

Abstract
The p53 tumor suppressor protein accumulates to very high concentrations in normal human fibroblasts infected by adenovirus type 5 mutants that cannot direct assembly of the viral E1B 55-kDa protein-containing E3 ubiquitin ligase that targets p53 for degradation. Despite high concentrations of nuclear p53, the p53 transcriptional program is not induced in these infected cells. We exploited this system to examine select post-translational modifications (PTMs) present on a transcriptionally inert population of endogenous human p53, as well as on p53 activated in response to etoposide treatment of normal human fibroblasts. These forms of p53 were purified from whole cell lysates by means of immunoaffinity chromatography and SDS-PAGE, and peptides derived from them were subjected to nano-ultra-high-performance LC-MS and MS/MS analyses on a high-resolution accurate-mass MS platform (data available via ProteomeXchange, PXD000464). We identified an unexpectedly large number of PTMs, comprising phosphorylation of Ser and Thr residues, methylation of Arg residues, and acetylation, ubiquitinylation, and methylation of Lys residues—for example, some 150 previously undescribed modifications of p53 isolated from infected cells. These modifications were distributed across all functional domains of both forms of the endogenous human p53 protein, as well as those of an orthologous population of p53 isolated from COS-1 cells. Despite the differences in activity, including greater in vitro sequence-specific DNA binding activity exhibited by p53 isolated from etoposide-treated cells, few differences were observed in the location, nature, or relative frequencies of PTMs on the two populations of human p53. Indeed, the wealth of PTMs that we have identified is consistent with a far greater degree of complex, combinatorial regulation of p53 by PTM than previously anticipated.
The p53 tumor suppressor protein, first discovered by virtue of its interaction with simian virus 40 large T antigen (1, 2), is an integral component of numerous cellular signaling pathways, including those that regulate apoptosis, cell cycle arrest, mitosis, and cellular metabolism (3–11). This protein has long been known to be a crucial determinant of cell fate in response to genotoxic and other forms of stress (3–7), and the p53 gene is frequently mutated in human cancers (12–15). The p53 protein is present at low concentrations in unstressed cells, primarily as a result of the action of the Mdm2 E3 ubiquitin ligase, which polyubiquitinylates p53 to promote its nuclear export and target it for proteasomal degradation (16, 17). The MdmX protein, which binds to both Mdm2 and p53, is also thought to ensure transcriptional inactivity of p53 and/or its rapid turnover, by increasing the efficiency of p53 ubiquitinylation (16–19). In response to particular stresses, Mdm2 and MdmX may be inactivated via phosphorylation or deubiquitinylation, targeted for degradation, or sequestered through the binding of proteins such as p14ARF (16, 17, 19). Post-translational modifications (PTMs)1 of p53 may also inhibit association with Mdm2 or MdmX and prevent p53 degradation (16, 17, 20, 21). In response to cellular stress, newly synthesized p53 undergoes a series of stabilizing and activating modifications and initiates a stressor-specific response that is dictated by the severity of the cellular damage and may be transcription-dependent or -independent (22–24). Modified p53 enters the nucleus to act as a transcriptional regulator, or it remains in the cytoplasm, where it inhibits autophagy or triggers apoptosis via mitochondrial membrane permeablization (17, 21, 22). As a transcriptional regulator, p53 modulates the expression of a broad range of target genes, including those associated with cell cycle progression, apoptosis, and the responses to DNA damage or the accumulation of reactive oxygen species (9, 11, 25–27). The precise biochemical mechanisms that govern the ability of p53 to activate or repress particular subsets of genes in response to a specific signal remain unclear, as do the characteristics of p53 that determine such cellular responses as apoptosis and cell cycle arrest.
In both stressed and unstressed cells, the p53 protein is subject to extensive PTMs (5, 20, 28–32) that regulate its stability, localization, and transcriptional activity in both a stress- and a cell-type-specific manner (21, 24). Although the signals, pathways, and enzymes that result in modification of p53 at a significant number of sites have been investigated extensively (20, 28, 29, 31, 32), the significance of particular modifications is not yet well understood, particularly in vivo (33–36). The p53 protein is organized into four primary functional domains, an N-terminal transactivation domain (TAD), a core DNA-binding domain (DBD), a tetramerization domain (TD) containing a nuclear localization sequence, and a C-terminal regulatory basic domain (BD) (28, 29, 37–40). PTMs identified within these domains include acetylation, methylation, ubiquitinylation, sumoylation and neddylation of Lys residues, methylation of Arg residues, phosphorylation of Ser or Thr residues, ADP-ribosylation of Asp or Glu residues, and O-glycosylation of Ser residues (28, 29). Of these, phosphorylation, acetylation, ubiquitinylation, and methylation have been best shown to modulate the transcriptional activity of p53 (17, 20, 28, 29). Phosphorylation of Ser or Thr residues in the TAD of p53 has been correlated with increasing p53 activity in response to cellular stress (10, 28, 29, 36, 41). Within the DBD, Ser and Thr phosphorylation induce alterations in local structure that direct sequence-specific DNA binding (42–44), affect the ability of p53 to interact with transcriptional coactivators such as 14–3-3 (45, 46), and contribute to the modulation of p53 transcriptional activity (47–49) or degradation (50, 51). Toward the C terminus, phosphorylation of specific Ser or Thr residues has been implicated in the regulation of p53 transcriptional activity, DNA binding, and conformational change within the p53 tetramer (44, 52–54). Acetylation of Lys residues within the DBD and TD enhances sequence-specific DNA binding and p53 transcriptional activity (55–60), and acetylation of the six Lys residues present within the BD has been shown to be required for tetramer formation, nuclear localization, and activation of p53-dependent transcription in response to stress (55, 61–64). Mono- and polyubiquitinylation of Lys residues within the DBD have been demonstrated to regulate p53 protein stability (65), whereas mono- and polyubiquitinylation of Lys residues within the TD have been implicated in targeting p53 for nuclear export, inhibiting p53 nuclear import, and down-regulating the apoptotic transcriptional response (41, 66–68). Monoubiquitinylation of Lys residues within the BD has also been reported to regulate the nuclear export of p53 (69), and the polyubiquitinylation of these same residues has been demonstrated to contribute to the targeting of p53 for proteasomal degradation (16, 17, 20, 41). However, the ubiquitinylation of any of these residues alone is not sufficient for proteasomal degradation, suggesting that constellations of mono- and/or polyubiquitinylated lysines may govern p53 protein stability (17, 20, 33, 34, 65). Finally, methylation of three Arg residues within the TD influences p53 binding activity and discrimination among transcriptional targets (70), and different combinations of mono- and dimethylation of the six Lys residues within the BD have been reported to activate or inhibit p53-dependent transcription (71–75). Thus, there is a substantial body of evidence indicating that PTMs of p53 provide complex and combinatorial regulation, enabling the protein to become activated rapidly in response to stress and coordinating the appropriate transcriptional response (5, 10, 74, 76, 77).
During infection with the prototypical species C human adenovirus, serotype 5 (Ad5), interactions of the viral immediate early E1A gene products with the pRb tumor suppressor protein and p300/CBP histone acetyltransferases promote the stabilization and accumulation of p53 (78–82). The two major proteins encoded by early gene E1B prevent this population of p53 from initiating either cell cycle arrest or an apoptotic, antiviral response within the host cell. The E1B 19-kDa protein is a mimetic of the anti-apoptotic Bcl2-type protein Mcl-1, which averts apoptosis by binding to and neutralizing the proapoptotic proteins Bax and Bak (81). The E1B 55-kDa protein assembles with the viral E4orf6 protein and the cellular proteins cullin 5, elongins B and C, and Rbx1 to form a virus-specific E3 ubiquitin ligase that targets p53 for proteasomal degradation (83–87). When this enzyme cannot assemble in cells infected by E1B 55-kDa-null or E4orf6-null mutants, the p53 protein accumulates to high concentrations (88, 89). This population of p53 appears to localize correctly within the nucleus and perinuclear regions of the host cell (90), but it does not induce apoptosis or expression of p53-responsive genes (90–93).
The great majority of previous analyses of p53 PTMs have relied upon the overproduction of exogenous p53 and/or the application of stressors to transformed mammalian cell lines to circumvent the normally rapid turnover and low steady-state concentrations of p53. The high concentrations of this protein that accumulate in cells infected with E1B 55-kDa null mutants of Ad5 provide a unique opportunity to characterize the PTMs of endogenous, wild-type p53 made in normal human cells and, furthermore, p53 that is not engaged in transcriptional regulation. We have therefore exploited this system to generate an extensive PTM profile of p53 isolated from normal human foreskin fibroblasts and to compare it to that of p53 derived from the same cells, activated in response to etoposide treatment.
EXPERIMENTAL PROCEDURES
Cells and Viruses
Human foreskin fibroblasts (HFFs) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 7.5% bovine growth serum (ThermoFisher Scientific. Hyclone, Logan, UT). HeLa and COS-1 cells were maintained in DMEM supplemented with 5% bovine growth serum and 5% calf serum (Invitrogen).
