

Background: TorsinB is an AAA+ ATPase of unknown function.
Results: ATPase-arrested TorsinB induces the formation of LULL1-enriched, luminally constricted ER membranes requiring a highly conserved C-terminal motif in TorsinB.
Conclusion: Membrane structures formed by the TorsinB dominant-negative mutant are dependent on association with ATPase-activating factor LULL1.
Significance: This study supports a role for TorsinB in membrane dynamics.
Keywords: ATPases, Electron Microscopy (EM), Endoplasmic Reticulum(ER), Enzyme Mechanisms, Membrane Structure, AAA+ ATPase, Torsin
Abstract
Torsins are membrane-tethered AAA+ ATPases residing in the nuclear envelope (NE) and endoplasmic reticulum (ER). Here, we show that the induction of a conditional, dominant-negative TorsinB variant provokes a profound reorganization of the endomembrane system into foci containing double membrane structures that are derived from the ER. These double-membrane sinusoidal structures are formed by compressing the ER lumen to a constant width of 15 nm, and are highly enriched in the ATPase activator LULL1. Further, we define an important role for a highly conserved aromatic motif at the C terminus of Torsins. Mutations in this motif perturb LULL1 binding, reduce ATPase activity, and profoundly limit the induction of sinusoidal structures.
Introduction
Torsins are AAA+ (ATPases associated with a variety of cellular activities)2 ATPases (1) that reside within the endoplasmic reticulum (ER) and the contiguous nuclear envelope (NE). Of the four Torsins present in humans (TorsinA, TorsinB, Torsin2A, and Torsin3A), TorsinA (TorA) is the best studied due to its association with the disease Early Onset Torsion Dystonia, a disorder caused by the deletion of a glutamate residue (TorAΔE) near the C terminus of the protein (2). This mutation leads to perinuclear inclusions in the cell (3–7), and strongly compromises TorA binding to lamina-associated polypeptide 1 (LAP1) and the related protein LULL1 (8, 9), which are type II transmembrane proteins that reside in the nuclear envelope and ER, respectively (10).
Considerably less is known about TorsinB (TorB), despite its similarity to TorsinA at the sequence level. TorsinA and TorsinB are both ubiquitously expressed in all cell types though TorsinA is more highly expressed in neurons (11). A homozygous knock-out of TorsinA in mice is lethal and causes blebbing of the nuclear membrane in the perinuclear space specifically in neurons (6). A similar phenotype was observed in the nuclear envelope of TorsinA-deficient Drosophila larvae (12). When TorsinB is also depleted in TorsinA-deficient mouse embryonic fibroblasts, the blebbing phenotype is observed in these non-neuronal cells (13). This suggests that TorsinA and TorsinB may be somewhat functionally redundant, with TorsinA being more important in neuronal cells and TorsinB being more important in other tissues (13).
A similar phenotype is seen in LAP1-deficient mice, however the nuclear blebbing phenotype is no longer restricted to neuronal tissues (13). Although both LAP1 and LULL1 function similarly to activate the dormant ATPase activity of TorsinA and TorsinB (9), only LAP1 localizes exclusively to the NE due to its association with the nuclear lamina (14). This leads us to speculate that a LAP1 knock-out might be equivalent to a TorA/TorB double knock-out, at least with respect to their putative function in nuclear envelope dynamics (9). Much less is known about the physiological significance of LULL1. Depletion of LULL1 via siRNA does not cause a NE blebbing phenotype (15), and a LULL1-deficient animal model has not been reported so far. LULL1 is the more potent ATPase activator in comparison to LAP1, in particular when engaging TorsinB (9). The fact that LULL1 is not enriched in the NE relative to the ER thus suggests a possible function for this ATPase/cofactor system in the ER.
Here we utilize a conditional TorsinB trap allele as a novel strategy to induce the formation of structures similar to those categorized as organized smooth ER (OSER) (16). This unusual ER expansion can be broadly categorized as sinusoidal ER, with unique topological properties that were not previously reported in OSER structures (16, 17). Importantly, the membrane-molding activity of TorsinB depends on LULL1, leading us to propose a novel role for this activating cofactor.
EXPERIMENTAL PROCEDURES
Construction of TorsinB E178Q Stable Cell Line
TorsinB E178Q stable cell line was derived from HeLa cells (ATCC) using the retroviral “Tet-On” system. The TorB E178Q gene was amplified by PCR and cloned via standard procedures into the pRetroX-Tight-Pur vector (Clontech). The vector was packaged into retrovirus particles by cotransfecting it with a VSV-G envelope plasmid and a Gag-Pol retroviral packaging plasmid from Murine Leukemia Virus into HEK293T cells. Virus was harvested by collecting the supernatant from infected cells. Supernatant containing virus and 8 μg/ml polybrene (Millipore) were added to HeLa cells already stably containing the transactivating “Tet-On” plasmid (created using a similar procedure) overnight at 37 °C with 5% CO2. Cells were then selected in DMEM supplemented with 10% FBS, geneticin, and puromycin to isolate those containing both the “Tet-On” transactivator and the TorsinB E178Q construct.
Constructs for Mammalian Cell Expression and Transfections
All constructs were cloned into pcDNA3.1+ using standard PCR-based procedures. LULL1 luminal domain used was cloned as in (9). TorsinB E178Q (used for transient transfections) was cloned with a C-terminal HA tag. HEK293T (ATCC) and HeLa cells (ATCC) were grown in DMEM supplemented with 10% FBS in a humidified environment with 5% (v/v) CO2 at 37 °C. HEK293T cells were transfected with Lipofectamine 2000 (Invitrogen). HeLa cells (and the HeLa-derived TorsinB E178Q stable cell line) were transfected with X-tremeGENE 9 (Roche). All experiments were performed 24 h post transfection unless otherwise stated.
Immunofluorescence
TorsinB E178Q overexpression from stable cell lines was induced with 500 ng/ml doxycycline for 24 h at 37 °C. Cells were fixed with 4% paraformaldehyde and permeablized with 0.1% Triton X-100 before being blocked in 4% BSA/PBS followed by incubation with primary antibodies. After washing, cells were incubated with Alexa 488/568 secondary antibodies (Invitrogen) followed by a wash with Hoechst33342 (Invitrogen) in PBS and mounting on slides using Fluoromount-G (Southern Biotech). Images were acquired at room temperature using a Zeiss Axio Observer D1 microscope with a 63×/1.4 oil immersion lens and an AxioCam MRm.
