptor. The enzyme contains five tryptophan residues distributed evenly over all four functional domains. This provides a unique opportunity to detect the movement of the functional domains relative to the substrate-binding catalytic region. FRET measurements indicate that NO brings tryptophan 22 in the αB helix of the β1 heme NO binding domain and tryptophan 466 in the second short helix of the α1 coiled-coil domain closer to the catalytic domain. We propose that the respective domains act as a pair of tongs forcing the catalytic domain into the nitric oxide-activated conformation.
Introduction
There are two pharmacologically important classes of guanylate cyclases that catalyze the reaction from GTP to cGMP; some are transmembrane receptors for extracellular peptides and others are intracellular receptors for the gaseous ligand NO (for reviews, see Refs. 1 and 2). Transmembrane and NO-sensitive guanylate cyclases (NOsGC)2 show significant homology not only in their dimeric catalytic domain but also in the directly preceding helices forming a dimeric coiled-coil. In transmembrane forms similar to GC-A, the extracellular binding of the ligand ANP to preformed homodimers is transmitted across the transmembrane helices and reorients the two intracellular domains into the active conformation (3). By analogy, it was assumed that binding of NO to the amino-terminal heme NO binding (HNOX) domain is communicated to the carboxyl-terminal catalytic domain through the coiled-coil domain (4). This linear model of NO activation has been questioned by findings that the isolated HNOX domain can directly interact with the isolated catalytic region of NOsGC (5). In support of this, we have shown that the amino termini of NOsGC are in close proximity to the carboxyl termini based on fluorescence resonance energy transfer using fluorescent fusion proteins (6). Fusion of the fluorescent protein to the critical HNOX domain of the β1 subunit led to a heme-free NO unresponsive enzyme. This proved to be a serious obstacle for the detection of conformational changes during NO activation. In the current paper, we have overcome this challenge by using naturally occurring tryptophans as FRET donors and the substrate analog 2′-Mant-3′-dGTP as FRET acceptor.
(N-Methyl)anthraniloyl-substituted nucleotides (Mant-NTPs) are fluorescently labeled nucleotides that compete with GTP or ATP for the substrate binding site of adenylate and guanylate cyclases (7, 8). Mant-GTP can be converted to Mant-cGMP and has been used to develop an assay of NOsGC activity based on direct fluorescence (9). In contrast, 3′-deoxy forms are competitive inhibitors of adenylate and guanylate cyclases (7, 8). When tryptophan residues are excited and a sufficient proximity to the Mant group is provided by the conformation of the enzyme, Mant-deoxynucleotides can act as FRET acceptors (10–12). This energy transfer can then be used to analyze the topical relationship of tryptophans to the substrate-binding site. Under optimal conditions, where a tryptophan moves during ligand-induced activation, conformational changes can be detected.
In the present work, we analyzed conformational changes of full-length NOsGC upon NO-activation using 2′-Mant-3′-dGTP as a FRET partner of naturally occurring tryptophan residues. NOsGC contains five tryptophan residues, which are evenly distributed over the four functional domains: one is located in the heme NO binding (HNOX) domain of the β1 subunit (β1, Trp-22), another resides in the Per-Arnt-Sim (PAS) domain of α1 (α1, Trp-352), one is in the coiled-coil domain of the α1 subunit (α1, Trp-466) and the last two tryptophan residues are located in the catalytic domain (α1, Trp-669; β1, Trp-602) (Fig. 1). Based on our results, NO occupancy of the amino-terminal HNOX domain is communicated to the catalytic domain both through the coiled-coil helix and by direct interaction between the HNOX and catalytic domain.
FIGURE 1.
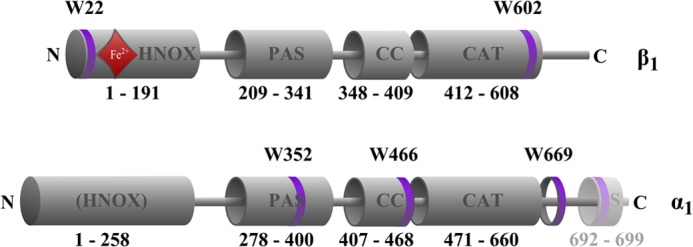
Positions of the five tryptophan residues in rat NOsGC subunits. Tryptophan residues are marked as purple rings. Amino acids 692–699 represent the Strep-tag II (S) containing an additional tryptophan residue. The red rhombus indicates position of the heme group. The schematic representation of functional domains is based on the crystallized region of the HNOX domain of Alr2278 from Nostoc sp. (PCC 7120) (32) on the crystallized region of PAS domain of signal transduction histidine kinase from Nostoc punctiforme (PCC 73102) (33) on the crystallized region of the coiled-coil domain of the NOsGC β1 subunit from Rattus norvegicus (24) and on the crystallized catalytic region of the guanylate cyclase CYG12 from Chlamydomonas reinhardtii (34).
EXPERIMENTAL PROCEDURES
Materials
2,2-Diethyl-1-nitroso-oxyhydrazine (DEA/NO) and all other chemicals were obtained from Sigma Aldrich in the highest grade of purity. 2′-Mant-3′-dGTP (2′-O-(N-methyl-anthraniloyl)-3′-deoxy-guanosine-5′-triphosphate, triethyl-ammonium salt) was obtained from Jena Bioscience (Jena, Germany). NOsGC stimulators BAY 41-2272 and BAY 41-8543 were a generous gift from Johannes-Peter Stasch (Bayer Pharma AG, Wuppertal, Germany).
