

Abstract
Sirtuin 6 (SIRT6), a member of the mammalian sirtuin family, is a nuclear deacetylase with substrate-specific NAD+-dependent activity. SIRT6 has emerged as a critical regulator of diverse processes, including DNA repair, gene expression, telomere maintenance, and metabolism. However, our knowledge regarding its interactions and regulation remains limited. Here, we present a comprehensive proteomics-based analysis of SIRT6 protein interactions and their dependence on SIRT6 catalytic activity. We also identify evolutionarily conserved SIRT6 phosphorylations, including four within a proline-rich disordered region, and show that the conserved S338 phosphorylation can modulate selected SIRT6 interactions. By integrating molecular biology tools, microscopy, immunoaffinity purifications, label-free quantitative mass spectrometry, and bioinformatic analyses, we have established the first large-scale SIRT6 interaction network. Relative protein abundances and gene ontology functional assessment highlighted proteins involved in transcription regulation, chromatin organization, nuclear transport, telomerase function, and RNA processing. Independent immunoisolations under increased stringency distinguished the most stable SIRT6 interactions. One prominent interaction with Ras-GTPase-activating protein-binding protein 1 (G3BP1) was further validated by microscopy, reciprocal purifications, and isolations in different cell types and of endogenous SIRT6. Interestingly, a subset of specific interactions, including G3BP1, were significantly reduced or abolished in isolations of catalytically deficient SIRT6 mutant, revealing previously unknown interplay between SIRT6 activity and its associations. Overall, our study reveals putative means of regulation of SIRT6 functions via interactions and modifications, providing an important resource for future studies on the molecular mechanisms underlying sirtuin functions.
Mammalian sirtuins, SIRT1–7, are a family of seven evolutionarily conserved enzymes with homologues throughout all kingdoms of life (1, 2). Members of this family share a common catalytic domain, requirement for the co-factor nicotinamide adenine dinucleotide (NAD+), and homology to the founding family member, the Saccharomyces cerevisiae transcription regulator Sir2p (1, 3–6). Yet human sirtuins differ in their subcellular localizations, biological functions, and N- and C-terminal protein sequences (1, 7, 8), likely important for their regulation and substrate specificity. Among the seven sirtuins, recent research has transformed the nuclear and ubiquitously expressed sirtuin 6 (SIRT6)1 (7, 9) from a virtually uncharacterized enzyme to a major point of attention in this field. Similar to other, more extensively studied sirtuins, SIRT6 was identified as a critical contributor to a wide range of cellular processes, including gene expression, DNA repair, telomeric chromatin modulation, and glucose homeostasis (10–16). Not surprisingly, SIRT6 disruption is linked to many human diseases (12–14, 17–19). SIRT6-deficient mice undergo premature death caused by an aging-like phenotype, metabolic defects, and genomic instability (20). Its biological impact has made SIRT6 a prime target for therapeutic development, and continuous effort is being made toward the discovery of small molecules that can specifically modulate SIRT6 and other SIRT activities (6, 21–23).
Despite the ever-growing body of research on SIRT6, knowledge of its substrates and interactions mediating its functions remains limited. Originally discovered as a self mono-ADP-ribosylase (24), SIRT6 was later characterized as an NAD+-dependent deacetylase, targeting lysines 9 and 56 on histone 3 (16, 25–28). SIRT6-mediated deacetylation of H3 plays a central role in its known functions, as the enzyme is targeted to specific chromatin regions (e.g. telomeres) to maintain chromatin integrity (25, 28, 29) or regulate gene expression via recruitment by transcription factors (e.g. NF-kB, HIF1α) (26, 30, 31). This catalytic activity was shown to be diminished upon the mutation of histidine 133 (H133), a residue conserved among all seven human sirtuins (1, 9, 25, 32). Interestingly, recent studies have demonstrated SIRT6 functions in the hydrolysis of long-chain fatty acyl groups (33, 34), highlighting a larger repertoire of SIRT6 enzymatic roles and target substrates than anticipated. Yet it remains to be established whether SIRT6 carries out its multiple biological functions primarily through its enzymatic activities or also through the establishment of interactions within different protein complexes. Studies on this have been limited to the roles of SIRT6 under specific biological conditions, such as stress-induced DNA damage or DNA repair (35–37). This has revealed the C-terminal binding protein interacting protein, involved in double-strand DNA break repair, and poly-[ADP-ribose] polymerase 1 (PARP1) as SIRT6 non-histone substrates. Other known interactions include DNA-PKcs (35), RelA/p65, and TDG, TSPYL2, and PIAS1 (38), suggesting SIRT6 functions in DNA repair, cell cycle regulation, and NF-kB transcription. However, further knowledge of SIRT6 protein associations and/or substrates is required in order for SIRT6 functions to be explored and relevant molecular mechanisms and regulatory factors to be characterized.
Here, we establish, to our knowledge, the first proteomics-based network of SIRT6 protein interactions. We identify SIRT6 phosphorylations, demonstrating enrichment of modifications within a C terminus proline-rich domain that we predict is a naturally disordered region, and we define the impact of the conserved S338 phosphorylation on selected SIRT6 interactions. Importantly, we utilize a catalytically deficient SIRT6 mutant to determine the dependence of protein interactions on SIRT6 enzymatic activity. We report and validate prominent SIRT6 interactions and demonstrate a previously unknown dependence of interactions on SIRT6 catalytic activity. Our findings provide a new perspective on the likely means of regulation of the diverse range of SIRT6 functions in health and disease processes.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies used were in-house-generated rabbit polyclonal anti-GFP (39); commercially available rabbit polyclonal antibodies for SIRT6 (Cell Signaling, Billerica, MA and Sigma), BCLAF1 (Abcam, Cambridge, MA), YLPM1 (Novus Biologicals, Littleton, CO), HSP90 (Cell Signaling), and PARP1 (Cell Signaling); and mouse monoclonal antibodies for GFP (Roche), G3BP1 (BD Transduction Laboratories), and IgG (Santa Cruz Biotechnology, Dallas, Texas). Protein A/G PLUS-Agarose beads were obtained from Santa Cruz. The DNA oligo primers generated for this research were obtained from Integrated DNA Technologies, San Diego, California and are shown in supplemental Table S1. Unless otherwise noted, all remaining reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cloning of SIRT6-EGFP Wild Type, H133Y and S338A Mutants, and EGFP Control and Generation of Cell Lines
Plasmids with full-length FLAG-tagged SIRT6 cDNA were purchased from Addgene, Cambridge, MA (#13817), and EGFP cDNA was purchased from Clontech (pEGFP-N1, Clontech, Mountain View, CA). EGFP cDNA was cloned into a pLXSN retroviral vector (Clontech). SIRT6 specific primers (supplemental Table S1) were used to amplify SIRT6 cDNA via PCR. The PCR product was purified and digested with BamHI and XhoI restriction enzymes and ligated to the 5′ end of EGFP, resulting in the retroviral plasmid pLXSN-SIRT6-EGFP-FLAG. Human embryonic kidney cells (HEK293) and human T-cell lymphoblasts (CEMT) were used to generate cell lines stably expressing SIRT6-EGFP. HEK293 cells were cultured in DMEM, and CEMT cells in RPMI media, both supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C and 5% CO2. To generate the cell lines, plasmid DNA was transfected into Phoenix cells using the FuGENE transfection system (Roche, Indianapolis, IN). The medium with resulting retrovirus was used for transductions of wild-type (wt) HEK293 and CEMT cells. Cells stably expressing the EGFP constructs were selected with 300 μg/ml G418 (EMD, Gibbstown, NJ) for 2 weeks and sorted via fluorescence-activated cell sorting (Vantage SE with TurboSort II, Becton Dickinson, Franklin Lakes, NJ). SIRT6 H133Y and S338A mutants were generated using QuikChange XL Site-Directed Mutagenesis (Stratagene, Agilent Technologies, Santa Clara, CA) and SIRT6 primers (supplemental Table S1). HEK293 cell lines expressing the SIRT6 mutants were generated as described above. We refer to all generated cell lines as follows: wt HEK293 for wild-type cells, HEK293-EGFP for cells expressing EGFP alone, HEK293-SIRT6-EGFP for cells expressing SIRT6-EGFP-FLAG, HEK293-SIRT6-H133Y-EGFP for cells expressing H133Y catalytically incompetent mutant, and HEK293-SIRT6-S338A-EGFP for cells expressing the S338A mutant.
Microscopy
Wild-type or HEK293 cell lines were plated on glass slides coated with poly-d-lysine (Sigma, St. Louis, MO) and cultured overnight. Cells were washed, fixed, permeabilized, and incubated with primary and secondary antibodies as described elsewhere (40). Images were obtained via confocal microscopy on a Leica SP5 and PerkinElmer RS3 Spinning Disk instrument using a 60× or 100× oil immersion lens. Immuno-EM analyses were conducted at the Princeton University Microscopy Core Facility using a Carl Zeiss LEO Omega 912 energy filtered transmission electron microscope. Sample preparations and ultrathin cryosections were prepared as described elsewhere (41), and SIRT6-EGFP was visualized using in-house-generated anti-GFP antibodies and secondary antibodies conjugated to 10-nm gold particles.
Real-time Quantitative PCR Analysis
The expression levels of endogenous and EGFP-tagged SIRT6 were assessed via real-time quantitative PCR (RT-qPCR). mRNA was isolated with Trizol (Invitrogen) and used as a template to synthesize cDNA following standard reverse-transcription procedures (Retroscript, Ambion, Life Technologies, Grand Island, NY). The obtained cDNA was used to run an RT-qPCR reaction (ABI 7900HT) in combination with primers designed to amplify SIRT6 cDNA (supplemental Table S1) and a DNA polymerase mixture containing SYBR green I dye (ABI, Life Technologies, Grand Island, NY). GAPDH was used as internal control to normalize gene expression levels, and the experiment was performed in biological triplicate.
Deacetylation Activity Assay
The enzymatic activity of purified SIRT6-EGFP was assessed using the SIRT6 Direct Fluorescent Screening Assay Kit (Cayman Chemicals, Ann Arbor, MI) following the manufacturer's instructions. Briefly, SIRT6-EGFP or EGFP as a control was immunoaffinity isolated on magnetic beads from 0.1 g cells using the protocol described below. Triplicate deacetylation assays were set up on the beads in 50-μl reactions containing 400 μm SIRT6 substrate, 3 mm NAD+, and 1x assay buffer and incubated for 45 min at 37 °C in the dark. Negative control reactions were set up with the general sirtuin inhibitor nicotinamide at 20 mm. Reactions were developed by separating the supernatants from the beads, adding 50 μl of developing solution (40 mm nicotinamide, 6 mg/ml developer in 1x assay buffer) to the supernatants, and incubating for 30 min at room temperature, protected from light. Samples were analyzed using a fluorescence reader (Tecan, Durham, NC) at 350 nm excitation and 450 nm emission wavelengths. Gen5 Microplate Data Collection & Analysis software was utilized for data analysis. Plots were generated using Prism 5 software (GraphPad Software, La Jolla, CA). The presence of SIRT6-EGFP or EGFP was assessed by eluting protein mixtures from the remaining beads and performing Western blot (WB) analysis.