The E1B 55-kDa null mutant AdEasyE1Δ2347 is a phenotypically wild-type derivative of AdEasy containing the adenoviral E1A and E1B transcription units and a single nucleotide deletion at bp 2347 within the E1B coding sequence. The construction and characterization of this mutant have been described previously (94, 95).
p53 Immunoaffinity Resin
p53 immunoaffinity resin was generated by incubating protein G–conjugated agarose beads (ThermoFisher Pierce, Rockford, IL) with a mixture of equal volumes of hybridoma supernatants containing mouse monoclonal antibodies 421, 1620, and 1801 (96–98) for 24 to 48 h at 4 °C with constant rotation. Antibody-bound beads were washed two to three times in 10- to 15-fold excess volumes of Tris-buffered saline (TBS) (50 mm Tris-HCl, pH 7.5, containing 150 mm NaCl) and equilibrated in 200 mm triethanolamine (pH 8.9). The beads were then crosslinked in 200 mm triethanolamine (pH 8.9) and 50 mm dimethyl pimelimidate, blocked in 100 mm ethanolamine (pH 8.9), and washed as described above in TBS containing 4 μm leupeptin, 1.7 μm antipain, 1.5 μm pepstatin, and 1 mm PMSF. The p53 resin was stored in TBS on ice at 4 °C until use.
p53 Purification and Immunoprecipitation
HFFs at 90% confluency were infected with 100 pfu/cell AdEasyE1Δ2347 for 44 h. Cells were then washed twice with cold phosphate-buffered saline (PBS) and incubated on ice for 20 min in lysis buffer (20 mm Tris-HCl (pH 7.5) containing 250 mm NaCl, 1% (v/v) ionic detergent (Triton X-100 or N-lauryl sarcosine), 4 μm leupeptin, 1.7 μm antipain, 1.5 μm pepstatin, 1 mm PMSF, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1.5 mm sodium butyrate, 1 mm β-glycerophosphate, and 5 μm microcystin). For preparations using Triton X-100, nuclei were lysed by the addition of NaCl to 500 mm. Upon a visible increase in sample viscosity, lysates were diluted 1:2 in lysis buffer lacking NaCl. MgCl2 was added to a final concentration of 1 mm, and genomic DNA was degraded by treatment with 250 units of Benzonase nuclease (Sigma Aldrich, St. Louis, MO) for 20 min at 37 °C. The lysates were centrifuged at 13.2 krpm for 10 min at 4 °C to pellet debris and then chilled on ice for 15 min. p53 immunoaffinity resin was preblocked in an equivalent volume of HeLa cell lysate, prepared as described above, for 1 h at room temperature with constant rocking. The resin was then washed in TBS and added to infected HFF cell lysates. For those preparations including N-lauryl sarcosine, cell lysates were centrifuged at 13.2 krpm for 15 min at 4 °C. Following the removal of pelleted genomic DNA, lysates were added directly to p53 immunoaffinity resin, without an intervening blocking step. For both preparations, p53 resin was added to infected cell lysates at a ratio of 25 μl of resin per 10-cm dish of lysed cells.
To isolate p53 from COS-1 cells, cells at 90% confluency were washed twice with cold PBS and lysates prepared via extraction in the presence of N-lauryl sarcosine and benzonase nuclease digestion of DNA exactly as described above. The lysates were centrifuged at 13.2 krpm for 10 min at 4 °C to pellet debris, chilled on ice for 15 min, and added to p53 immunoaffinity resin at a ratio of 50 μl of resin per 10-cm dish of cells.
In all cases, the lysates were rotated with p53 resin for 4 h at 4 °C prior to washing of the resin once in TBS, four times in TBS with [NaCl] increasing from 350 to 500 mm, and once with TBS. All wash buffers were added to at least 50 bead volumes and contained 4 μm leupeptin, 1.7 μm antipain, 1.5 μm pepstatin, and 1 mm PMSF. For preparations using Triton X-100, p53 was eluted from the resin in two bead volumes of 200 mm triethanolamine, pH 11.75, and immediately neutralized with one bead volume of 1 m Tris, pH 7. For those obtained with N-lauryl sarcosine, p53 was eluted by washing the resin in 10 bead volumes of 50 mm Tris, pH 7.5, followed by boiling for 5 min at 95 °C in 1 bead volume of 4X LDS sample buffer (Novex, Life Technologies, Hercules, CA).
Immunoprecipitation efficiency and p53 protein concentration were assessed via immunoblotting as described previously (99). p53 was visualized with mouse monoclonal antibody D01-HRP (sc-126, Santa Cruz Biotechnologies, Santa Cruz, CA), actin with mouse monoclonal antibody AC-15 HRP (Abcam, Cambridge, MA), and IgG heavy chain with goat anti-mouse HRP (AP124P, Chemicon, EMD Millipore, Billerica, MA). Densitometric measurements were performed using Image J (100), with the β-actin signal as an internal control. For the comparison of p53 protein concentrations in infected and etoposide-treated cells, etoposide (Sigma) was diluted to 50 mm in dimethyl sulfoxide and added to medium at a final concentration of 125 μm. Cells were maintained in the presence of etoposide for 44 h, washed twice in PBS, harvested, and incubated in lysis buffer containing N-lauryl sarcosine for analysis via immunoblotting.
Assignment of p53 Post-translational Modifications via Mass Spectrometry
Eluted p53 samples were resolved via SDS-PAGE on 4–12% NuPAGE Novex® Bis-Tris gels (Invitrogen) and colloidal Coomassie stained (GelCode Blue, Thermo Pierce) according to the manufacturers' protocols. Processing and MS analysis of samples were performed at the Princeton Proteomics and Mass Spectrometry Core. Stained gel bands of ∼50 kDa (which typically comprised a doublet with a third band of slightly slower migration) were excised, diced into 1-mm3 cubes, and dehydrated in acetonitrile. Gel pieces were destained with ammonium bicarbonate buffer, washed extensively, and subjected to thiol reduction by Tris (2-carboxyethyl)phosphine hydrochloride; iodoacetamide alkylation; overnight digestion with trypsin, chymotrypsin, or Lys-C endoproteinases; and peptide elution, following the methods of Shevchenko et al. (101). Peptides were desalted using StageTip micro-scale reversed-phase chromatography (102). They were then subjected to reversed-phase nano-LC-MS and MS/MS performed on a nano-flow capillary ultra-high-performance liquid chromatography (UPLC) system (Nano Ultra 2D Plus, Eksigent, Redwood City, CA) coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (ThermoFisher Scientific, Bremen, Germany) outfitted with a Triversa NanoMate ion source robot (Advion, Ithaca, NY) or the same UPLC system coupled to an LTQ-Orbitrap Velos hybrid mass spectrometer (Thermo Fisher Scientific) outfitted with a Flex ion source (ThermoFisher Proxeon, Odense, Denmark). Sample concentration and washing were performed online using a trapping capillary column (150 μm × ∼40 mm, packed with 3-μm 100-Å Magic AQ C18 resin (Bruker Michrom, Auburn, CA)) at a flow rate of 4 μl/min for 4 min. Separation was achieved using an analytical capillary column (75 μm × ∼15 cm, packed with 1.7-μm 100-Å BEH C18 resin (Waters, Milford, MA)) and a linear gradient of solutions A and B (solution A: 3% acetonitrile/0.1% formic acid/0.1% acetic acid; solution B: 97% acetonitrile/0.1% formic acid/0.1% acetic acid) applied over 180 min at a flow rate of ∼250 nl/min. Nanoelectrospray ionization was carried out using the NanoMate ion source at 1.74 kV, with the LTQ heated capillary set to 200 °C. Alternatively, nanoelectrospray was accomplished on the Orbi Velos platform at 2 to 2.4 kV using a capillary temperature of 275 °C. Full-scan mass spectra were acquired in the Orbitrap in positive ion mode over the m/z range of 335–1800 at a resolving power of 100,000. MS/MS spectra were simultaneously acquired using collision-induced dissociation in the LTQ for the 7 (Orbi XL) or 15 (Orbi Velos) most abundant multiply charged species in the full-scan spectrum having signal intensities of >1000 NL. All spectra were acquired in profile mode. Dynamic exclusion was set such that MS/MS scans were acquired only once for each species over a period of 120 s.
The resulting LC-MS/MS data were subjected to preprocessing into peaklist files (mgf) using ProteomeDiscoverer (v. 1.3, Thermo Fisher). These files were searched against the Swiss-Prot Human database and the human p53 sequence determined via direct sequencing of the p53 gene from cDNA derived from the specific strain of HFFs used in this study (identical to the consensus sequence for UniProt P04637). The Mascot search engine (v. 2.2.7, Matrix Science, London, UK) was employed, allowing for an initial mass error of 8 ppm for the precursor and 1.2 Da for fragment ion species, no more than five missed trypsin cleavages, carbamidomethylation of cysteines as a fixed modification, and methionine oxidation and N-terminal protein acetylation as initial variable modifications. Results from Mascot Error Tolerant searching, which employed a customized unimod.xml file from which irrelevant chemical modification classes had been removed, were used to guide the iterative inclusion of PTMs as variable modifications in subsequent variable modification searches. Phosphorylation of Ser and Thr; acetylation of Lys; methylation, dimethylation, and trimethylation of Lys and Arg; diglycine modification of Lys (ubiquitinylation); and deamidation of Asn and Gln were used iteratively as variable modifications. Aggregate search results for each sample were collated and consolidated using the Scaffold software (Proteome Software, Portland, OR) according to the PeptideProphet and ProteinProphet parsimony algorithms (ISB) and filtered to the 90% peptide confidence level and a precursor mass accuracy within 2 to 3 ppm (complete sets of annotated, mass-labeled peptide spectral matches, as well as raw data files, have been deposited at the ProteomeXchange Consortium (proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000464 and DOI 10.6019/PXD000464).
Ascore-derived PTM site localization probabilities (103) were calculated for each PTM using the implementation of this algorithm in the ScaffoldPTM software (v. 2.0, Proteome Software). Fragmentation spectral assignments were then subject to manual inspection and validation using the original tandem mass spectra acquired in profile mode using Xcalibur software (Thermo Fisher). Spectral counting-based quantification of PTMs by residue was performed by ScaffoldPTM upon the peptide spectral matches with ≥90% peptide ID confidence levels and ≥95% PTM site Ascore localization probabilities. Equal concentrations of the p53 samples were loaded on the LC-MS instrument.
Sequencing of p53 cDNA
HFFs from the same strain as those infected for p53 purification and analysis were washed twice in PBS and homogenized in TRI Reagent (Sigma). Whole-cell RNA was isolated according to the manufacturer's protocol. Purified RNA was resuspended in DNase I buffer (Roche Biosciences, Palo Alto, CA), digested with 10 U/μl DNAseI (Roche) for 30 min at 37 °C, extracted with Tris-buffered phenol:chloroform (1:1), precipitated with 100% ethanol, washed with 75% ethanol, dried, and resuspended in RNase-free water. RNA concentrations were determined from A260 readings measured using a NanoDrop ND-1000 spectrophotometer. HFF cDNA was prepared from the purified RNA using the AccuScript High Fidelity 1st Strand cDNA Synthesis Kit (Stratagene, Agilent, La Jolla, CA) and 0.5 μg of random nonamer primers, according to the manufacturer's protocol. An amplicon within human glyceraldehyde-3 phosphate dehydrogenase was used as an internal control to assess the resulting cDNA quality. The forward (5′-CTGTTGCTGTAGCCAAATTCGT-3′) and reverse (5′-ACCCACTCCTCCACCTTTGAC-3′) primers for this amplicon were a kind gift from E. O'Keefe (Princeton University).