Cloning and Site-directed Mutagenesis of Tryptophan Residues
The β1 subunit of human guanylate cyclase was cloned into the pFASTBAC vector as described by Koglin and Behrends (13). Cloning of the α1 subunit with Strep-tag II (S) (α1S) into the pFASTBAC vector was described in Ref. 14. They served as template for site-directed mutagenesis with the QuikChange® Lightning mutagenesis kit (Agilent Technologies, Böblingen, Germany).
Expression and Purification of NOsGC
NOsGC subunits were recombinantly co-expressed using the baculovirus/Sf9 system. Recombinant baculoviruses of the respective subunits were generated according to the Bac-To-Bac® baculovirus expression system manual (Invitrogen). For cultivation of Sf9 insect cells (Invitrogen catalog no. 11496-015), Sf-900 II serum-free medium was supplemented with 1% penicillin/streptomycin and 10% fetal calf serum. Spinner cultures were grown at 27 °C at 140 rpm on a shaking incubator and diluted to 2 × 106 cells/ml for infection. 500 ml of cell suspension were co-infected with the recombinant baculovirus stock of the carboxyl-terminally Strep-tagged α1 subunit or a corresponding mutant and with either the wild-type or a mutant β1 subunit. Afterward, the enzyme was purified to apparent homogeneity as described previously (15).
Spectrofluorimetric Measurements
FRET was measured with a Varian spectrofluorometer at 37 °C using a quartz UV microcuvette from Hellma (Mühlheim, Germany) in a volume of 50 μl. Purified NOsGC and 2′-Mant-3′-dGTP were used at equimolar concentrations of 3 μm. First, spectra of NOsGC were detected, and then 2′-Mant-3′-dGTP was added. The measurements were performed in a buffer containing 50 mm triethanolamine/HCl, 1 mm EDTA, and 5 mm MnCl2 (pH 7.4). Previous studies with adenylate cyclase showed that FRET signals were much larger in the presence of Mn2+ than in the presence of Mg2+ (11). The sensitized emission method with three channels was used for the determination of FRET (16). The excitation slit for all excitation wavelengths was 5 nm. In the donor channel, NOsGC was excited at 280 nm and at 295 nm as the tryptophan-specific excitation (17). The emission was detected at 335 nm. In the acceptor channel, 2′-Mant-3′-dGTP was excited at 335 nm, and the emission wavelength was 430 nm. Therefore, the FRET channel was selected with an excitation wavelength of 335 nm and an emission wavelength of 430 nm. FRET efficiencies were calculated according to Wallrabe and Periasamy (18). In addition, spectra for both excitation wavelengths were recorded for an emission from 300 to 500 nm. Experiments with purified NOsGC were performed in the presence and absence of DEA/NO (100 μm).
Photometric Measurements
Absorption measurements were carried out on a Varian photometer at 37 °C. The same buffer and concentration of purified NOsGC were used as for the spectrofluorometric measurements. Absorption was recorded from 200–600 nm.
NOsGC Activity Assay
NOsGC activity was quantified by the formation of [α-32P]cGMP from [α-32P]GTP, as described by Ref. 19. The activity of 50 ng of purified enzyme was determined in a total volume of 100 μl in the presence of 50 mm triethanolamine/HCl buffer, pH 7.4, 1 mm 3-isobutyl-1-methylxanthine, 3 mm MgCl2, 3 mm dithiothreitol, 5 mm creatine phosphate, 1 mm cyclic GMP, 500 μm GTP, [α-32P]GTP, and 0.025 mg creatine phosphokinase. The reaction was started by the incubation at 37 °C and was stopped after 10 min by adding 500 μl of 125 mm zinc acetate and 500 μl of 120 mm sodium carbonate leading to co-precipitation of zinc carbonate and 5′nucleotides. The purification of the enzyme-formed cGMP was performed on short alumina columns as described by Ref. 19. Stimulated measurements were performed in the presence of BAY 41-8543 (100 μm) and DEA/NO (100 μm). For the dose-response curves, DEA/NO was used at a concentration ranging from 1 nm to 100 μm. BAY 41-8543 was used at concentrations ranging from 1 nm to 100 μm in the presence of DEA/NO (100 μm).
SDS-PAGE Analysis
The SDS-PAGE analysis was performed as described in Ref. 15.
Statistical Analysis
The results are expressed as means ± S.E. of at least three independent experiments. All results were controlled for their statistical significance by Student's t test. A value of p < 0.05 was considered to be statistically significant.
RESULTS
Fluorescence Resonance Energy Transfer between Wild-type NOsGC and Mant
To confirm that 2′-Mant-3′-dGTP binds to NOsGC and can be used as a conformation-sensitive detector, purified NOsGC (α1S/β1, 3 μm) was excited at 280 nm (Fig. 2A) and 295 nm, which represents the tryptophan-specific wavelength (Fig. 2B) (17). The excitation of NOsGC with both wavelengths (280 and 295 nm) resulted in an emission with a maximum at 335 nm (Fig. 2, black lines). But in the presence of 2′-Mant-3′-dGTP (3 μm), the maximum at 335 nm decreased, and a new maximum at 430 nm appeared (Fig. 2, dark gray lines). The appearance of this new maximum at 430 nm indicates that tyrosine and tryptophan residues are able to serve as FRET donors, whereas 2′-Mant-3′-dGTP acts as FRET acceptor. The resulting basal FRET efficiency reached 12.1 ± 1.0% for an excitation at 280 nm and 5.0 ± 0.4% for the tryptophan-specific excitation at 295 nm. NOsGC contains only five tryptophan residues. They are distributed over the four functional domains, providing the opportunity to study FRET between the substrate analog 2′-Mant-3′-dGTP and the specific tryptophan residues of the HNOX, PAS, coiled-coil, and catalytic region. Therefore, we decided to use tryptophan-specific excitation at 295 nm in all further FRET experiments.
