

Abstract
More than 170 million people worldwide are infected with the hepatitis C virus (HCV), for which future therapies are expected to rely upon a combination of oral antivirals. For a rapidly evolving virus like HCV, host-targeting antivirals are an attractive option. To decipher the role of novel HCV–host interactions, we used a proteomics approach combining immunoprecipitation of viral–host protein complexes coupled to mass spectrometry identification and functional genomics RNA interference screening of HCV partners. Here, we report the proteomics analyses of protein complexes associated with Core, NS2, NS3/4A, NS4B, NS5A, and NS5B proteins. We identified a stringent set of 98 human proteins interacting specifically with one of the viral proteins. The overlap with previous virus–host interaction studies demonstrates 24.5% shared HCV interactors overall (24/98), illustrating the reliability of the approach. The identified human proteins show enriched Gene Ontology terms associated with the endoplasmic reticulum, transport proteins with a major contribution of NS3/4A interactors, and transmembrane proteins for Core interactors. The interaction network emphasizes a high degree distribution, a high betweenness distribution, and high interconnectivity of targeted human proteins, in agreement with previous virus–host interactome studies. The set of HCV interactors also shows extensive enrichment for known targets of other viruses. The combined proteomic and gene silencing study revealed strong enrichment in modulators of HCV RNA replication, with the identification of 11 novel cofactors among our set of specific HCV partners. Finally, we report a novel immune evasion mechanism of NS3/4A protein based on its ability to affect nucleocytoplasmic transport of type I interferon-mediated signal transducer and activator of transcription 1 nuclear translocation. The study revealed highly stringent association between HCV interactors and their functional contribution to the viral replication cycle and pathogenesis.
Infecting almost 170 million people worldwide, hepatitis C virus (HCV)1 is a leading cause of chronic hepatitis, liver fibrosis, cirrhosis, and hepatocellular carcinoma (1). Major improvements in patient treatment were achieved with the approval of the first direct-acting antivirals in 2011, namely, Incivek (Vertex Pharmaceuticals) and Victrelis (Merck & Co) (2). However, these drugs co-administered with pegylated interferon (IFN)-α and ribavirin inflict a heavy burden of side effects and have relatively low effectiveness in IFN-experienced nonresponder patients. Unlike chronic HIV infection, HCV does not integrate its genome into the host chromosomes and replicate in the cytoplasm of the cell. This suggests that eradicating HCV from all infected individuals is an achievable goal.
HCV is a Hepacivirus member of the Flaviviridae family. Following attachment of the virus to host factors at the surface of hepatocytes, virions are internalized in a clathrin-dependent manner (3). Subsequent to membrane fusion, the 9.6-kb viral RNA genome is released into the cytoplasm and translated into a single 3,000-amino-acid polyprotein precursor via an internal ribosome entry site (4). Host and viral peptidases cleave the polyprotein precursor to form 10 mature viral proteins: Core, E1, E2, P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B. The nonstructural (NS) proteins are important for the intracellular life cycle. Briefly, NS2 is a serine protease required for polyprotein cleavage at the NS2–NS3 junction. NS3 forms a complex with NS4A, and this complex possesses two main enzymes, an ATPase/helicase and a serine protease. NS4B induces the formation of an HCV specific membrane structure, the membranous web, which creates a microdomain within the cell where viral replication occurs. NS5A is a phosphoprotein that contributes to the membranous web by inducing double-membrane vesicles and controls RNA replication (5). Last but not least, the RNA-dependent RNA polymerase NS5B generates a negative RNA strand-template for viral RNA (vRNA) replication, from which it produces the genomic strands. P7 forms an ion channel of the viporin family and is important for virion assembly, together with NS2, NS3, and NS5A. Together with host proteins acquired during the release process, the mature virion is composed of the vRNA and the structural proteins Core, E1, and E2 (6).