HFF p53 cDNA was amplified using primers corresponding to the following positions in p53 cDNA: forward 152–172 and reverse 686–704; forward 491–510 and reverse 1157–1176; forward 973–990 and reverse 1579–1597; forward 1458–1476 and reverse 2366–2388; and forward 2197–2217 and reverse 2564–2587. All primers were designed against the published sequence for human TP53 mRNA transcript variant 1 (NCBI RefSeq NM_000546.5), and the amplification conditions were optimized separately for each primer pair. Amplified products were gel-purified and sequenced on both strands (Genewiz, South Plainfield, NJ). Primers used in sequencing include those listed above, as well as forward 2404–2424 and reverse 2039–2059. The resulting sequences were aligned and compared using NCBI BLAST.
Analysis of DNA Binding by p53
HFFs at 90% confluency were infected with 100 pfu/cell AdEasyE1Δ2347 or treated with medium containing 125 μm etoposide for 44 h. Cells were harvested, washed twice with cold PBS, and incubated on ice for 20 min in 20 mm Tris-HCl (pH 7.5) containing 250 mm NaCl, 1% (v/v) N-lauryl sarcosine, 4 μm leupeptin, 1.7 μm antipain, 1.5 μm pepstatin, 1 mm PMSF, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1.5 mm sodium butyrate, and 1 mm β-glycerophosphate. Cell lysates were centrifuged at 13.2 krpm for 15 min at 4 °C. The p53 protein concentrations in these lysates were assessed and compared via immunoblotting as described above. Densitometric measurements were performed using Image J (100), with β-actin as an internal control. The ability of the p53 populations to bind DNA was determined using the p53 Transcription Factor Assay Kit (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer's protocol. Briefly, serial 2-fold dilutions of lysates containing equal starting concentrations of p53 were allowed to bind to an immobilized, synthetic consensus p53 DNA-binding sequence. The quantity of bound p53 was then determined via an ELISA-based method, and signals were measured at 450 nm using a Synergy Mx spectrophotometer (Biotek, Winooski, VT). Data analysis was performed using Gen5 software (v2.0, Biotek) and Microsoft Excel.
RESULTS
Isolation of p53 from Human Foreskin Fibroblasts
In order to examine in detail the PTMs of endogenous human p53 that is not transcriptionally engaged, we exploited normal HFFs infected with an adenovirus mutant that cannot direct synthesis of the E1B 55-kDa protein, AdEasyE1Δ2347 (94). As the virus-specific E3 ubiquitin ligase that targets p53 for proteasomal degradation is not formed in the absence of the E1B protein (85, 86, 88, 89), high concentrations of p53 accumulate in HFFs infected with this mutant (Fig. 1A). The initiation of p53 transcription and translation at multiple sites coupled with alternative splicing can produce nine distinct isoforms of p53, which retain all or most of the central DBD but differ in the extent of truncation of the N-terminal TAD and in C-terminal sequences (104, 105). Isoforms that lack the activation domain act as dominant-negative repressors of full-length p53-dependent transcription, whereas those that do not include the C-terminal TD and BDs are transcriptionally inactive (104, 105). We initially examined p53 accumulation in ΔE1B-infected cells using a monoclonal antibody that recognizes an N-terminal epitope of p53, and this antibody could not detect all dominant negative or inactive isoforms. However, exactly the same p53 species were observed using both this antibody and MAb240, which detects an epitope present in all denatured p53 isoforms, amino acids 210–226 (106) (Fig. 1B). Electrophoresis was carried out under conditions that would resolve p53 from isoforms with alternative C-terminal sequences (p53β or p53γ), both 46 kDa (104). We therefore conclude that neither dominant negative nor inactive p53 isoforms accumulate to significant concentrations in AdEasyE1Δ2347-infected HFFs.
Fig. 1.
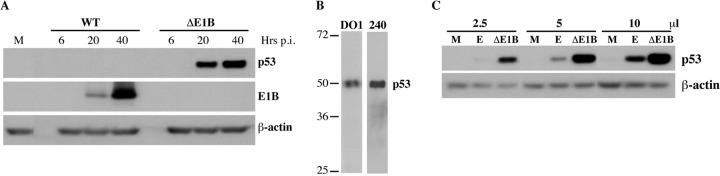
Properties of p53 isolated from normal human fibroblasts. A, HFFs were infected with 100 pfu/cell AdEasy E1 (WT) or AdEasyE1Δ2347 (ΔE1B) for the periods indicated, or mock infected (M). Whole cell extracts were prepared and the proteins listed were examined via immunoblotting as described under “Experimental Procedures.” B, p53 present in HFFs infected with AdEasyE1Δ2347 for 44 h was examined using the antibodies indicated at the top. C, Whole cell extracts were prepared from cells infected with AdEasyE1Δ2347 (ΔE1B) or exposed to 125 μm etoposide (E) for 44 h, and the quantity of p53 present in the increasing extract volumes indicated at the top was compared via immunoblotting.
In order to compare the steady-state concentration of p53 in such infected HFFs to that attained following stabilization and activation in response to DNA damage, HFFs were infected with AdEasyE1Δ2347 at 100 pfu/cell or exposed to the well-characterized topoisomerase II inhibitor etoposide (107, 108). Forty-four hours after infection or treatment, cells were lysed and p53 protein concentrations were examined via immunoblotting. Steady-state p53 concentrations were observed to be significantly higher in AdEasyE1Δ2347-infected HFFs than in the etoposide-treated cells (Fig. 1C)—greater than 10-fold higher, as measured by densitometric quantification of the immunoblot signals. As one measure of the activities of these p53 populations, we examined sequence-specific DNA-binding activity. The relative concentrations of p53 in whole cell lysates prepared in parallel from AdEasyE1Δ2347-infected and etoposide-treated cells were determined via immunoblotting. The binding of equal quantities of the two p53 proteins to a consensus p53 DNA binding site was then measured as a function of p53 concentration, as described under “Experimental Procedures.” Results from two independent experiments are shown in Fig. 2. At the highest concentrations of p53 tested, very similar quantities of p53 isolated from etoposide-treated HFFs (subsequently referred to as E p53) or from AdEasyE1Δ2347-infected HFFs (subsequently referred to as ΔE1B p53) bound to DNA (Fig. 2A). However, at lower concentrations, E p53 exhibited greater DNA binding activity, with increases of some 4- to 6-fold over the activity exhibited by ΔE1B p53 (Figs. 2B and 2C).
Fig. 2.
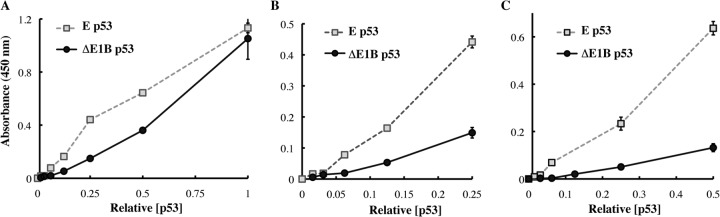
Analysis of DNA binding by p53. The relative concentrations of p53 present in whole cell extracts of HFFs infected with 100 pfu/cell AdEasyE1(Δ)2347 (ΔE1B p53) or exposed to 125 μm etoposide (E p53) for 44 h were determined via immunoblotting and quantification of signals, with β-actin as an internal control. The binding of equal quantities of the two forms of p53 to a consensus DNA binding site was then examined as a function of p53 concentration, as described under “Experimental Procedures.” All panels show the means and standard deviations of triplicate technical replicates. The results of independent experimental replicates are shown in panels A and C, and the initial portion of the binding curve shown in panel A is expanded in panel B.
To isolate p53 for the characterization of PTM, we used a custom immunoaffinity matrix, which proved more efficient than commercially available anti-p53 affinity resin or previously described metal ion affinity-based column chromatography (109). We observed that the lysis and solubilization conditions had a major effect on whether the p53 was amenable or refractory to purification. Nevertheless, ΔE1B p53 was purified successfully following lysis in the presence of either Triton X-100 or N-lauryl sarcosine, as described under “Experimental Procedures.” Representative results displaying the efficiency of p53 recovery and the removal of abundant cellular proteins are shown in Figs. 3A and 3B. As the sarkosyl-based method resulted in consistently higher yields of p53, we utilized it to isolate sufficient quantities of purified ΔE1B p53 to permit detailed PTM analysis via mass spectrometry.
Fig. 3.
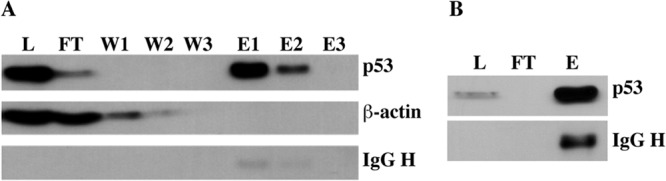
Examples of immunoaffinity purification of p53. HFFs were infected with 100 p.f.u./cell AdEasyE1Δ2347 for 44 h, and following lysate preparation, p53 was purified via immunoaffinity chromatography. A, lysates were prepared in buffer containing 1% (v/v) Triton X-100, and p53 was eluted in 200 mm TEA. The relative concentrations of p53, β-actin, and IgG heavy chain (IgG) in the lysate (L), flow-through (F), wash fractions (W1–W3), and eluates (E1 and E2) were determined via immunoblotting. B, lysates were prepared in buffer containing 1% (v/v) N-laurylsarcosine, and p53 was eluted from the immunoaffinity matrix via heat treatment. p53 and IgG heavy chain were visualized as described in panel A.