FIGURE 2.

Emission of NOsGC in absence and presence of 2′-Mant-3′-dGTP. Normalized emission curves of wild-type NOsGC at 280 nm excitation (A) and at 295 nm excitation (B) in the absence of 2′-Mant-3′-dGTP (black line) and in the presence of 2′-Mant-3′-dGTP (3 μm) (gray line) are presented. For comparison, 2′-Mant-3′-dGTP autofluorescence is shown (light gray line). Graphs show emission curves of one of four representative measurements (a.u., arbitrary unit).
Increase in FRET Efficiency between Tryptophans and the Mant Group upon Addition of DEA/NO
The addition of the NO-donor DEA/NO to NOsGC and 2′-Mant-3′-dGTP increased FRET efficiency by 2.7 ± 0.1% (Fig. 3). The increase in FRET efficiency was significant. As a negative control, we used a mutant isoform of the β1 subunit (α1S/β1H105A), which does not increase its catalytic activity in the presence of NO (20). This catalytically NO-insensitive mutant was also NO-insensitive with respect to the observed increase in FRET efficiency (see Fig. 3). The data indicate that binding of NO to NOsGC leads to a conformational change that increases FRET efficiency between the substrate analog 2′-Mant-3′-dGTP and endogenous tryptophans. FRET efficiency increases when the distance between the donor fluorophore and the acceptor fluorophore decreases (21). Therefore, we hypothesized that one or more tryptophan residues move toward the substrate binding site upon activation by NO.
FIGURE 3.

FRET efficiency of NOsGC under basal and NO-stimulated conditions. FRET efficiencies (%) of wild-type NOsGC and NO-insensitive NOsGC mutant at 295 nm excitation in the presence of 2′-Mant-3′-dGTP (black columns) and in the presence of 2′-Mant-3′-dGTP and DEA/NO 100 μm (gray columns) are shown. An asterisk marks p value < 0.05 compared with values in the absence of DEA/NO. The results are expressed as means ± S.E. of at least four independent experiments.
Mutations of Tryptophan
There are five tryptophan residues within rat NOsGC, and each could contribute to FRET under basal and NO-stimulated conditions (see Fig. 1). For purification purposes, each mutant contains a Strep-tag carboxyl-terminally to the α1 subunit that has an additional tryptophan residue. To identify the tryptophan residues involved in FRET, we mutated each of them to either the amino acids alanine (Ala) or phenylalanine (Phe).
Comparison of FRET Efficiency of Wild-type NOsGC and Mutants
Each of the generated five mutants lacked a single tryptophan and therefore a potential donor for FRET. Without this FRET donor, the basal FRET efficiency should be reduced if the corresponding tryptophan takes part in basal FRET. As expected, the FRET efficiency under basal conditions decreased in some tryptophan mutants (Fig. 4A).
FIGURE 4.

Comparison of FRET efficiencies of NOsGC mutants. A, FRET efficiency (%) under basal conditions for NOsGC wild-type enzyme and NOsGC mutants are compared. B, change in FRET efficiency (%) after the addition of DEA/NO (100 μm) for NOsGC wild-type enzyme and NOsGC mutants. Values were calculated as the difference of basal FRET efficiency to NO-stimulated FRET efficiency. An asterisk marks p value < 0.05 compared with wild-type enzyme. The results are expressed as means ± S.E. of at least three independent experiments.
The increase in FRET efficiency of the mutants after activation by NO should be considerably reduced if the corresponding tryptophan is responsible for the increase in FRET efficiency seen for the wild-type enzyme. The mutant of Trp-22 (W22F) of the β1 subunit and the mutants of Trp-669 (W669A) and Trp-352 (W352F) of the α1 subunit showed no significant difference, whereas two mutants stood out in comparison with the wild-type (Fig. 4B): the substitution of a phenylalanine for Trp-466 (W466F) of the α1 subunit led to a significant reduction in the presence of DEA/NO. This indicates that Trp-466 is likely responsible for the observed increase in FRET efficiency of the wild-type enzyme. The substitution of a phenylalanine for Trp-602 (W602F) in the β1 subunit led to a significant increase in FRET under NO-stimulated conditions. This was unexpected and will be discussed after the following paragraph.