Isolation of SIRT6-EGFP, SIRT6-H133Y, SIRT6-S338A, or Endogenous SIRT6 for Analysis of Protein Interactions
Immunoaffinity purification (IP) of SIRT6 or control EGFP was performed on magnetic beads via the EGFP tag, as described elsewhere (39). The isolations via the EGFP tag for wt or H133Y SIRT6 or for GFP alone were performed in two biological replicates, plus one technical replicate to test consistency. The isolations of S338A SIRT6 mutant were done in three biological replicates. HEK293-EGFP, -SIRT6-EGFP, -SIRT6-H133Y-EGFP, or -SIRT6-S338A-EGFP cells were grown to confluency in 10 × 15 cm plates, washed with ice-cold Dulbecco's PBS (Invitrogen), harvested via scraping, subjected to centrifugation at 1500 rpm for 5 min, and frozen as described elsewhere (39). The cells were cryogenically disrupted using a Retsch MM 301 Mixer Mill (10 cycles for 2 min each at 30 Hz) (Retsch, Newtown, PA), and the cellular powder was lysed in 5 ml per IP ice-cold lysis buffer optimized for effective SIRT6 isolation (supplemental Fig. S1, IP6). The composition of the optimized buffer was 20 mm HEPES-KOH, pH 7.4, containing 0.1 m potassium acetate, 2 mm MgCl2, 0.1% Tween-20, 1 μm ZnCl2, 1 μm CaCl2, 0.5% Triton X-100, 200 mm NaCl, 4 μg/ml DNaseI, 1/100 (v/v) phosphatase inhibitor mixture II (Sigma), 1/100 (v/v) phosphatase inhibitor mixture III (Sigma), and 1/100 (v/v) protease inhibitor mixture. To disrupt most interactions and identify those tightly associated with SIRT6, additional protein isolations were performed using a high-stringency buffer containing, in addition to the components of the optimized buffer, 1% Triton X-100 and 0.5% deoxycholate. Cellular lysates were further homogenized for 15 s using a Polytron (Kinematica, Bohemia, NY) and then subjected to centrifugation at 8000 × g for 10 min. Supernatants were used for IP by incubation for 1 h with 8 mg of magnetic beads (M270 Epoxy Dynabeads, Invitrogen) conjugated with in-house-generated rabbit anti-GFP antibodies, as described elsewhere (40). The beads were washed six times with the lysis buffer and twice with Dulbecco's PBS (Invitrogen). Proteins were eluted with 30 μl of a mixture of 1X LDS sample buffer (Invitrogen) containing 1x reducing agent (Invitrogen) via heating for 10 min at 70 °C and shaking again for 5 min at room temperature. Eluted proteins were processed for mass spectrometry analysis or stored at −80 °C. The protocol outlined above was also used to isolate endogenous SIRT6 via antibody against SIRT6 and using IgG as control.
Sample Preparation for Mass Spectrometry Analysis
Eluted proteins were subjected to either one-dimensional SDS-PAGE separation and in-gel protein digestion, as described elsewhere (42, 43), or in-solution digestion using the filter-aided sample preparation method (44). In the first case, protein eluates were resolved on a 4–12% Bis-Tris NuPAGE gel (Invitrogen) and stained with SimplyBlue Coomassie (Invitrogen), and the gel lanes were cut into 1-mm slices and pooled into nine total fractions. Gel pieces were destained, washed, dehydrated, and digested with 12.5 ng/μl trypsin (Promega, Madison, WI) as described elsewhere (45). The resulting peptides were extracted from the gel in 0.5% formic acid for 4 h at room temperature and once again in 0.5% formic acid containing 50% acetonitrile for 2 h. The extractions were pooled and further centrifuged under vacuum to ∼10 μl. For in-solution digestion, IP samples were mixed with urea buffer (8 m urea in 0.1 m TrisHCl, pH 8.0) and applied to Vivacon 500 centrifugal filters (Sartorius Stedim Biotech, Goettingen, Germany) following the filter-aided sample preparation method (44, 45). Retained proteins were washed once with urea buffer, alkylated by treatment with urea buffer containing 0.05 m iodoacetamide, and washed three times with 50 mm ammonium bicarbonate. Proteins were subjected to 37 °C overnight in-solution digestion with 100 μl of 50 mm ammonium bicarbonate containing 5 ng/μl trypsin (Promega). Peptides were obtained via filter centrifugation, and the peptides were desalted using StageTips and vacuum centrifuged to ∼10 μl, as described elsewhere (40).
Mass Spectrometry Analysis
Peptides (∼4 μl) were analyzed via reverse-phase nano-liquid chromatography (nano-LC) using the Dionex Ultimate 3000 nanoRSLC system (Dionex Corp., Sunnyvale, CA) coupled online to an LTQ-Orbitrap Velos or an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Briefly, sample peptides were separated via reverse-phase chromatography (Acclaim PepMap RSLC, 1.8 μm, 75 μm × 25 cm) at a flow rate of 250 nl/min, applying a 90-min discontinuous acetonitrile gradient in the following order: 4% to 20% B over 50 min, 20% to 40% B over 40 min (mobile phase A: 0.1% formic acid and 0.1% acetic acid in water; mobile phase B: 0.1% formic acid and 0.1% acetic acid in 97% acetonitrile). The mass spectrometers were set to data-dependent acquisition mode, operating with Fourier transform preview scan disabled and predictive automatic gain control and dynamic exclusion enabled (repeat count: 1; exclusion duration: 70 s). Further instrument parameters were as follows: Fourier transform MS1 and Ion trap MS2 target values of 1E6 and 5E3, respectively; and Fourier transform MS1 and Ion trap MS2 maximum injection times of 300 ms and 100 ms, respectively. Individual acquisition cycles included a single full-scan mass spectrum (m/z = 350–1700) in the Orbitrap (resolution = 30,000 at m/z = 400) followed by collision-induced dissociation (CID) fragmentation on the top 15 or 20 most intense precursor ions (minimum signal = 1E3 and isolation width = 2.0 Th) in the dual-pressure linear ion trap. The CID activation time and normalized collision energy were 10 ms and 30, respectively. Targeted MSn was performed for further validation of phosphosite assignment. Full-scan MS2 or MS3 spectra were acquired for phosphopeptides previously identified via data-dependent acquisition (described above). Phosphopeptides were fragmented by either CID or electron transfer dissociation in the ion trap using the same instrument parameters as above, except that for CID, the IT MSn target value and maximum injection time were 1E4 and 150 ms, respectively. In addition, the G3BP1 phosphopeptide containing Ser149 was also analyzed via targeted CID on a MALDI LTQ-Orbitrap XL as detailed in Ref. 46. For targeted electron transfer dissociation performed on an electrospray ionization LTQ Orbitrap Velos, instrument parameters were as follows: maximum anion injection time and target value of 100 ms and 2E5, respectively, and activation time of 100 ms with supplemental collision energy enabled.
Processing of Mass Spectrometry Data
Raw files with MS/MS spectra from individual IPs were subjected to data extraction using Proteome Discoverer (version 1.3, Thermo Fisher Scientific) and subsequent SEQUEST (version 1.20) analysis to search against the UniProt Swiss-Prot sequence database (downloaded November 2010). The latter contained 21,570 entries comprising the subset of human and common contaminant sequences, and spectra were searched against peptide databases generated from the forward and reverse protein sequence entries. Search settings included parent and fragment ion mass tolerances (10 ppm and 0.5 Da, respectively), full enzyme specificity, and a maximum of two missed cleavages. Post-translational modifications were fixed for carbamidomethylcysteine (+57 Da) and variable for phosphoserine, threonine, tyrosine (+80 Da), acetyl-lysine (+42 Da), and methionine (+16 Da). Scaffold (version 3.2; Proteome Software, Inc., Portland, OR) was further used to analyze the resulting peptide spectrum matches by rescoring SEQUEST peptide spectrum matches with PeptideProphet and ProteinProphet, then re-searching with X!Tandem (GPM 2010.12.1.1). For the latter, high mass accuracy search and the subset database parameter were used, together with modification of pyro-Glu, formed from peptide N-terminal glutamine (−17 Da) and deamidation of asparagine and glutamine (+1 Da). The resulting peptide spectrum matches were grouped into protein categories following parsimony rules. Peptide and protein global false discovery rates were controlled to <1% by selecting 95% peptide confidence and 99% protein confidence, and as a minimum two unique protein peptides within at least one biological sample. The amino acid position of unique phosphorylation sites was assessed by means of individual examination of MS/MS spectra and independently validated using the phosphoRS algorithm (v3.1 in Proteome Discoverer, v1.3, Thermo Fisher Scientific) (supplemental Table S2). Filtered proteins were exported as “unweighted spectrum counts” to Excel, and a spectral counting approach was applied to assess the specificity of SIRT6 interactions in comparison to parallel EGFP or IgG control isolations. Data from two biological replicates, plus one technical replicate for wt and H133Y mutant IPs, were analyzed for interaction specificity using the SAINT algorithm (47). All interactions with a probability score of >0.85 and with predicted nuclear localization (from the UniProt database) were subjected to subsequent functional analyses while excluding proteins with strictly cytoplasmic reported localization (supplemental Table S3). For IPs of endogenous SIRT6 or SIRT6-EGFP under highly stringent isolation conditions, average spectral counts from biological replicates were calculated, and only proteins with more than three spectral counts in at least one condition and with ≥5-fold enrichment relative to the appropriate IP control isolations were considered for further analysis (supplemental Tables S4, S6, and S7). For comparison of wt to mutant H133Y or S338A interactions, spectral count values were normalized to the SIRT6 protein spectral count ratio between SIRT6 wt and the mutant (supplemental Table S3). These analyses were performed only for interactions previously assessed as specific after SAINT analysis (supplemental Table S3). Final lists of spectral counts from all IPs analyzed in this study are provided in supplemental Tables S3–S7.
Functional Network Analyses of Interactions
To build a comprehensive SIRT6 interactions network, we clustered interactions according to known protein complex associations and biological function and assessed relative protein abundances using the normalized spectral abundance factor (NSAF) method (48). Briefly, SIRT6 binding partners were imported into the STRING database of functional protein associations (v9.05) (49). Identified protein interaction networks were visualized with Cytoscape (50), representing proteins as nodes and functional connections as black edges. Gene ontology annotations were obtained from the UniProt protein database and incorporated into Cytoscape for network analyses. NSAF values for SIRT6 interactions were calculated as described elsewhere (45) to assess relative protein abundances within different functional groups (supplemental Tables S3, S4, S6, and S7). In short, protein spectral counts (averaged from the multiple biological replicates) were first normalized by protein length (spectral abundance factor), and then NSAF values were calculated by dividing individual spectral abundance factor values by the sum of all spectral abundance factor values. These were imported into Cytoscape and visualized with node color gradients. WT, H133Y, and S338A sets of average spectral counts were also directly compared to assess catalytically and phosphorylation-dependent interactions. This was achieved using Prism 5 software.