Electron Microscopy
Electron microscopy was carried out by the Yale Biological Electron Microscopy facility using the following procedure: cells were fixed in 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4, for 1 h at room temperature. After rinsing, the cells were incubated with 0.1% tannic acid in 0.1 m sodium cacodylate for 30 min, then rinsed 3 times in sodium cacodylate rinse buffer and pelleted in 2% agar. Trimmed blocks were postfixed in 1% osmium tetroxide for 1 h, en bloc stained in 2% aqueous uranyl acetate at pH 5.2 for another hour then rinsed, dehydrated and infiltrated with Embed 812 (Electron Microscopy Science) and baked overnight at 60 °C. Hardened blocks were cut using a Leica UltraCut UC7. Sixty nanometer sections were collected on formvar/carbon-coated nickel grids and stained using 2% uranyl acetate and lead citrate. For immunolabeling of resin sections, grids were placed section side down on drops of 1% hydrogen peroxide for 5 min, rinsed, and blocked for nonspecific binding with 3% bovine serum albumin in Tris-buffered saline (TBS) containing 1% Triton X-100 for 30 min. Grids were incubated with rabbit anti-TorB (Covance) primary antibody 1:100 overnight, rinsed in TBS then incubated with 10 nm protein A gold (UtrechtUMC) for 30 min. The grids were rinsed in PBS, fixed using 1% glutaraldehyde for 5 min, rinsed again, dried, and heavy metal stained using 2% aqueous uranyl acetate and lead citrate. Samples were all viewed on a FEI Tencai Biotwin TEM at 80 kV. Images were taken using Morada CCD and iTEM (Olympus) software.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting were carried out as described in (9) with the following changes: samples were immunoprecipitated overnight at 4 °C and washed three times before dissociation. Additionally, immunoblots were detected with Amersham Biosciences Hyperfilm ECL (GE Healthcare).
Metabolic Labeling Pulse-Chase
TorsinB E178Q and parental Tet-On HeLa cells were grown in 10-cm dishes with DMEM containing 10% FBS, 100 μg/ml geneticin, and 1 μg/ml puromycin (TorsinB E178Q cells only). Twenty-four hours before the experiment the cells were induced with 500 ng/ml doxycycline. For the metabolic labeling the cells were washed, trypsinized, and starved for 30 min in DMEM containing 10% cysteine/methionine-free FBS. The cells were then pulse-labeled for 10 min with 1000 μCi/ml [35S]cysteine/methionine and then chased for 0, 30, or 60 min in DMEM containing 10% FBS and an excess of unlabeled cysteine/methionine. The cells were lysed in 1% SDS, and the lysates were immunoprecipitated with anti-HLA-A antibody (abcam ab52922) on protein A beads. The beads were then washed and treated with EndoH, PNGaseF, or buffer at 37 °C for 1 h. The supernatant from the MHC-I immunoprecipitation was re-immunoprecipitated with polyclonal anti-TorsinB (Covance) on protein A beads.
siRNA Knockdown
A LULL1 siRNA SMARTpool (Fisher Scientific) was transfected into TorsinB E178Q stable cell line using Lipofectamine RNAiMAX (Invitrogen). After 24 h the transfection agent was removed and fresh DMEM supplemented with 10% FBS was added. Cells were incubated at 37 °C for another 48 h (a total of 72 h post-transfection) before being harvested for immunoblotting and immunofluorescence.
Antibodies
Antibodies were purchased from the following sources: Anti-HA (3F10), Roche. Anti-Calnexin, anti-protein-disulfide isomerase (PDI), anti-LaminB1, anti-HLA-A, Abcam. Anti-Sec61β, Millipore. Anti-Emerin, Santa Cruz Biotechnology. Anti-HA conjugated to horseradish peroxidase (HRP), Roche. Anti-rabbit HRP, Southern Biotech. Anti-chicken, anti-rabbit, and anti-rat conjugated to Alexa fluors were purchased from Invitrogen. Chicken anti-LAP1 and chicken anti-LULL1 were raised against the N-terminal portion of each protein (Covance). Rabbit anti-LULL1 was raised against the luminal domain of LULL1 (Covance). Anti-TorsinB was raised using purified TorsinB lacking the signal sequence and N-terminal hydrophobic domain (Covance).
Measurement of ATPase Activity
TorsinB purification and measurement of ATPase activity were performed as previously described (9). For mixing of TorB WT with TorB E178Q, 3 μm WT was mixed with 30 μm TorB E178Q and 2 mm ATP for 5 min at 37 °C before adding 3 μm LULL1LD to start the reaction. For non-mixing reactions 3 μm WT was incubated with 2 mm ATP for 5 min at 37 °C before adding 3 μm LULL1LD or LAP1LD to start the reaction. Data analysis was performed in Microsoft Excel.
Single Cell Analysis of Fluorescence Intensity
Cells were processed for immunofluorescence as described above. Random fields of view were imaged using AxioVision release 4.8 with identical exposure times for every image. Individual cells were then outlined using the corresponding brightfield image of the cell and average fluorescence intensity in the green channel was measured using ZEN 2012 lite. Cells were binned by average fluorescence intensity and assessed as foci-positive or foci-negative. Chi-squared analysis to determine significance was performed in Microsoft Excel.
RESULTS
Generation of Stable Cell Lines Expressing a Dominant-negative Torsin Variant
We set out to investigate the putative function of TorsinB in membrane dynamics using a dominant-negative approach. This strategy was chosen since four Torsins are encoded in the human genome, and we expect that the established redundancy between TorsinA and TorsinB (13) would complicate a knockdown or knock-out approach. Moreover, dominant-negative strategies are of proven efficacy in studying the function of AAA+ ATPases and GTPases in membrane dynamics, in particular when redundancy complicates the analysis of phenotypes (18, 19).
We used our previously established in vitro system (9) to determine how much of the TorsinB dominant-negative mutant (TorsinB E178Q) is required to inactivate wild type (WT) TorsinB. To this end, we incubated TorsinB WT in the presence and absence of LULL1. Consistent with our previous observations (9), ATPase activity was strictly dependent on the presence of the ATPase activator LULL1 (Fig. 1A). As expected, TorsinB E178Q did not display ATPase activity in the same setting (Fig. 1A), since the Walker B mutation arrests AAA ATPases in the ATP-bound state (20). We then mixed TorsinB WT and TorsinB E178Q and determined the remaining ATPase activity of WT TorsinB. As judged by monitoring ATPase activity, it takes a 10-fold excess of TorsinB E178Q to reproducibly see a greater than 95% reduction in wild type TorsinB ATPase activity (Fig. 1A). Given this requirement to achieve the dominant-negative effect, we set out to create stable cell lines that mimicked that level of high overexpression.
FIGURE 1.

Overexpression of a TorsinB dominant-negative “trap” mutant induces cytoplasmic foci. A, ATPase activity of TorsinB wild type in the absence or presence of activating cofactor LULL1 and the dominant-negative TorsinB E178Q mutant. Phosphate release was measured using a malachite green assay after 1 h as described previously (9) B, TorsinB E178Q dominant-negative mutant expressed from HeLa-derived stable cell lines by addition of 500 ng/ml doxycycline. Immunoblot was probed with anti-TorsinB antibody. Control cells are parental cells of the protein-producing stable cell line containing only the “Tet-On” plasmid. Bands corresponding to TorsinB and its glycosylated forms are indicated. Arrowhead: TorsinB degradation product. Numbers on the left refer to molecular mass in kilodaltons. C, immunofluorescent images of same stable cell line as in B uninduced or induced with 500 ng/ml doxycycline. D, single cell analysis of foci formation in TorsinB E178Q overexpressing cells (black bars) compared with TorsinB wild type-overexpressing cells (gray bars) after 24 h treatment with doxycycline to overexpress their respective proteins. (*): p < 0.05. E, immunofluorescent images of TorsinB E178Q cell line induced with 500 ng/ml doxycycline for 24 h and costained with antibodies against endogenous LULL1 or LAP1. All cells were stained with anti-TorsinB antibody. Scale bar: 10 μm.