Tryptophan 466 Is Necessary and Sufficient for NO-induced FRET Increase of Wild-type NOsGC
The previous experiments show that substitution of a phenylalanine for Trp-466 (W466F) leads to a significant loss of the NO-induced increase in FRET efficiency between tryptophans and 2′-Mant-3′-dGTP. To positively identify Trp-466 as the tryptophan responsible for the NO-induced increase in FRET efficiency, we left Trp-466 intact and mutated all other endogenous NOsGC tryptophan residues to either the amino acids alanine or phenylalanine (α1S-W352F,W669A/β1-W22F,W602F). After adding DEA/NO to the quadruple mutant, we were able to detect an increase in FRET efficiency, which equals the increase of the wild-type enzyme (see Fig. 4). Because Trp-466 is the only endogenous tryptophan in the quadruple mutant, the visible increase in FRET efficiency of this mutant and the wild-type enzyme can likely be traced back to Trp-466. A considerable participation and movement of the tryptophan in the Strep-tag after NO stimulation cannot be completely ruled out but is unlikely: the mutation of α1 Trp-669 only 23 amino acids upstream of the tryptophan in the Strep-tag does not show significant differences in FRET efficiency under basal or under NO-stimulated conditions. The data resulting from analysis of the quadruple mutant indicate that Trp-466 is not only required but is also sufficient for the visible increase in FRET efficiency. Because FRET efficiency increases, when the distance between the participating fluorophores decreases (21), it follows that Trp-466 of the α1 subunit moves toward the substrate binding site upon NO stimulation.
Interaction between Tryptophan 602 and Tryptophan 22
Even though the substitution of Trp-602 led to a decrease of FRET efficiency in comparison with the wild-type enzyme under basal conditions (see Fig. 4A), FRET efficiency increased under NO-stimulated conditions, much to our surprise. We therefore postulated that Trp-602 masks the movement of a tryptophan other than Trp-466 toward the Mant group. A candidate for this act is Trp-22 of β1 HNOX. We and others (5, 6) recently reported a spatial proximity of the catalytic domain and the HNOX domain, suggesting that Trp-22 in the β1 HNOX and Trp-602 in the β1 catalytic domain could indeed be close enough for an interaction. To positively identify Trp-22 as the tryptophan masked by Trp-602, we left Trp-466 and Trp-22 intact and mutated all other endogenous tryptophan residues to the amino acids alanine or phenylalanine (α1S-W352F,W669A/β1-W602F). In the presence of NO, the FRET efficiency of this triple mutant exceeded the value of the wild-type enzyme and reached the value of α1S/β1-W602F. These results indicate that in the triple mutant, Trp-22 and Trp-466 move toward the substrate binding site upon activation by NO and cause an increase in FRET efficiency by 3.9 ± 0.3% (see Fig. 4).
Comparison of the tryptophan emission maxima under basal conditions showed a shift to shorter wavelengths in the triple mutant containing only Trp-22 and Trp-466 (336.7 ± 0.8 nm) opposed to the wild-type enzyme (340.5 ± 1.0 nm). The emission of the quadruple mutant containing only Trp-466 revealed a small shift in the opposite direction to a maximum at 342.5 ± 1.5 nm. From this, it follows that the tryptophan emission maximum of Trp-22 is significantly blueshifted in comparison with the other tryptophans. This is an important prerequisite for homo-FRET and provides a likely explanation for the observed interaction between Trp-22 and Trp-602 (see “Discussion”).
Preserved NO Heme Binding in All Tryptophan Mutants, Including W22F and W466F Mutants
The heme absorbance spectra for the enzyme lacking Trp-22 (α1S/β1-W22F) or lacking Trp-466 (α1S-W466F/β1) were measured in the absence and also in the presence of DEA/NO (Fig. 5A). The Soret peak of heme undergoes the typical shift from 430 nm under basal conditions to 399 nm under NO-stimulated conditions (BAY 41-8543). Thus, the absence or presence of Trp-22 or Trp-466 neither influences the binding of heme to NOsGC nor interferes with the binding of NO to the heme group. The same is true for all other tryptophan mutants used in this study (data not shown).
FIGURE 5.

Absorbance spectra and SDS-PAGE analysis of NOsGC mutants. A, presented are the normalized absorbance spectra for α1S/β1-W22F and α1S W466F/β1 in the absence and presence of DEA/NO (100 μm). Graphs show absorbance spectra of one of three representative measurements. B, Coomassie staining of SDS-PAGE analysis for α1S/β1-W22F and α1S-W466F/β1. C indicates lanes with cytosolic fractions (50 μg), and P marks lanes with purified enzymes (1 μg). The marker M is given in kDa.
Influence of Mutations on Enzyme Activity and Regulation by NO and Ciguates
Fig. 5B shows SDS-PAGE analysis of the W22F and W466F mutants. There were no apparent differences in purity between the wild-type enzyme and all other enzyme mutants used in this study (data not shown). Table 1 provides information on enzyme activity measurements under basal, NO-stimulated and NO plus ciguat-stimulated conditions. All tryptophan mutants used in this study showed significant activation by NO and significant activation in the presence of ciguat plus NO. Activity per mg protein under basal and NO-stimulated conditions was lowest for the W466F mutant. This was not due to lower purity of the enzyme preparations as controlled by SDS-PAGE analysis (see Fig. 5B). Much to our surprise, the addition of BAY 41-8543 in addition to DEA/NO had the greatest effect on the W466F mutant. As seen in Fig. 6A, NO activated both the α1S-W352F,W669A/β1-W22F,W602F mutant and wild-type NOsGC by 80–100-fold, whereas the α1S-W466F/β1 mutant is activated only 15-fold. Addition of BAY 41-8543 in the presence of DEA/NO (100 μm) (Fig. 6B), increased enzyme activity 2-fold in the wild-type enzyme and in the α1S-W352F,W669A/β1-W22F,W602F mutant, whereas BAY 41-8543 increased the activity of α1S-W466F/β1 >6-fold over NO-stimulated activity. This approaches the level of activation elicited by DEA/NO in the wild-type enzyme.