Reciprocal Isolations
HEK293-SIRT6-EGFP cells were harvested from one confluent 15-cm plate (∼0.1-g cell pellet), washed, lysed in 1 ml of optimized lysis buffer, and homogenized as described above. Cell lysates were incubated for 1.5 h at 4 °C with 2 μg of anti-G3BP1 antibody or control IgG per 1 ml of lysate. Protein A/G PLUS-Agarose beads (30 μl per IP) were incubated for 1 h at 4 °C in lysis buffer with gentle rotation and washed twice before their addition to cell lysates. IPs were carried out for 2.5 h at 4 °C and were followed by bead washing with lysis buffer and 2x Dulbecco's PBS and elution of proteins via heating at 70 °C for 10 min in 30 μl of 1x LDS sample buffer containing reducing agent. Co-isolated proteins were detected via WB analysis.
RESULTS
SIRT6-EGFP Localizes to the Nucleus and Is Functionally Active
To characterize SIRT6 protein interactions and post-translational modifications, we constructed EGFP-tagged SIRT6 (Fig. 1A) and generated human HEK293 cell lines stably expressing the protein chimera (HEK293-SIRT6-EGFP) or only EGFP (HEK293-EGFP) as control. HEK293 cells were selected because SIRT6 is endogenously expressed in mammalian kidney tissue (20, 51, 52), and these cells have been commonly used in SIRT6 studies (9, 36, 51, 53). Furthermore, similar to other recent studies on SIRT6 (9), we selected to focus on the full-length isoform of SIRT6, as the shorter isoform misses amino acids from the catalytic domain (amino acids 179–205). To confirm that the EGFP-tagged SIRT6 was functionally active, first its subcellular localization was assessed via confocal microscopy. As expected, the tagged protein localized to the nucleus, similar to endogenous SIRT6 (Fig. 1B) and in agreement with previous reports (9, 20). RT-qPCR analysis showed ∼3-fold overexpression of SIRT6-EGFP relative to endogenous SIRT6 mRNA levels in wt or EGFP control cells (Fig. 1C), indicating that overexpression did not dramatically increase its mRNA levels in the generated cell lines. Finally, an in vitro deacetylation activity assay confirmed that immunoaffinity-purified SIRT6-EGFP had preserved catalytic properties, displaying ∼70-fold more enzymatic activity than control EGFP (Fig. 1D). WB analysis of these samples verified the isolation of both SIRT6-EGFP and EGFP alone (Fig. 1D). Together, these results demonstrate that the EGFP-tagged SIRT6 maintains its nuclear localization and its ability to deacetylate substrates.
Fig. 1.
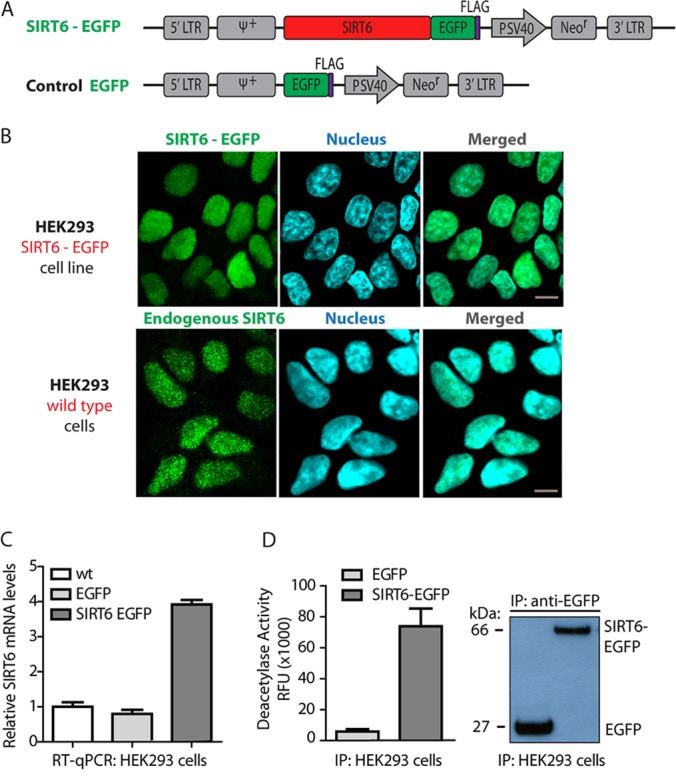
EGFP-tagged SIRT6 retains nuclear localization and is functionally active in HEK293 cells stably expressing the fusion protein. A, diagram of retroviral plasmids used to generate HEK239 cells stably expressing SIRT6-EGFP or control EGFP. B, confocal microscopy by direct fluorescence demonstrates nuclear localization of SIRT6-EGFP in the generated cells, consistent with the localization pattern of endogenous SIRT6 in wt HEK293 cells, observed using an anti-SIRT6 antibody; endogenous or EGFP-tagged SIRT6 (green), nucleus (blue, DAPI staining), 60× oil immersion lens, 2× optical zoom, scale bar = 10 μm. C, RT-qPCR analysis (from three biological replicates) shows moderate (∼3-fold) overexpression of SIRT6-EGFP relative to endogenous SIRT6 mRNA levels in wt or EGFP control cells. D, EGFP-tagged SIRT6 has preserved deacetylation activity in stably expressing HEK293 cells as assessed by in vitro deacetylation activity assays of immunoaffinity-purified tagged protein in parallel to control EGFP isolations (left). Deacetylation assays were performed in two biological replicates, each with three technical replicates. Western blot analysis demonstrated successful isolation of EGFP or SIRT6-EGFP in these functional activity assays (right).
Complementary Mass Spectrometry Approach for Analysis of SIRT6 Interactions
Having generated the necessary tools to perform proteomics-based analyses of SIRT6, we next optimized the lysis buffer conditions for efficient isolations of SIRT6-EGFP (supplemental Fig. S1). We performed a series of SIRT6-EGFP and control EGFP IPs testing a range of mild to stringent isolation conditions (IP1–IP6 in supplemental Fig. S1). Using this strategy, we selected a balance between detergent stringency and salt concentrations to achieve efficient isolations of SIRT6-EGFP while minimizing the amount of nonspecific associations (IP6 in supplemental Fig. S1). This optimal isolation condition was selected for the analysis of SIRT6 interactions and modifications throughout the rest of this study. To identify SIRT6 interactions, we used a mass-spectrometry-based approach (Fig. 2A) implementing cryogenic cell disruption followed by lysis in the optimized buffer. Immunoaffinity purifications via the EGFP tag (39) resulted in efficient isolations for both SIRT6-EGFP and EGFP control, as evidenced by Western blotting and the prominence of the corresponding Coomassie Blue–stained bands (Figs. 2B and 2C). To identify interactions, co-isolated proteins were further analyzed via nano-LC-MS/MS using an LTQ-Orbitrap Velos or an LTQ-Orbitrap XL. This workflow allowed for the identification of both SIRT6 putative protein interactions and post-translational modifications.
Fig. 2.
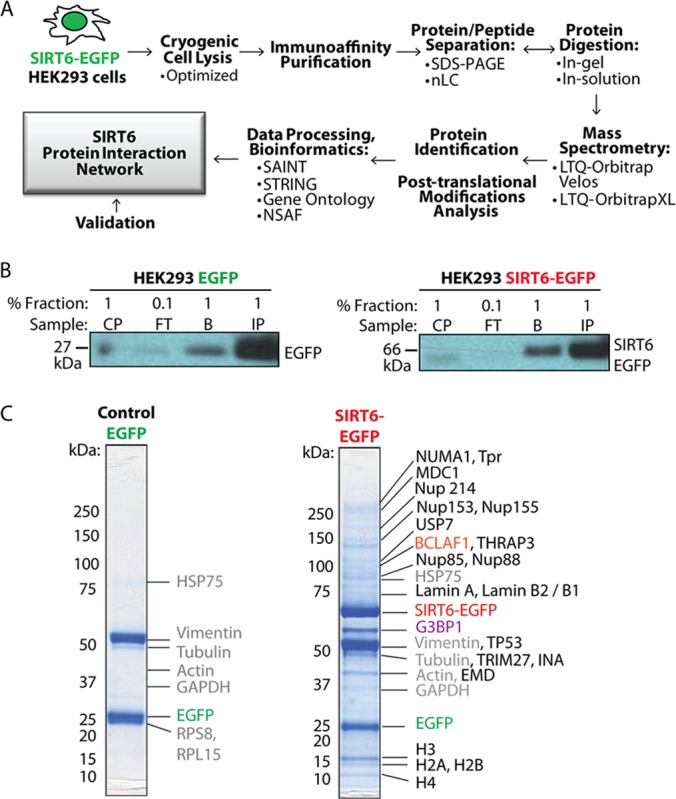
Proteomics-based workflow for analysis of SIRT6 interactions. A, workflow of isolation and analysis of SIRT6 interactions involving immunoaffinity purification (IP) of EGFP-tagged SIRT6 from HEK293 cell lines, mass spectrometry analysis, and data processing. B, efficient protein isolation is demonstrated by WB analysis of fractions collected at progressive IP stages (CP, insoluble cell pellet; FT, flow through; IP, primary protein elution; B, secondary protein elution) using anti-EGFP antibody. C, IP proteins are resolved via SDS-PAGE and visualized by means of Coomassie Blue staining, confirming isolation of the bait SIRT6-EGFP and EGFP proteins. Subsequent mass spectrometry analysis highlighted known and putative SIRT6 interactions. Green, EGFP; red, SIRT6-EGFP; purple, Ras-GTPase-activating protein-binding protein 1 (G3BP1); orange, Bcl-2-associated transcription factor 1 (BCLAF1); gray, contaminants.
SIRT6-EGFP Interacts with G3BP1, Transcription Regulators, and Chromatin Organizing Factors
Having confirmed the efficient isolation of SIRT6-EGFP, we analyzed the co-isolated proteins via mass spectrometry and assessed their consistency in replicate experiments using the SAINT algorithm (47). With our focus on proteins with nuclear annotations, a total of 74 proteins passed our stringent criteria (i.e. present in all biological replicates and SAINT scores of >0.85) and were further subjected to functional network analyses using STRING (49) and Gene Ontology clustering. To highlight prominent interactions within each functional group, relative protein abundances were assessed using NSAF (48) values. Visualization with Cytoscape (50) revealed a broad network of SIRT6 interactions falling within distinct functional groups (Fig. 3A). As expected, we identified previously known SIRT6 binding partners, such as core histone proteins known to co-isolate with SIRT6 in HEK293T cells (35). Several recently reported SIRT6 interactions (e.g. SMARCA5 and MYBBP1A (38)) were also detected but did not pass our stringent specificity criteria, and thus were excluded from further analysis. Interestingly, the most prominent SIRT6 interaction, identified consistently throughout our analyses, was Ras-GTPase activating protein SH3-domain-binding protein 1 (G3BP1) (Fig. 3A). G3BP1 is known to bind DNA and RNA duplexes and to shuttle between the cytoplasm and nucleus dependent on its phosphorylation state (54). Early studies identified this protein as a member of stress granules formed in the cytoplasm (55), but upon phosphorylation the protein can localize to the nucleus (54) and mediates other processes (e.g. DNA repair as a component of promyelocytic leukemia nuclear bodies) (56). Consistent with the nuclear localization of SIRT6, we detected G3BP1 as phosphorylated at Ser149 and Ser232 (supplemental Fig. S2; additional phosphorylations at S230 and S231 cannot be excluded), with Ser149 phosphorylation being shown to mediate its nuclear redistribution (54). Therefore, the SIRT6–G3BP1 interaction seems to occur within the nuclear compartment.
