

Background: The TACC3-chTOG protein complex is essential for mitotic spindle assembly.
Results: TACC3-chTOG binding is directed and mediated by specific intradomain and interdomain interactions that are not affected by Aurora-A kinase.
Conclusion: Formation of the TACC3-chTOG complex is Aurora-A-independent, in contrast to its recruitment to the spindle apparatus.
Significance: Novel insight into regulation and domain specificity of TACC3-chTOG interaction is provided.
Keywords: Centrosome, Microtubules, Mitosis, Mitotic Spindle, Protein Domains, Aurora-A, Coiled-Coil, Protein Interaction, TACC3, chTOG
Abstract
The cancer-associated, centrosomal adaptor protein TACC3 (transforming acidic coiled-coil 3) and its direct effector, the microtubule polymerase chTOG (colonic and hepatic tumor overexpressed gene), play a crucial function in centrosome-driven mitotic spindle assembly. It is unclear how TACC3 interacts with chTOG. Here, we show that the C-terminal TACC domain of TACC3 and a C-terminal fragment adjacent to the TOG domains of chTOG mediate the interaction between these two proteins. Interestingly, the TACC domain consists of two functionally distinct subdomains, CC1 (amino acids (aa) 414–530) and CC2 (aa 530–630). Whereas CC1 is responsible for the interaction with chTOG, CC2 performs an intradomain interaction with the central repeat region of TACC3, thereby masking the TACC domain before effector binding. Contrary to previous findings, our data clearly demonstrate that Aurora-A kinase does not regulate TACC3-chTOG complex formation, indicating that Aurora-A solely functions as a recruitment factor for the TACC3-chTOG complex to centrosomes and proximal mitotic spindles. We identified with CC1 and CC2, two functionally diverse modules within the TACC domain of TACC3 that modulate and mediate, respectively, TACC3 interaction with chTOG required for spindle assembly and microtubule dynamics during mitotic cell division.
Introduction
The centrosome represents the main microtubule (MT)4 organizing center in all metazoans and is thereby responsible for equal partitioning of chromosomes to daughter cells during the mitotic phase of the cell cycle (1–5). Numerical and structural abnormalities of centrosomes are associated with aneuploidy, chromosomal instability and transformation, developmental defects, apoptotic cell death, and cell cycle arrest through induction of premature senescence (6–12). Cancer cells e.g. counteract extra centrosomes and, therefore, the danger of multipolar divisions and excess aneuploidy/cell death through centrosome clustering (13–15). This process became an attractive pharmacological tumor target (16, 17).
During the cell cycle centrosomes undergo a division and maturation process called the centrosome cycle (4, 18, 19). Mitotic centrosomes are structurally made up of one pair of centrioles surrounded by the pericentriolar matrix (20). More than two hundred proteins are involved in centrosome assembly, organization, and function (19, 21–23). These proteins have structural, functional/enzymatic, and regulatory/signaling roles in MT nucleation and spindle dynamics, mitotic progression, and cytokinesis (24). Recent work by the Mitocheck consortium (25, 26) provided a global confirmation of known and identification of novel cell division genes and their protein complexes that require biochemical and functional elucidation in greater detail.
Members of the centrosomal TACC (transforming acidic coiled-coil) family of proteins are important structural components of the mitotic spindle apparatus (27–29). TACCs are conserved in all metazoans and play a vital role as adaptor proteins in the regulation of centrosomal integrity and spindle MT stability and dynamics (27, 28, 30–34). Vertebrates express three TACC isoforms, TACC1, TACC2, and TACC3, of which the latter is typically found at high levels in proliferative and regenerative cell types and tissues (35–37). During the cell cycle TACC3 expression increases strongly in the G2/M phase (38) followed by Cdh1-dependent degradation of TACC3 during mitotic exit (39). TACC3 deficiency leads to growth retardation and embryonic lethality (38, 40), in line with the anti-proliferative impact of shRNA mediated gene silencing of TACC3 (41, 42).
A crucial regulator of TACC3 is the mitotic kinase Aurora-A that phosphorylates TACC3 (pTACC3) and thereby determines its differential centrosomal/proximal spindle (pTACC3) versus distal spindle MT (TACC3) localization during (pro)metaphase (43–46). Interestingly, recent findings expand the function of TACC3 and the Aurora-A-TACC3 axis to the regulation of kinetochore-microtubule connections (47) and central spindle assembly at later stages of mitosis (48), respectively. Other known TACC3 binding partners with regulatory/effector functions include the endocytic and vesicle trafficking protein clathrin (i.e. clathrin heavy chain) (49, 50) that binds to the clathrin interaction domain of pTACC3 to ensure intermicrotubule bridging and mitotic spindle organization (51, 52). Moreover, the evolutionary conserved interaction between TACCs and MT polymerases of the XMAP215 family is crucial for spindle pole stabilization and growth of centrosomal MTs (43, 46, 53). Family members, which comprise XMAP215 in Xenopus laevis, Msps in Drosophila melanogaster, and chTOG/CKAP5 (cytoskeleton associated protein 5) in Homo sapiens, are identified by the presence of several “TOG” domains involved in MT binding.
TACC proteins are structurally characterized by a rather variable N-terminal region of which the approximately first 100 residues are uniquely conserved among vertebrate TACC3 isoforms. Further features include a central serine-proline-glutamate-rich repeat region (28, 33) that in the case of murine TACC3 is characterized by seven perfect repeats of 24 amino acids each (thereafter referred to as “7R”) (33, 38) as well as a highly conserved, coiled-coil-containing C terminus (thereafter referred to as “CC” or “TACC domain”). This signature domain is composed of ∼200 amino acids (aa) (27, 33, 55), required for centrosomal localization, and known to be involved in protein-protein interaction (28). Here, TACC proteins interact from yeast to human through their TACC domain with the C terminus of XMAP215 family members (28, 46, 56, 57), thereby targeting them to spindle poles. In contrast, the functional role of the N-terminal part of TACC3 outside of the TACC domain is rather undefined besides being a substrate for Aurora-A-mediated phosphorylation that is required for centrosomal and proximal spindle localization of TACC3 (28).
From the analysis of X. laevis TACC3, it has been proposed that the N-terminal part masks the TACC domain and thereby inhibits its function (58, 59). Aurora-A mediated phosphorylation of TACC3 was implicated to “unmask” and thereby expose the TACC domain to intermolecular interaction with XMAP215 (46, 59). However, the molecular basis/details of the masking/unmasking mechanism of the TACC domain and its interaction with the C terminus of XMAP215 remained enigmatic. Here, we subjected recombinant murine TACC3 and the C-terminal part of the murine XMAP215 homologue chTOG to a deletion and biochemical interaction analysis. We identify within the TACC domain two functionally distinct subdomains, CC1 (aa 414–530) and CC2 (aa 530–630), which are involved in interdomain and intradomain protein interaction, respectively. We demonstrate that TACC3 forms a stable intramolecular complex through the interaction of 7R with CC2 (TACC domain “masked”). Interestingly, the C terminus of chTOG (aa 1806–2032) right hand to the putative MT-interacting TOG6 domain (52) binds selectively to the CC1 module and thereby disrupts the intramolecular CC2–7R complex, thereby giving rise to the effector-bound state of the TACC domain (TACC domain “unmasked”). Neither intradomain interaction of TACC3 nor its binding to chTOG was affected by Aurora-A kinase. Thus, consecutive intra- and intermolecular protein interactions direct and determine TACC3-chTOG protein complex formation before its Aurora-A-regulated centrosomal and proximal spindle recruitment required for MT growth and mitotic spindle assembly.
EXPERIMENTAL PROCEDURES
In Silico Analysis of TACC3
Protein sequences of TACC family members were retrieved from the NCBI database and used for further analysis. For sequence alignment and evolutionary analysis of conserved domains, the ClustalW multiple sequence alignment algorithm was used (60), and alignment was analyzed with JALVIEW. Further analysis of the coiled-coil boundary in the TACC domain of murine TACC3 was performed using the COILS server in 14, 21, and 28 residue scan mode (61). Algorithms and tools from the EXPASY proteomic server were employed for sequence-based protein characterization.
Cloning of Expression Constructs
Coding sequences for murine TACC3 and its variants were amplified using sequence-specific primer and cloned into the pGEX-4T1-NTEV expression vector. The following constructs were created: full-length TACC3 (aa 1–630); TACC3-ΔN (Δ1–118); TACC3-ΔR (Δ141–308), lacking the serine-proline-glutamate-rich repeat region; TACC3-ΔNΔR (Δ1–118 and Δ141–308); 7R (aa 119–324) comprising the serine-proline-glutamate-rich repeat region; CC (TACC domain; aa 414–630); CC1 (aa 414–530); CC2 (aa 530–630) (62). Moreover, pGEX-4T1-NTEV-based expression constructs for C-terminal fragments of human chTOG were created: chTOG-Cterm (aa 1574–2032), chTOG-A (aa 1544–1805), and chTOG-B (aa 1806–2032). To generate TACC3 deletion mutants fused at the C terminus to GFP, the following constructs were cloned in a pEGFP-N1 (Clontech)-based vector: full-length TACC3; TACC-ΔCC1 and TACC-ΔCC2 lacking the CC1 or CC2 subdomains, respectively; TACC-ΔCC lacking the entire TACC domain. All constructs were validated by DNA sequencing.