To this end, we established stable cell lines using the “Tet-On” system to produce TorsinB E178Q in HeLa cells under control of the tetracycline responsive element (see “Experimental Procedures”). To confirm expression of the TorsinB E178Q mutant in this stable cell line, cells were treated with 500 ng/ml doxycycline for 24 h, lysed, and subjected to SDS-PAGE and immunoblotting using anti-TorsinB antiserum. The TorsinB E178Q cell line is “leaky” in that it overproduces the protein of interest to some extent without doxycycline treatment compared with the control cells, which are HeLa cells containing only the “Tet-On” transgene (Fig. 1B). Upon induction with doxycycline, the TorB E178Q cell line significantly up-regulates the production of TorsinB E178Q, which has a slightly lower electrophoretic mobility (equivalent to a higher position on the immunoblot) than WT due to the charge difference resulting from the mutation (Fig. 1B). The double band can be attributed to differentially glycosylated versions of TorB E178Q which partially overlap with the endogenous protein also being recognized by our antibody. The overexpression of TorsinB E178Q also results in the appearance of a minor band of lower molecular mass, which is likely a degradation product of TorsinB (Fig. 1B, arrowhead).
TorsinB E178Q Induces Foci That Are Derived from the ER
Next, the TorsinB E178Q cell line was analyzed by indirect immunofluorescence to assess the subcellular localization of TorsinB E178Q. In the absence of doxycycline, TorsinB is partitioned between the ER and nuclear envelope (Fig. 1C). However, upon doxycycline addition TorsinB E178Q relocalizes to distinct, brightly fluorescent, punctate structures at the nuclear periphery and in the cytoplasm (Fig. 1C), which are reminiscent of the structures seen upon expression of the TorsinA dystonia mutant (TorAΔE) (3, 21–23).
To confirm the specificity of these structures to the E178Q mutant, we tested whether TorsinB WT also forms these structures. To this end, we did a single cell analysis of percent foci-positive cells compared with level of protein expression (quantified by measuring average cellular fluorescence) (Fig. 1D). Overexpression of wild type TorsinB does result in the formation of somewhat similar foci, but there are significantly fewer foci-positive cells at equivalent protein expression levels (using a chi-squared test: <500: p = 0.03, 500–749: p = 0.0002, 750–1000: p = 0.02, >1000: p = 0.03). This intermediate phenotype is not surprising given that Torsins require stoichiometric amounts of their activating cofactors LAP1 or LULL1 to hydrolyze ATP (9). Overexpression of wild type TorsinB results in cofactor levels being limiting since we are not co-overexpressing them and most of the TorsinB will be locked in an ATP-bound state similar to the E178Q mutant. Therefore, it is likely still the ATP-bound TorsinB that is responsible for these structures.
Given that the related ATPase TorsinA preferentially interacts with LAP1 and LULL1 in the ATP-bound state (8, 9), we next tested whether these regulatory cofactors are also present in these structures. Surprisingly, only LULL1 was found to colocalize with TorsinB, whereas the bulk of LAP1 remained confined to the nuclear envelope (Fig. 1E).
As a first step in further characterizing the foci seen upon overexpression of TorsinB E178Q, we used indirect immunofluorescence to determine if suitable marker proteins colocalized to these foci. Given that TorsinB is a luminal, membrane-associated glycoprotein that localizes to the ER and perinuclear space (24), it is likely that these foci are associated with membranes, and the two most likely origins of the membranes are the endoplasmic reticulum or the nuclear membrane.
To investigate the identity of the foci, TorsinB E178Q-HA was transiently transfected into HeLa cells or untagged TorsinB E178Q was overexpressed via doxycycline induction from the HeLa-derived stable cell line. Both tagged and untagged TorsinB were used because the antibody against TorsinB is of the same serotype as the majority of the compartmental marker antibodies, necessitating use of an anti-HA antibody as a TorsinB marker for most experiments. Both transient transfection and overexpression from the stable cell line produce identical foci as judged by immunofluorescence (see Figs. 1C and 2). As ER markers we chose the proteins calnexin (Cxn), protein-disulfide isomerase (PDI), Sec61, and a general ER marker (anti-KDEL) while inner nuclear membrane markers included LaminB1, Sun2, and Emerin.
FIGURE 2.

Characterization of TorsinB E178Q foci with compartmental markers. A, immunofluorescent images of transiently transfected TorsinB E178Q-HA (TorBEQ-HA) or TorsinB E178Q overexpressed from doxycycline-induced stable cell lines (TorBEQ) stained with standard ER markers calnexin, PDI, Sec61, and KDEL. B, immunofluorescent images of transiently transfected TorsinB E178Q-HA (TorBEQ-HA) stained with antibodies against inner nuclear membrane proteins LaminB1, Sun2, and Emerin. Green: TorsinB or HA; Red: compartmental marker proteins; Scale bar: 10 μm.
Calnexin is a membrane-associated resident ER protein, and as such, normally gives a reticular staining characteristic of the ER. In cells overexpressing TorsinB E178Q, calnexin is relocalized into punctate foci at the nuclear periphery and in the cytoplasm. These calnexin-positive foci overlay with some (though not all) of the TorsinB-positive foci in those cells. A low level of calnexin is also localized outside of the foci (Fig. 2A, top panels). The colocalization of calnexin with TorsinB in these foci is not wholly surprising because, in addition to both being residents of the ER under normal cellular conditions, calnexin is a known interaction partner of the related ATPase TorsinA (8, 9). In any case, the overlapping localization of calnexin and TorsinB in these foci suggests that the foci are derived from the ER.
To confirm the proposed identity, we chose PDI as a soluble ER-resident protein that yields a reticular ER stain under normal cellular conditions. Upon overexpression of TorsinB E178Q, PDI still gives a nearly typical ER stain, but there are also isolated patches of staining in the cytoplasm. A small subset of dimmer isolated patches colocalize with the TorsinB foci (Fig. 2A, middle panels). Therefore, similar to Calnexin, PDI partially colocalizes to the TorsinB foci, although the extent of colocalization is considerably less pronounced possibly because PDI is not known to interact with TorsinB.
We also tested the ER marker Sec61, a constituent of the translocon (25). When TorsinB E178Q is overexpressed, Sec61 relocalizes from a typical ER stain to punctate structures that colocalize with the TorsinB foci (Fig. 2A, middle panels), again suggesting that the TorsinB foci are indeed derived from the ER.
Lastly, as a more general ER marker, we chose to use an antibody against the ER retrieval signal (anti-KDEL), which should stain for many ER proteins. Upon overexpression of TorsinB E178Q the anti-KDEL antibody staining looks very similar to a typical ER stain (Fig. 2A, bottom panels). KDEL-containing proteins do not appear to be enriched or excluded from the TorsinB E178Q foci, which is similar to the PDI staining (Fig. 2A, middle panels). Since KDELs are found on many soluble ER proteins (26), and PDI is also a soluble ER protein, the enrichment of ER proteins in these TorsinB E178Q-induced structures may be specific to membrane proteins.
Given that TorsinB is located in the perinuclear space and nuclear membranes due to their continuity with the ER, we also examined if inner nuclear membrane proteins colocalized with the TorsinB foci. We first examined LaminB1 as a marker of the nucleoskeletal lamin network that underlies the inner nuclear membrane. In normal cells LaminB1 gives a very distinct nuclear rim stain. This staining pattern is unaffected by the overexpression of TorsinB E178Q (Fig. 2B, top panels), implying that overall nuclear architecture is left intact despite TorsinB foci formation.