Characterization of Post-translationally Modified p53 Peptides of Endogenous Human p53
Three separate biological preparations of ΔE1B p53, as well as three preparations of E p53, were processed as described under “Experimental Procedures.” Briefly, cells were lysed in situ in the presence of multiple protease and phosphatase inhibitors and then p53 enriched rapidly via immunoaffinity purification. Purified p53 was subjected to SDS-PAGE and colloidal Coomassie staining prior to the in-gel digestion of protein bands of an approximate molecular mass of 50 kDa. To broaden the biochemical repertoire of peptides thus generated, increase sequence coverage, and maximize the chances of the recovery of modified peptides, we utilized trypsin, chymotrypsin, or Lys-C endoproteinases and employed high enzyme-to-substrate weight ratios (1:10). To obtain the highest sensitivity of detection, we then submitted the gel-eluted peptides to reversed-phase nano-LC-MS and MS/MS using UPLC Orbitrap hybrid MS platforms. High-resolution, high-mass-accuracy measurements of the intact peptide species were made in the Orbitrap, and the high-speed and ultra-high-sensitivity analyses of their fragmentation spectra were made in the instrument's linear ion trap. This approach was critical for deep exploration of low-abundance, substoichiometric modifications.
The p53 gene is notorious as a hotspot of nucleotide polymorphisms and disease-associated mutations (3, 110–112), catalogued, for example, in the IARC TP53 Mutation Database. The p53 coding sequence specified by the p53 gene present within the strain of HFFs used in these experiments was therefore determined through the sequencing of p53 cDNA as described in “Experimental Procedures.” This sequence was identical to the canonical human sequence specified in the UniProt database (data not shown). It was therefore possible to eliminate DNA sequence variation from consideration during the assignment of tandem mass spectra to modified p53 peptides. We performed an iterative, variable modification search strategy using the Mascot and X!Tandem search engines and both the Swiss-Prot Human and a p53-specific database, initially selecting the most commonly reported modifications, then adding additional modification classes iteratively, to determine whether additional high-confidence peptide matches could be made. We also performed “open” PTM searching using Mascot's Error Tolerant search feature, constrained by the use of a customized unimod.xml file from which entries associated with single amino acid substitutions and representing irrelevant chemical derivatizing agents (e.g. stable isotope labeling reagents) had been eliminated. These Error Tolerant results were used to indicate whether any readily detectable, significant classes of modification had been overlooked and to inform further variable modification searches. Aggregate database search results were consolidated and filtered to the 90% peptide confidence level using the Scaffold software implementation of the ProteinProphet parsimony algorithms (113, 114). These peptides were then further screened to include only those within a mass precision of 2 to 3 ppm of the highest scoring unmodified peptide assignments (which served as internal controls), and for the recurrence of identical PTM assignments on distinct but overlapping peptides across biological replicates. Finally, Ascore-derived localization probabilities (103) were used to designate the residue on which each assigned PTM was most likely located (supplemental Table S1) and to indicate the degree of confidence in that localization. Examples of high-confidence assignments of MS/MS spectra to PTM-bearing ΔE1B p53 and E p53 peptides are shown in Figs. 4 and 5, respectively.
Fig. 4.

Representative MS/MS spectra of ΔE1B p53. Peptides prepared from p53 isolates were subjected to reversed-phase nano-LC-MS and MS/MS on a UPLC-Orbitrap Velos platform as described under “Experimental Procedures.” Shown are representative examples of tandem mass spectra displaying their PTM-bearing peptide fragment ion assignments. Prominent fragment ions are labeled with their empirical m/z values and b- and y-ion designations or annotations indicating neutral losses from precursor species. The peptide sequences are displayed above the spectra, with all of the fragment ions matched in the spectra indicated by flags. Modified residues are color-coded by PTM in the following manner: phosphorylation, red; acetylation, green; ubiquitinylation, purple; monomethylation, navy; dimethylation, cerulean; trimethylation, turquoise; oxidation, tan; and carbamidomethylation, orange.
Fig. 5.

Representative MS/MS spectra of Ε p53. Peptides prepared from p53 isolated from HFFs treated with etoposide were subjected to reversed-phase nano-LC-MS and MS/MS on a UPLC-Orbitrap Velos platform as described under “Experimental Procedures.” Shown are representative examples of tandem mass spectra displaying their PTM-bearing peptide fragment ion assignments, labeled as described in the legend to Fig. 4.
Our analysis both confirmed many previously described p53 modifications and characterized a large number of modifications at novel sites. When the analysis of ΔE1B p53 was restricted to PTM classes best known to modulate p53 stability and transcriptional activity (phosphorylation, acetylation, ubiquitinylation, and methylation), we identified 99 modified residues bearing 222 PTMs. The complete catalogue of these sites of PTM and their classification is given in supplemental Table S2.
The previously recognized PTMs of ΔE1B p53 were present in all domains of the protein and were particularly dense within the TD and BD (Fig. 6A). Many of the modifications of these residues affect specific functions of the p53 protein. Notable examples include phosphorylation of Thr 150, Thr 155, Ser 183, Ser 215, Ser 269, and Thr 284 in the DBD, which has been implicated in targeting p53 for ubiquitin-dependent proteasomal degradation (47, 49–51, 115) and inducing a conformational change that destabilizes p53 and impairs DNA-binding and transcriptional activity (43, 48). Ubiquitinylation of Lys 351 and 357 in the TD by Msl2 results in increased cytoplasmic localization of p53 (67), as does ubiquitinylation of Lys 319, Lys 320, and Lys 321 by Mdm2 (68), whereas ubiquitinylation of Lys 291 and Lys 292 in the DBD/TD by Mkrn1 targets the protein for proteasomal degradation (116). Methylation of Arg 333 and dimethylation of Arg 335 and 337 within the TD by protein arginine methyltransferase 5 have been demonstrated to affect p53 transcriptional target site specificity (70). Six Lys residues (Lys 370, Lys 372, Lys 373, Lys 381, Lys 382, and Lys 386) in the BD were observed to be particularly heavily modified (Figs. 4 and 6A). Acetylation of these residues is known to contribute to the modulation of p53-mediated apoptosis and cell cycle arrest, inhibition of the interaction of Mdm2 with p53, and inhibition of Mdm2 recruitment to certain p53-dependent promoters (55–59), whereas ubiquitinylation of these same residues by Mdm2 results in the destabilization and proteasome-mediated degradation of p53 (34, 61, 117–121).
Fig. 6.

Post-translational modification of p53 isolated from AdEasyE1Δ2347-infected HFFs. The modifications reproducibly identified across three independent biological replicate samples of ΔE1B p53 are summarized, classified according to whether they have been previously described (A) or are novel (B). AD, activation domain; DBD, DNA-binding domain; NLS, nuclear localization signal; TD, tetramerization domain; BD, basic domain.
The 49 sites and 147 PTMs that were classified as novel were also distributed across all functional domains of ΔE1B p53 (Fig. 6B). These modifications included phosphorylation of Ser and Thr residues; acetylation and mono-, di-, or trimethylation of Lys residues; and these same methylations of numerous Arg residues (Fig. 6B). The great majority of the novel phosphorylation sites that we mapped were within, or close to, the DBD. Additional sites were present within the TAD, where the phosphorylation of particular residues has been correlated with the activation of p53 and the up-regulation of p53-dependent transcription in response to cellular stress (10, 29, 36, 67), and in the TD and BD, where phosphorylation has been shown to regulate the efficiency of p53-dependent transcription (44, 52–54). We identified trimethylation sites on the six C-terminal Lys residues (Lys 370, Lys 372, Lys 373, Lys 381, Lys 382, and Lys 386) previously identified as important in the control of p53 stability, localization, and transcriptional activity (55–59, 71–75). An extensive set of modifications were also detected on 13 additional Lys residues within the DBD, NLS, and TD. Notable examples include mono- and dimethylation of Lys 120, Lys 164, and Lys 320; acetylation of Lys 120 by Tip60/hMOF in response to DNA damage is required for the induction of apoptosis (56, 57, 122), whereas acetylation of Lys 164, by p300/Cbp, and of Lys 320, by Pcaf, enhances sequence-specific DNA-binding activity (55, 123–125). We also identified acetylation, methylation, and dimethylation sites on Lys 24 in the TAD, which is in close proximity to several well-characterized phosphorylation sites (e.g. Ser 15, Ser 20) (10, 29, 36, 67), as well as to an Mdm2 binding site (65, 126, 127). Twenty Arg residues, fifteen of which are in the DBD, were observed to be mono-, di-, and/or trimethylated. Significantly, these included Arg 158, Arg 175, Arg 248, Arg 249, Arg 273, and Arg 282, all of which correspond to mutation “hotspots” in human colorectal, breast, and lung cancers (110–112). Arg 174 and Arg 181 have been implicated in maintaining the folded structure of the p53 monomer (128), Arg 202 contributes to dimer structural stability (129), Arg 280 is essential for DNA binding (128, 130), and Arg 342 is necessary for p53-dependent transcription (131). The location and apparent extent of these modified Arg residues indicate a previously unrecognized degree of potential p53 regulation.
Of the 101 modified residues identified on E p53, 52 sites and 77 PTMs have been described previously, leaving a total of 49 sites and 153 PTMs classified as novel (supplemental Table S3). Both the locations of the modified residues and the PTMs identified on E p53 correspond closely to those assigned to ΔE1B p53 (Fig. 6; compare supplemental Tables S2 and S3).
Post-translational Modifications of COS-1 p53
COS-1 cells are monkey kidney epithelial cells that have been transformed with simian virus 40 DNA containing the viral early transcription unit (132). These cells constitutively produce the simian virus 40 large T antigen, which binds to and sequesters p53 (1, 2, 133), leading to the accumulation of transcriptionally inert p53 protein (133). This stable population of endogenous p53 has been examined previously via mass spectrometry for the presence of PTMs (134). In order to compare the nature and extent of PTM that we observed on HFF p53 to that of COS-1 p53, we isolated p53 from COS-1 cells via immunoaffinity purification and subjected it to MS analysis as described above. Examples of high-confidence assignments of tandem mass spectra to PTM-bearing COS-1 p53 peptides are shown in Fig. 7.