TABLE 1.
Activity data of NOsGC mutants
The first three columns of the table present the specific activity μmol × min−1 × mg−1 of NOsGC wild-type enzyme and the mutants under basal conditions and in the presence of DEA/NO (100 μm) or BAY 41–8543 and DEA/NO (both 100 μm). The results are expressed as means ± S.E. of at least three independent experiments. The next two columns give the values for the x-fold stimulation of basal activity by DEA/NO or BAY 41–8543 plus DEA/NO. Additionally, the x-fold activation of NO-stimulated activity by BAY 41–8543 is shown. The last three columns present percent activity of the mutants compared with wild-type enzyme.
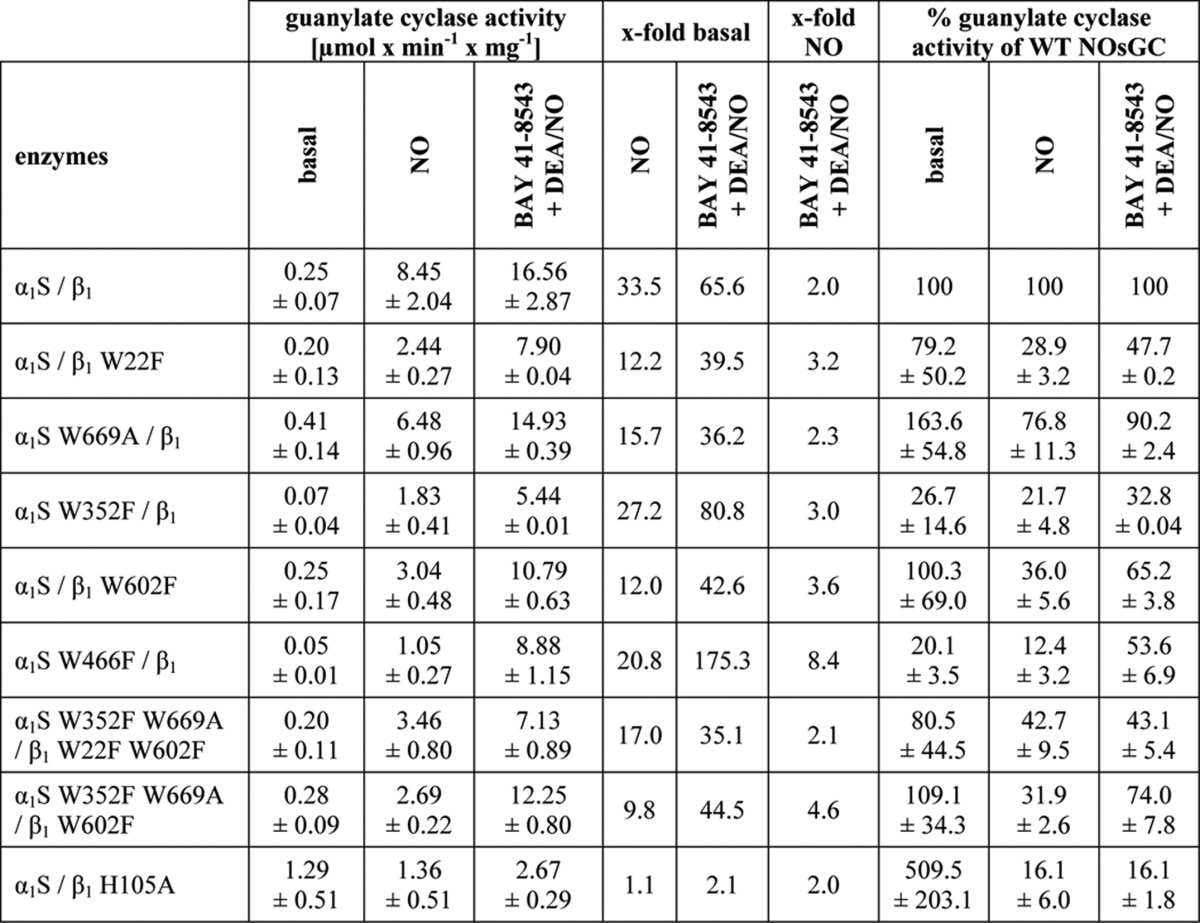
FIGURE 6.

Dose-response curves of NOsGC wild-type enzyme and NOsGC mutants. Concentration-dependent activation of α1S/β1, α1S-W352F,W669A/β1-W22F,W602F and α1S-W466F/β1 was measured in a range of 1 nm to 100 μm DEA/NO or 0.1 nm to 100 μm BAY 41-8543 in the presence of DEA/NO (100 μm). A, fold stimulation by DEA/NO. B, fold stimulation by BAY 41-8543 in the presence of DEA/NO. C and D, same data as in A and B normalized to the maximum. The results are expressed as means ± S.E. of at least three independent experiments.
DISCUSSION
In the current study, we measured fluorescence resonance energy transfer from tryptophan residues of full-length guanylate cyclase to the substrate analog 2′-Mant-3′-dGTP under basal and NO-stimulated conditions. Our results indicate that NO binding to the amino-terminal β1 HNOX domain is transmitted to the carboxyl-terminal catalytic domain both through the coiled-coil helices and by direct interdomain signal transmission between the β1 HNOX domain and the catalytic domain.