Next we determined the localization of INM protein Sun2, which is part of the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex that bridges the inner nuclear membrane and the outer nuclear membrane connecting the lamin network in the nucleus with the cellular cytoskeletal filaments (27). Another component of these complexes (Nesprin3) has been shown to interact with TorsinA (28). Upon overexpression of TorsinB E178Q, the normal nuclear rim staining seen for Sun2 changes profoundly. The nuclear rim staining becomes less pronounced and some of the protein is relocalized to small punctae in the cytoplasm (Fig. 2B, middle panels). However, neither the rim staining nor these small punctae colocalized with the much larger TorsinB E178Q foci, indicating that these foci are not necessarily derived from the inner nuclear membrane, though the overexpression of TorsinB E178Q does have some effect on the localization of Sun2.
Lastly, we observed the localization of INM resident protein Emerin in the presence of the TorsinB mutant. Similar to Sun2, the normal nuclear rim staining for Emerin becomes less pronounced upon overexpression of TorsinB E178Q and the protein appears to relocalize to punctae in the cytoplasm. However, unlike Sun2, these punctae of Emerin colocalize strongly with the TorsinB foci (Fig. 2B, bottom panels). Given that Emerin is the only inner nuclear membrane protein tested that colocalizes with the TorsinB foci, these foci are not necessarily derived from nuclear membranes, but instead, TorsinB has some effect on Emerin specifically.
Overexpression of TorsinB E178Q Leads to Formation of Sinusoidal ER
To further scrutinize the structure of the foci that appear upon overexpression of TorsinB E178Q, we employed electron microscopy (EM). The TorsinB E178Q cell line or corresponding control cell line was induced with doxycyline for 24 h, fixed, and embedded in resin. Osmium tetroxide was used as the stain, allowing us to examine ultrastructural differences between these thin-sectioned samples and in particular, changes in membrane morphology.
Control cells show normal cellular morphology with an evenly-spaced perinuclear space (Fig. 3A). Canonical rough ER can also be seen just outside of the nucleus in Fig. 3A. There are several profound differences in cellular organization upon overexpression of TorsinB E178Q compared with the control cells. First, TorsinB E178Q-expressing cells display a perinuclear vesicle phenotype (Fig. 3C, arrowhead) that was previously observed in TorsinA and LAP1 “knock-out” mouse models (13), thus validating our dominant-negative approach. Even more striking is the additional presence of organized undulating membranes at the nuclear periphery in the cytoplasm (Fig. 3, B and C). Upon closer examination these structures appear to be connected to the outer nuclear membrane in a manner similar to normal ER, again suggesting that these structures are ER-derived. However, in contrast to canonical rough ER, these structures are largely devoid of ribosomes (see Fig. 3, A and C). These highly unusual membranes have a repeating unit or minimal building block consisting of a double-membrane bleb that, depending on the bleb, appears to be at a different stage of being pinched off. This periodicity is the hallmark of Tor E178Q-overexpressing cells, best seen in isolated cytoplasmic foci (Fig. 3D). Most of the repeating structures also share the common feature of an electron-dense, fuzzy patch at the neck (Fig. 3D). These structures are morphologically similar to a subclass of organized smooth ER (OSER) structure termed sinusoidal ER (17). As we shall discuss in more detail below, it appears that it is the ER lumen that is constricted between the two tightly apposed, undulating membranes (Fig. 3, E and F). This constriction of the ER lumen may explain why only membrane-bound ER resident proteins are enriched in these structures and not soluble luminal proteins.
FIGURE 3.

Reorganization of the endoplasmic reticulum by a TorsinB trap variant. A, EM image of control parental “Tet-On” cells showing normal ER and nuclear envelope morphology. B, low magnification image of HeLa-derived stable cell line overexpressing TorsinB E178Q showing a reorganization of cytoplasmic membranes into sinusoidal ER structures. C, high magnification image of section depicted in B showing a perinuclear vesicle (arrowhead). D, EM image of a representative isolated cytosolic focus from TorsinB E178Q overexpressing cells induced with 500 ng/ml doxycycline for 24 h. E, EM image showing connectivity of TorB E178Q-induced structures. Sinusoidal ER membranes can be seen forming as branches emanating from ER tubules (arrows). F, schematic representation of sinusoidal ER structures. The arrow marks an analogous position to the arrow in panel E. N: nucleus. NPC, nuclear pore complex.
TorsinB Is Highly Enriched in Sinusoidal ER Structures
Despite the obvious morphological differences between TorsinB E178Q overexpressing cells and the control cells, it was still unclear whether these patches of sinusoidal ER were the same as the TorsinB E178Q-positive foci seen in indirect immunofluorescence. To confirm the identity of the structures, we used immunogold labeling in TorsinB E178Q-overexpressing cells to confirm the enrichment of TorsinB in these structures.
TorsinB E178Q was overexpressed from the HeLa-derived stable cell line by treating the cells with doxycycline for 24 h before fixing and embedding the cells. The cells were embedded in resin before immunolabeling instead of cryo-freezing in an attempt to better preserve the membrane morphology of the cells. Cells were labeled with the same anti-TorsinB antibody used for immunofluorescence, followed by incubation with proteinA-conjugated gold beads.
As expected, the gold particles are highly enriched in the sinusoidal ER structures (Fig. 4, A–C) indicating that these structures contain a high concentration of TorsinB E178Q. We thus conclude that these structures are identical to the TorsinB-enriched foci observed via immunofluorescence. In addition to confirming the identity of these structures, immunolabeling also shows that TorsinB is distributed relatively uniformly throughout the structures and does not accumulate at any particular point. Outside of the structures, TorsinB immunostaining is also present on the outer nuclear membrane (Fig. 4B), which is consistent with its known localization (24).
FIGURE 4.