Overexpression and Purification
GST fusion proteins were overexpressed in Escherichia coli BL21 Rosetta strain (Novagen). Protein extraction was carried out by incubating cells at 4 °C with DNase I (10 μg/ml) followed by cell lysis in a microfluidizer (model M110S, Microfluidics Corp.) at a pressure of 10.000 p.s.i. Bacterial lysates were centrifuged to collect soluble fractions, and GST-tagged proteins were isolated from the supernatant via GST affinity purification. Upon cleavage of the GST tag with tobacco etch virus protease (4 units/mg, 4 °C, overnight) or thrombin (2 units/mg, 4 °C, overnight) proteins were subjected to gel filtration using a standard buffer containing 30 mm Tris-HCl, pH 7.5, 200 mm NaCl, 3 mm DTT, and 2 mm EDTA. The GST tag from the TACC3-ΔN and TACC3-ΔNΔR deletion mutants could not be cleaved by tobacco etch virus protease for unknown reasons (data not shown), thus employing thrombin for both GST tag removal and cleavage at the internal site (supplemental Fig. S4). The final purity was analyzed on SDS-PAGE, proteins were concentrated using centrifugal ultrafiltration devices (Amicon Ultra; Millipore), and protein concentration was determined by the Bradford assay. For mass spectrometric analysis of thrombin-cleaved TACC3, the protein was desalted by passing through NAP-25 columns (GE Healthcare) and analyzed by MALDI-TOF at the central BMFZ facility of the Heinrich-Heine-University Düsseldorf.
Immunoblotting
Proteins were separated using SDS-PAGE gels and transferred to nitrocellulose membranes (Hybond C, GE Healthcare). Blots were probed overnight with primary antibodies: αTACC3 (N18) and αTACC3 (C18), both generated in rabbits (38), are specific for the N and C terminus of murine TACC3, respectively; αGST (Abcam); αchTOG (Abcam, QED Bioscience/Acris, and Novus Biologicals); αGFP (Roche Applied Science). After three washing steps membranes were incubated with horseradish peroxidase-coupled secondary antibodies for 1 h. Signals were visualized by the ECL detection system (GE Healthcare), and images were collected using the INTAS chemostar imager.
Analytical Gel Filtration/Size Exclusion Chromatography (SEC)
Gel filtration was performed using a Superose 6 10/300 GL column connected to an ÄKTATM purifier (GE Healthcare) and UV900 detector. For molecular weight determination, the column was calibrated with standard proteins of known molecular mass: thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), ovalbumin (43 kDa), carbonic anhydrase (29 kDa), ribonuclease A (13.7 kDa), and aprotinin (6.5 kDa). Protein samples (50–200 μg) were injected onto the preequilibrated column, and elution fractions of 0.5 ml were collected. Elution profiles were recorded using UNICORN4.11 software, and peak fractions were analyzed by SDS-PAGE followed by Coomassie Brilliant Blue (CBB) staining and immunoblotting.
GST Pulldown Assay
GST, GST-fused TACC3 variants, and fragments of the C terminus of chTOG (chTOG-Cterm) were expressed in E. coli and purified using standard protocols. To obtain prey proteins, the GST tag was cleaved off with tobacco etch virus protease and cleared by reverse GSH affinity purification. GSH-Sepharose beads (GE Healthcare) in a 100-μl volume were washed 3 times with standard buffer. GST and GST-fused proteins (10–20 μm) were added and incubated in a final volume of 200 μl at 4 °C for 1 h. Blocking with 5% BSA (2 h at 4 °C) was performed followed by three washing steps with standard buffer (30 mm Tris-HCl, pH 7.5, 200 mm NaCl, 3 mm DTT, 2 mm EDTA). Finally, samples were incubated at an equimolar ratio with prey proteins at 4 °C for 2 h. After five washing steps, 100 μl of 2× Laemmli buffer was added, and samples were heat-denatured (5 min at 95 °C) and analyzed by SDS-PAGE followed by CBB staining and immunoblotting.
Isothermal Titration Calorimetry (ITC)
Purified TACC3 variants and chTOG-Cterm were first subjected to gel filtration (Superose 6 XK16/60) using ITC buffer (30 mm Tris-HCl, pH 7.5, 200 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine, 2 mm EDTA). For the 7R fragment, buffer was exchanged by overnight dialysis against ITC buffer using Slide-A-Lyzer dialysis cassettes (Thermo Scientific). All ITC measurements were carried out at 20 °C using a VP-ITC microcalorimeter (Microcal) (63, 64). Proteins were loaded into the sample cell and titrated with their putative interaction partners (10–15-fold higher protein concentration in the syringe compared with the concentration in the cell; titration volume 8, 10, or 15 μl; spacing of 150–180 s; reference power of 13 μcal s−1, stirring at 310 rpm). The final data analysis was carried out using Origin software (Microcal Software). The experimental data were evaluated using Origin 7.0 software (Microcal Software) to determine the binding parameters, including association constant (Ka), number of binding sites (N), and enthalpy (ΔH). Control measurements were performed by titrating buffer to the protein and vice versa.
Immunoprecipitation
Purified TACC3 proteins before and after thrombin cleavage (∼10–15 μg) were mixed with 2 μl of rabbit antisera (αTACC3 N18 or αTACC3 C18) (38) and incubated overnight at 4 °C. Thereafter, 25 μl of protein A/G-agarose (Santa Cruz Biotechnology) preabsorbed with BSA was added, and the volume was adjusted to 100 μl with IP buffer (30 mm Tris-HCl, 200 mm NaCl, 2 mm EDTA). After an incubation period of 1 h the beads were washed with IP buffer, and protein complexes were eluted with 2× Laemmli loading buffer and analyzed by SDS-PAGE and immunoblotting using primary antibodies (N18, C18) and horseradish peroxidase-conjugated secondary antibodies. Moreover, total cell lysates from HEK293 cells, which were transfected with expression vectors for TACC3 or TACC3 deletion mutants fused C-terminally to GFP, were prepared essentially as described (32) and thereafter subjected to immunoprecipitation using αGFP antibodies (Roche Applied Science). chTOG was detected in the co-immunoprecipitates using an αchTOG antibody from QED Bioscience/Acris.
Protein Kinase Assay
Purified human Aurora-A kinase (Signal Chem) was employed according to the manufacturer's instructions. In brief, kinase assays were performed in a volume of 25 μl by mixing 5 μl (0.1 μg/μl) of Aurora-A kinase (diluted in kinase assay buffer: 5 mm MOPS, pH 7.2, 2.5 mm β-glycerol phosphate, 5 mm MgCl2, 1 mm EGTA, 0.4 mm EDTA, 50 ng/μl BSA) with 1 μg of purified TACC3 protein (before and after thrombin cleavage) or 1 μg of the TACC3-chTOG complex as a substrate. The final volume was adjusted to 20 μl with double distilled H2O, and reactions were started by adding 5 μl of 10 mm ATP (dissolved in 25 mm MOPS, pH 7.2, 12.5 mm β-glycerol phosphate, 25 mm MgCl2, 5 mm EGTA, 0.4 mm EDTA). Reactions were incubated at 30 °C for 15 min and stopped by the addition of 10 μl of 2× Laemmli loading buffer. Samples were separated by SDS-PAGE, and phosphorylated proteins were subsequently detected by Pro-Q Diamond (Molecular Probes, Invitrogen) and CBB staining.
Eukaryotic Cell Culture
HEK293 and HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (Invitrogen), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. GFP-fused TACC3 expression vectors were transfected into HEK293 cells using the TurboFect transfection reagent (ThermoScientific). The Aurora-A kinase inhibitor MLN8237 (Selleckchem) was applied in cell culture at a concentration of 0.5 μm for 2 h (65).
Confocal Laser Scanning Microscopy
Cells were seeded at densities of 8 × 103 cells/cm2 on coverslips and fixed with ice-cold methanol/acetone (1:1) for 20 min at −20 °C upon MLN8237 treatment. Subsequently, cells were incubated in IF buffer (4% bovine serum albumin, 0.05% saponin in PBS) for 1 h and stained in IF buffer with the following primary antibodies at the indicated dilutions: anti-α-tubulin (DM1a,1:500, Sigma, or YOL1/34, Acris Antibodies, Hiddenhausen, Germany); αTACC3 (H300 or D2, Santa Cruz Biotechnology); anti-γ-tubulin (GTU-88, 1:100, Sigma); αchTOG (1:500, Acris); αpT288 Aurora-A (1:500; Cell Signaling). DNA was detected using 4,6-diamidino-2-phenylindole (DAPI, 1 mg/ml; Sigma). Analyses were performed with a LSM510-Meta confocal microscope (Zeiss) equipped with 40/1.3 or 63/1.4 immersion objectives and excitation wavelengths of 364, 488, 543, and 633 nm.