Fig. 7.

Representative MS/MS spectra of COS-1 p53. Peptides prepared from p53 isolated from COS-1 cells were subjected to reversed-phase nano-LC-MS and MS/MS on a UPLC-Orbitrap Velos platform as described under “Experimental Procedures.” Shown are representative examples of tandem mass spectra displaying their PTM-bearing peptide fragment ion assignments, labeled as described in the legend to Fig. 4.
The peptides generated from COS-1 p53 appear to be as extensively modified as those from HFF p53, with dense combinations of modified residues evident (compare Figs. 4 and 5 to Fig. 7). We identified a total of 71 modified residues on COS-1 p53, 63 of which had not been described previously, with a total of 145 PTMs, 139 of which were classified as novel (supplemental Table S4). The locations of the modified residues and the types of PTMs identified recapitulated the vast majority of the previously described and the novel PTMs that we detected on both ΔE1B p53 and E p53 isolated from HFFs. Such recapitulation confirms and extends previous COS-1 p53 PTM mapping studies and demonstrates that the extensive modification that we observed for p53 populations isolated from HFFs is not a unique property of that normal cell system.
Comparison of ΔE1B p53 and E p53 PTM Profiles
The identification of co-occurring PTMs in p53, and of mutually exclusive PTMs on particular residues, indicated that the p53 protein is likely to be subject to combinatorial regulation by PTM, in a manner analogous that established for histone proteins (5, 10, 74, 76, 77). As only a few differences in the nature or localization of PTMs were readily apparent in ΔE1B p53 compared with E p53, it seemed possible that a difference in relative degrees of modification at specific residues might underlie the observed differences in functional properties of these two p53 populations. Spectral counting of the high-confidence ΔE1B p53 and E p53 peptides was therefore used to provide rough comparative estimates of each type of PTM at each modified residue. The comparative PTM profiles for the sites of phosphorylation and Arg modifications present on ΔE1B p53 and E p53 are shown in Figs. 8A and 8B, respectively, and profiles for sites of Lys modification are provided in supplemental Fig. S1. This analysis indicated some differences in the relative degrees to which specific residues bear particular PTMs.
Fig. 8.

Spectral count profiles of modified Ser, Thr, and Arg residues. Peptides from equivalent populations of ΔE1B p53 and E p53 isolated in parallel were subjected to nano-UPLC-MS and MS/MS analyses on the Orbitrap Velos platform. Modification profiles were generated from technical triplicate LC-MS runs for each sample by means of spectral counting–based quantification of matched PTMs per residue, as described under “Experimental Procedures.” Shown are the modification profiles for (A) phosphorylation of Ser and Thr residues and (B) mono-, di-, and trimethylation of Arg residues present on both p53 populations.
ΔE1B p53 exhibited a slightly higher degree of phosphorylation within the TAD and the N-terminal region of the DBD, whereas E p53 appeared phosphorylated to a somewhat greater degree at sites in the C-terminal half of the protein (Fig. 8A). The most striking differences were seen at Ser 183 and Ser 185, which are clearly more likely to be phosphorylated than other residues subject to this modification in E p53 than in ΔE1B p53 (Fig. 8A). Although Ser 185 has not been described previously, phosphorylation of Ser 183 by the Aurora B kinase has been implicated in the targeting of p53 for proteasomal degradation (49, 51). The extensive phosphorylation of these two residues in E p53 might indicate additional roles of these sites in governing the activity of p53. E p53 appeared to exhibit a higher degree of mono-, di-, and trimethylation of Arg residues than ΔE1B p53. However, there was striking concordance between the two p53 populations in both the identities of the modified residues and the relative degrees of the various modifications of individual amino acids (Fig. 8B); this was also the case for the patterns of acetylation, ubiquitinylation, and methylation of Lys residues (supplemental Fig. S1). A notable feature of the patterns of Arg methylation is the extensive modification of four of the six Arg residues identified as mutation hotspots in human cancers (Arg 158, Arg 248, Arg 249, and Arg 273) (110–112) in both ΔE1B p53 and E3p53, but little to no modification of Arg residues in the DBD and TD previously described as essential for maintaining global p53 structure or transcriptional function (Arg 174, Arg 181, Arg 202, Arg 280, and Arg 342) (128–131).
DISCUSSION
The relatively high concentrations of p53 that accumulate in normal human fibroblasts infected by an E1B 55-kDa protein null mutant of Ad5 or exposed to etoposide (Fig. 1C) facilitated purification of these populations of p53 for the identification of PTMs. In fact, our analysis of wild-type, endogenous p53 identified an unexpectedly large number of such modifications on both ΔE1B p53 and E p53 (Fig. 6, supplemental Tables S1–S3) and extensively modified peptides of both forms of the protein (Figs. 4 and 5). This set included the great majority of previously reported sites of phosphorylation, acetylation, methylation, and ubiquitinylation (Fig. 6, supplemental Tables S2 and S3). However, we detected an even greater number of previously unknown PTMs consistently across the three biological replicates included in this study. The profusion of such novel PTMs of p53 could not be attributed to differences in the repertoires of relevant modifying enzymes in the normal human cells used in these experiments and the transformed cells employed in many previous studies: p53 isolated from simian virus 40–transformed COS-1 cells was also observed to carry a large number of PTMs that have not been reported previously (supplemental Table S4), with the great majority corresponding to those detected on p53 from HFFs. Rather, several methodological differences seem likely to account for the wealth of previously unknown PTMs that we observed. These include the isolation of p53 from whole cell lysates, a population that should include multiple functional forms (see the Introduction), rather than from nuclear fractions. In addition, immunoaffinity purification exploited three monoclonal antibodies that recognize distinct epitopes in the TAD, DBD, and BD (135, 136), a strategy that should facilitate the recovery of differentially modified forms of p53. For example, Arg209 is essential for the recognition of unmodified p53 synthesized in E. coli by one of these antibodies (1620) (136). Methylation of this residue, as observed in both ΔE1B p53 and E p53 (Fig. 6, supplemental Tables S2 and S3), would preclude binding of this antibody, but not of the other two, which recognize epitopes mapped to residues 45–91 and 371–380 (135, 136). However, the most important difference seems likely to be the increased sensitivity and specificity of peptide identification and PTM assignment conferred by the utilization of current LC-MS instrumentation, including a nano-UPLC employing sub-2-μm chromatographic resin and an accurate-mass mass spectrometer with improved ion-source brightness enabled by ion-funnel-like technology and with high-speed tandem MS capabilities. Furthermore, the purification of the p53 protein to near homogeneity prior to LC-MS analysis minimized the complexity of p53 peptide-containing mixtures and consequently facilitated both the sensitive detection of low-abundance species and the bioinformatics of PTM assignment.
We were able to confirm the previously described and unknown PTM assignments initially detected in these experiments in three independent biological replicate preparations of both p53 populations, as well as in an additional, orthologous population of endogenous p53 isolated from COS-1 cells, which we determined to be modified to the same degree as the p53 populations isolated from HFFs (Fig. 7, supplemental Table S4). We were also able to rule out any potential misassignments due to mutation(s) in the p53 coding sequence present within the strain of HFFs employed. Consequently, we are confident in the authenticity of these previously unknown PTMs, despite their unanticipated profusion. However, we make no claim to have characterized exhaustively the PTMs possible on p53 or indeed even all those present in the populations we analyzed; many classes of recently reported p53 modifications, including O-glycosylation, ADP-ribosylation, sumoylation, and neddylation, which have been shown to affect p53 function (28, 29), were not observed at all, because of abundances below the thresholds of detection or technical biases against such observations, or both. Furthermore, rigorous stoichiometric quantification of the modifications that we did characterize and the site occupancy at any given residue are beyond the scope of this work. Future mass spectrometric studies employing stable isotope labeling at the protein or peptide level, together with labeled internal standards of fixed absolute concentration, may help to elucidate the relative and absolute stoichiometries of these PTMs. The PTMs we have identified are clearly not co-occurring species present on the same molecules of p53; rather, they are likely to be distributed among many different subpopulations of p53 molecules—that is, many of these PTMs are likely to be highly substoichiometric. Indeed, this interpretation is consistent with our observation that p53 was confined to a broad band migrating at ∼50 kDa in SDS-PAGE (Fig. 1B). Additionally, although we were able to specify a great many of the PTM positional assignments with 100% localization probability, many remained specified to some degree less than 100%. Still, this information is valuable in confining PTMs to short stretches of p53 sequences that may be targeted by any future analyses.
As noted in the Introduction, it is well established that the structural and functional properties of p53 are governed by the extensive PTMs to which the protein is subject. A major conclusion of these studies is that the panoply of such PTMs is even more considerable than appreciated, or perhaps anticipated. We identified 28 previously unrecognized sites of Ser or Thr phosphorylation and mono-, di, and/or trimethylation of 26 Arg residues, and we demonstrated that every Lys residue is potentially acetylated, ubiquitinylated, or methylated (Fig. 6, supplemental Tables S2 and S3). Although the functions or specific downstream effects of the novel PTMs remain to be investigated, we can draw on a trove of findings from decades of research on the regulation of the properties of p53 by PTM to propose possible consequences of a few of them. For example, the majority of previously unknown phosphorylation sites are located within the DBD (Fig. 6). As previously identified sites of phosphorylation within this domain have been linked to modulation of the DNA binding and transcriptional activity of p53 (42–49), the previously unknown sites of phosphorylation within this region may have similar regulatory potential. In the same vein, the previously described effects of phosphorylation of specific residues in the TD and BD (44, 52–54, 137) suggest that the additional sites of phosphorylation identified in these experiments may also modulate p53 conformation, and hence DNA binding and transcriptional activity. The previously unknown Arg residues observed to be mono-, di-, and/or trimethylated include the five Arg residues that are most frequently altered as a result of the mutations in human cancers (Arg 158, Arg 175, Arg 248, Arg 249, Arg 273, and Arg 282) (14, 111, 112) and five additional residues that modulate either the structural stability (Arg 174, Arg 181, Arg 202) or transcriptional activity (Arg 280, Arg 342) of the p53 protein (128–131). It is noteworthy that the majority of the residues identified as mutation hotspots, including three residues (Arg 248, Arg 249, and Arg 273) essential for both general and sequence-specific DNA binding (128–130), are subject to a moderate to high degree of modification, whereas those that contribute to structural stability are not (Fig. 8B). It is possible that the extensive methylation of p53 Arg residues, in conjunction with the phosphorylation of Ser and Thr residues and acetylation or methylation of lysines, serves as part of a charge-based regulatory rheostat that governs global and local p53 structure, DNA-binding activity and specificity, and transcriptional activity.