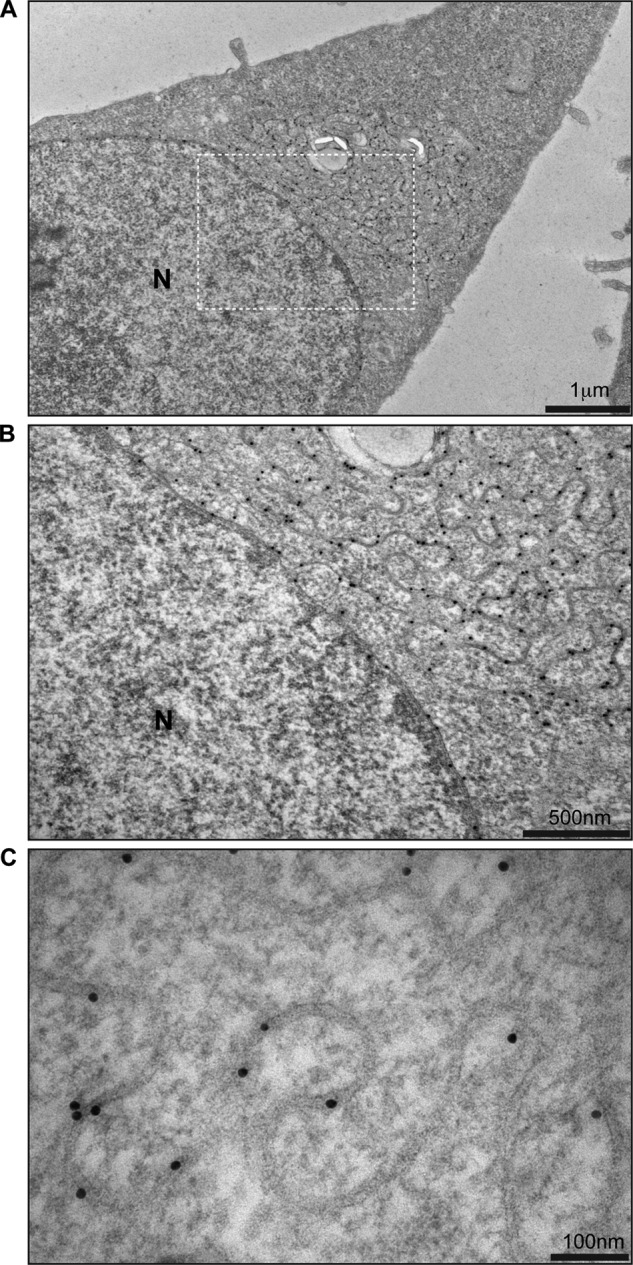
TorsinB E178Q immunostaining. A, low magnification image of HeLa-derived stable cell line overexpressing TorsinB E178Q immunostained with anti-TorsinB antibody and proteinA conjugated to gold. B, higher magnification image of section depicted in A. C, high magnification image of membrane structures associated with the overexpression of TorsinB E178Q. Gold particle diameter is 10 nm.
The immunogold labeling also allows us to measure the approximate distance between the two apposed membranes in the structures as seen in Fig. 4C. Given that the diameter of each gold particle is 10 nm, we estimate that the membranes are ∼15 nm apart, which, by comparison, is approximately one-third the width of the perinuclear space (29). This distance between membranes is fairly uniform along the length of the structures (Fig. 4C).
Overexpression of TorsinB E178Q Does Not Affect MHC-I Trafficking
Does the membrane proliferation and deformation found in cells overexpressing TorsinB E178Q interfere with the secretory pathway? We used major histocompatibility complex I (MHC-I) glycosylation as an indicator of proper functioning of the secretory pathway since its maturation is easily monitored using glycan processing as readout (30). Each cell line (the TorB E178Q expressing cell line and the control parental cell line) was induced for 24 h with 500 ng/ml doxycycline before being metabolically labeled with [35S]cysteine/methionine. After 24 h induction, the TorB E178Q foci are observed as a penetrant phenotype. Postlabeling, cells were “chased” with unlabeled cysteine and methionine. Lysates were prepared at distinct time points and subjected to IP using anti-HLA-A antibodies, followed by SDS-PAGE and autoradiography (Fig. 5A). TorsinB was immunoprecipitated from supernatants of the MHC-I IP samples to confirm that TorsinB E187Q was evenly expressed in those samples (Fig. 5B). At both 30 and 60 min after the chase in the untreated condition the band for MHC-I becomes wider relative to the zero time point (Fig. 5A), indicative of glycan processing. This assessment is confirmed by treatment of an aliquot of these immunoprecipitates with EndoH revealing that the glycans are becoming increasingly resistant to EndoH activity over time, consistent with the expected glycan maturation from the ER-resident high-mannose form at the zero time point to the complex forms that result from passage through the Golgi apparatus at later time points (30). As expected, all MHC-I species are sensitive to PNGase F, which removes all N-linked glycans from proteins irrespective of their precise composition (Fig. 5A). Importantly, the pattern of modification after treatment with EndoH is identical for cells overexpressing TorsinB E178Q or control cells, which indicates that the kinetics of MHC-I trafficking are not affected by overexpression of TorsinB E178Q despite profound changes in ER morphology.
FIGURE 5.

Effect of overexpressing TorsinB E178Q on MHC-I processing. A, autoradiogram of MHC-I immunoprecipitated from TorsinB E178Q or control cell lines at 0, 30, or 60 min post radiolabeling with 35S. Each sample was treated to remove high mannose N-glycans (EndoH) or all glycans (PNGaseF) as acquisition of EndoH resistance is a measure of transport of MHC-I through the golgi. B, supernatants from A were immunoprecipitated with anti-TorsinB antibody to confirm overexpression. Numbers on the left refer to molecular mass in kilodaltons.
Foci Formation Correlates with Endogenous LULL1 Levels
There are several reports of proteins whose overexpression causes changes in ER morphology (Ref. 17 and references cited therein). Much remains to be learned about cellular factors that might contribute to the formation of these induced structures. Given that LULL1 is the only known regulator of TorsinB activity that is found in TorB-positive foci (Fig. 1E), we tested whether LULL1 is implicated in foci formation. To this end, an anti-LULL1 siRNA SMARTpool was transfected into the TorsinB E178Q-inducible stable cell line 48 h before induction for 24 h with doxycycline. After 24 h of induction, cells from the same tissue culture well were either processed for immunofluorescence or harvested and lysed to assess the efficacy of the LULL1 depletion.
As expected, no TorsinB-containing foci were observed in uninduced cells, whereas one-third of the cells have TorsinB-positive foci after doxycycline induction (Fig. 6, A and B). Notably, when LULL1 was reduced to ∼30% of the normal cellular level via siRNA (Fig. 6C), the percentage of cells that are foci-positive significantly decreases to 17.3% of cells (Fig. 6, A and B). This indicates that foci formation is at least partially dependent on the presence of LULL1. It seems reasonable to propose that this difference would become even more pronounced upon complete depletion of LULL1.
FIGURE 6.

Dependence of TorsinB E178Q foci on the presence of LULL1. A, representative images of cells from TorsinB E178Q stable cell lines either uninduced or induced with 500 ng/ml doxycycline for 24 h. siLULL1 is treated with siRNA against LULL1 for 48 h pre-induction with doxycycline. B, quantitation of number of foci-positive cells in the conditions used in A. Each bar represents the mean of three separate samples of 100–110 cells each. Error bars: ± one S.D. C, immunoblot of samples from conditions as in A and B showing knockdown of LULL1 and overexpression TorsinB E178Q. (*): nonspecific bands
Aromatic C-terminal Residues of TorsinB Are Required for Efficient Binding to LULL1
If LULL1 is truly required for foci formation, then a TorsinB mutant that has a greatly reduced capacity for binding LULL1 should also decrease foci formation. To create such a mutant, we focused on the C termini of Torsins, since related AAA+ ATPases, e.g. components of the 19S proteasome and p97, interact with binding partners by virtue of C-terminal aromatic motifs (31, 32). Such aromatic residues are indeed conserved in the C termini of individual members of the Torsin family (Fig. 7A). We made two different types of C-terminal mutations in TorsinB. One mutation removes the last three amino acids at the C terminus (TorsinBΔ334), which removes all aromatic amino acids from that region (Fig. 7A, vertical line). The other mutation is a single point mutation that exchanges F335 for an alanine (Fig. 7A, box).
FIGURE 7.