RESULTS
The TACC Domain of TACC3 Consists of Two Distinct Coiled-coil Subdomains
To examine the primary structure of murine TACC3 and thereby define functional modules in TACC3, sequence alignments of vertebrate TACC family members were performed. Consistent with previous findings (28, 33, 55), the N termini of TACC3 isoforms are characterized by a variable length and different amino acid composition as compared with other vertebrate TACC family members. Interestingly, the first 100 amino acids of TACC3 isoforms display a sequence identity of up to 75% (data not shown) followed by the central repeat region that in the case of murine TACC3 comprises seven conserved serine-proline-glutamate-rich repeats (7R) each consisting of 24 amino acids. Interestingly, coiled-coil prediction analysis indicated the presence of one breaking region that divides the C-terminal TACC domain of mammalian TACC3 proteins into two coiled-coil-containing subdomains, CC1 and CC2 (supplemental Fig. S1). Here, CC2 clearly revealed a higher amino acid sequence identity than the CC1 subdomain (supplemental Fig. S2). Overall, the domain organization of vertebrate TACC3 proteins emphasizes their isoform-specific functional roles as compared with TACC1 and TACC2. That is exemplified by the embryonic lethality caused by TACC3 deficiency, which is not observed for TACC2 deficiency (37, 38, 40), as well as by the selective interaction of the aryl hydrocarbon receptor nuclear translocator (ARNT) with TACC3 but not with TACC1 and TACC2 (66).
Moreover, we identified a unique and functional thrombin cleavage site (410LEPR/GL415) close to the TACC domain of murine TACC3 (Fig. 1A) that is absent in all other TACC3 proteins and TACC isoforms. This cleavage site resembles a bona fide recognition site for thrombin (410LVPR/GS415). Although the thrombin site present in murine TACC3 might not have any physiological relevance, it was used as a tool in this study to analyze the intra- and intermolecular interaction of murine TACC3.
FIGURE 1.

Thrombin cleavage of murine TACC3 generates two N- and C-terminal fragments staying in one intramolecular complex. A, a novel and unique thrombin site divides TACC3 into two separate parts, TACC3-N7R and TACC3-CC. N specifies the conserved ∼100 amino acid residues at the N terminus. CC1 and CC2 indicate two distinct coiled-coil subdomains in the C terminus. r1 to r7 denotes the central region in TACC3 consisting of seven serine-proline-glutamate-rich repeats (7R). B, determination of the apparent molecular mass of TACC3 by aSEC (Superpose 6, 10/300). Elution profiles of TACC3 before (solid line) and after thrombin cleavage (dashed line) are indicated. Vo, void volume. mAu, milliabsorbance units. C, peak elution fractions were analyzed by SDS-PAGE followed by CBB staining (left panel) and immunoblotting (central and right panels employing N18 and C18 antibodies directed against the N- or C-terminal end of TACC3, respectively) (38). D, co-immunoprecipitation analysis of TACC3 before (upper panel) and after thrombin cleavage (lower panel) was performed either without antibody input (beads control, lane 2) or by using N18 and C18 antibodies (lanes 3 and 4). Thr., thrombin.
Purification of Deletion Mutants and Fragments of TACC3 and chTOG
Based on the in silico analysis we cloned murine TACC3 and its variants (supplemental Fig. S3) as well as C-terminal fragments of chTOG (supplemental Fig. S5A) into the prokaryotic expression vector pGEX-4T1-NTEV. Proteins fused N-terminally to glutathione S-transferase (GST) were purified, cleaved using tobacco etch virus protease to remove the GST tag, and finally separated by gel filtration and/or reverse glutathione affinity chromatography (supplemental Figs. S4 and S5, B and C). Thrombin cleavage of TACC3 resulted in two fragments, a larger N-terminal part (TACC3-N7R, aa 1–413) and a smaller C-terminal fragment (CC, aa 414–630) containing the TACC domain (Fig. 1A and supplemental Fig. S4A). The identity and size of the CC fragment (25.2 kDa) was corroborated by mass spectrometry using MALDI-TOF. The adaptability of the thrombin site was also confirmed for the deletion mutants TACC3-ΔR, TACC3-ΔN, and TACC3-ΔNΔR (supplemental Fig. S3 and S4).
Intradomain Association Leads to an Intramolecular Masked State of TACC3
We next subjected TACC3 to analytical size exclusion chromatography (aSEC) using a Superose 6 10/300 column. Interestingly, TACC3 eluted before and after thrombin cleavage in peak fractions with apparent molecular masses of ∼1200 and ∼630 kDa, respectively (Fig. 1B), the latter being unexpectedly the only peak detected after thrombin cleavage. Subsequent peak fraction analysis by SDS-PAGE and CBB staining as well as immunoblotting demonstrated the co-elution of both fragments, TACC3-N7R and CC (Fig. 1C). These findings strongly suggested the formation of a tight complex between the N- and C-terminal parts of TACC3. We confirmed this intramolecular interaction by co-immunoprecipitation of TACC3 before and after thrombin cleavage using antibodies specific for the N or C termini of murine TACC3 (38). As indicated in Fig. 1D, TACC3-N7R and CC was co-immunoprecipitated in both directions. Of note, thrombin cleavage of TACC3 did not result in changes in its secondary structure as indicated from circular dichroism measurements (supplemental Fig. S11A).
The Intramolecular Interaction of TACC3 Is Mediated between the 7R and CC2 Domains
To identify the domains involved in intradomain TACC3 interaction, we deleted the conserved N-terminal region (Δ1–118; TACC-ΔN) and the central 7R region (Δ141–308; TACC3-ΔR) (supplemental Fig. S3). The respective purified TACC3 variants were analyzed before and after thrombin cleavage by aSEC. The absence of the first 118 amino acids did not have any detectable effect on the elution pattern of TACC3-ΔN that was comparable to full-length TACC3 (Fig. 2B). Also, upon thrombin cleavage, both protein fragments (named TACC3–7R and TACC3-CC in Fig. 2) still co-eluted in one peak, suggesting that the conserved N-terminal region is not essentially involved in the intramolecular interaction of TACC3. In contrast, deletion of the 7R region abolished intradomain binding and resulted after thrombin cleavage in the separation of the fragments (named TACC3-N and TACC3-CC in Fig. 3) in two distinct peaks with apparent molecular masses of 160 and 630 kDa, respectively). Their identity was reconfirmed by immunoblot analysis (Fig. 3C). Thus, the central serine-proline-glutamate-rich repeat region (7R) is required for the intramolecular interaction with the C-terminal CC domain.
FIGURE 2.

The N-terminal 118 amino acids are not involved in intramolecular complex formation of murine TACC3. A, primary structure of TACC3-ΔN lacking the N-terminal part of 118 aa. B, aSEC (Superpose 6, 10/300) elution profiles of GST-TACC3-ΔN before (solid line) and after (dashed line) thrombin cleavage. Vo, void volume. mAu, milliabsorbance units. C, peak elution fractions were analyzed by SDS-PAGE followed by CBB staining (upper panel) and immunoblotting using anti-TACC3 C18 (middle panel) or anti-GST antibodies (lower panel). GST was used as a control.
FIGURE 3.

Deletion of the central repeat (7R) domain prevents intramolecular TACC3 complex formation. A, primary structure of TACC3-ΔR lacking the 7R domain. B, elution profile of TACC3-ΔR on analytical gel filtration (Superpose 6, 10/300) before (solid line) and after (dashed line) thrombin cleavage. Vo, void volume. C, peak elution fractions were analyzed on SDS-PAGE (4–15% gradient gel) followed by CBB staining (upper panel) and immunoblotting using the indicated antibodies (middle and lower panels). Thr., thrombin. D, the interaction between the isolated repeat region (7R) and the TACC domain (CC) was analyzed by pulldown assays and immunoblotting using N18 and C18 antibodies against TACC3. The asterisk indicates bovine albumin used to reduce unspecific binding to GSH-Sepharose beads. E, analysis of the interaction between 7R and subdomains of the TACC domain (CC1, CC2) employing ITC. Heat changes after association of the indicated protein fragments indicate that 7R selectively interacts with CC2 (upper panel) but not CC1 (lower panel). See supplemental Fig. S6 for experimental ITC controls.
We further validated this finding by subjecting the isolated domains, i.e. GST-7R (bait) and CC (prey) (supplemental Fig. S3) to pulldown-based interaction analysis. GST alone was used as the control. Although CC showed an unspecific binding to the beads, we could still detect a clearly stronger binding of CC when GST-7R was used as bait and analyzed by immunoblotting (Fig. 3D). Last, to determine the region within the TACC domain that binds to the central repeat region, we purified 7R and the CC subdomains CC1 and CC2 (supplemental Fig. S3 and S4E) and analyzed their interaction by employing ITC. As indicated in Fig. 3E, significant calorimetric changes as well as changes in the temperature as a function of the molar ratio of the interacting proteins were observed when 7R was titrated onto CC2. In contrast, binding of 7R to CC1 could not be detected (Fig. 3E, lower panel). We conclude that the central repeat region, i.e. 7R, binds selectively to CC2 and thereby mediates intramolecular TACC3 binding potentially masking the C terminus of TACC3 before intermolecular protein interaction.