The two populations of endogenous p53 examined in these experiments represent forms that differ in transcriptional activity. Exposure of cells to the DNA-damaging agent etoposide induces the stabilization and activation of p53 and the transcription of numerous p53-regulated genes (107, 108). In contrast, the p53 accumulating in the nucleus of cells infected by E1B 55-kDa null mutants of Ad5 is stable but does not trigger the p53 transcriptional program (91–93). Consistent with these reported differences, E p53 bound more efficiently in vitro to a synthetic, consensus p53 DNA-binding sequence than did ΔE1B p53 (Fig. 2). Nevertheless, the sites of PTM and the nature of the modifications identified at individual sites were virtually identical in two p53 populations (supplemental Tables S2 and S3). Indeed, we detected only 11 and 4 modifications unique to E p53 and ΔE1B p53, respectively. These observations underscore the view that the absence or presence of individual PTMs is not the primary determinant of the properties and biological activity of p53. Furthermore, although our methods did not permit rigorous normalization between the p53 populations, with the exception of two sites of Ser phosphorylation in the DBD, E p53 and ΔE1B p53 exhibited no striking differences in the relative degrees to which individual amino acids carried particular modifications (Fig. 8, supplemental Fig. S1). Indeed, despite variation in extent, their PTM profiles displayed very similar overall patterns. However, it is readily apparent from the distributions of these PTMs that both the ΔE1B p53 and the E p53 populations comprise numerous subpopulations, defined by specific combinations of modified residues that dictate localization, stability, and function.
The precise biochemical mechanisms by which p53 is stabilized and activated in response to multiple molecular stresses are still not fully understood; focused studies of the modifications of specific residues of p53 have illuminated the roles of particular pathways in p53-regulated responses to different signals, but an integrative model for how the combined presence of many PTMs drives the spatial and temporal activity of p53 during those responses has yet to be developed. Previously identified p53 PTMs have been demonstrated to engage in combinatorial regulation and signaling crosstalk in a manner similar to that of histones, with the presence of one or more modifications on specific residues governing the presence or location of others (5, 10, 74, 76, 77, 138, 139). The large number of modified residues that we have now identified, although minute in comparison to the total set of PTMs that are theoretically possible on the p53 sequence, results in a staggeringly large number of possible regulatory combinations. Clearly, elucidating the patterns and extent of co-occurrence of these PTMs—in other words, defining the repertoire of p53 proteoforms (140) in the cell—remains a critical objective for future studies: such information will be essential to delineate the functional interplay of these constellations of modifications in the context of the full-length protein. Recent developments in top-down and middle-down proteomic strategies and instrumentation (141–143) should facilitate these pursuits. Nevertheless, a bottom-up proteomics approach can provide considerable insight by defining the PTM landscape occupied by endogenous p53 in multiple functional states. In so doing, studies such as this help to constrain the enormously vast combinatorial permutations of PTMs that could theoretically be possible on p53 to a more bioinformatically manageable repertoire of those that are likely, thereby substantially limiting the search space for future proteomic analyses.
Supplementary Material
Acknowledgments
We thank Arnold J. Levine for the generous gift of anti-p53 hybridomas, Henry Shwe and Saw Kyin (Princeton Proteomics and Mass Spectrometry Core) for their expert assistance in sample processing and MS instrumentation, Daniel B. Maselli for his valuable assistance with the isolation of E p53, and Ellen Brindle-Clark for her expert assistance in manuscript preparation. We thank Attila Csordas and the other members of the PRIDE team at the EMBL-EBI for their assistance in making the data used in this manuscript publicly accessible via the PRIDE repository.
Footnotes
* This study was funded by grants from NIAID, National Institutes of Health (RO1AI1058172 and R56A111091785) to S.J.F. C.J.D. was partially supported by a pre-doctoral fellowship from the New Jersey Commission on Cancer Research (NJCCR 10-249-CCR-E0).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PTM
- post-translational modification
- BD
- basic domain
- DBD
- DNA-binding domain
- HFF
- human foreskin fibroblast
- TD
- tetramerization domain
- TAD
- transactivation domain
- TBS
- Tris-buffered saline
- UPLC
- ultra-high performance liquid chromatography.
REFERENCES
- 1. Lane D. P., Crawford L. V. (1979) T antigen is bound to a host protein in SY40-transformed cells. Nature 278, 261–263 [DOI] [PubMed] [Google Scholar]
- 2. Linzer D. I., Levine A. J. (1979) Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52 [DOI] [PubMed] [Google Scholar]
- 3. Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 4. Harris S. L., Levine A. J. (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24, 2899–2908 [DOI] [PubMed] [Google Scholar]
- 5. Meek D. W., Anderson C. W. (2009) Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1(6), a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levine A. J., Oren M. (2009) The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gottlieb E., Vousden K. H. (2010) p53 regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2(4), a001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green D. R., Chipuk J. E. (2006) p53 and metabolism: inside the TIGAR. Cell 126, 30–32 [DOI] [PubMed] [Google Scholar]
- 9. Riley T., Sontag E., Chen P., Levine A. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9(5), 402–412 [DOI] [PubMed] [Google Scholar]
- 10. Vousden K. H., Prives C. (2009) Blinded by the light: the growing complexity of p53. Cell 137, 413–431 [DOI] [PubMed] [Google Scholar]
- 11. Menendez D., Inga A., Resnick M. A. (2009) The expanding universe of p53 targets. Nat. Rev. Cancer 9, 724–737 [DOI] [PubMed] [Google Scholar]
- 12. Vogelstein B., Fearon E. R., Kern S. E., Hamilton S. R., Preisinger A. C., Nakamura Y., White R. (1989) Allelotype of colorectal carcinomas. Science 244, 207–211 [DOI] [PubMed] [Google Scholar]
- 13. Hollstein M., Sidransky D., Vogelstein B., Harris C. C. (1991) p53 mutations in human cancers. Science 253, 49–53 [DOI] [PubMed] [Google Scholar]
- 14. Joerger A. C., Fersht A. R. (2007) Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 97, 1–23 [DOI] [PubMed] [Google Scholar]
- 15. Olivier M., Eeles R., Hollstein M., Khan M. A., Harris C. C., Hainaut P. (2002) The IARC TP53 database: new online mutation analysis and recommendation to users. Hum. Mutat. 19, 607–614 [DOI] [PubMed] [Google Scholar]
- 16. Hock A., Vousden K. H. (2010) Regulation of the p53 pathway by ubiquitin and related proteins. Int. J. Biochem. Cell Biol. 42, 1618–1621 [DOI] [PubMed] [Google Scholar]
- 17. Brooks C. L., Gu W. (2011) p53 regulation by ubiquitin. FEBS Lett. 585, 2803–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okamoto K., Taya Y., Nakagama H. (2009) Mdmx enhances p53 ubiquitination by altering the substrate preference of the Mdm2 ubiquitin ligase. FEBS Lett. 583, 2710–2714 [DOI] [PubMed] [Google Scholar]
- 19. Wade M., Wang Y. V., Wahl G. M. (2010) The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 20, 299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brooks C. L., Gu W. (2011) The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2, 456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horn H. F., Vousden K. H. (2007) Coping with stress: multiple ways to activate p53. Oncogene 26, 1306–1316 [DOI] [PubMed] [Google Scholar]
- 22. Green D. R., Kroemer G. (2009) Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moll U. M., Wolff S., Speidel D., Deppert W. (2005) Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell. Biol. 17, 631–636 [DOI] [PubMed] [Google Scholar]
- 24. Murray-Zmijewski F., Slee E. A., Lu X. (2008) A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 9, 702–712 [DOI] [PubMed] [Google Scholar]
- 25. Kannan K., Amariglio N., Rechavi G., Jakob-Hirsch J., Kela I., Kaminski N., Getz G., Domany E., Givol D. (2001) DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20, 2225–2234 [DOI] [PubMed] [Google Scholar]
- 26. Fridman J. S., Lowe S. W. (2003) Control of apoptosis by p53. Oncogene 22, 9030–9040 [DOI] [PubMed] [Google Scholar]
- 27. Wei C. L., Wu Q., Vega V. B., Chiu K. P., Ng P., Zhang T., Shahab A., Yong H. C., Fu Y., Weng Z., Liu J., Zhao X. D., Chew J. L., Lee Y. L., Kuznetsov V. A., Sung W. K., Miller L. D., Lim B., Liu E.T., Yu Q., Ng H. H., Ruan Y. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219 [DOI] [PubMed] [Google Scholar]
- 28. Kruse J. P., Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dai C., Gu W. (2010) p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 16, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loewer A., Batchelor E., Gaglia G., Lahav G. (2010) Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell 142, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taira N., Yoshida K. (2012) Post-translational modifications of p53 tumor suppressor: determinants of its functional targets. Histol. Histopathol. 27, 437–443 [DOI] [PubMed] [Google Scholar]
- 32. Gu B., Zhu W. G. (2012) Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 8, 672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feng L., Lin T., Uranishi H., Gu W., Xu Y. (2005) Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol. 25(13), 5389–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krummel K. A., Lee C. J., Toledo F., Wahl G. M. (2005) The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc. Natl. Acad. Sci. U.S.A. 102(29), 10188–10193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toledo F., Wahl G. M. (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 6, 909–923 [DOI] [PubMed] [Google Scholar]
- 36. Olsson A., Manzl C., Strasser A., Villunger A. (2007) How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 14, 1561–1575 [DOI] [PubMed] [Google Scholar]