Effect of C-terminal aromatic residue of TorsinB on LULL1 binding. A, domain diagram of TorsinB. Sequence comparison showing very C-terminal amino acids with boxed aromatic amino acid conserved between all human Torsins. Line demarcates beginning of truncation for TorBΔ334 construct. B, immunoprecipitation of LULL1LD-HA with various TorsinB constructs including the C-terminal mutants. Numbers on the left refer to molecular mass in kilodaltons. C, ATPase activity of TorsinB WT (circles) versus TorsinB-GGG (triangles) in the presence of varying concentrations of LULL1. Phosphate release was measured using a malachite green assay as described previously (9). D, ATPase activity of TorsinB WT (circles) versus TorsinB-GGG (triangles) in the presence of varying concentrations of LAP1. Phosphate release was measured using a malachite green assay as described previously (9). Kinetic parameters are adjacent to their corresponding curves.
To determine if any of the mutations altered the affinity of TorsinB for LULL1, we cotransfected the luminal domain of LULL1 (LULL1LD-HA) into 293T cells with TorsinB WT or TorsinB E178Q containing the various C-terminal mutations for 24 h and then performed a co-immunoprecipitation (Fig. 7B). LULL1LD-HA is able to retrieve TorsinB WT, while TorsinB WT Δ334 is retrieved less efficiently, and the TorsinB F335A has an affinity intermediate to WT and Δ334 (Fig. 7B). TorsinB E178Q binds much more strongly to LULL1LD-HA than TorsinB WT. This is expected since the E178Q mutation locks the ATPase into the ATP-bound state, which is the high affinity state for LULL1 in TorA (8, 9). In the TorsinB E178Q background, the Δ334 mutation drastically reduces the amount of TorsinB E178Q retrieved by LULL1LD-HA. The single point mutation of F335A in the TorsinB E178Q background again shows an intermediate amount of retrieval, similar to what is seen in the TorsinB WT background (Fig. 7B).
As a further validation of the importance of the C-terminal aromatic motif in association of TorsinB with LULL1, we expressed and purified a TorsinB mutant that is similar to the TorB Δ334 except that instead of truncating the last three amino acids, they have been replaced with glycines (TorB-GGG). We took this approach to further enforce the idea that it is the identity (not the presence) of the C-terminal residues that matter for this interaction.
To determine the effect of the TorB-GGG mutation on the association with LULL1, we tested the ability of LULL1 to stimulate the ATPase activity of TorB-GGG. Compared with TorB WT, the ATPase activity of an identical amount of TorB-GGG with LULL1 is considerably reduced (Fig. 7C). The apparent Km is approximately twice as high as that of wild type and its Vmax is reduced (Fig. 7C). Interestingly, the mutation has a similar effect on activation by TorsinB's other cofactor LAP1 as well (Fig. 7D). Taken together, these data indicate that the C-terminal aromatic motif of TorsinB is important for LULL1 binding.
Having established a mutant that is largely deficient in LULL1 binding, we next created a doxycycline-inducible cell line that expresses TorsinB E178Q Δ334 in the same manner as described for the TorsinB E178Q mutant. We then used this cell line to examine whether this mutant that binds LULL1 poorly still causes the same foci when overexpressed.
Upon addition of doxycycline the TorsinB staining in the TorsinBΔ334 cells becomes significantly brighter than in uninduced cells, which confirms that they properly induce (Fig. 8A). Since the formation of TorsinB E178Q foci is dependent on expression level of the protein (Fig. 1C), and we could not guarantee equivalent expression levels between cell lines, we did a single cell analysis of fluorescence on individual cells from each sample to compare cells expressing equivalent amounts of protein and determine if the Δ334 mutation had an effect on foci formation.
FIGURE 8.
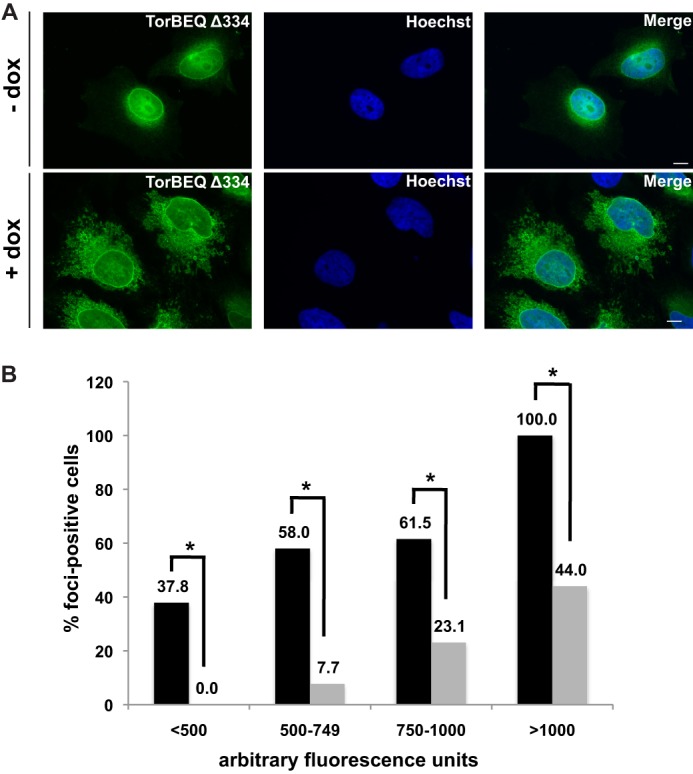
Effect of TorsinB C-terminal deletion on foci formation. A, immunofluorescent images of TorB E178Q Δ334 in the absence or presence of 500 ng/ml doxycycline for 24 h. B, single cell analysis of foci formation in TorB E178Q cell line (black bars) versus TorB E178Q Δ334 cell line (gray bars) after 24 h treatment with 500 ng/ml doxycycline to induce overexpression of their respective proteins. (*): p < 0.05.
For the single cell analysis, greater than 100 cells from each sample were imaged and analyzed for average fluorescence intensity. Cells were then binned according to their average fluorescence and assessed as foci-positive or foci-negative. For each bin, the percent of cells that were foci-positive for each sample was calculated and the percent of foci-positive cells for the TorB E178Q and TorB E178Q Δ334 were compared using a Chi squared test. While some foci do form in the TorB E178Q Δ334 cells at a higher level of expression, across all bins the difference between the TorB E178Q and TorB E178Q Δ334 samples was statistically significant (<500: p = 0.0002, 500–749: p = 9 × 10−7, 750–1000: p = 0.005, >1000: p = 0.0005), showing that the TorB E178Q Δ334 mutation has a strong effect on foci formation. This data, together with the decrease in foci formation upon LULL1 knockdown, leads us to conclude that the interaction between TorsinB and its activating factor LULL1 is important for formation of sinusoidal ER structures.
DISCUSSION
The shape of the cellular endomembrane system is dynamically controlled by the concerted action of proteins that impose or recognize membrane curvature, as well as by activities that promote the assembly or disassembly of the corresponding protein-protein or protein-lipid complexes (29, 33). The formation of Organized Smooth ER (OSER) structures (more specifically, the whorl and sinusoidal ER subclasses) is largely driven by homotypic interactions between the cytoplasmic domains of overexpressed ER membrane proteins (16, 34–37). These presumably anti-parallel interactions that keep the membranes tightly apposed are surprisingly weak and the proteins can freely diffuse within the structures (16). Nonetheless these interactions are important as evidenced by the lack of OSER formation when non-dimerizing versions of proteins that normally do form OSER structures were overexpressed (16, 37).