Fig. 3.

Network of putative SIRT6 interactions. A, SIRT6 co-isolates with proteins from diverse functional groups, including transcription regulators, chromatin organization factors, and the prominent G3BP1. The network was generated by Cytoscape and comprises 74 putative SIRT6 interactions identified via our proteomics-based approach, confirmed in multiple biological replicates, and assessed for specificity using the SAINT algorithm (scores > 0.85). Node color indicates relative protein abundance and correlates with calculated NSAF values. Interaction partners are clustered in functional groups and subgroups based on STRING and Gene Ontology analyses assessing functional protein associations and annotation in biological processes, respectively. Edge lines connecting individual nodes represent previously reported or predicted protein–protein interactions within distinct protein complexes, as imported from the STRING database. B, Proteomic analysis of SIRT6-EGFP interactions under highly stringent isolation conditions defines tight associations, including G3BP1 and proteins linked to transcription regulation and nuclear transport. Node colors indicate NSAF values.
Two of the biggest functional categories identified in our proteomic analysis comprised interactions that link SIRT6 to gene expression regulation and chromatin maintenance (Fig. 3A). The transcription regulation group included proteins involved in apoptosis and protein degradation (e.g. TP53, USP7, BAZ1B), DNA repair (e.g. BLCAF1 and THRAP3), and NF-kB-mediated regulation (e.g. NKRF and PDCD11) (Fig. 3A). These associations comprised diverse transcription factors, such as the major transcription regulator tumor suppressor p53, Bcl-2-associated transcription factor 1 (BCLAF1), tyrosine-protein kinase BAZ1B, tripartite motif-containing protein 27 (TRIM27), and NF-κ-B-repressing factor (NKRF). While SIRT6 is known to regulate DNA damage repair (35–37) and NF-κ-B-mediated gene expression (26, 30), our results provide evidence of previously unknown SIRT6 associations with factors involved in these processes. Moreover, interactions observed in the absence of stress stimulation highlight the importance of characterizing SIRT6 binding partners under normal conditions. In addition to transcription regulators, we also identified SIRT6 interactions involved in chromatin organization, including nuclear organization factors (e.g. LMNA, LMNB1, LMNB2, and EMD) and proteins regulating mitotic assembly (e.g. MKI67 and NUMA1). Other smaller groups of SIRT6 interactions pointed to links to RNA polymerase activity, telomerase regulation, and the formation of neurofilament networks (Fig. 3A). These binding partners included coilin (COIL) and nucleolar and coiled-body phosphoprotein 1 (NOLC1), linked to polymerase I activity (57), and 5′-3′ exoribonuclease 2 (XRN2), associated with polymerase II–mediated transcription (58). We also observed an association with YLP motif-containing protein 1 (YLPM1), also known as nuclear protein ZAP3, a protein involved in telomerase regulation (59), suggesting a putative regulatory role for SIRT6 in telomerase activity. Finally, SIRT6 also associated with α-internexin (INA) and neurofilament light polypeptide (NEFL), proteins linked to the establishment of neurofilament networks (60).
Our functional network analysis also highlighted proteins involved in nuclear transport (Fig. 3A). These comprised a group of highly interconnected nuclear pore complex proteins (Nups), known to mediate the traffic of molecules between the nucleus and the cytoplasm (61). The nuclear pore complex (NPC) is composed of Nups assembled toward the cytoplasmic, transmembrane, or nuclear side (62). Among the identified Nups were members of the central FG Nups (e.g. Nup54), outer ring (e.g. Nup160), linker Nups (e.g. Nup93), and the nuclear basket (e.g. TPR and Nup153). These interactions fall within the transmembrane or nuclear side of the NPC, in agreement with the nuclear localization of SIRT6. Consistent with the nuclear transport group, SIRT6 was also found to associate with proteins involved in mRNA and rRNA processing. As these often involve RNA transport from the nucleus to the cytoplasm and transport-dependent gene expression (63), it comes as no surprise that SIRT6 interacts with proteins from all three categories.
We expect the observed SIRT6 associations to represent both direct and indirect interactions, which was necessary in order for us to gain an understanding of protein complexes that SIRT6 may associate with. However, to distinguish the tight interactions, we repeated the SIRT6-EGFP isolations using a high-stringency lysis buffer (Fig. 3B). As expected, this increased stringency disrupted numerous SIRT6 interactions, allowing only a few associations to remain. Importantly, the remaining binding partners still reflected the major categories previously outlined, including transcription regulation, nuclear transport, and RNA processing. G3BP1 and transcription regulators, such as BAZ1B, TRIM27, and NKRF, constituted a group of associations, together with interactions involved in mRNA processing (e.g. BRIX, WDR36, UTP15 and -18, and FTSJ3). Nuclear transport protein members were also identified, along with the neurofilament protein NEFL. NSAF analysis indicated that mRNA processing proteins and expression restriction factors (e.g. H2AFY2) were prominent in this subset of SIRT6 stringent IP interactions (Fig. 3B and supplemental Table S4). These results pointed to stronger, and some possibly direct, SIRT6 interactions. Overall, our findings strengthen the hypothesis that SIRT6 is involved in multiple, highly interconnected processes.
SIRT6-EGFP Localizes within the Nucleoplasm and in the Vicinity of Nuclear Pores
Our results demonstrated that SIRT6 associates with Nups and nuclear organization factors that localize to the nuclear periphery. The interaction of SIRT6 with the nuclear basket protein TPR and Nups persisted even after most interactions had been disrupted by a highly stringent lysis buffer, suggesting tight associations. To further investigate the possible localization of SIRT6-EGFP to the nuclear periphery, we performed immunogold labeling for detection via electron microscopy. As expected, gold particles were observed throughout the nucleoplasm (Fig. 4A, supplemental Fig. S3). Additionally, a subset of SIRT6-EGFP was observed to localize close to NPCs, in agreement with the identified interactions with Nups. It is tempting to speculate that Nups may play some roles in the organization of chromatin, and possibly in the transcriptional regulatory functions of SIRT6.
Fig. 4.

SIRT6-EGFP localizes within the nucleoplasm and near nuclear pores. Immunogold electron microscopy analyses were performed on HEK293 cells to detect the localization of SIRT6-EGFP. SIRT6 was visualized using anti-GFP antibodies and secondary antibodies conjugated to 10-nm gold particles (observed as black dense dots). Arrows point to nuclear pores. Gold particles are distributed within the nucleoplasm (panels A and C) and within the vicinity of nuclear pores (B).
SIRT6 Is Phosphorylated at Conserved Residues within a C-terminal Naturally Disordered Region
As the current knowledge regarding SIRT6 post-translational modifications is limited, the enrichment of SIRT6 via immunoaffinity purification provided the opportunity to characterize its modification status. We found SIRT6 to be phosphorylated at multiple sites (Fig. 3A and supplemental Fig. S4) within the 79% protein sequence coverage obtained following protein digestion (supplemental Fig. S4A). One phosphorylated serine residue (S10) localized to the N terminus, while the rest of the modifications at three serines (S303, S330, and S338) and one threonine (T294) were clustered at the C terminus (Fig. 5A). Phylogenetic analyses revealed conservation among mammalian organisms for all SIRT6 phosphorylations, with the N-terminal S10 being the most highly conserved (Fig. 5C). Interestingly, the C-terminal phosphorylations were present within a proline-rich region, which, using the PONDR software, we determined to be likely naturally disordered (Fig. 5B). While this is the first demonstration that SIRT6 is phosphorylated within a disordered area, such regions are generally known to modulate protein functions by forming interactions or helping to recognize substrates (64). Therefore, we tested the possible impact of phosphorylation on SIRT6 localization and interactions. We focused on S338, as this site was the most conserved within the C-terminal sites and the closest to the nuclear localization signal. Although this Pro-rich region has been shown to be important for the nuclear localization of SIRT6 (9), the S338 phosphorylation is not required, as our results demonstrated that a serine-to-alanine mutant (S338A) retains its nuclear localization (supplemental Fig. S5). To further assess its possible effect on SIRT6 localization, we analyzed the interactions of the S338A mutant specifically with proteins involved in nuclear transport. We observed reduced associations with Nups for the S338A mutant relative to the wt SIRT6, suggesting that this phosphorylation might have an effect on certain SIRT6 interactions and, indirectly, its localization within the nucleoplasm. This indicates that SIRT6 phosphorylation can be an important factor in the regulation of its interactions and related functions. As the phosphorylation of other mammalian sirtuins (SIRT1 (65), SIRT2 (66), and SIRT7 (67)) is known to modulate their enzymatic activities and functions, the identification of these phosphorylation sites on SIRT6 provides a starting point for assessing their putative roles in regulating its activity and/or protein interactions.
Fig. 5.
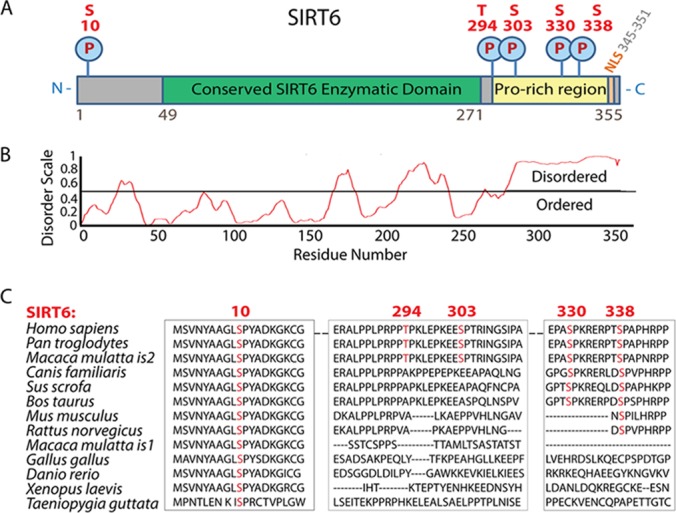
SIRT6 is phosphorylated at conserved residues within a C-terminal naturally disordered region. A, SIRT6 is phosphorylated at multiple sites localized to the N and C terminus of the protein: serine 10 (S10), serine 303 (S303), threonine 294 (T294), serine 330 (S330), and serine 338 (S338). C-terminal sites fall within a proline-rich amino acid sequence. B, SIRT6 is predicted to be natively disordered at its C-terminal region as analyzed by the PONDR algorithm. C, phosphorylated SIRT6 residues (red) are evolutionarily conserved among mammalian organisms. Sequence alignment was generated using NCBI Standard Protein BLAST with the following NCBI protein gi numbers for SIRT6 homologs: Homo sapiens (human) 38258612, Pan troglodytes (chimp) 114674699, Macaca mulatta is2 (monkey is2) 109122957, Canis familiaris (dog) 73987415, Sus scrofa (wild boar) 217331637, Bos taurus (cattle) 296485693, Mus musculus (mouse) 31712018, Rattus norvegicus (rat) 72255525, Macaca mulatta is1 (monkey is1) 109122959, Gallus gallus (chicken) 8612955, Danio rerio (zebrafish) 50344796, Xenopus laevis (frog) 148238219, and Taeniopygia guttata (zebra finch) 224052096.