TACC3 Interacts with chTOG via Its CC1 Domain
We next characterized the interdomain binding between TACC3 and its major effector, the MT polymerase chTOG. We employed the C terminus of chTOG as bait (GST-chTOG-Cterm; supplemental Fig. S5) in pulldown assays demonstrating a strong interaction between chTOG-Cterm and the CC domain of TACC3 (Fig. 4A). Interestingly, when analyzing the relative contribution of the two coiled-coil-containing subdomains of the TACC domain (i.e. CC1 and CC2; supplemental Fig. S3) to chTOG-Cterm binding in aSEC experiments, we observed a prominent peak shift of CC1 mixed with chTOG-Cterm that did not occur in the case of CC2 (Fig. 4, B and C). This selective CC1-chTOG-Cterm interaction was still observable in the presence of equimolar amounts of CC2, i.e. when mixing and analyzing all three fragments together (Fig. 4D). Of note, CC1 and CC2 did not interact with each other (supplemental Fig. S7). Subsequent ITC-based binding measurements revealed a strong association between CC1 and chTOG-Cterm with a Kd of 0.7 μm and a binding stoichiometry (n = 0.83) of nearly 1:1 (Fig. 4E). Consistent with the aSEC data, we did not detect any interaction between CC2 and chTOG-Cterm in ITC experiments (Fig. 4E). Importantly, we confirmed these findings also in vivo by expressing deletion mutants of TACC3 lacking either the TACC domain (CC) or one of its subdomains (i.e. CC1 or CC2) in HEK293 cells as C-terminally tagged GFP fusion proteins. Transfected cells were subjected to co-immunoprecipitation analysis using GFP-specific antibodies. As indicated in Fig. 5, deletion of CC1, but not of the CC2 subdomain, abrogated interaction with endogenous chTOG, strongly indicating that also under in vivo conditions the CC1 domain specifically determines binding of TACC3 to chTOG.
FIGURE 4.

The CC1 subdomain of TACC3 mediates chTOG binding. A, interaction between the C terminus of chTOG (chTOG-Cterm; aa 1574–2032; supplemental Fig. S6) and the CC domain of TACC3 was analyzed by pulldown assays and immunoblotting using antibodies against the C-terminal end of TACC3 (C18) and GST. B and C, binding of purified TACC subdomains (CC1 or CC2) to chTOG-Cterm was analyzed by aSEC (superpose 6, 10/300) followed by SDS-PAGE and CBB staining of the respective peak fractions. D, chTOG-Cterm interacts in a competition experiment selectively with CC1 when mixed with both CC1 and CC2 fragments. Samples were analyzed by aSEC followed by SDS-PAGE and CBB staining of the respective peak fractions. The dotted box indicates elution fractions from the analysis of the CC1+CC2+chTOG-Cterm mixture employing anti-chTOG and anti-TACC3 (C18 recognizing CC2, but not CC1) antibodies. E, analysis of binding of chTOG-Cterm to CC1 (left panel) and CC2 (right panel) using ITC confirms selective protein complex formation between chTOG-Cterm and CC1. See supplemental Fig. S6 for experimental ITC controls.
FIGURE 5.

The CC1 subdomain is required for binding of TACC3 to chTOG in vivo. HEK293 cells were transfected with expression constructs for TACC3 or the indicated C-terminal deletion mutants all C-terminally fused to GFP. After 48 h, total cell lysates were prepared and subjected to co-immunoprecipitation analysis using a GFP-specific antibody followed by SDS-PAGE analysis and detection of chTOG in the immunoprecipitates. mAu, milliabsorbance units; IP, immunoprecipitation. The asterisk indicates an unspecific band.
chTOG Binds to TACC3 via a C-terminal Region after the MT-interacting TOG Domains
Having localized CC1 as part in the TACC domain that binds to the C terminus of chTOG, we now narrowed down the TACC3 interacting part within the C terminus of chTOG. For this, we purified and analyzed two subdomains, chTOG-A (aa 1544–1805), which contains the putative, MT-interacting TOG6 domain (52), and chTOG-B (aa 1806–2032) within the C terminus of chTOG (supplemental Fig. S5). As indicated in the pulldown-based interaction analysis in Fig. 6, chTOG-B, but not chTOG-A, efficiently bound to TACC3 or its isolated CC1 domain. Consistent with this, employing aSEC and subsequent peak fraction analysis on SDS-PAGE, we observed a clear complex formation for chTOG-B-CC1 but no interaction and peak shift when chTOG-A and CC1 were mixed together (supplemental Fig. 10, A and B). Thus, chTOG binds specifically via a C-terminal fragment adjacent to the putative MT-binding TOG6 domain to the CC1 domain of TACC3.
FIGURE 6.
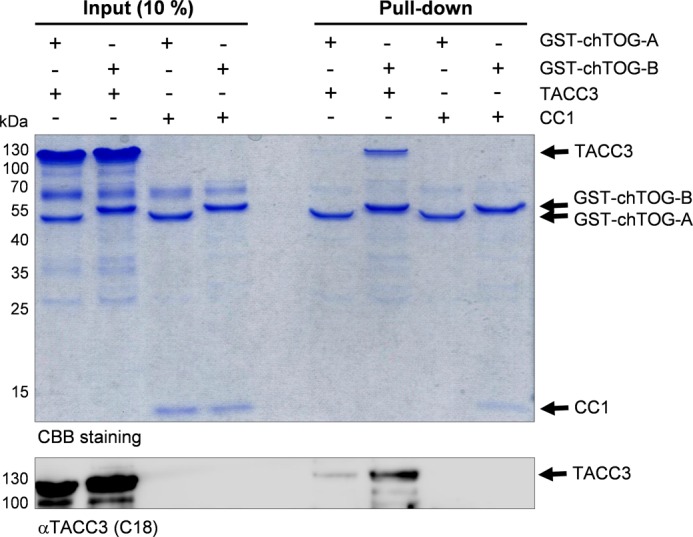
The C terminus of chTOG following the six TOG domains binds to the CC1 subdomain of TACC3. The interaction between the C terminus of chTOG (divided into the subfragments chTOG-A and chTOG-B; supplemental Fig. S5) with full-length TACC3 or its CC1 subdomain was analyzed by pulldown assays and subsequently detected by CBB staining and immunoblotting using antibodies against the C-terminal end of TACC3 (C18). chTOG-B, but not the TOG6 domain containing fragment chTOG-A (supplemental Fig. S5), displays a selective interaction with TACC3.
Interaction of chTOG with TACC3 Abrogates Intradomain Masking of TACC3
Based on the findings above, we next addressed the relation between the intradomain TACC3 interaction (mediated through 7R and CC2) and intermolecular CC1-chTOG-Cterm binding thereby analyzing the “directionality” of formation of these protein complexes. As indicated in Fig. 7A, GST-fused chTOG-Cterm was able to pull down full-length TACC3 or its C terminus (CC) produced upon thrombin cleavage of TACC3. However, in the latter case, the N-terminal part of TACC3 (TACC3-N7R) could not be detected in GST-chTOG-Cterm pulldown complexes by immunoblotting (Fig. 7A, last lane) strongly indicating that chTOG-Cterm binding uncouples the intramolecular TACC3 interaction. These findings were further validated by subjecting the TACC3-chTOG-Cterm protein complex before and after thrombin cleavage to aSEC. Fig. 7B shows that both TACC3 alone and TACC3 bound to chTOG-Cterm eluted on aSEC comparably at a peak fraction equivalent to an apparent molecular mass of ∼1200 kDa. In contrast, when TACC3 was prebound to chTOG-Cterm and then subjected to thrombin cleavage, a shift of the TACC3 complex (TACC3-N7R-CC; apparent mass of ∼630 kDa) toward an earlier elution volume corresponding to an apparent mass of ∼800 kDa was detected (Fig. 7C). SDS-PAGE analysis confirmed that this peak shift was due to the interaction of chTOG-Cterm with the TACC domain, once more indicating that the intramolecular interaction between 7R and CC2 is relieved upon chTOG-Cterm binding to CC1. We conclude that 7R-CC2 and CC1-chTOG-Cterm are consecutive and mutually exclusive interactions representing the two distinct masked and unmasked states of the C-terminal TACC domain of TACC3.
FIGURE 7.

Binding of chTOG-Cterm to TACC3 disrupts the intradomain interaction of TACC3. A, the interaction between chTOG-Cterm and thrombin-cleaved TACC3 was analyzed by pulldown assays and immunoblotting using the indicated antibodies. Intramolecular interaction of TACC3-N7R with CC is thereby abrogated upon chTOG-Cterm binding, as chTOG-Cterm pulls down CC but not TACC3-N7R (last lane). Thr., thrombin. B and C, aSEC-based analysis of the TACC3 - chTOG-Cterm complex was performed employing uncleaved TACC3 (B) or thrombin-cleaved TACC3 either alone or prebound to chTOG-Cterm (C). Peak fractions were analyzed by SDS-PAGE and CBB staining. The dashed lines within the CBB-stained SDS-PAGE gels highlight the peak shift of the chTOG-Cterm-CC complex (but not of TACC3-N7R) after thrombin cleavage. mAu, milliabsorbance units.
Aurora-A Kinase Does Not Interfere with TACC3-chTOG Complex Formation
Aurora-A kinase phosphorylates and targets TACC3 to centrosomes and proximal mitotic spindles as a prerequisite for TACC3-chTOG protein complex-dependent centrosomal MT assembly and dynamics (43–46). Sequence alignment and phosphorylation prediction revealed that murine TACC3 displays three putative Aurora-A phosphorylation sites (Ser-34, -341, and -347) that are localized in the N-terminal part outside of the CC domain (Fig. 1A) and are conserved in human TACC3 (Ser-34, -552, and -558) as well as X. laevis TACC3 (Ser-33, -620, and -626) (28). Our in vitro data presented above already indicated that Aurora-A-mediated phosphorylation of TACC3 is not required to expose the TACC domain for intermolecular protein interaction. However, the time point when Aurora-A phosphorylates TACC3, i.e. before or after chTOG binding, remained unclear. We, therefore, subjected (i) TACC3 before and after thrombin cleavage, (ii) TACC3-ΔR lacking the central repeat region required for intramolecular masking, and (iii) TACC3 prebound to chTOG-Cterm to in vitro kinase assays. As shown in Fig. 8, under all these conditions the N terminus of TACC3 outside of the TACC domain was efficiently phosphorylated by Aurora-A kinase. Thus, Aurora-A phosphorylates TACC3 independent from the masked or unmasked status of the TACC domain and does thereby not discriminate between the unbound or chTOG-bound state.