- 37. Scoumanne A., Chen X. (2008) Protein methylation: a new mechanism of p53 tumor suppressor regulation. Histol. Histopathol. 23, 1143–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Candau R., Scolnick D. M., Darpino P., Ying C. Y., Halazonetis T. D., Berger S. L. (1997) Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 15, 807–816 [DOI] [PubMed] [Google Scholar]
- 39. Zhu J., Zhang S., Jiang J., Chen X. (2000) Definition of the p53 functional domains necessary for inducing apoptosis. J. Biol. Chem. 275(51), 39927–39934 [DOI] [PubMed] [Google Scholar]
- 40. Ayed A., Mulder F. A., Yi G. S., Lu Y., Kay L. E., Arrowsmith C. H. (2001) Latent and active p53 are identical in conformation. Nat. Struct. Biol. 8, 756–760 [DOI] [PubMed] [Google Scholar]
- 41. Lee J. T., Gu W. (2010) The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 17, 86–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y., Prives C. (1995) Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature 376, 88–91 [DOI] [PubMed] [Google Scholar]
- 43. Fraser J. A., Madhumalar A., Blackburn E., Bramham .J, Walkinshaw M. D., Verma C., Hupp T. R. (2010) A novel p53 phosphorylation site within the MDM2 ubiquitination signal: II. A model in which phosphorylation at SER269 induces a mutant conformation to p53. J. Biol. Chem. 285(48), 37773–37786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maclaine N. J., Hupp T. R. (2010) The regulation of p53 protein function by phosphorylation. In p53 (Ayed A., Hupp T. R., eds.) Landes Bioscience, Austin, TX: 53–64 [Google Scholar]
- 45. Waterman M. J., Stavridi E. S., Waterman J. L., Halazonetis T. D. (1998) ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat. Genet. 19, 175–178 [DOI] [PubMed] [Google Scholar]
- 46. Rajagopalan S., Jaulent A. M., Wells M., Veprintsev D. B., Fersht A. R. (2008) 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 36, 5983–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu Q., Kaneko S., Yang L., Feldman R. I., Nicosia S. V., Chen J., Cheng J. Q. (2004) Aurora-A abrogation of p53 binding and transactivation activity by phosphorylation of serine 215. J. Biol. Chem. 279(50), 52175–52182 [DOI] [PubMed] [Google Scholar]
- 48. Fraser J. A., Vojtesek B., Hupp T. R. (2010) A novel p53 phosphorylation site within the MDM2 ubiquitination signal: I. Phosphorylation at SER269 in vivo is linked to inactivation of p53 function. J. Biol. Chem. 285, 37762–37772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu L., Ma C. A., Zhao Y., Jain A. (2011) Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA-binding domain and subsequent functional suppression. J. Biol. Chem. 286(3), 2236–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bech-Otschir D., Kraft R., Huang X., Henklein P., Kapelari B., Pollmann C., Dubiel W. (2001) COP9 signalsome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 20, 1630–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gully C. P., Velazquez-Torres G., Shin J. H., Fuentes-Mattei E., Wang E., Carlock C., Chen J., Rothenberg D., Adams H. P., Choi H. H., Guma S., Phan L., Chou P. C., Su C. H., Zhang F., Chen J. S., Yang T. Y., Yeung S. C., Lee M. H. (2012) Aurora B kinase phosphorylates and instigates degradation of p53. Proc. Natl. Acad. Sci. U.S.A. 109(24), E1513–E1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ullrich S. J., Sakaguchi K., Lees-Miller S. P., Fiscella M., Mercer W. E., Anderson C. W., Appella E. (1993) Phosphorylation at Ser-15 and Ser-392 in mutant p53 molecules from human tumors is altered compared to wild-type p53. Proc. Natl. Acad. Sci. U.S.A. 90(13), 5954–5958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hupp T. R., Lane D. P. (1994) Allosteric activation of latent p53 tetramers. Curr. Biol. 4, 865–875 [DOI] [PubMed] [Google Scholar]
- 54. Prives C., Hall P. A. (1999) The p53 pathway. J. Pathol. 187, 112–126 [DOI] [PubMed] [Google Scholar]
- 55. Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. (2008) Acetylation is indispensable for p53 activation. Cell 133, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mellert H., Sykes S. M., Murphy M. E., McMahon S. B. (2007) The ARF/oncogene pathway activates p53 acetylation within the DNA binding domain. Cell Cycle 6, 1304–1306 [DOI] [PubMed] [Google Scholar]
- 57. Sykes S. M., Mellert H. S., Holbert M. A., Li K., Marmorstein R., Lane W. S., McMahon S. B. (2006) Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 59. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mellert H. S., Stanek T. J., Sykes S. M., Rauscher F. J., 3rd, Schultz D. C., McMahon S. B. (2011) Deacetylation of the DNA-binding domain regulates p53-mediated apoptosis. J. Biol. Chem. 286(6), 4264–4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luo J., Li M., Tang Y., Laszkowska M., Roeder R. G., Gu W. (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 101(8), 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kawaguchi Y., Ito A., Appella E., Yao T. P. (2006) Charge modification at multiple C-terminal lysine residues regulates p53 oligomerization and its nucleus-cytoplasm trafficking. J. Biol. Chem. 281(3), 1394–1400 [DOI] [PubMed] [Google Scholar]
- 63. Yamaguchi H., Woods N. T., Piluso L. G., Lee H. H., Chen J., Bhalla K. N., Monteiro A., Liu X., Hung M. C., Wang H. G. (2009) p53 acetylation is crucial for its transcription-independent proapoptotic functions. J. Biol. Chem. 284(17), 11171–11183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Itahana Y., Ke H., Zhang Y. (2009) p53 oligomerization is essential for its C-terminal lysine acetylation. J. Biol. Chem. 284, 5158–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chan W. M., Mak M. C., Fung T. K., Lau A., Siu W. Y., Poon R. Y. (2006) Ubiquitination of p53 at multiple sites in the DNA-binding domain. Mol. Cancer Res. 4, 15–25 [DOI] [PubMed] [Google Scholar]
- 66. Le Cam L., Linares L. K., Paul C., Julien E., Lacroix M., Hatchi E., Triboulet R., Bossis G., Shmueli A., Rodriguez M. S., Coux O., Sardet C. (2006) E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 127, 775–788 [DOI] [PubMed] [Google Scholar]
- 67. Kruse J. P., Gu W. (2009) MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J. Biol. Chem. 284, 3250–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Marchenko N. D., Hanel W., Li D., Becker K., Reich N., Moll U. M. (2010) Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ. 17, 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nie L., Sasaki M., Maki C. G. (2007) Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J. Biol. Chem. 282, 14616–14625 [DOI] [PubMed] [Google Scholar]
- 70. Jansson M., Durant S. T., Cho E. C., Sheahan S., Edelmann M., Kessler .B, La Thangue N. B. (2008) Arginine methylation regulates the p53 response. Nat. Cell Biol. 10, 1431–1439 [DOI] [PubMed] [Google Scholar]
- 71. Chuikov S., Kurash J. K., Wilson J. R., Xiao B., Justin N., Ivanov G. S., McKinney K., Tempst P., Prives C., Gamblin S. J., Barlev N. A., Reinberg D. (2004) Regulation of p53 activity through lysine methylation. Nature 432, 353–360 [DOI] [PubMed] [Google Scholar]
- 72. Huang J., Perez-Burgos L., Placek B. J., Sengupta R., Richter M., Dorsey J. A., Kubicek S., Opravil S., Jenuwein T., Berger S. L. (2006) Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632 [DOI] [PubMed] [Google Scholar]
- 73. Shi X., Kachirskaia I., Yamaguchi H., West L. E., Wen H., Wang E. W., Dutta S., Appella E., Gozani O. (2007) Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ivanov G. S., Ivanova T., Kurash J., Ivanov A., Chuikov S., Gizatullin F., Herrera-Medina E. M., Rauscher F., 3rd, Reinberg D., Barlev N. A. (2007) Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell. Biol. 27(19), 6756–6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huang J., Dorsey J., Chuikov S., Pérez-Burgos L., Zhang X., Jenuwein T., Reinberg D., Berger S. L. (2010) G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 285(13), 9636–9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Young N. L., Dimaggio P. A., Garcia B. A. (2010) The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 67, 3983–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Altelaar A. F., Munoz J., Heck A. J. (2013) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48 [DOI] [PubMed] [Google Scholar]
- 78. Debbas M., White E. (1993) Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 7, 546–554 [DOI] [PubMed] [Google Scholar]
- 79. Lowe S. W., Ruley H. E. (1993) Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 7, 535–545 [DOI] [PubMed] [Google Scholar]
- 80. Zhang X., Turnell A. S., Gorbea C., Mymryk J. S., Gallimore P. H., Grand R. J. (2004) The targeting of the proteasomal regulatory subunit S2 by adenovirus E1A causes inhibition of proteasomal activity and increased p53 expression. J. Biol. Chem. 279(24), 25122–25133 [DOI] [PubMed] [Google Scholar]
- 81. White E. (2001) Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene 20, 7836–7846 [DOI] [PubMed] [Google Scholar]
- 82. Frisch S. M., Mymryk J. S. (2002) Adenovirus-5 E1A: paradox and paradigm. Nat. Rev. Mol. Cell Biol. 3, 441–452 [DOI] [PubMed] [Google Scholar]
- 83. Querido E., Marcellus R. C., Lai A., Charbonneau R., Teodoro J. G., Ketner G., Branton P. E. (1997) Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 71, 3788–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Querido E., Morrison M. R., Chu-Pham-Dang H., Thirlwell S. W., Boivin D., Branton P. E. (2001) Identification of three functions of the adenovirus E4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. J. Virol. 75, 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Harada J. N., Shevchenko A., Shevchenko A., Pallas D. C., Berk A. J. (2002) Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76, 9194–9206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Luo K., Ehrlich E., Xiao Z., Zhang W., Ketner G., Yu X. F. (2007) Adenovirus E4orf6 assembles with Cullin5-ElonginB-ElonginC E3 ubiquitin ligase through an HIV/SIV Vif-like BC-box to regulate p53. FASEB J. 21, 1742–1750 [DOI] [PubMed] [Google Scholar]