SIRT6 Interactions Are Affected by SIRT6 Catalytic Activity
Protein deacetylases (HDACs), such as the better understood class I histone deacetylases, are known to exert their functions both by deacetylating substrates and forming functional complexes with proteins that are not their substrates (45). One important aspect that is not yet well understood is whether the catalytic activity of these enzymes modulates their ability to form interactions. Having established the network of SIRT6 interactions, we aimed to address this question and determine whether SIRT6 enzymatic activity modulates the formation of certain interactions. For this purpose, we generated cell lines stably expressing a catalytically deficient SIRT6 mutant (Fig. 6A). The evolutionarily conserved H133 residue falls within the catalytic domain, and its mutation to tyrosine (H133Y) is known to disrupt SIRT6 activity (9, 25, 36, 68). We introduced this mutation in our EGFP-tagged SIRT6 construct and generated HEK293 cell lines stably expressing the mutant protein. Confocal microscopy confirmed that, consistent with previous reports (9), H133Y mutation does not affect the nuclear localization of SIRT6 (Fig. 6B). Additionally, in vitro deacetylation activity assays verified that the mutant, isolated from HEK293 cells, had diminished enzymatic activity, similar to that observed after treatment with the sirtuin inhibitor nicotinamide (Fig. 6C). IPs of wt SIRT6-EGFP or EGFP tag alone served as positive and negative controls, respectively, and WB analysis confirmed the isolation of equivalent amounts of SIRT6 for these deacetylation assays (Fig. 6C). We next performed IPs of mutant EGFP-tagged SIRT6 and compared the identified interactions to those observed for wt SIRT6-EGFP. These experiments followed the same workflow, conditions, and replicates as described for the wt protein. Interestingly, our analysis revealed that the H133Y mutation led to complete abolishment or significant down-regulation (>50-fold decrease of average spectrum counts) of prominent interactions observed for the wt SIRT6 (Fig. 6D). We eliminated the possibility that this change in interactions was due to less efficient isolations of the mutant SIRT6-EGFP, as spectral count numbers for both the wt and the H133Y mutant were similar and, hence, comparable (supplemental Table S3). Most strikingly, the prominent interaction with G3BP1 was disrupted by the H133Y mutation, further emphasizing the likely functional significance of this association. Other interactions that were absent or significantly down-regulated in H133Y isolations were YLPM1, the NF-κ-B transcription regulator USP7 (69), several Nup family members, and the uncharacterized protein WD repeat-containing protein 74. The H133Y mutation also led to the enrichment of selected interactions, such as NEFL involved in neurofilament formation and the DNA sensor probable ATP-dependent RNA helicase DDX41 (70). Additionally, a subset of interactions remained constant, being present at similar levels in both wt and H133Y-mutant SIRT6 isolations (Fig. 6D). These consistent interactions included lamins (i.e. LMNA, LMNB1, and LMNB2), histones (i.e. H2AFX and H2AFY), the transcription regulator TP53, and the protein MKI67 (antigen KI-67) (Fig. 6D). In summary, disruption of SIRT6 catalytic activity and proteomic analysis demonstrated that certain SIRT6 associations are dependent on SIRT6's intact enzymatic status. These findings highlight the dynamic nature of SIRT6 associations and provide a foundation for mechanistic studies on the activity-dependent regulation of cellular pathways.
Fig. 6.
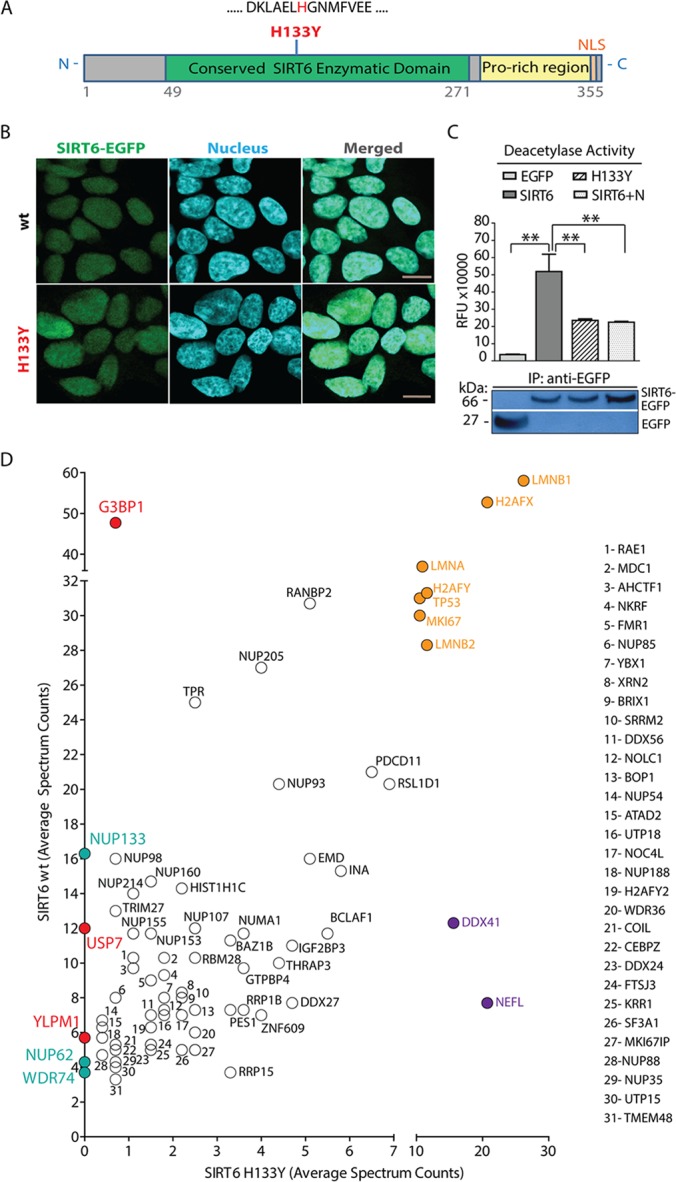
SIRT6 interactions are affected by H133Y activity mutation. A, diagram of SIRT6 protein domains showing localization of the mutated histidine 133 (H133) residue within a conserved enzymatic region. B, mutated SIRT6-H133Y-EGFP protein stably expressed in HEK293 cells retains nuclear localization as demonstrated by confocal microscopy and comparison to the localization of endogenous SIRT6 in wt cells; endogenous or EGFP-tagged H133Y SIRT6 (green), nucleus (blue, DAPI staining), 60× oil immersion lens, 2× optical zoom, scale bar = 10 μm. C, deacetylation activity assays of immunoaffinity-purified wt (SIRT6) and H133Y SIRT6-EGFP (H133Y) confirmed reduced enzymatic activity for the mutant protein, measured in relative fluorescence units (RFU). Control EGFP tag isolations (EGFP) and treatment of immunoaffinity-purified wt SIRT6-EGFP with the sirtuin inhibitor nicotinamide (SIRT6+N) served as negative controls (top). Deacetylation assays were performed in two biological replicates with two technical replicates; p values were calculated using Student's t test. **p < 0.01 (n = 3). Western blot with anti-EGFP antibodies showed successful isolation of EGFP or SIRT6-EGFP in these assays (bottom). D, specific interactions, including with G3BP1, USP7, and YLPM1, were significantly reduced or completely abolished in deacetylase inactive SIRT6 mutant isolations relative to wt SIRT6 (red and blue). Direct comparison of protein average spectrum counts revealed SIRT6 putative interactions unique to the mutant protein (purple), common (black), and highly enriched for both wt and H133Y SIRT6 (orange).
SIRT6 Interaction with G3BP1 Is Conserved in Different Cell Types and Dependent on Catalytic Activity
Our analyses identified G3BP1 as one of the most prominent SIRT6 interactions, being consistently present with over 45 average spectral counts (supplemental Table S3). Strikingly, the association with G3BP1 was effectively disrupted by the H133Y mutation. This prompted us to further validate and investigate this interaction. First, we assessed the subcellular localization of this protein and the possibility that it co-localized with SIRT6 in the nucleus of cells. G3BP1 is known to be primarily cytoplasmic under stress conditions but to have both nuclear and cytoplasmic localizations in nonstressed environments (54). Indeed, we observed that the two proteins co-localized within the nuclei of HEK293 cells, in which G3BP1 was distributed throughout the nucleus and the cytoplasm (Fig. 7A). This was consistent with our earlier observations that G3BP1 bore phosphorylations mediating nuclear compartmentalization (supplemental Fig. S3). Second, we validated this interaction by performing reciprocal isolation of endogenous G3BP1 in HEK293 cell lines expressing SIRT6-EGFP or EGFP tag alone (Fig. 7E). Western blot analysis demonstrated the co-isolation of SIRT6-EGFP (at ∼66 kDa) in the G3BP1 IP, but not in the control IgG and EGFP isolations (Fig. 7E). As G3BP1 is a highly abundant protein involved in numerous interactions and complexes that do not contain SIRT6, we anticipated that only a subset of G3BP1 would interact with SIRT6. This is likely the reason that only a relatively small amount of SIRT6 was isolated in the reciprocal isolation of G3BP1. Third, as an additional confirmation, we tested whether the SIRT6–G3BP1 interaction is only observed in HEK293 cells or common among diverse cell types. We chose to test the presence of this interaction in human T cell lymphoblasts (CEMT), in which deacetylases are known to play biologically and clinically significant roles (45). We generated CEMT cell lines stably expressing SIRT6-EGFP or the EGFP tag and performed SIRT6 isolation following the optimized proteomic workflow described above. SIRT6-EGFP was efficiently isolated from these cells, as illustrated by WB analysis (Fig. 7F), and mass spectrometry analysis demonstrated that G3BP1, and not its close relative protein G3BP2, co-isolated with SIRT6, as demonstrated by the identification of sequences unique to G3BP1 (SSSPAPADIAQTVQEDLR) (Fig. 7G). Therefore, the SIRT6–G3BP1 interaction is conserved in different cell types.