FIGURE 8.
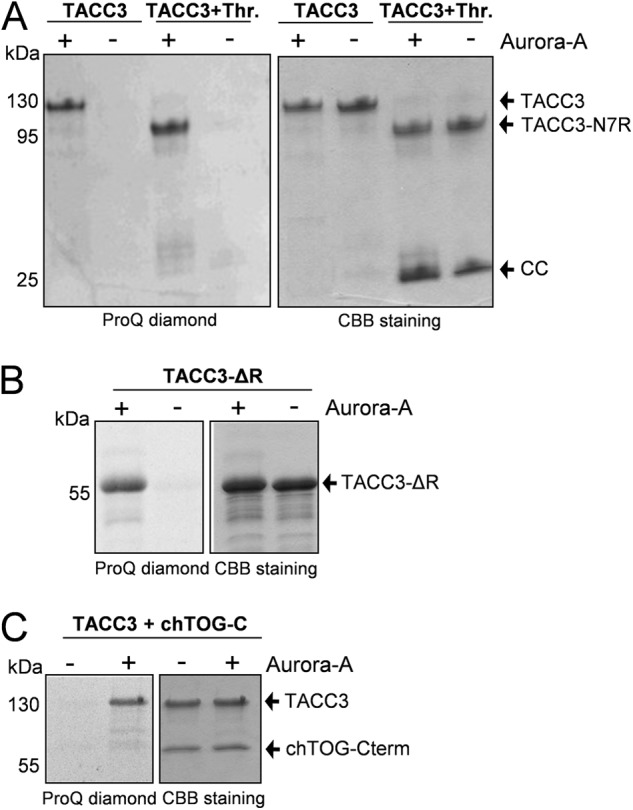
Aurora-A kinase phosphorylates murine TACC3 in vitro independent from its intra- or intermolecular binding status. Kinase assays were performed in the presence and absence of purified Aurora-A kinase employing uncleaved and thrombin-cleaved TACC3 (A), TACC3-ΔR (B), and the TACC3-chTOG-Cterm complex (C). Phosphorylated protein fragments were visualized by ProQ diamond staining. Thr., thrombin.
These in vitro findings were also tested in vivo by employing the Aurora-A kinase inhibitor MLN8237 in cell culture under conditions where Aurora-A kinase activity (as monitored by autophosphorylation at Thr-288) was abrogated (supplemental Fig. S8A). As indicated in supplemental Fig. 8, B–D, MLN8237-mediated inhibition of Aurora-A kinase impaired spindle formation and colocalization of TACC3 and chTOG to microtubules and spindle poles. However, centrosomal colocalization of TACC3 and chTOG was still detectable despite the occurrence of fragmented centrosomes. Consistent with this, employing co-immunoprecipitation analysis, interaction of TACC3 with chTOG was still detectable in MLN8237-treated cells (supplemental Fig. S9). Taken together, these findings emphasize that Aurora-A functions solely as a recruitment factor of the TACC3-chTOG complex to centrosomes and proximal spindle microtubules (43–46) without affecting its formation and protein interaction.
DISCUSSION
This study provides novel molecular insight into the basis of spindle MT stability and dynamics during mitosis by determining the interaction between the centrosomal adaptor protein TACC3 and the MT polymerase chTOG. The main findings of our work are as follows. 1) The C-terminal TACC domain of TACC3 consists of two functionally distinct modules, CC1 and CC2. 2) CC2 performs an intradomain interaction with the central repeat region (7R), a complex that masks intermolecular interaction of TACC3. 3) chTOG directly binds CC1 via a C-terminal fragment adjacent to N-terminal MT binding TOG domains. 4) Aurora-A kinase, a major regulator of TACC3, does not interfere with TACC3-chTOG complex formation either in vitro or in vivo. 5) Thus, Aurora-A solely acts as a centrosomal/proximal spindle recruitment factor for the TACC3-chTOG complex consistent with previous findings (43–46).
Our data argue against the possibility that the evolutionary conserved interaction between TACC3 and chTOG family members, as observed by several groups (31, 53, 58, 59, 67), requires the complete TACC domain. By analyzing the TACC3-chTOG protein complex, we define CC1 as an chTOG interacting domain. Moreover, we show that the deletion mutant TACC3-ΔCC1, in contrast to TACC3-ΔCC2, fails to co-immunoprecipitate/interact with chTOG in vivo (Fig. 5). Our findings are consistent with recent work of Hood et al. (52) that has analyzed the interaction of human TACC3 and chTOG isoforms using a deletion mapping approach. The authors narrowed down the corresponding human CC1 domain to a short region of 12 amino acids (aa 673–684) that appears to be sufficient for chTOG binding and chTOG localization on spindle MTs in vivo (52). Interestingly, centrosomal localization of chTOG was apparently reduced but still detectable, further indicating that chTOG may be recruited to centrosomes via both TACC3-dependent and -independent mechanisms.
As indicated in our model (Fig. 9), the mutually exclusive intradomain 7R-CC2 and interdomain CC1-chTOG interactions, respectively, provide novel functional insight into the subdomain selectivity and directionality of TACC3-chTOG complex formation. Our findings obtained by ITC analysis (Figs. 3E and 4E) are of particular relevance by providing clear insights into differential binding affinities for a strong chTOG-Cterm-CC1 interaction versus a weak 7R-CC2 interaction. Accordingly, we propose that chTOG binding to CC1 results in a conformational change of the CC2 subdomain, which is in turn released from its intramolecular complex with 7R and hence unmasks both CC2 and the central repeat region of TACC3. As a consequence, not only CC2, but also 7R may become available for further interactions with other downstream binding partners. However, in the latter case, no protein is currently known that binds to the central repeat domain of TACC3 despite the presence of bona fide PXXP binding motifs known to interact with SH3 domain-containing proteins in intracellular signaling processes. This is different for the TACC domain that has been identified by yeast two hybrid-based screening as well as pulldown and immunoprecipitation assays as major binding partner for various, functionally rather diverse proteins. These include factors involved in cortical neurogenesis (Cep192, DOCK7) (68, 69), hematopoietic development (FOG-1) (70), hypoxia response and gene expression (ARNT) (66), transcriptional regulation (MBD2) (71), and regulation of mTOR signaling (TSC2) (55). Interestingly, FOG-1 and ARNT have been proposed to bind to a region containing the last 20 residues of the CC2 subdomain (66, 72). Consistently, CC2 may be involved not only in intradomain but also in intermolecular protein interactions, whereas CC1 may only undergo intermolecular effector binding.
FIGURE 9.
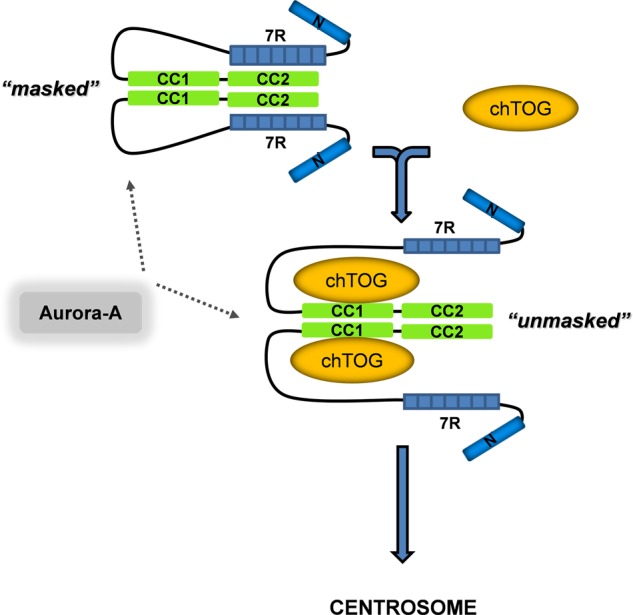
Integrative model for the domain specificity and directionality of TACC3-chTOG complex formation. We propose that the intradomain (7R-CC2; TACC3 masked) and intermolecular (CC1-chTOG; TACC3 unmasked) binding states of TACC3 are mutually exclusive, thereby defining a directionality of TACC3 regulated adaptor function toward chTOG binding and function. TACC3 can be phosphorylated by Aurora-A kinase in both binding states, indicating that Aurora-A acts as a centrosomal recruitment factor but is not involved in exposing the TACC domain for intermolecular interaction with chTOG or TACC3-chTOG complex formation. Moreover, based on biophysical characterization of murine TACC3 (supplemental Fig. 11, B and C, and supplemental Table S1; Ref. 34) we conclude that TACC3 displays an dimeric to oligomeric state. Thr., thrombin.