Overexpression of the dominant-negative, WalkerB “trap” mutant of TorsinB (TorB E178Q) leads to the formation of brightly fluorescent foci when viewed by immunofluorescence. Further examination via electron microscopy shows that, at an ultrastructural level, these foci are composed of sinusoidal ER membranes that feature two tightly apposed membranes separated by ∼15 nm (Fig. 4C). This type of sinusoidal ER is similar to one of the five subclasses of Organized Smooth ER (OSER) structures, which are generally characterized by an expansion of the smooth ER that organizes into membrane stacks or symmetric arrays (38).
TorsinB fits the profile of proteins that could cause OSER structures in that it is an ER resident membrane protein that is thought to homo-oligomerize (15). However, TorsinB does not fit the model of cytoplasmic dimerization because by analogy to TorA (39), it is expected to be a monotopic membrane protein that resides entirely within the ER lumen; it has no cytoplasmic domain. This raises the question of whether our sinusoidal ER structures induced by TorsinB E178Q are topologically identical to those OSER structures previously observed (34–36, 40). In the case of OSER formed by cytoplasmic dimerization, the space between the two tightly apposed membranes is the cytoplasm (16). However in the TorsinB-induced structures, that space is topologically equivalent to the ER lumen instead. Via electron microscopy, it is possible to see the two membranes come together as a branch emanating from an ER tubule, clearly sandwiching the lumen inside (Fig. 3, E and F). The immunogold labeling pattern for TorsinB (Fig. 4), which is itself a luminal protein, supports this interpretation. This topology could also explain why a soluble ER protein (PDI) is less abundant than other ER markers that are membrane proteins (Fig. 2A). Therefore, we believe that the sinusoidal structures induced by TorB E178Q are of inverse topology, i.e. “inside out” compared with most of those previously reported (34–36, 40). Notably, Lingwood et al. found that using ER membrane proteins that dimerize in the ER lumen created immunofluorescent structures indicative of OSER formation, although their topology was not scrutinized via EM (37). We suspect that these structures may more closely resemble the topology reported here.
Given that the formation of the TorB E178Q-induced structures drastically constricts the ER lumen, it would seem plausible that these structures interfere with normal ER function, in particular given their abundance relative to canonical ER in cells expressing the TorB trap (see Fig. 3, A and B). Surprisingly, the synthesis and trafficking of MHC-I as measure for the “kinetic fitness” of the early secretory pathway was unaffected by these structures (Fig. 5). It is possible that these structures result from an active mechanism that serves to ensure proper functioning of the ribosome-bound ER by spatial separation from a sinusoidal sub-compartment. It is noteworthy that the structures contain only non-translating ER as evidenced by their near-complete lack of ribosomes when examined by electron microscopy (Figs. 3, C and D), which means that the spatially separated translating ER may be unperturbed. It is interesting to note, however, that the ribosome docking channel Sec61 is not excluded from the TorsinB E178Q-induced structures (Fig. 2A) despite the fact that they are devoid of ribosomes.
An unresolved question about OSER structures in general is whether the formation of the structures is due to the dimerizing proteins themselves or the accompanying ER membrane expansion. Anti-parallel protein dimerization seems to explain the tightly apposed membranes characteristic of these structures; however, it is not obvious how dimerization of proteins alone would cause sinusoidal ER morphology. Given that these OSER structures can form from protein dimerization on either side of the ER membrane (this report, (37)) and that these dimerizing proteins are relatively evenly spaced throughout the structures (at many different types of curvature) (Fig. 4, B and C), it seems unlikely that the dimerizing proteins themselves are the sole cause of membrane curvature. Instead, the overall morphology of the structures may also be related to the properties of the lipids themselves, with characteristic segregation according to their overall shape in higher or lower curvature areas. The sinusoidal ER morphology may present the most efficient way of packing a tightly constricted ER into a cell, as was suggested previously (37).
Given that these sinusoidal ER and other OSER structures have been found to form in response to overexpression of various unrelated ER membrane proteins, it is impossible to conclude definitively that the formation of sinusoidal ER in presence of TorsinB E178Q has distinct implications for TorsinB's cellular activities. However, given that a “knockdown” of TorsinB in a TorsinA−/− background causes blebbing in the perinuclear space (13) and our own observation of analogous perinuclear blebs when dominant-negative TorsinB E178Q is overexpressed (Fig. 3C), our proposal that the formation of these OSER structures is related to TorsinB function remains reasonable.
Another argument for the possible functional relevance of OSER structure formation to TorsinB function is that we found that our sinusoidal ER structures were not only dependent on the levels of TorB E178Q but were also at least partially dependent on the levels of TorB's ATPase activating cofactor LULL1 (Fig. 6). It is possible that the interaction of the luminal domain of TorB with LULL1 (9) contributes to a higher order hetero-oligomer that is no longer present when LULL1 is depleted leading to fewer structures because the structures are highly dependent on protein-protein interactions. However, a more interesting possibility is that LULL1 modulates the oligomerization state of TorB and lowering the amount of LULL1 in the cell leads to dysregulation of this process. Alternatively, the cytosolic domain of LULL1 may contribute to the generation of curvature, which in turn may be influenced or regulated by association of TorsinB on the luminal side of the membrane. Finally, it is possible that a yet to be identified protein complex that acts as molecular ruler or membrane tether is either up-regulated or stabilized in the presence of TorB E178Q.
While formation of OSER structures by overexpression of ER-resident membrane proteins is not unprecedented, the TorsinB luminal topology and the resulting “inverted topology” of two tightly apposed membranes containing the ER lumen qualify the TorB E178Q-induced structures reported here as distinct from previously characterized OSER structures (34–36, 40). The appearance of these structures also supports the idea that TorsinB is involved in regulating membrane dynamics in the cell, possibly by modulating membrane spacing.
Acknowledgments
We thank Morven Graham for help with electron microscopy, Walther Mothes for reagents, and members of the Schlieker laboratory for comments on the manuscript.
This work was funded, in whole or in part, by National Institutes of Health Grant DP2 OD008624-01 and Ellison Medical Foundation Grant AG-NS-0662-10.
- AAA+
- ATPases associated with a variety of cellular activities
- ER
- endoplasmic reticulum
- NE
- nuclear envelope
- Tor
- Torsin
- PDI
- protein-disulfide isomerase
- OSER
- organized smooth ER.