Fig. 7.

SIRT6–G3BP1 interaction occurs in different cell types and is dependent on the catalytic activity of SIRT6. A, confocal microscopy revealed the nuclear and cytoplasmic distribution of G3BP1 and demonstrated nuclear co-localization with SIRT6-EGFP; EGFP-tagged SIRT6 (green), G3BP1 (red, anti-G3BP1 antibody), nucleus (blue, DAPI staining), 60× oil immersion lens, 2× optical zoom, scale bar = 10 μm. B, endogenous SIRT6 is isolated from whole cell lysates of wt HEK293 cells as shown by WB analysis (FT, flow through; IP, primary protein elution; B, secondary protein elution). Control IgG isolations were used as a negative control. C, IP of endogenous SIRT6 performed after nuclear fractionation. WB analysis showed bait isolation using antibodies against SIRT6 and IgG (left). Successful nuclear/cytoplasmic fractionation was demonstrated by WB with anti-PARP1 and -HSP90 antibodies, used as nuclear and cytoplasmic markers, respectively (right). D, mass spectrometry and bioinformatics analyses of endogenous SIRT6 isolations. Cytoscape network generated for overlapping whole cell and nuclear fractionation interactions comprised proteins with more than five spectrum counts and at least 3-fold enrichment relative to IgG. Node color reflects NSAF values. White nodes indicate interactions identified only in the nuclear fractionation endogenous IP (set not analyzed by NSAF). Edge lines represent known protein clusters. E, reciprocal IP using anti-G3BP1 antibody revealed co-isolation of EGFP-tagged SIRT6 in HEK293 cell lines expressing the tagged protein. Parallel IgG isolations and IPs from HEK293-EGFP cells served as negative controls. WB analyses of reciprocal IP samples were performed with antibodies against G3BP1 (top) and SIRT6 (bottom). F, G, association between SIRT6 and G3BP1 occurred in CEMT cells. F, SIRT6-EGFP was efficiently isolated (CP, insoluble cell pellet; FT, flow through; IP, primary protein elution; B, secondary protein elution). G, association between SIRT6 and G3BP1 was observed in CEMT cells. Mass spectrometry analysis revealed G3BP1 as a constant SIRT6 binding partner (CID MS/MS using nano-LC LTQ Orbitrap Velos). H, YLPM1 co-localizes with SIRT6-EGFP in the nucleus of HEK293 cells. Confocal microscopy was performed using direct fluorescence for SIRT6-EGFP (green), antibodies against YLPM1 (red), and nuclear staining (blue, DAPI), with a 60× oil immersion lens and 2× optical zoom; scale bar = 10 μm. I, isolation of wt and H133Y SIRT6-EGFP confirmed the dependence on the SIRT6 catalytic activity for the associations with G3BP1 and YLPM1.
Finally, to further validate the interaction with G3BP1, we performed IPs of endogenous SIRT6 protein in wt HEK293 cells from either whole cell lysates or nuclear fractions. The latter served to enrich for nuclear proteins and acted as an alternative, localization-specific confirmation of interactions. The efficiency of the isolation (using the optimized lysis buffer, as in Fig. 3A) and of the nuclear/cytoplasmic fractionation was confirmed by Western blotting (Figs. 7B and 7C). The two bands observed for the endogenous SIRT6 were in agreement with its known two isoforms (36 and 39.1 kDa), and for the SIRT6-EGFP studies we focused on the full-length isoform that has an intact catalytic domain. The number of SIRT6 spectrum counts was ∼2-fold less than those from the SIRT6-EGFP isolations (supplemental Tables S6 and S7), which also led to a reduced number of spectra for interactions. This was an expected caveat for these experiments, as the used commercially available anti-SIRT6 antibody does not have the high affinity of our in-house-generated anti-EGFP antibody. However, despite this less efficient isolation of endogenous SIRT6, we argued that these experiments could provide additional confirmation of some of the prominent SIRT6 interactions, such as that with G3BP1. Indeed, IPs of endogenous SIRT6 from both whole-cell lysate and nuclear fractions reflected SIRT6 associations within the biological categories discussed above (Fig. 3A). Importantly, the interaction with G3BP1 was also the most prominent for endogenous SIRT6, as shown by the NSAF analysis (Fig. 7D and supplemental Table S6). Additionally, endogenous SIRT6 still associated with the transcription regulators NKRF, BCLAF1, and THRAP3; the telomerase modulator YLPM1; and the RNA polymerase activity factors XRN2 and COIL. Of particular interest is the interaction with YLPM1, which was completely abolished in the isolation of the catalytically deficient H133Y mutant. We confirmed that YLPM1 and SIRT6 co-localize within the nucleus (Fig. 7H). Additionally, in separate immunoisolation experiments, we used Western blotting to confirm that the SIRT6 associations with G3BP1 and YLPM1 are disrupted for the H133Y mutant.
Collectively, our results demonstrate that SIRT6 interacts and co-localizes with G3BP1 and YLPM1 within the nucleoplasm, and that these interactions are observed for the endogenous SIRT6 and are dependent on its enzymatic activity. We anticipate that these observations can form an important basis for defining the mechanisms of regulation of SIRT6 functions in health and disease processes.
DISCUSSION
In recent years SIRT6 has been established as a modulator of a range of critical processes and cellular pathways. Several reports have provided insights into SIRT6 substrates and/or interactions (e.g. H3, DNA-PKcs, C-terminal binding protein interacting protein, PARP1 (35–37)), expanding our understanding of SIRT6 functions. Nevertheless, knowledge of SIRT6 interactions or means of regulation remains limited. Here, we present a proteomics-based analysis of SIRT6 protein interactions and phosphorylations. We built a network of SIRT6 wt binding partners and demonstrated a previously unknown interplay between formation of SIRT6 interactions and its catalytic activity that can help define central mechanisms in SIRT6 regulation.
SIRT6 Phosphorylations
The importance of phosphorylation in regulating the functions of other sirtuins has begun to be recognized (71). CyclinB/Cdk1-mediated phosphorylation of SIRT1 was shown to modulate its deacetylase activity and affect cell proliferation (72), while CK2 acts to enhance its substrate-binding affinity, deacetylase activity, and cellular response to DNA damage (73). SIRT2 and SIRT7 are also phosphorylated by cyclinB/Cdk1, possibly modulating their activities during mitosis (66, 67). In agreement with these reports, our enrichment of SIRT6 led to the identification of several phosphorylation sites. Previous large-scale phosphoproteome studies have reported two SIRT6 phosphorylations (S303 and S330) (74, 75). Here we confirmed these sites, and expanded the list with three previously unknown ones—S10, T294, and S338. A phylogenetic analysis showing evolutionary conservation among mammalian organisms suggests biological relevance of these sites in regulating SIRT6 functions. The observed clustering of these modifications at the N or C termini of the protein, regions thought to bring specificity to the functions of the seven mammalian sirtuins (1), further supports this hypothesis. The C terminus of SIRT6 is known to affect its subcellular localization, and the N terminus is linked to chromatin association and enzymatic activity (9). Thus, it is tempting to speculate that these phosphorylations might regulate SIRT6 conformation changes that, in turn, could modulate SIRT6 interactions or targeting of substrates. Indeed, our studies show that mutation of the conserved S338 site can disrupt selected SIRT6 interactions. This modification localizes within a region predicted by our analyses to be naturally disordered, and reported by others as resistant to crystallization attempts (68). Further analyses will be required in order to determine the effect of these phosphorylation sites on the roles of SIRT6 in transcription regulation and other processes.
SIRT6 Protein Interaction Network
Consistent with SIRT6 roles in numerous cellular processes, our network of SIRT6 interactions comprised multiple functional categories spanning diverse nuclear processes. Our observations suggest that SIRT6 likely executes its multiple functions through the formation of interactions within numerous protein complexes. As anticipated, the array of SIRT6 associations included several of the limited number of previously known binding partners (e.g. HIST1H1C) and excluded interactions reported during DNA damage or other stress conditions (e.g. PARP1 and C-terminal binding protein interacting protein). We also detected the recently reported MYBBP1A (known RelA/p65 interactor) and SMARCA5 (linked to chromatin remodeling) SIRT6 associations (38); however, these interactions did not pass our specificity criteria and were excluded from the final network.
One of the most prominent SIRT6 interactions was with G3BP1, observed even under stringent lysis conditions, in endogenous isolations, and in different cell types, suggesting a strong and persistent binding to SIRT6. Early studies identified G3BP1 as a member of cytoplasmic stress granules (55), but upon phosphorylation at Ser149 the protein localizes to the nucleus (54) and mediates other processes (e.g. DNA repair) (56). Further reports link G3BP1 to cell cycle progression (76), targeted deubiquitination (77), cancer development (76, 78), and viral infection (41, 79), suggesting a putative effect of the identified SIRT6–G3BP1 association on diverse cellular processes. As we observed the G3BP1 co-isolated with SIRT6 to be phosphorylated at Ser149 and to share nuclear localization with SIRT6, it is likely that this interaction occurs within the nucleus. We previously observed G3BP1 interactions with members of the NPC (41); however, in our isolation of endogenous SIRT6, the presence of Nups was reduced. Therefore, it is unlikely that the SIRT6–G3BP1 interaction is mediated by NPC proteins. Importantly, our results establish a strong interaction between SIRT6 and G3BP1 and its dependence on the enzymatic activity of SIRT6 or on the structural integrity of the SIRT6 catalytic pocket. Recent studies showed that G3BP1 might not be a direct substrate of SIRT6 (80), leading us to hypothesize that G3BP1 might be linked to SIRT6 activation or deregulation of its functions as an interaction partner. The SIRT6–G3BP1 association might be biologically relevant to the activity of p53 tumor suppressor. G3BP1 is known to regulate p53 degradation by triggering its relocation from the nucleus to the cytoplasm (81), and down-regulation of G3BP1 expression was demonstrated to cause p53 activation (81). Interestingly, p53 has been reported as a positive SIRT6 regulator, stabilizing its protein and mRNA levels under normal conditions (51), and is not thought to be a SIRT6 substrate (7). Thus, our observation that p53 co-isolates with SIRT6 together with several of its known protein associations (e.g. USP7 and MDC1 involved in DNA repair) indicates a potential connection between p53 activity and the SIRT6–G3BP1 interaction. However, a direct link between these three factors remains to be established, and determining the range of processes regulated by this SIRT6 interaction will require further investigation.