Aurora-A-mediated phosphorylation of TACC3 seems not to interfere with TACC3 intradomain and TACC3-chTOG interdomain interactions under in vitro conditions (Fig. 8). Accordingly, in vivo, TACC3-chTOG interaction and centrosomal colocalization was still detectable in HeLa cells that have been subjected to treatment with the Aurora-A kinase inhibitor MLN8237 (supplemental Figs. S8 and S9). These findings also contradict the previous model proposing that Aurora-A-mediated phosphorylation of X. laevis TACC3 triggers unmasking of the TACC domain and thereby exposes it for intermolecular interaction (i.e. XMAP215 binding) and centrosomal targeting (46, 59). In fact, Aurora-A-mediated phosphorylation of TACC3 seems to be solely required for targeting of the TACC3-chTOG complex to centrosomes and spindle MTs (28, 44). In the latter case, pTACC-chTOG interacts with another key effector in mitotic spindle assembly, the clathrin heavy chain, thereby cross-linking and stabilizing MT bundles (31, 51, 52).
Based on this study a sequential function of TACC3-chTOG effector complexes in the course of mitosis can be proposed. TACC3 interacts with the C terminus of chTOG thereby targeting it in an Aurora-A-dependent manner to spindle poles. On the other hand, the evolutionary conserved N terminus of chTOG likely comprises MT-stabilizing activity as demonstrated for XMAP215. In particular, XMAP215/chTOG proteins contain a variable number of TOG domains that bind to αβ-tubulin heterodimers, load them as MT polymerase (73) to the plus ends of MTs, and thereby inhibit “MT catastrophes.” In contrast, the C-terminal part of XMAP215 (and likely also chTOG) suppresses MT growth by promoting MT catastrophes (74). Therefore, the engagement of chTOG-Cterm by the CC1 subdomain of TACC3 during G2/M transition and metaphase might be a vital step in shifting the equilibrium toward MT polymerization. Upon mitotic exit, Cdh1 and ubiquitin-dependent degradation of TACC3 (39) then “disengages” the MT catastrophe promoting activity of the C terminus of XMAP215/chTOG. As a consequence, a shift of the equilibrium occurs toward MT “shrinkage” and disassembly of the spindle apparatus. Thus, TACC3 family members may function as “engagement factors” for the C terminus of XMAP215/chTOG to ensure a dynamic balance between MT rescue and catastrophe during the course of mitosis.
Besides a better molecular understanding regarding the mechanism and directionality of TACC3-chTOG interaction, we furthermore obtained novel insight into the unusual biophysical properties of TACC3. Analysis by aSEC (e.g. Fig. 1) clearly demonstrated that TACC3 displays a higher oligomeric mass and/or an elongated rod-like structure, obviously due to the presence of the coiled-coil containing TACC domain that elutes inherently at an apparent molecular mass of ∼630 kDa (Fig. 3B). Moreover, endogenous TACC3 or FLAG-tagged TACC3 from transfected eukaryotic cells behaves on aSEC comparable to purified TACC3 (data not shown). These findings are in accordance with the observation that TACC isoforms overexpressed in HeLa cells form in a TACC domain-dependent manner punctuate-like structures resembling cytoplasmic polymers (data not shown) (27). Employing further analytical methods including multiangle light scattering and analytical ultracentrifugation allowed us to conclude that TACC3 is characterized by a oligomeric (i.e. dimeric to hexameric) structure and a highly extended shape (supplemental Fig. 11, B and C, and supplemental Table S1). These findings are consistent with data from electron microscopic analysis where TACC3 depicts an elongated, fiber-like appearance (34). Another abnormality of murine TACC3 represents its migration in SDS-PAGE gels at 120–130 kDa (Fig. 1C) as compared with its theoretical molecular mass of 70.5 kDa. Interestingly, this unusual “gel shifting” is not based on the presence of the coiled-coil containing TACC domain (data not shown) but is rather caused by the central repeat region (supplemental Fig. S4, B versus A). As proof, deletion of the 7R domain restored normal gel migration of TACC3 (Fig. 3C and supplemental Fig. S4B). Of note, abnormal SDS-PAGE migration of acidic proteins can be caused by an altered binding of surfactants (like SDS) (75), a possibility that remains to be clarified for TACC3 and in particular the 7R domain.
Biological and pathobiological roles of TACC3 are underlined by several observations. TACC3 deficiency leads to severe growth retardation and embryonic lethality (38, 40). This is in line with the anti-proliferative and cell cycle arrest/senescence-inducing impact of shRNA-mediated gene silencing of TACC3 (41, 42). Moreover, it could be shown that TACC3 depletion sensitizes cells to the apoptotic and senescence-inducing effects of mitotic spindle poisons. Accordingly, inducible gene disruption of TACC3 in vivo in the p53−/− sarcolymphoma model is highly effective in causing apoptotic tumor regression (76). Interestingly, besides quantitative deregulation of gene expression of TACC isoforms in several tumor types, TACC1 and TACC3 point mutants have been identified in melanoma and ovarian cancer patients (77, 78). Moreover, oncogenic fusions between TACC and fibroblast growth factor receptor genes have been recently described in glioblastoma multiforme and bladder cancer patients (79, 80). The impact of these structural and tumor-associated alterations on Aurora-A-mediated regulation and function of TACCs is currently unknown and requires a more in-depth molecular understanding of TACC-effector interactions. Irrespective, it is tempting to speculate that these TACC mutants translate through loss-of-function or gain-of-function mechanisms into chromosomal instability and aneuploidy and thereby support cellular transformation (81, 82). Taken all these points into account, TACC3 represents an attractive antitumor target that may be at least indirectly drug-treatable at the level of its interactome. This assumption is supported by the recent identification of small drugs that act as inhibitors of protein-protein interaction and thereby impair the half-life and stability of TACC3 (KSH101), disrupt the TACC3-ARNT complex (KG-548), or inhibit the function of the TACC3-chTOG complex (spindlactone) (54, 66, 83, 84).
Acknowledgments
We thank Britta Tschapek for help in setting up the multiangle light scattering method and Jürgen Scheller and members of the Institute of Biochemistry and Molecular Biology II for input and fruitful comments during the course of this work and on the manuscript.
This work was supported by a fellowship of the NRW (North Rhine-Westphalia) graduate school “BioStruct; Biological Structures in Molecular Medicine and Biotechnology” (to H. C. T.), the Deutsche Forschungsgemeinschaft (SFB 728/TP A5;to R. P. P.), the research commission of the medical faculty of the Heinrich-Heine-University (grants to R. P. P. and M. R. A.), the strategic research fund of the Heinrich-Heine-University (grants to R. P. P. and M. R. A.), and the International Graduate School of Protein Science and Technology (iGRASP; to K. N. and M. R. A.).

This article contains supplemental Table S1 and Figs. S1–S11.
- MT
- microtubule
- aa
- amino acid
- SEC
- size exclusion chromatography
- aSEC
- analytical SEC
- CBB
- Coomassie Brilliant Blue
- CC
- coiled-coil
- chTOG
- colonic and hepatic tumor overexpressed gene
- ITC
- isothermal titration calorimetry
- 7R
- serine-proline-glutamate-rich repeat region
- TACC
- transforming acidic coiled-coil
- XMAP215
- Xenopus microtubule associated protein 215 kDa
- ARNT
- aryl hydrocarbon receptor nuclear translocator.