The Role of Trp-22 in the β1 HNOX Domain
Trp-22 of the β1 subunit is located in the αB helix of the HNOX domain according to the crystal structure of Nostoc sp. PCC 7120, a cyanobacterial HNOX homolog (22). The authors show that the binding of NO initially leads to a six-coordinated NO-heme state, which finally passes into a five-coordinated NO-heme state. This is accompanied by a severe distortion of the heme leading to an extensive conformational change of the amino terminus. In this process, helix αB containing Trp-22 rotates for ∼20° (22). In the crystal structure of the HNOX domain derived from Thermoanaerobacter tengcongenesis, Olea et al. (23) show an NO-induced shift of the αB helix of 4.9 Å. Our data indicate that this very movement of the helix containing Trp-22 brings it closer to the catalytic domain and the Mant group. This is consistent with the idea first suggested by Winger and Marletta that the NO activation signal is transmitted via a direct interaction between the β1 HNOX domain and the catalytic domain (5).
Nature of the Trp-22 and Trp-602 Interaction
The movement of Trp-22 in the wild-type enzyme as detected by FRET is masked by an interaction with Trp-602. A likely explanation for the quenching effect is the occurrence of homo-FRET (17). Homo-FRET occurs when there is considerable overlap between the emission and absorbance of the same type of fluorophore. This is generally not the case for tryptophans unless the microenvironment of one tryptophan causes a shift in the emission spectrum. Our data confirm that Trp-22 shows a blueshifted emission in comparison with the other tryptophan residues (see “Results”). This is consistent with a more unpolar microenvironment within the heme-binding HNOX domain. The blueshift leads to greater overlap of the respective spectra that is a prerequisite for homo-FRET of tryptophans. This strongly supports the idea that homo-FRET occurs between tryptophans 22 and 602. The homo-FRET interaction between Trp-602 of the catalytic region and Trp-22 of the HNOX domain provides additional evidence for an interaction between the β1 HNOX domain and the catalytic domain.
The Coiled-coil Functions as Signaling Helix
In analogy to the crystallized β1 coiled-coil, Trp-466 is located in a short helix (αB) behind the long amphipathic helix (αA) of the coiled-coil domain (24). Based on comparative genomics, this domain has been suggested to act as a signaling helix that transmits the flow of signals between modules in diverse multi-domain signaling proteins (25). Our experimental data fully support the prediction that the coiled-coil functions as a signaling helix.
Comparison with Existing Models of NOsGC
Fritz et al. (26) examined a carboxyl-terminally truncated NOsGC from Manduca sexta (tobacco hornworm) using analytical ultracentrifugation, small angle x-ray scattering, cross-linking, and tandem mass spectrometry. They found 20 intramolecular contacts and hypothesized a model with a central parallel coiled-coil domain upon which the HNOX and PAS domains are assembled. Our data are consistent with the model proposed by Fritz et al. Fig. 7 shows the resulting model where we fitted the catalytic domain beneath the central coiled-coil domain in close proximity to the β1 HNOX and the carboxyl terminus of α1 coiled-coil domain.
FIGURE 7.
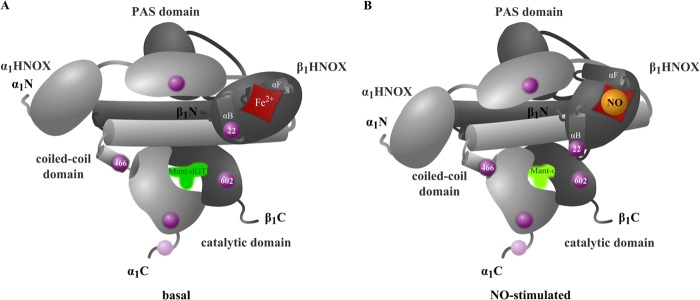
Model of NOsGC signal transmission. The model was developed on basis of NOsGC structure postulated by Fritz et al. (26). The α1 subunit is shown in light gray, and the β1 subunit is shown in dark gray.
Model of NO Signal Transmission Based on our FRET Data
Based on the crystal structure of the free and NO-bound HNOX domain from a cyanobacterial homolog, it has been suggested that NO binding induces a rotational shift of the helix αB containing Trp-22 due to heme bending and a shift of the helix αF away from the heme due to breakage of the bond between its histidine residue 105 and the heme iron (22). The NO-induced movement of helix αB containing Trp-22 can easily be applied to our findings because we provide evidence for a direct approximation of Trp-22 in the αB helix to the catalytic domain. The NO-induced movement of the helix αF containing histidine 105 is likely transferred to the second short helix of the coiled-coil domain of the α1 subunit, which contains Trp-466. This transfer could be mediated via the α1 PAS domain. In agreement with this thought, Underbakke et al. (27) propose an interaction between the αF helix of the HNOX domain with the PAS domain, and Fritz et al. (26) found that the α1 PAS domain is in contact with the β1 HNOX domain. The central importance of the α1 PAS domain for the signal transmission of NO is also underlined by previous findings from our group that in contrast to the α1 HNOX domain, α1 PAS is necessary for the intramolecular transmission of the NO activation signal (28).