Not surprisingly, a central functional group in our network of SIRT6 associations was linked to transcription regulation and comprised multiple transcription factors and related proteins (e.g. BCLAF1, tumor suppressor p53, BAZ1B, TRIM27, NKRF). Previous studies have demonstrated roles for SIRT6 in gene expression regulation via H3 lysine deacetylation at HIF-1α (82) and NF-κ-B (26) target gene promoters, indicating SIRT6 links to glycolytic gene expression and NF-κ-B signaling pathways. Our observation that SIRT6 associates with PDCD11 and NKRF, a known interaction of the transcription factor NF-κ-B (83), provides further evidence in support of SIRT6 roles in NF-κ-B-mediated transcription regulation. Importantly, we identified previously unknown SIRT6 connections to other transcription regulatory mechanisms. For example, BCLAF1 is a transcriptional repressor known to interact with the histone variant H2AX and promote DNA repair upon DNA damage induced by ionizing radiation (84). The presence of both BCLAF1 and H2AX in our network suggests that SIRT6 may deacetylate H3 or other histones at DNA break sites to facilitate recovery from DNA damage. BCLAF1 is also a known component of the SNIP1/SkIP-associated RNA-processing (SNARP) complex, regulating cyclin D1 expression and cellular proliferation (85). Given that we observed THRAP3, another member of this complex, it is possible that SIRT6 might play a role in SNARP-mediated gene expression regulation.
Our results also pointed to associations of SIRT6 with proteins involved in chromatin organization and nuclear transport, including lamin proteins and Nups. These proteins are known to be enriched at the nuclear periphery and participate in nuclear assembly and organization (86), or act as building blocks of the nuclear pore, mediating transport of molecules in and out of the nucleus (61). While recent reports also point to lamin and Nup roles in gene expression regulation (87, 88), our immunogold electron microscopy results demonstrate that a subset of SIRT6 is present at nuclear pores. Therefore, this is a likely a site for these interactions. Recent reports on Caenorhabditis elegans indicate that the nematode homolog of SIRT6, SIR-2.4, is also enriched near the nuclear pore and periphery, as observed by confocal microscopy (80), supporting our observations for human SIRT6 localization and interactions with Nups and lamins. It remains to be determined whether Nups and/or lamin proteins can also help recruit SIRT6 to certain regions of the nucleoplasm to regulate gene expression in target chromatin regions or mediate chromatin remodeling.
Finally, we observed several SIRT6 interactions linked to RNA polymerase activity, telomere regulation, and mRNA and rRNA processing. To the best of our knowledge, this is the first report suggesting a link between SIRT6 and RNA polymerase I and II activity via its interactions with COIL, NOLC1, and XRN2. COIL was recently reported to participate in the suppression of RNA polymerase I in response to DNA damage (57), and XRN2 was proposed to terminate transcription mediated by RNA polymerase II through exoribonuclease activity (58, 89). It is encouraging that these interactions were also observed with endogenous SIRT6. Another intriguing interaction is the YLPM1 protein (i.e. ZAP3) linked to the reduction of telomerase activity by down-regulating gene expression of the murine reverse transcriptase unit of telomerase (59). Previous reports have linked SIRT6 to telomere maintenance (25, 28, 29), and this interaction might represent a new means for SIRT6 roles in regulating telomere stability. Collectively, our findings significantly expand the previous knowledge of SIRT6 interactions, providing the possibility for exploration of new SIRT6 functions.
Impact of Catalytic Activity on SIRT6 Interactions
In addition to characterizing SIRT6 interactions, we evaluated their dependence on the enzymatic activity of SIRT6 and its intact catalytic pocket. One striking finding was that one of the most prominent SIRT6 interactions from our study, that with G3BP1, was almost completely abolished when we isolated the catalytically deficient SIRT6 (H133Y) mutant. Similarly, the interactions with several Nups, YLPM1, and USP7 (involved in ubiquitination and protein degradation) were absent, suggesting direct dependence on SIRT6 enzymatic activity. To our knowledge, this is the first proteomic study to demonstrate such interplay between SIRT6 catalytic activity and the formation of SIRT6 interactions, identifying a concrete small number of critical associations for future functional analyses. Our finding suggests that mutation of the catalytic H133 residue can prevent the interaction and recruitment of SIRT6 binding partners. In support of this is the observation that mutation of the SIRT6 H133 residue leads to lower affinity binding for ADP-ribose, a SIRT6 substrate important in the mediation of its catalytic activity, relative to binding with wt SIRT6 (68). Additionally, this mutation could cause a slight conformational change that disrupts interaction bindings, such that G3BP1 or other interactions are simply not needed when SIRT6 is catalytically nonfunctional. If this turns out to be true, it would suggest a direct and interdependent role for these binding partners in SIRT6 activity. It is also important to consider the range of SIRT6 enzymatic activities currently known (24, 33). It would be informative in future studies to assess whether alternative mutations, unique to each activity type, would trigger comparable changes in protein interactions. A starting point would be to explore the recently reported SIRT6 single point mutations proposed to abolish the deacetylation or mono-ADP-ribosylation activity of SIRT6, or both (37). Our observations provide an important first step in determining the regulatory role of SIRT6 activity in the formation of its interactions.
In summary, recent studies have established SIRT6 as a central regulator of numerous biological processes. However, the current knowledge of SIRT6 interactions that regulate SIRT6 activity and/or functions remained limited. Our proteomics-based study presents a comprehensive network of SIRT6 interactions, together with evidence regarding post-translational modifications. We also demonstrate dependence of the formation of SIRT6 interactions on SIRT6 catalytic activity. Collectively, we expect our findings to provide important knowledge for further elucidation of SIRT6-mediated functions and means of regulation.
Supplementary Material
Acknowledgments
We express our gratitude to Todd Greco for help with mass spectrometry analyses of phosphorylated peptides, to Hanna Budayeva and Todd Greco for critical reading of the manuscript, and to Fang Yu for help with the cloning of SIRT6-EGFP and EGFP into retroviral expression vectors. We appreciate the technical help provided by Woo Jung Cho, J. Goodhouse, and C. DeCoste (Microscopy and Flow Cytometry Core Facilities, Princeton University).
Footnotes
Author contributions: Y.V.M. and I.M.C. designed research; Y.V.M. performed research; I.M.C. contributed new reagents or analytic tools; Y.V.M. and I.M.C. analyzed data; Y.V.M. and I.M.C. wrote the paper.
* This work was supported by grants from the National Institute on Drug Abuse (Grant Nos. DP1DA026192, R21AI102187, and R21HD073044) and HFSPO award RGY0079/2009-C to I.M.C.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SIRT6
- sirtuin 6
- IP
- immunoaffinity purification
- CID
- collision-induced dissociation
- G3BP1
- Ras-GTPase activating protein SH3-domain-binding protein 1
- BCLAF1
- Bcl-2-associated transcription factor 1
- NKRF
- NF-κ-B-repressing factor
- HEK293
- human embryonic kidney cell line
- H133Y
- point mutation on SIRT6 amino acid residue 133 altering histidine to tyrosine and disrupting its catalytic activity
- NAD+
- nicotinamide adenine dinucleotide
- PARP1
- poly-[ADP-ribose] polymerase 1
- NPC
- nuclear pore complex
- NSAF
- normalized spectral abundance factor
- Nup
- nuclear pore complex protein
- SIRT
- sirtuin
- wt
- wild type
- USP7
- Ubiquitin carboxyl-terminal hydrolase 7
- WB
- Western blot
- YLPM1
- YLP motif-containing protein 1.
REFERENCES
- 1. Frye R. A. (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273, 793–798 [DOI] [PubMed] [Google Scholar]
- 2. Greiss S., Gartner A. (2009) Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol. Cell. 28, 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanny J. C., Dowd G. J., Huang J., Hilz H., Moazed D. (1999) An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99, 735–745 [DOI] [PubMed] [Google Scholar]
- 4. Landry J., Sutton A., Tafrov S. T., Heller R. C., Stebbins J., Pillus L., Sternglanz R. (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Imai S., Armstrong C. M., Kaeberlein M., Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 6. Lavu S., Boss O., Elliott P. J., Lambert P. D. (2008) Sirtuins—novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 7, 841–853 [DOI] [PubMed] [Google Scholar]
- 7. Michishita E., Park J. Y., Burneskis J. M., Barrett J. C., Horikawa I. (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. North B. J., Verdin E. (2004) Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 5, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tennen R. I., Berber E., Chua K. F. (2010) Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Age. Dev. 131, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michan S., Sinclair D. (2007) Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finkel T., Deng C. X., Mostoslavsky R. (2009) Recent progress in the biology and physiology of sirtuins. Nature 460, 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haigis M. C., Sinclair D. A. (2010) Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toiber D., Sebastian C., Mostoslavsky R. (2011) Characterization of nuclear sirtuins: molecular mechanisms and physiological relevance. Handb. Exp. Pharmacol. 206, 189–224 [DOI] [PubMed] [Google Scholar]
- 14. Beauharnois J. M., Bolivar B. E., Welch J. T. (2013) Sirtuin 6: a review of biological effects and potential therapeutic properties. Mol. Biosyst. 9, 1789–1806 [DOI] [PubMed] [Google Scholar]
- 15. Jia G., Su L., Singhal S., Liu X. (2012) Emerging roles of SIRT6 on telomere maintenance, DNA repair, metabolism and mammalian aging. Mol. Cell. Biochem. 364, 345–350 [DOI] [PubMed] [Google Scholar]
- 16. Tennen R. I., Chua K. F. (2011) Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 36, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morris B. J. (2013) Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 56, 133–171 [DOI] [PubMed] [Google Scholar]
- 18. Houtkooper R. H., Pirinen E., Auwerx J. (2012) Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roth M., Chen W. Y. (2013) Sorting out functions of sirtuins in cancer. Oncogene 32, 0950–9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mostoslavsky R., Chua K. F., Lombard D. B., Pang W. W., Fischer M. R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M. M., Mills K. D., Patel P., Hsu J. T., Hong A. L., Ford E., Cheng H. L., Kennedy C., Nunez N., Bronson R., Frendewey D., Auerbach W., Valenzuela D., Karow M., Hottiger M. O., Hursting S., Barrett J. C., Guarente L., Mulligan R., Demple B., Yancopoulos G. D., Alt F. W. (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 [DOI] [PubMed] [Google Scholar]
- 21. Moniot S., Weyand M., Steegborn C. (2012) Structures, substrates, and regulators of Mammalian sirtuins—opportunities and challenges for drug development. Front. Pharmacol. 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang W. J. (2008) Sirtuins: novel targets for metabolic disease in drug development. Biochem. Biophys. Res. Commun. 373, 341–344 [DOI] [PubMed] [Google Scholar]
- 23. Chakrabarty S. P., Balaram H., Chandrasekaran S. (2011) Sirtuins: multifaceted drug targets. Curr. Mol. Med. 11, 709–718 [DOI] [PubMed] [Google Scholar]
- 24. Liszt G., Ford E., Kurtev M., Guarente L. (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 280, 21313–21320 [DOI] [PubMed] [Google Scholar]