- 87. Woo J. L., Berk A. J. (2007) Adenovirus ubiquitin-protein ligase stimulates viral late mRNA nuclear export. J. Virol. 81, 575–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Blanchette P., Cheng C. Y., Yan Q., Ketner G., Ornelles D. A., Dobner T., Conaway R. C., Conaway J. W., Branton P. E. (2004) Both BC-box motifs of adenovirus protein E4orf6 are required to efficiently assemble an E3 ligase complex that degrades p53. Mol. Cell. Biol. 24(21), 9619–9629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schwartz R. A., Lakdawala S. S., Eshleman H. D., Russell M. R., Carson C. T., Weitzman M. D. (2008) Distinct requirements of adenovirus E1b55K protein for degradation of cellular substrates. J. Virol. 82, 9043–9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cardoso F. M., Kato S. E., Huang W., Flint S. J., Gonzalez R. A. (2008) An early function of the adenoviral E1B 55 kDa protein is required for the nuclear relocalization of the cellular p53 protein in adenovirus-infected normal human cells. Virology 378, 339–346 [DOI] [PubMed] [Google Scholar]
- 91. O'Shea C. C., Johnson L., Bagus B., Choi S., Nicholas C., Shen A., Boyle L., Pandey K., Soria C., Kunich J., Shen Y., Habets G., Ginzinger D., McCormick F. (2004) Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6, 611–623 [DOI] [PubMed] [Google Scholar]
- 92. Hobom U., Dobbelstein M. (2004) E1B-55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J. Virol. 78, 7685–7697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Miller D. L., Rickards B., Mashiba M., Huang W., Flint S. J. (2009) The adenoviral E1B 55-kilodalton protein controls expression of immune response genes but not p53-dependent transcription. J. Virol. 83, 3591–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kato S. E., Huang W., Flint S. J. (2011) Role of the RNA recognition motif of the E1B 55 kDa protein in the adenovirus type 5 infectious cycle. Virology 417, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. He T. C., Zhou S., da Costa L. T., Yu J., Kinzler K. W., Vogelstein B. (1998) A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U.S.A. 95(5), 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Harlow E., Crawford L. V., Pim D. C., Williamson N. M. (1981) Monoclonal antibodies specific for simian virus 40 tumor antigens. J. Virol. 39, 861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ball R. K., Siegl B., Quellhorst S., Brandner G., Braun D. G. (1984) Monoclonal antibodies against simian virus 40 nuclear large T tumor antigen: epitope mapping, papova virus cross reaction and cell surface staining. EMBO J. 3, 1485–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Banks L., Matlashewski G., Crawford L. (1986) Isolation of human-p53-specific monoclonal antibodies and their use in the studies of human p53 expression. Eur. J. Biochem. 159, 529–534 [DOI] [PubMed] [Google Scholar]
- 99. Gonzalez R. A., Flint S. J. (2002) Effects of mutations in the adenoviral E1B 55 kDa protein coding sequence on viral late mRNA metabolism. J. Virol. 76, 4507–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rasband W. S. (1997–2013) ImageJ. U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/ [Google Scholar]
- 101. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 102. Rappsilber J., Ishihama Y., Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 103. Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 104. Jänicke R. U., Graupner V., Budach W., Essmann F. (2009) The do's and don'ts of p53 isoforms. Biol. Chem. 390, 951–963 [DOI] [PubMed] [Google Scholar]
- 105. Hollstein M., Hainaut P. (2010) Massively regulated genes: the example of TP53. J. Pathol. 220, 164–173 [DOI] [PubMed] [Google Scholar]
- 106. Gannon J. V., Greaves R., Iggo R., Lane D. P. (1990) Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J. 9(5), 1595–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Arriola E. L., Lopez A. R., Chresta C. M. (1999) Differential regulation of p21waf-1/cip-1 and Mdm2 by etoposide: etoposide inhibits the p53-Mdm2 autoregulatory feedback loop. Oncogene 18, 1081–1091 [DOI] [PubMed] [Google Scholar]
- 108. Karpinich N. O., Tafani M., Rothman R. J., Russo M. A., Farber J. L. (2002) The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J. Biol. Chem. 277(19), 16547–16552 [DOI] [PubMed] [Google Scholar]
- 109. Chalkley G. E., Knowles P. P., Whitehead P. C., Coffer A. I. (1994) Biochemical characterisation of purified human wild-type p53 overexpressed in insect cells. Eur. J. Biochem. 221, 167–175 [DOI] [PubMed] [Google Scholar]
- 110. Joerger A. C., Fersht A. R. (2007) Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26, 2226–2242 [DOI] [PubMed] [Google Scholar]
- 111. Olivier M., Hollstein M., Hainaut P. (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2(1), a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rivlin N., Brosh R., Oren M., Rotter V. (2011) Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 114. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 115. Uhle S., Medalia O., Waldron R., Dumdey R., Henklein P., Bech-Otschir D., Huang X., Berse M., Sperling J., Schade R., Dubiel W. (2003) Protein kinase CK2 and protein kinase D are associated with the COP9 signalsome. EMBO J. 22, 1302–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lee E. W., Lee M. S., Camus S., Ghim J., Yang M. R., Oh W., Ha N. C., Lane D. P., Song J. (2009) Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis, EMBO J. 28, 2100–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rodriguez M. S., Desterro J. M., Lain S., Lane D. P., Hay R. T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin proteasome-mediated degradation. Mol. Cell. Biol. 20(22), 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nakamura S., Roth J. A., Mukhopadhyay T. (2000) Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol. Cell. Biol. 20, 9391–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Li M., Luo J., Brooks C. L., Gu W. (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 277(52), 50607–50611 [DOI] [PubMed] [Google Scholar]
- 120. Ito A., Kawaguchi Y., Lai C. H., Kovacs J. J., Higashimoto Y., Appella E., Yao T. P. (2002) MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 21, 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Li M., Brooks C. L., Wu-Baer F., Chen D., Baer R., Gu W. (2003) Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302, 1972–1975 [DOI] [PubMed] [Google Scholar]
- 122. Tang Y., Luo J., Zhang W., Gu W. (2006) Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839 [DOI] [PubMed] [Google Scholar]
- 123. Gu W., Roeder R. G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 [DOI] [PubMed] [Google Scholar]
- 124. Sakaguchi K., Herrera J. E., Saito S., Miki T., Bustin M., Vassilev A., Anderson C. W., Appella E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12, 2831–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liu L., Scolnick D. M., Trievel R. C., Zhang H. B., Marmorstein R., Halazonetis T. D., Berger S. L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19(2), 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Moll U. M., Petrenko O. (2003) The MDM2-p53 interaction. Mol. Cancer Res. 1, 1001–1008 [PubMed] [Google Scholar]
- 127. Ferreon J. C., Lee C. W., Arai M., Martinez-Yamout M. A., Dyson H. J., Wright P. E. (2009) Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. U.S.A. 106(16), 6591–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kitayner M., Rozenberg H., Kessler N., Rabinovich D., Shaulov L., Haran T. E., Shakked Z. (2006) Structural basis of DNA recognition by p53 tetramers. Mol. Cell 22, 741–753 [DOI] [PubMed] [Google Scholar]
- 129. Chen Y., Dey R., Chen L. (2010) Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Structure 18, 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cho Y., Gorina S., Jeffrey P. D., Pavletich N. P. (1994) Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346–355 [DOI] [PubMed] [Google Scholar]
- 131. Kato S., Han S. Y., Liu W., Otsuka K., Shibata H., Kanamaru R., Ishioka C. (2003) Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. U.S.A. 100(14), 8424–8429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hancock J. F. (1992) COS cell expression. Methods Mol. Biol. 8, 153–158 [DOI] [PubMed] [Google Scholar]
- 133. Reich N. C., Oren M., Levine A. J. (1983) Two distinct mechanisms regulate the levels of a cellular tumor antigen, p53. Mol. Cell. Biol. 3, 2143–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Joubel A., Chalkley R. J., Medzihradszky K. F., Hondermarck H., Burlingame A. L. (2009) Identification of new p53 acetylation sites in COS-1 cells. Mol. Cell. Proteomics 8, 1167–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Danks M. K., Whipple D. O., McPake C. R., Lu D., Harris L. C. (1998) Differences in epitope accessibility of p53 monoclonal antibodies suggest at least three conformations or states of protein binding of p53 protein in human tumor cell lines. Cell Death Differ. 5, 678–686 [DOI] [PubMed] [Google Scholar]
- 136. Wang P. L., Sait F., Winter G. (2001) The “wildtype” conformation of p53: epitope mapping using hybrid proteins. Oncogene 20, 2318–2324 [DOI] [PubMed] [Google Scholar]
- 137. Sakaguchi K., Sakamoto H., Lewis M. S., Anderson C. W., Erickson J. W., Appella E., Xie D. (1997) Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 36, 10117–10124 [DOI] [PubMed] [Google Scholar]
- 138. Sims R. J., 3rd, Reinberg D. (2008) Is there a code embedded in proteins that is based on post-translational modifications? Nat. Rev. Mol. Cell Biol. 9, 815–820 [DOI] [PubMed] [Google Scholar]
- 139. West L. E., Gozani O. (2011) Regulation of p53 function by lysine methylation. Epigenomics 3, 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Smith L. M., Kelleher N. L.; Consortium for Top Down Proteomics (2013) Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ahlf D. R., Compton P. D., Tran J. C., Early B. P., Thomas P. M., Kelleher N. L. (2012) Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J. Proteome Res. 11, 4308–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tipton J. D., Tran J. C., Catherman A. D., Ahlf D. R., Durbin K. R., Lee J. E., Kellie J. F., Kelleher N. L., Hendrickson C. L., Marshall A. G. (2012) Nano-LC FTICR tandem mass spectrometry for top-down proteomics: routine baseline unit mass resolution of whole cell lysate proteins up to 72 kDa. Anal. Chem. 84, 2111–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Müller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J. J., Cox J., Horning S., Mann M., Makarov A. (2012) Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11(3), O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.