Ciguat Hypersensitivity of the α1S-W466F/β1 Mutant
In contrast to the deletion mutant of the α1 PAS domain that abrogates both stimulation by NO and ciguat stimulators (28), the α1S-W466F mutation had a peculiar differential influence on NO versus NO plus ciguat stimulation. Yoo et al. (29) suggested that the influence of the ciguat stimulator, BAY 41-2272, is the same for full-length NOsGC and a truncated β1(1–200) HNOX domain. Consequently, it was concluded that NOsGC stimulators bind to the HNOX domain of β1. Recent evidence indicates that ciguat stimulators relieve allosteric inhibition of the HNOX domain of β1 by the α1 PAS domain thereby enhancing NO binding (26, 30). The differential influence of the α1S-W466F mutation on ciguat/NO stimulation can be accommodated in such an allostery model where signal transmission from the HNOX domain of β1 via the α1 PAS domain and the coiled-coil region of α1 to the catalytic domain is not unidirectional. This is consistent with data that argue in favor of reciprocal communication between the catalytic and NO-binding domain (31).
Summary
In summary, we propose a model of NO activation where the movement of the αB helix of the β1 HNOX domain containing Trp-22 interacts directly with the catalytic domain, whereas the movement of the signaling helix containing His-105 is indirectly transmitted to the catalytic domain via the α1 PAS domain and α1 coiled-coil domain. This is proposed to allow the short helix of the coiled-coil domain in α1 containing Trp-466 and the αB helix of the β1 HNOX domain containing Trp-22 to form a pair of tongs forcing the catalytic domain into the nitric oxide-activated conformation.
Acknowledgments
We thank Dr. Johannes-Peter Stasch for critical reading of the manuscript and the generous gift of NOsGC stimulators and Janine Watts for the linguistic support in preparing the manuscript. The expert technical assistance of Ines Thomsen, Gerlind Henze-Wittenberg, and Anja Stieler is gratefully acknowledged.
This work was supported by the Deutsche Forschungsgemeinschaft (BE 1865/5-1) and the European Cooperation in Science and Technology Program BM1005.
- NOsGC
- nitric oxide-sensitive guanylate cyclase(s)
- HNOX
- heme-nitric oxide/oxygen binding
- DEA/NO
- 2,2-diethyl-1-nitroso-oxyhydrazine
- Mant-NTP
- (N-methyl)anthraniloyl-substituted nucleotide.
REFERENCES
- 1. Misono K. S., Philo J. S., Arakawa T., Ogata C. M., Qiu Y., Ogawa H., Young H. S. (2011) Structure, signaling mechanism and regulation of the natriuretic peptide receptor guanylate cyclase. FEBS J. 278, 1818–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Derbyshire E. R., Marletta M. A. (2012) Structure and regulation of soluble guanylate cyclase. Annu. Rev. Biochem. 81, 533–559 [DOI] [PubMed] [Google Scholar]
- 3. Ogawa H., Qiu Y., Ogata C. M., Misono K. S. (2004) Crystal structure of hormone-bound atrial natriuretic peptide receptor extracellular domain: rotation mechanism for transmembrane signal transduction. J. Biol. Chem. 279, 28625–28631 [DOI] [PubMed] [Google Scholar]
- 4. Padayatti P. S., Pattanaik P., Ma X., van den Akker F. (2004) Structural insights into the regulation and the activation mechanism of mammalian guanylyl cyclases. Pharmacol. Ther. 104, 83–99 [DOI] [PubMed] [Google Scholar]
- 5. Winger J. A., Marletta M. A. (2005) Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry 44, 4083–4090 [DOI] [PubMed] [Google Scholar]
- 6. Haase T., Haase N., Kraehling J. R., Behrends S. (2010) Fluorescent fusion proteins of soluble guanylyl cyclase indicate proximity of the heme nitric oxide domain and catalytic domain. PLoS One 5, e11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gille A., Lushington G. H., Mou T. C., Doughty M. B., Johnson R. A., Seifert R. (2004) Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J. Biol. Chem. 279, 19955–19969 [DOI] [PubMed] [Google Scholar]
- 8. Gille A., Seifert R. (2003) 2′(3′)-O-(N-methylanthraniloyl)-substituted GTP analogs: a novel class of potent competitive adenylyl cyclase inhibitors. J. Biol. Chem. 278, 12672–12679 [DOI] [PubMed] [Google Scholar]
- 9. Newton M., Niewczas I., Clark J., Bellamy T. C. (2010) A real time fluorescent assay of the purified nitric oxide receptor, guanylyl cyclase. Anal. Biochem. 402, 129–136 [DOI] [PubMed] [Google Scholar]
- 10. Göttle M., Dove S., Steindel P., Shen Y., Tang W. J., Geduhn J., König B., Seifert R. (2007) Molecular analysis of the interaction of Bordetella pertussis adenylyl cyclase with fluorescent nucleotides. Mol. Pharmacol. 72, 526–535 [DOI] [PubMed] [Google Scholar]
- 11. Mou T. C., Gille A., Fancy D. A., Seifert R., Sprang S. R. (2005) Structural basis for the inhibition of mammalian membrane adenylyl cyclase by 2′(3′)-O-(N-methylanthraniloyl)-guanosine 5′-triphosphate. J. Biol. Chem. 280, 7253–7261 [DOI] [PubMed] [Google Scholar]
- 12. Mou T. C., Gille A., Suryanarayana S., Richter M., Seifert R., Sprang S. R. (2006) Broad specificity of mammalian adenylyl cyclase for interaction with 2′,3′-substituted purine and pyrimidine nucleotide inhibitors. Mol. Pharmacol. 70, 878–886 [DOI] [PubMed] [Google Scholar]