- 25. Michishita E., McCord R. A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T. L., Barrett J. C., Chang H. Y., Bohr V. A., Ried T., Gozani O., Chua K. F. (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawahara T. L., Michishita E., Adler A. S., Damian M., Berber E., Lin M., McCord R. A., Ongaigui K. C., Boxer L. D., Chang H. Y., Chua K. F. (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang B., Zwaans B. M., Eckersdorff M., Lombard D. B. (2009) The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 8, 2662–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michishita E., McCord R. A., Boxer L. D., Barber M. F., Hong T., Gozani O., Chua K. F. (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8, 2664–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tennen R. I., Bua D. J., Wright W. E., Chua K. F. (2011) SIRT6 is required for maintenance of telomere position effect in human cells. Nat. Commun. 2, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu S. S., Cai Y., Ye J. T., Pi R. B., Chen S. R., Liu P. Q., Shen X. Y., Ji Y. (2013) Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. Br. J. Pharmacol. 168, 117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawahara T. L., Rapicavoli N. A., Wu A. R., Qu K., Quake S. R., Chang H. Y. (2011) Dynamic chromatin localization of Sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet. 7, e1002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith B. C., Denu J. M. (2006) Sir2 protein deacetylases: evidence for chemical intermediates and functions of a conserved histidine. Biochemistry 45, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R., Hang H. C., Hao Q., Lin H. (2013) SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feldman J. L., Baeza J., Denu J. M. (2013) Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 43, 31350–31356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCord R. A., Michishita E., Hong T., Berber E., Boxer L. D., Kusumoto R., Guan S., Shi X., Gozani O., Burlingame A. L., Bohr V. A., Chua K. F. (2009) SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 1, 109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaidi A., Weinert B. T., Choudhary C., Jackson S. P. (2010) Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329, 1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37. Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., Gorbunova V. (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Polyakova O., Borman S., Grimley R., Vamathevan J., Hayes B., Solari R. (2012) Identification of novel interacting partners of Sirtuin6. PloS One 7, e51555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cristea I. M., Williams R., Chait B. T., Rout M. P. (2005) Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 4, 1933–1941 [DOI] [PubMed] [Google Scholar]
- 40. Guise A. J., Greco T. M., Zhang I. Y., Yu F., Cristea I. M. (2012) Aurora B-dependent regulation of class IIa histone deacetylases by mitotic nuclear localization signal phosphorylation. Mol. Cell. Proteomics 11, 1220–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cristea I. M., Carroll J. W., Rout M. P., Rice C. M., Chait B. T., MacDonald M. R. (2006) Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem. 281, 30269–30278 [DOI] [PubMed] [Google Scholar]
- 42. Tsai Y. C., Greco T. M., Boonmee A., Miteva Y., Cristea I. M. (2012) Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell. Proteomics 11, 60–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greco T. M., Yu F., Guise A. J., Cristea I. M. (2011) Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol. Cell. Proteomics 10, M110.004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 45. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo Y., Li T., Yu F., Kramer T., Cristea I. M. (2010) Resolving the composition of protein complexes using a MALDI LTQ Orbitrap. J. Am. Soc. Mass Spectrom. 21, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 49. von Mering C., Huynen M., Jaeggi D., Schmidt S., Bork P., Snel B. (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kanfi Y., Shalman R., Peshti V., Pilosof S. N., Gozlan Y. M., Pearson K. J., Lerrer B., Moazed D., Marine J. C., de Cabo R., Cohen H. Y. (2008) Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 582, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim H. S., Xiao C., Wang R. H., Lahusen T., Xu X., Vassilopoulos A., Vazquez-Ortiz G., Jeong W. I., Park O., Ki S. H., Gao B., Deng C. X. (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grimley R., Polyakova O., Vamathevan J., McKenary J., Hayes B., Patel C., Smith J., Bridges A., Fosberry A., Bhardwaja A., Mouzon B., Chung C. W., Barrett N., Richmond N., Modha S., Solari R. (2012) Over expression of wild type or a catalytically dead mutant of Sirtuin 6 does not influence NFkappaB responses. PloS One 7, e39847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tourriere H., Gallouzi I. E., Chebli K., Capony J. P., Mouaikel J., van der Geer P., Tazi J. (2001) RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell. Biol. 21, 7747–7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tourriere H., Chebli K., Zekri L., Courselaud B., Blanchard J. M., Bertrand E., Tazi J. (2003) The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56. Liu J., Song Y., Tian B., Qian J., Dong Y., Liu B., Sun Z. (2010) Functional proteomic analysis of promyelocytic leukaemia nuclear bodies in irradiation-induced MCF-7 cells. J. Biochem. 148, 659–667 [DOI] [PubMed] [Google Scholar]
- 57. Gilder A. S., Do P. M., Carrero Z. I., Cosman A. M., Broome H. J., Velma V., Martinez L. A., Hebert M. D. (2011) Coilin participates in the suppression of RNA polymerase I in response to cisplatin-induced DNA damage. Mol. Biol. Cell 22, 1070–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. West S., Gromak N., Proudfoot N. J. (2004) Human 5′ → 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432, 522–525 [DOI] [PubMed] [Google Scholar]
- 59. Armstrong L., Lako M., van Herpe I., Evans J., Saretzki G., Hole N. (2004) A role for nucleoprotein Zap3 in the reduction of telomerase activity during embryonic stem cell differentiation. Mech. Dev. 121, 1509–1522 [DOI] [PubMed] [Google Scholar]
- 60. Gentil B. J., Mushynski W. E., Durham H. D. (2013) Heterogeneity in the properties of NEFL mutants causing Charcot-Marie-Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol. Int. J. Biochem. Cell Biol. 45, 1499–1508 [DOI] [PubMed] [Google Scholar]
- 61. Wente S. R., Rout M. P. (2010) The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2, a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Rout M. P., Sali A. (2007) Determining the architectures of macromolecular assemblies. Nature 450, 683–694 [DOI] [PubMed] [Google Scholar]
- 63. Kohler A., Hurt E. (2007) Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 8, 761–773 [DOI] [PubMed] [Google Scholar]
- 64. Schreiber G., Keating A. E. (2011) Protein binding specificity versus promiscuity. Curr. Opin. Struct. Biol. 21, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nasrin N., Kaushik V. K., Fortier E., Wall D., Pearson K. J., de Cabo R., Bordone L. (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PloS One 4, e8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. North B. J., Verdin E. (2007) Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J. Biol. Chem. 282, 19546–19555 [DOI] [PubMed] [Google Scholar]
- 67. Grob A., Roussel P., Wright J. E., McStay B., Hernandez-Verdun D., Sirri V. (2009) Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J. Cell Sci. 122, 489–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pan P. W., Feldman J. L., Devries M. K., Dong A., Edwards A. M., Denu J. M. (2011) Structure and biochemical functions of SIRT6. J. Biol. Chem. 286, 14575–14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Colleran A., Collins P. E., O'Carroll C., Ahmed A., Mao X., McManus B., Kiely P. A., Burstein E., Carmody R. J. (2013) Deubiquitination of NF-kappaB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 618–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Z., Yuan B., Bao M., Lu N., Kim T., Liu Y. J. (2011) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Flick F., Luscher B. (2012) Regulation of sirtuin function by posttranslational modifications. Front. Pharmacol. 3, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sasaki T., Maier B., Koclega K. D., Chruszcz M., Gluba W., Stukenberg P. T., Minor W., Scrable H. (2008) Phosphorylation regulates SIRT1 function. PloS One 3, e4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kang H., Jung J. W., Kim M. K., Chung J. H. (2009) CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PloS One 4, e6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dephoure N., Zhou C., Villen J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rigbolt K. T., Prokhorova T. A., Akimov V., Henningsen J., Johansen P. T., Kratchmarova I., Kassem M., Mann M., Olsen J. V., Blagoev B. (2011) System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 4, rs3. [DOI] [PubMed] [Google Scholar]
- 76. Guitard E., Parker F., Millon R., Abecassis J., Tocque B. (2001) G3BP is overexpressed in human tumors and promotes S phase entry. Cancer Lett. 162, 213–221 [DOI] [PubMed] [Google Scholar]
- 77. Wang C. Y., Wen W. L., Nilsson D., Sunnerhagen P., Chang T. H., Wang S. W. (2012) Analysis of stress granule assembly in Schizosaccharomyces pombe. RNA 18, 694–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. French J., Stirling R., Walsh M., Kennedy H. D. (2002) The expression of Ras-GTPase activating protein SH3 domain-binding proteins, G3BPs, in human breast cancers. Histochem. J. 34, 223–231 [DOI] [PubMed] [Google Scholar]
- 79. Katsafanas G. C., Moss B. (2004) Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J. Biol. Chem. 279, 52210–52217 [DOI] [PubMed] [Google Scholar]
- 80. Jedrusik-Bode M., Studencka M., Smolka C., Baumann T., Schmidt H., Kampf J., Paap F., Martin S., Tazi J., Muller K. M., Kruger M., Braun T., Bober E. (2013) The sirtuin SIRT6 regulates stress granules formation in C. elegans and in mammals. J Cell Sci. 2013 September 6 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 81. Kim M. M., Wiederschain D., Kennedy D., Hansen E., Yuan Z. M. (2007) Modulation of p53 and MDM2 activity by novel interaction with Ras-GAP binding proteins (G3BP). Oncogene 26, 4209–4215 [DOI] [PubMed] [Google Scholar]
- 82. Zhong L., D'Urso A., Toiber D., Sebastian C., Henry R. E., Vadysirisack D. D., Guimaraes A., Marinelli B., Wikstrom J. D., Nir T., Clish C. B., Vaitheesvaran B., Iliopoulos O., Kurland I., Dor Y., Weissleder R., Shirihai O. S., Ellisen L. W., Espinosa J. M., Mostoslavsky R. (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nourbakhsh M., Hauser H. (1999) Constitutive silencing of IFN-beta promoter is mediated by NRF (NF-kappaB-repressing factor), a nuclear inhibitor of NF-kappaB. EMBO J. 18, 6415–6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lee Y. Y., Yu Y. B., Gunawardena H. P., Xie L., Chen X. (2012) BCLAF1 is a radiation-induced H2AX-interacting partner involved in gammaH2AX-mediated regulation of apoptosis and DNA repair. Cell Death Dis. 3, e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bracken C. P., Wall S. J., Barre B., Panov K. I., Ajuh P. M., Perkins N. D. (2008) Regulation of cyclin D1 RNA stability by SNIP1. Cancer Res. 68, 7621–7628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dechat T., Pfleghaar K., Sengupta K., Shimi T., Shumaker D. K., Solimando L., Goldman R. D. (2008) Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zullo J. M., Demarco I. A., Pique-Regi R., Gaffney D. J., Epstein C. B., Spooner C. J., Luperchio T. R., Bernstein B. E., Pritchard J. K., Reddy K. L., Singh H. (2012) DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 149, 1474–1487 [DOI] [PubMed] [Google Scholar]
- 88. Capelson M., Liang Y., Schulte R., Mair W., Wagner U., Hetzer M. W. (2010) Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gromak N., West S., Proudfoot N. J. (2006) Pause sites promote transcriptional termination of mammalian RNA polymerase II. Mol. Cell. Biol. 26, 3986–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.