REFERENCES
- 1. Bornens M. (2012) The centrosome in cells and organisms. Science 335, 422–426 [DOI] [PubMed] [Google Scholar]
- 2. Compton D. A. (2000) Spindle assembly in animal cells. Annu. Rev. Biochem. 69, 95–114 [DOI] [PubMed] [Google Scholar]
- 3. Delgehyr N., Sillibourne J., Bornens M. (2005) Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J. Cell Sci. 118, 1565–1575 [DOI] [PubMed] [Google Scholar]
- 4. Mattison C., Winey M. (2006) The Centrosome Cycle/Cell Cycle Regulation (Kaldis P., ed.) pp. 925–925, Springer, Berlin [Google Scholar]
- 5. Nigg E. A. (2004) Centrosomes in Development and Disease, Wiley-VCH Verlag, Weinheim [Google Scholar]
- 6. Anderhub S. J., Krämer A., Maier B. (2012) Centrosome amplification in tumorigenesis. Cancer Lett. 322, 8–17 [DOI] [PubMed] [Google Scholar]
- 7. Basto R., Brunk K., Vinadogrova T., Peel N., Franz A., Khodjakov A., Raff J. W. (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133, 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bettencourt-Dias M., Hildebrandt F., Pellman D., Woods G., Godinho S. A. (2011) Centrosomes and cilia in human disease. Trends Genet. 27, 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan J. Y. (2011) A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 7, 1122–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mardin B. R., Schiebel E. (2012) Breaking the ties that bind. new advances in centrosome biology. J. Cell Biol. 197, 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidt S., Essmann F., Cirstea I. C., Kuck F., Thakur H. C., Singh M., Kletke A., Jänicke R. U., Wiek C., Hanenberg H., Ahmadian M. R., Schulze-Osthoff K., Nürnberg B., Piekorz R. P. (2010) The centrosome and mitotic spindle apparatus in cancer and senescence. Cell Cycle 9, 4469–4473 [DOI] [PubMed] [Google Scholar]
- 12. Zyss D., Gergely F. (2009) Centrosome function in cancer. Guilty or innocent? Trends Cell Biol. 19, 334–346 [DOI] [PubMed] [Google Scholar]
- 13. Fielding A. B., Lim S., Montgomery K., Dobreva I., Dedhar S. (2011) A critical role of integrin-linked kinase, ch-TOG, and TACC3 in centrosome clustering in cancer cells. Oncogene 30, 521–534 [DOI] [PubMed] [Google Scholar]
- 14. Kwon M., Godinho S. A., Chandhok N. S., Ganem N. J., Azioune A., Thery M., Pellman D. (2008) Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 22, 2189–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marthiens V., Piel M., Basto R. (2012) Never tear us apart. The importance of centrosome clustering. J. Cell Sci. 125, 3281–3292 [DOI] [PubMed] [Google Scholar]
- 16. Krämer A., Maier B., Bartek J. (2011) Centrosome clustering and chromosomal (in)stability. A matter of life and death. Mol. Oncol. 5, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raab M. S., Breitkreutz I., Anderhub S., Rønnest M. H., Leber B., Larsen T. O., Weiz L., Konotop G., Hayden P. J., Podar K., Fruehauf J., Nissen F., Mier W., Haberkorn U., Ho A. D., Goldschmidt H., Anderson K. C., Clausen M. H., Krämer A. (2012) GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res. 72, 5374–5385 [DOI] [PubMed] [Google Scholar]
- 18. Meraldi P., Nigg E. A. (2002) The centrosome cycle. FEBS Lett. 521, 9–13 [DOI] [PubMed] [Google Scholar]
- 19. Nigg E. A., Stearns T. (2011) The centrosome cycle. Centriole biogenesis, duplication, and inherent asymmetries. Nat. Cell Biol. 13, 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunha-Ferreira I., Bento I., Bettencourt-Dias M. (2009) From zero to many. Control of centriole number in development and disease. Traffic 10, 482–498 [DOI] [PubMed] [Google Scholar]
- 21. Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 [DOI] [PubMed] [Google Scholar]
- 22. Jakobsen L., Vanselow K., Skogs M., Toyoda Y., Lundberg E., Poser I., Falkenby L. G., Bennetzen M., Westendorf J., Nigg E. A., Uhlen M., Hyman A. A., Andersen J. S. (2011) Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 30, 1520–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Müller H., Schmidt D., Steinbrink S., Mirgorodskaya E., Lehmann V., Habermann K., Dreher F., Gustavsson N., Kessler T., Lehrach H., Herwig R., Gobom J., Ploubidou A., Boutros M., Lange B. M. (2010) Proteomic and functional analysis of the mitotic Drosophila centrosome. EMBO J. 29, 3344–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doxsey S., McCollum D., Theurkauf W. (2005) Centrosomes in cellular regulation. Annu. Rev. Cell Dev. Biol. 21, 411–434 [DOI] [PubMed] [Google Scholar]
- 25. Hutchins J. R., Toyoda Y., Hegemann B., Poser I., Hériché J. K., Sykora M. M., Augsburg M., Hudecz O., Buschhorn B. A., Bulkescher J., Conrad C., Comartin D., Schleiffer A., Sarov M., Pozniakovsky A., Slabicki M. M., Schloissnig S., Steinmacher I., Leuschner M., Ssykor A., Lawo S., Pelletier L., Stark H., Nasmyth K., Ellenberg J., Durbin R., Buchholz F., Mechtler K., Hyman A. A., Peters J. M. (2010) Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neumann B., Walter T., Hériché J. K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U., Cetin C., Sieckmann F., Pau G., Kabbe R., Wünsche A., Satagopam V., Schmitz M. H., Chapuis C., Gerlich D. W., Schneider R., Eils R., Huber W., Peters J. M., Hyman A. A., Durbin R., Pepperkok R., Ellenberg J. (2010) Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gergely F., Karlsson C., Still I., Cowell J., Kilmartin J., Raff J. W. (2000) The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc. Natl. Acad. Sci. U.S.A. 97, 14352–14357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peset I., Vernos I. (2008) The TACC proteins. TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 18, 379–388 [DOI] [PubMed] [Google Scholar]
- 29. Raff J. W. (2002) Centrosomes and cancer. Lessons from a TACC. Trends Cell Biol. 12, 222–225 [DOI] [PubMed] [Google Scholar]
- 30. Gergely F. (2002) Centrosomal TACCtics. Bioessays 24, 915–925 [DOI] [PubMed] [Google Scholar]
- 31. Hood F. E., Royle S. J. (2011) Pulling it together. The mitotic function of TACC3. Bioarchitecture 1, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schneider L., Essmann F., Kletke A., Rio P., Hanenberg H., Wetzel W., Schulze-Osthoff K., Nürnberg B., Piekorz R. P. (2007) The transforming acidic coiled coil 3 protein is essential for spindle-dependent chromosome alignment and mitotic survival. J. Biol. Chem. 282, 29273–29283 [DOI] [PubMed] [Google Scholar]
- 33. Still I. H., Vettaikkorumakankauv A. K., DiMatteo A., Liang P. (2004) Structure-function evolution of the transforming acidic coiled coil genes revealed by analysis of phylogenetically diverse organisms. BMC Evol. Biol. 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thakur H. C., Singh M., Nagel-Steger L., Prumbaum D., Fansa E. K., Gremer L., Ezzahoini H., Abts A., Schmitt L., Raunser S., Ahmadian M. R., Piekorz R. P. (2013) Role of centrosomal adaptor proteins of the TACC family in the regulation of microtubule dynamics during mitotic cell division. Biol. Chem. 394, 1411–1423 [DOI] [PubMed] [Google Scholar]
- 35. Hao Z., Stoler M. H., Sen B., Shore A., Westbrook A., Flickinger C. J., Herr J. C., Coonrod S. A. (2002) TACC3 expression and localization in the murine egg and ovary. Mol. Reprod. Dev. 63, 291–299 [DOI] [PubMed] [Google Scholar]
- 36. Sadek C. M., Pelto-Huikko M., Tujague M., Steffensen K. R., Wennerholm M., Gustafsson J. A. (2003) TACC3 expression is tightly regulated during early differentiation. Gene Expr. Patterns 3, 203–211 [DOI] [PubMed] [Google Scholar]
- 37. Schuendeln M. M., Piekorz R. P., Wichmann C., Lee Y., McKinnon P. J., Boyd K., Takahashi Y., Ihle J. N. (2004) The centrosomal, putative tumor suppressor protein TACC2 is dispensable for normal development, and deficiency does not lead to cancer. Mol. Cell. Biol. 24, 6403–6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piekorz R. P., Hoffmeyer A., Duntsch C. D., McKay C., Nakajima H., Sexl V., Snyder L., Rehg J., Ihle J. N. (2002) The centrosomal protein TACC3 is essential for hematopoietic stem cell function and genetically interfaces with p53-regulated apoptosis. EMBO J. 21, 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jeng J. C., Lin Y. M., Lin C. H., Shih H. M. (2009) Cdh1 controls the stability of TACC3. Cell Cycle 8, 3529–3536 [DOI] [PubMed] [Google Scholar]
- 40. Yao R., Natsume Y., Noda T. (2007) TACC3 is required for the proper mitosis of sclerotome mesenchymal cells during formation of the axial skeleton. Cancer Sci. 98, 555–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmidt S., Schneider L., Essmann F., Cirstea I. C., Kuck F., Kletke A., Jänicke R. U., Wiek C., Hanenberg H., Ahmadian M. R., Schulze-Osthoff K., Nürnberg B., Piekorz R. P. (2010) The centrosomal protein TACC3 controls paclitaxel sensitivity by modulating a premature senescence program. Oncogene 29, 6184–6192 [DOI] [PubMed] [Google Scholar]
- 42. Schneider L., Essmann F., Kletke A., Rio P., Hanenberg H., Schulze-Osthoff K., Nürnberg B., Piekorz R. P. (2008) TACC3 depletion sensitizes to paclitaxel-induced cell death and overrides p21WAF-mediated cell cycle arrest. Oncogene 27, 116–125 [DOI] [PubMed] [Google Scholar]
- 43. Barros T. P., Kinoshita K., Hyman A. A., Raff J. W. (2005) Aurora A activates D-TACC-Msps complexes exclusively at centrosomes to stabilize centrosomal microtubules. J. Cell Biol. 170, 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kinoshita K., Noetzel T. L., Pelletier L., Mechtler K., Drechsel D. N., Schwager A., Lee M., Raff J. W., Hyman A. A. (2005) Aurora A phosphorylation of TACC3/maskin is required for centrosome-dependent microtubule assembly in mitosis. J. Cell Biol. 170, 1047–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. LeRoy P. J., Hunter J. J., Hoar K. M., Burke K. E., Shinde V., Ruan J., Bowman D., Galvin K., Ecsedy J. A. (2007) Localization of human TACC3 to mitotic spindles is mediated by phosphorylation on Ser-558 by Aurora A. A novel pharmacodynamic method for measuring Aurora A activity. Cancer Res. 67, 5362–5370 [DOI] [PubMed] [Google Scholar]