REFERENCES
- 1. Neuwald A. F., Aravind L., Spouge J. L., Koonin E. V. (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 [PubMed] [Google Scholar]
- 2. Ozelius L. J., Hewett J. W., Page C. E., Bressman S. B., Kramer P. L., Shalish C., de Leon D., Brin M. F., Raymond D., Corey D. P., Fahn S., Risch N. J., Buckler A. J., Gusella J. F., Breakefield X. O. (1997) The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 17, 40–48 [DOI] [PubMed] [Google Scholar]
- 3. Hewett J., Gonzalez-Agosti C., Slater D., Ziefer P., Li S., Bergeron D., Jacoby D. J., Ozelius L. J., Ramesh V., Breakefield X. O. (2000) Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum. Mol. Genet. 9, 1403–1413 [DOI] [PubMed] [Google Scholar]
- 4. Gonzalez-Alegre P., Paulson H. L. (2004) Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J. Neurosci. 24, 2593–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodchild R. E., Dauer W. T. (2004) Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. U. S. A. 101, 847–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goodchild R. E., Kim C. E., Dauer W. T. (2005) Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48, 923–932 [DOI] [PubMed] [Google Scholar]
- 7. Naismith T. V., Heuser J. E., Breakefield X. O., Hanson P. I. (2004) TorsinA in the nuclear envelope. Proc. Natl. Acad. Sci. U. S. A. 101, 7612–7617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naismith T. V., Dalal S., Hanson P. I. (2009) Interaction of torsinA with its major binding partners is impaired by the dystonia-associated DeltaGAG deletion. J. Biol. Chem. 284, 27866–27874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao C., Brown R. S., Chase A. R., Eisele M. R., Schlieker C. (2013) Regulation of Torsin ATPases by LAP1 and LULL1. Proc. Natl. Acad. Sci. U. S. A. 110, E1545–E1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goodchild R. E., Dauer W. T. (2005) The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 168, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jungwirth M., Dear M. L., Brown P., Holbrook K., Goodchild R. (2010) Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum. Mol. Genet. 19, 888–900 [DOI] [PubMed] [Google Scholar]
- 12. Jokhi V., Ashley J., Nunnari J., Noma A., Ito N., Wakabayashi-Ito N., Moore M. J., Budnik V. (2013) Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Reports 3, 988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim C. E., Perez A., Perkins G., Ellisman M. H., Dauer W. T. (2010) A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. U. S. A. 107, 9861–9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foisner R., Gerace L. (1993) Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 73, 1267–1279 [DOI] [PubMed] [Google Scholar]
- 15. Vander Heyden A. B., Naismith T. V., Snapp E. L., Hodzic D., Hanson P. I. (2009) LULL1 retargets TorsinA to the nuclear envelope revealing an activity that is impaired by the DYT1 dystonia mutation. Mol. Biol. Cell 20, 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Snapp E. L., Hegde R. S., Francolini M., Lombardo F., Colombo S., Pedrazzini E., Borgese N., Lippincott-Schwartz J. (2003) Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Borgese N., Francolini M., Snapp E. (2006) Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hurley J. H., Hanson P. I. (2010) Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nature Reviews 11, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferguson S. M., De Camilli P. (2012) Dynamin, a membrane-remodelling GTPase. Nature Reviews. Molecular Cell Biology 13, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanson P. I., Whiteheart S. W. (2005) AAA+ proteins: have engine, will work. Nature Reviews. Molecular Cell Biology 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 21. Bragg D. C., Camp S. M., Kaufman C. A., Wilbur J. D., Boston H., Schuback D. E., Hanson P. I., Sena-Esteves M., Breakefield X. O. (2004) Perinuclear biogenesis of mutant torsin-A inclusions in cultured cells infected with tetracycline-regulated herpes simplex virus type 1 amplicon vectors. Neuroscience 125, 651–661 [DOI] [PubMed] [Google Scholar]
- 22. Kustedjo K., Bracey M. H., Cravatt B. F. (2000) Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J. Biol. Chem. 275, 27933–27939 [DOI] [PubMed] [Google Scholar]
- 23. Bragg D. C., Kaufman C. A., Kock N., Breakefield X. O. (2004) Inhibition of N-linked glycosylation prevents inclusion formation by the dystonia-related mutant form of torsinA. Mol. Cell Neurosci. 27, 417–426 [DOI] [PubMed] [Google Scholar]
- 24. Hewett J. W., Kamm C., Boston H., Beauchamp R., Naismith T., Ozelius L., Hanson P. I., Breakefield X. O., Ramesh V. (2004) TorsinB–perinuclear location and association with torsinA. J. Neurochem. 89, 1186–1194 [DOI] [PubMed] [Google Scholar]
- 25. Görlich D., Prehn S., Hartmann E., Kalies K. U., Rapoport T. A. (1992) A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell 71, 489–503 [DOI] [PubMed] [Google Scholar]
- 26. Munro S., Pelham H. R. (1987) A C-terminal signal prevents secretion of luminal ER proteins. Cell 48, 899–907 [DOI] [PubMed] [Google Scholar]
- 27. Crisp M., Liu Q., Roux K., Rattner J. B., Shanahan C., Burke B., Stahl P. D., Hodzic D. (2006) Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 172, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nery F. C., Zeng J., Niland B. P., Hewett J., Farley J., Irimia D., Li Y., Wiche G., Sonnenberg A., Breakefield X. O. (2008) TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J. Cell Sci. 121, 3476–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voeltz G. K., Prinz W. A. (2007) Sheets, ribbons and tubules - how organelles get their shape. Nature Reviews. Molecular Cell Biology 8, 258–264 [DOI] [PubMed] [Google Scholar]
- 30. Jones T. R., Wiertz E. J., Sun L., Fish K. N., Nelson J. A., Ploegh H. L. (1996) Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. U. S. A. 93, 11327–11333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao G., Zhou X., Wang L., Li G., Schindelin H., Lennarz W. J. (2007) Studies on peptide:N-glycanase-p97 interaction suggest that p97 phosphorylation modulates endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. U. S. A. 104, 8785–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith D. M., Fraga H., Reis C., Kafri G., Goldberg A. L. (2011) ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144, 526–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shibata Y., Hu J., Kozlov M. M., Rapoport T. A. (2009) Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 25, 329–354 [DOI] [PubMed] [Google Scholar]
- 34. Fukuda M., Yamamoto A., Mikoshiba K. (2001) Formation of crystalloid endoplasmic reticulum induced by expression of synaptotagmin lacking the conserved WHXL motif in the C terminus. Structural importance of the WHXL motif in the C2B domain. J. Biol. Chem. 276, 41112–41119 [DOI] [PubMed] [Google Scholar]
- 35. Takei K., Mignery G. A., Mugnaini E., Südhof T. C., De Camilli P. (1994) Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron 12, 327–342 [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto A., Masaki R., Tashiro Y. (1996) Formation of crystalloid endoplasmic reticulum in COS cells upon overexpression of microsomal aldehyde dehydrogenase by cDNA transfection. J. Cell Sci. 109, 1727–1738 [DOI] [PubMed] [Google Scholar]
- 37. Lingwood D., Schuck S., Ferguson C., Gerl M. J., Simons K. (2009) Generation of cubic membranes by controlled homotypic interaction of membrane proteins in the endoplasmic reticulum. J. Biol. Chem. 284, 12041–12048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mullins C., and SpringerLink (Online service) (2005) The Biogenesis of Cellular Organelles, in Molecular Biology Intelligence Unit, Eurekah.com and Kluwer Academic, Plenum Publishers, Boston, MA [Google Scholar]
- 39. Vander Heyden A. B., Naismith T. V., Snapp E. L., Hanson P. I. (2011) Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum. EMBO J. 30, 3217–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anderson R. G., Orci L., Brown M. S., Garcia-Segura L. M., Goldstein J. L. (1983) Ultrastructural analysis of crystalloid endoplasmic reticulum in UT-1 cells and its disappearance in response to cholesterol. J. Cell Sci. 63, 1–20 [DOI] [PubMed] [Google Scholar]