- 13. Koglin M., Behrends S. (2000) Cloning and functional expression of the rat α(2) subunit of soluble guanylyl cyclase. Biochim. Biophys. Acta. 1494, 286–289 [DOI] [PubMed] [Google Scholar]
- 14. Haase N., Haase T., Kraehling J. R., Behrends S. (2010) Direct fusion of subunits of heterodimeric nitric oxide sensitive guanylyl cyclase leads to functional enzymes with preserved biochemical properties: evidence for isoform specific activation by ciguates. Biochem. Pharmacol. 80, 1676–1683 [DOI] [PubMed] [Google Scholar]
- 15. Kraehling J. R., Busker M., Haase T., Haase N., Koglin M., Linnenbaum M., Behrends S. (2011) The amino-terminus of nitric oxide sensitive guanylyl cyclase alpha(1) does not affect dimerization but influences subcellular localization. PLoS One 6, e25772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Youvan D. C., Silva C. M., Bylina E. J., Coleman W. J., Dilworth M. R., Yang M. M. (1997) Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads. Biotechnol. Alia 3, 1–18 [Google Scholar]
- 17. Lakowicz J. R. (1999) Principles of Fluorescence Spectroscopy, 2nd Ed., pp. 63–95, 443–475, Kluwer Academic/Plenum, New York [Google Scholar]
- 18. Wallrabe H., Periasamy A. (2005) Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 16, 19–27 [DOI] [PubMed] [Google Scholar]
- 19. Schultz G., Böhme E. (1984) Guanylate cyclase. GTP pyrophosphate-lyase (cyclizing) EC 4.6.1.2 in Method of Enzymatic Analysis (Bergmeyer H. U., Bergmeyer J., Grassl M., eds.), 3rd Ed., Verlag Chemie, pp 379–389, Weihheim, Germany [Google Scholar]
- 20. Wedel B., Humbert P., Harteneck C., Foerster J., Malkewitz J., Böhme E., Schultz G., Koesling D. (1994) Mutation of His-105 in the β1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 91, 2592–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miyawaki A., Llopis J., Heim R., McCaffery J. M., Adams J. A., Ikura M., Tsien R. Y. (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887 [DOI] [PubMed] [Google Scholar]
- 22. Ma X., Sayed N., Beuve A., van den Akker F. (2007) NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 26, 578–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olea C., Boon E. M., Pellicena P., Kuriyan J., Marletta M. A. (2008) Probing the function of heme distortion in the H-NOX family. ACS Chem. Biol. 3, 703–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma X., Beuve A., van den Akker F. (2010) Crystal structure of the signaling helix coiled-coil domain of the β-1 subunit of the soluble guanylyl cyclase. BMC Struct. Biol. 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anantharaman V., Balaji S., Aravind L. (2006) The signaling helix: a common functional theme in diverse signaling proteins. Biol. Direct 1, 10.1186/1745-6150-1-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fritz B. G., Roberts S. A., Ahmed A., Breci L., Li W., Weichsel A., Brailey J. L., Wysocki V. H., Tama F., Montfort W. R. (2013) Molecular model of a soluble guanylyl cyclase fragment determined by small-angle X-ray scattering and chemical cross-linking. Biochemistry 52, 1568–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Underbakke E. S., Iavarone A. T., Marletta M. A. (2013) Higher-order interactions bridge the nitric oxide receptor and catalytic domains of soluble guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 110, 6777–6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koglin M., Behrends S. (2003) A functional domain of the alpha1 subunit of soluble guanylyl cyclase is necessary for activation of the enzyme by nitric oxide and YC-1 but is not involved in heme binding. J. Biol. Chem. 278, 12590–12597 [DOI] [PubMed] [Google Scholar]
- 29. Yoo B. K., Lamarre I., Rappaport F., Nioche P., Raman C. S., Martin J. L., Negrerie M. (2012) Picosecond to Second Dynamics Reveals a Structural Transition in Clostridium botulinum NO-Sensor Triggered by the Activator BAY-41–2272. ACS Chem. Biol. 7, 2046–2054 [DOI] [PubMed] [Google Scholar]
- 30. Purohit R., Weichsel A., Montfort W. R. (2013) Crystal structure of the Alpha subunit PAS domain from soluble guanylyl cyclase. Protein Sci. 22, 1439–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Russwurm M., Koesling D. (2004) NO activation of guanylyl cyclase. EMBO J. 23, 4443–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martin F., Baskaran P., Ma X., Dunten P. W., Schaefer M., Stasch J. P., Beuve A., van den Akker F. (2010) Structure of cinaciguat (bay 58-2667) bound to nostoc H-NOX domain reveals insights into heme-mimetic activation of the soluble guanylyl cyclase. J. Biol. Chem. 285, 22651–22657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma X., Sayed N., Baskaran P., Beuve A., van den Akker F. (2008) PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J. Biol. Chem. 283, 1167–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winger J. A., Derbyshire E. R., Lamers M. H., Marletta M. A., Kuriyan J. (2008) The crystal structure of the catalytic domain of a eukaryotic guanylate cyclase. BMC Struct. Biol. 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]