- 46. Peset I., Seiler J., Sardon T., Bejarano L. A., Rybina S., Vernos I. (2005) Function and regulation of Maskin, a TACC family protein, in microtubule growth during mitosis. J. Cell Biol. 170, 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fu W., Chen H., Wang G., Luo J., Deng Z., Xin G., Xu N., Guo X., Lei J., Jiang Q., Zhang C. (2013) Self-assembly and sorting of acentrosomal microtubules by TACC3 facilitate kinetochore capture during the mitotic spindle assembly. Proc. Natl. Acad. Sci. U.S.A. 110, 15295–15300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lioutas A., Vernos I. (2013) Aurora A kinase and its substrate TACC3 are required for central spindle assembly. EMBO Rep. 14, 829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fu W., Jiang Q., Zhang C. (2011) Novel functions of endocytic player clathrin in mitosis. Cell Res. 21, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Royle S. J. (2012) The role of clathrin in mitotic spindle organisation. J. Cell Sci. 125, 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Booth D. G., Hood F. E., Prior I. A., Royle S. J. (2011) A TACC3/ch-TOG/clathrin complex stabilises kinetochore fibres by inter-microtubule bridging. EMBO J. 30, 906–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hood F. E., Williams S. J., Burgess S. G., Richards M. W., Roth D., Straube A., Pfuhl M., Bayliss R., Royle S. J. (2013) Coordination of adjacent domains mediates TACC3-ch-TOG-clathrin assembly and mitotic spindle binding. J. Cell Biol. 202, 463–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gergely F., Draviam V. M., Raff J. W. (2003) The ch-TOG/XMAP215 protein is essential for spindle pole organization in human somatic cells. Genes Dev. 17, 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yao R., Kondoh Y., Natsume Y., Yamanaka H., Inoue M., Toki H., Takagi R., Shimizu T., Yamori T., Osada H., Noda T. (2013) A small compound targeting TACC3 revealed its different spatiotemporal contributions for spindle assembly in cancer cells. Oncogene DOI: 10.1038/onc.2013.382 [DOI] [PubMed] [Google Scholar]
- 55. Gómez-Baldó L., Schmidt S., Maxwell C. A., Bonifaci N., Gabaldón T., Vidalain P. O., Senapedis W., Kletke A., Rosing M., Barnekow A., Rottapel R., Capellá G., Vidal M., Astrinidis A., Piekorz R. P., Pujana M. A. (2010) TACC3-TSC2 maintains nuclear envelope structure and controls cell division. Cell Cycle 9, 1143–1155 [DOI] [PubMed] [Google Scholar]
- 56. Lauffart B., Howell S. J., Tasch J. E., Cowell J. K., Still I. H. (2002) Interaction of the transforming acidic coiled-coil 1 (TACC1) protein with ch-TOG and GAS41/NuBI1 suggests multiple TACC1-containing protein complexes in human cells. Biochem. J. 363, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sato M., Vardy L., Angel Garcia M., Koonrugsa N., Toda T. (2004) Interdependency of fission yeast Alp14/TOG and coiled coil protein Alp7 in microtubule localization and bipolar spindle formation. Mol. Biol. Cell 15, 1609–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Albee A. J., Tao W., Wiese C. (2006) Phosphorylation of maskin by Aurora-A is regulated by RanGTP and importin β. J. Biol. Chem. 281, 38293–38301 [DOI] [PubMed] [Google Scholar]
- 59. Albee A. J., Wiese C. (2008) Xenopus TACC3/maskin is not required for microtubule stability but is required for anchoring microtubules at the centrosome. Mol. Biol. Cell 19, 3347–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thompson J. D., Higgins D. G., Gibson T. J. (1994) ClustalW. Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 62. Thakur H. C. (2012) Biochemical and biophysical characterization of the centrosomal protein TACC3. Doctoral dissertation, Faculty of Mathematics and Natural Sciences of the Heinrich Heine University, Düsseldorf, Germany [Google Scholar]
- 63. Eberth A., Lundmark R., Gremer L., Dvorsky R., Koessmeier K. T., McMahon H. T., Ahmadian M. R. (2009) A BAR domain-mediated autoinhibitory mechanism for RhoGAPs of the GRAF family. Biochem. J. 417, 371–377 [DOI] [PubMed] [Google Scholar]
- 64. Risse S. L., Vaz B., Burton M. F., Aspenström P., Piekorz R. P., Brunsveld L., Ahmadian M. R. (2013) SH3-mediated targeting of Wrch1/RhoU by multiple adaptor proteins. Biol. Chem. 394, 421–432 [DOI] [PubMed] [Google Scholar]
- 65. Sardon T., Pache R. A., Stein A., Molina H., Vernos I., Aloy P. (2010) Uncovering new substrates for Aurora A kinase. EMBO Rep. 11, 977–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Partch C. L., Gardner K. H. (2011) Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc. Natl. Acad. Sci. U.S.A. 108, 7739–7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cassimeris L., Morabito J. (2004) TOGp, the human homolog of XMAP215/Dis1, is required for centrosome integrity, spindle pole organization, and bipolar spindle assembly. Mol. Biol. Cell 15, 1580–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xie Z., Moy L. Y., Sanada K., Zhou Y., Buchman J. J., Tsai L.-H. (2007) Cep120 and TACCs control interkinetic nuclear migration and the neural progenitor pool. Neuron 56, 79–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang Y. T., Wang C. L., Van Aelst L. (2012) DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nat. Neurosci. 15, 1201–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Garriga-Canut M., Orkin S. H. (2004) Transforming acidic coiled-coil protein 3 (TACC3) controls friend of GATA-1 (FOG-1) subcellular localization and regulates the association between GATA-1 and FOG-1 during hematopoiesis. J. Biol. Chem. 279, 23597–23605 [DOI] [PubMed] [Google Scholar]
- 71. Angrisano T., Lembo F., Pero R., Natale F., Fusco A., Avvedimento V. E., Bruni C. B., Chiariotti L. (2006) TACC3 mediates the association of MBD2 with histone acetyltransferases and relieves transcriptional repression of methylated promoters. Nucleic Acids Res. 34, 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Simpson R. J., Yi Lee S. H., Bartle N., Sum E. Y., Visvader J. E., Matthews J. M., Mackay J. P., Crossley M. (2004) A classic zinc finger from friend of GATA mediates an interaction with the coiled-coil of transforming acidic coiled-coil 3. J. Biol. Chem. 279, 39789–39797 [DOI] [PubMed] [Google Scholar]
- 73. Brouhard G. J., Stear J. H., Noetzel T. L., Al-Bassam J., Kinoshita K., Harrison S. C., Howard J., Hyman A. A. (2008) XMAP215 is a processive microtubule polymerase. Cell 132, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Popov A. V., Pozniakovsky A., Arnal I., Antony C., Ashford A. J., Kinoshita K., Tournebize R., Hyman A. A., Karsenti E. (2001) XMAP215 regulates microtubule dynamics through two distinct domains. EMBO J. 20, 397–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shi Y., Mowery R. A., Ashley J., Hentz M., Ramirez A. J., Bilgicer B., Slunt-Brown H., Borchelt D. R., Shaw B. F. (2012) Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci. 21, 1197–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yao R., Natsume Y., Saiki Y., Shioya H., Takeuchi K., Yamori T., Toki H., Aoki I., Saga T., Noda T. (2012) Disruption of Tacc3 function leads to in vivo tumor regression. Oncogene 31, 135–148 [DOI] [PubMed] [Google Scholar]
- 77. Hodis E., Watson I. R., Kryukov G. V., Arold S. T., Imielinski M., Theurillat J. P., Nickerson E., Auclair D., Li L., Place C., Dicara D., Ramos A. H., Lawrence M. S., Cibulskis K., Sivachenko A., Voet D., Saksena G., Stransky N., Onofrio R. C., Winckler W., Ardlie K., Wagle N., Wargo J., Chong K., Morton D. L., Stemke-Hale K., Chen G., Noble M., Meyerson M., Ladbury J. E., Davies M. A., Gershenwald J. E., Wagner S. N., Hoon D. S., Schadendorf D., Lander E. S., Gabriel S. B., Getz G., Garraway L. A., Chin L. (2012) A landscape of driver mutations in melanoma. Cell 150, 251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lauffart B., Vaughan M. M., Eddy R., Chervinsky D., DiCioccio R. A., Black J. D., Still I. H. (2005) Aberrations of TACC1 and TACC3 are associated with ovarian cancer. BMC Women's Health 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Singh D., Chan J. M., Zoppoli P., Niola F., Sullivan R., Castano A., Liu E. M., Reichel J., Porrati P., Pellegatta S., Qiu K., Gao Z., Ceccarelli M., Riccardi R., Brat D. J., Guha A., Aldape K., Golfinos J. G., Zagzag D., Mikkelsen T., Finocchiaro G., Lasorella A., Rabadan R., Iavarone A. (2012) Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Williams S. V., Hurst C. D., Knowles M. A. (2013) Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 22, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vitre B. D., Cleveland D. W. (2012) Centrosomes, chromosome instability (CIN) and aneuploidy. Curr. Opin. Cell Biol. 24, 809–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Holland A. J., Cleveland D. W. (2012) Losing balance. The origin and impact of aneuploidy in cancer. EMBO Rep. 13, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Guo Y., Partch C. L., Key J., Card P. B., Pashkov V., Patel A., Bruick R. K., Wurdak H., Gardner K. H. (2013) Regulating the ARNT/TACC3 axis. Multiple approaches to manipulating protein/protein interactions with small molecules. ACS Chem. Biol. 8, 626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wurdak H., Zhu S., Min K. H., Aimone L., Lairson L. L., Watson J., Chopiuk G., Demas J., Charette B., Halder R., Weerapana E., Cravatt B. F., Cline H. T., Peters E. C., Zhang J., Walker J. R., Wu C., Chang J., Tuntland T., Cho C. Y., Schultz P. G. (2010) A small molecule accelerates neuronal differentiation in the adult rat. Proc. Natl. Acad. Sci. U.S.A. 107, 16542–16547 [DOI] [PMC free article] [PubMed] [Google Scholar]