As an obligatory intracellular parasite, HCV relies on its host to complete its viral life cycle. Furthermore, HCV encodes only 10 mature proteins, which emphasizes the dependence of viral replication steps on co-opted host factors. Probably the most characterized example of such an indispensable interaction is the dependence of vRNA replication on phosphatidylinositol-4-kinase α (7–11). The enzymatic activity of phosphatidylinositol-4-kinase α host cofactor is essential for viral replication through the enrichment of phosphatidylinositol-4-phosphate at the membranous web. Cyclophilin A interaction with NS5A is also necessary, and drugs targeting cyclophilin A are currently being tested in clinical trials for HCV treatment (12–14). Virus virulence determinants are also based on virus–host interactions that inhibit the host's immune system defenses. As an example, the adaptor molecules TIR-domain-containing adapter-inducing interferon-β (TRIF) and mitochondrial antiviral-signaling protein (MAVS), involved in the Toll-like receptor 3 (TLR3) and RIG-I-like receptor pathogen recognition signaling pathways, respectively, are targeted by the NS3/4A protease to block interferon regulatory factor 3 (IRF3)–dependent expression of IFNB1 and IFN-mediated cellular antiviral response (15). Thus HCV–host protein–protein interactions (PPI) are essential for viral propagation and viral strategies of immune evasion in order for a chronic infection to be established.
Because HCV is thought to replicate and mutate at a high rate, therapies directly targeting viral proteins with direct-acting antiviral agents are subject to the fast appearance of resistance mutation. Consequently, therapies targeting the host are an attractive alternative. Host-targeting antiviral therapies possess a greater genetic barrier to resistance than direct-acting antiviral therapies. Thus, unraveling the host–virus interactome is essential for novel antiviral target identification and for a better understanding of the cellular and viral biology. The majority of previous studies were done using a yeast two-hybrid system (16, 17). However, this system is not optimal for membrane-bound proteins like those of HCV. Genome-wide RNA interference (RNAi) studies also have yielded a load of valuable information on HCV biology, but these studies did not provide a link between viral protein and host gene products (7, 18–21).
In an effort to better understand the HCV life cycle, we used a systems biology approach combining viral protein immunoprecipitation (IP) coupled to tandem mass spectrometry identification and functional genomics RNAi screening to elucidate the role of novel HCV–host interactions. From 426 host proteins detected in the IP-MS/MS of Core, NS2, NS3/4A, NS4B, NS5A, and NS5B, 98 statistically enriched HCV interactors specific to a viral protein were identified, including 24 previously characterized in HCV–host interactions. The interacting proteins were further assessed via gene silencing in lentiviral-based short hairpin RNA (shRNA) screening in relevant HCV cell models. The combined proteomic and gene silencing approaches identified a total of 37 host proteins modulating HCV RNA replication and provided strong evidence for the following 11 HCV partners as previously uncharacterized HCV cofactors: HSD17B12 (Core); EXOC7, KPNB1, TARDBP, TNPO1, and MTHFD1 (NS3/4A); WNK1 (NS4B); BIN1, EGLN1, and SMYD3 (NS5A); and FKBP5 (NS5B). We also confirmed the two cofactors COPB1 and RAN among our enriched set of HCV partners that were previously identified in genomic screens of HCV replication (7). Finally, a novel immune evasion mechanism of NS3/4A is described that blocks the nucleocytoplasmic transport of STAT1, a phenotype reminiscent of the one obtained upon silencing of its partner protein KPNB1. Overall, the study identified HCV interactors with a high degree of stringency and provides an important association between some HCV interactors and their functional contribution to the viral life cycle and pathogenesis.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK 293T (human embryonic kidney), Huh7.5 (human hepatoma-derived), and Con1b (human hepatoma containing an HCV subgenomic replicon) cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) (Wisent, St-Bruno, Québec, Canada) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine (all from Wisent), and 1% nonessential amino acids (Invitrogen) at 37 °C in an atmosphere of 5% CO2. Huh7 cells harboring the Con1b replicon were submitted to a 500 μg/ml G418 selection pressure to maintain HCV replication during culturing but not during screening. For screening, Con1b/Huh7 and Huh7.5 (for infection) cells were seeded at a concentration of 5,000 cells per 96-well plate in white plates for replication assays and 96-well black plates for AlamarBlue assays. For lentivirus production, cells were seeded at 3,000,000 cells per 100-mm dish. Transfections for protein expression or for lentivirus production were performed with linear 25-kDa polyethylenimine (PEI) (Polysciences, Inc., Warrington, PA) (3 μg of PEI to 1 μg of DNA).
Plasmids and Antibody Reagents
A 3xFLAG cassette was inserted into the pcDNA3.1 expression vector using AflII/AgeI enzymes. This vector was used to clone NS2, NS3/4A, and NS5A. Core, NS4B, and NS5B were cloned by inserting 3xFLAG cassette into pcDNA3.1 vector containing the viral protein. FLAG-tagged versions of host protein interactors (FLAG-eYFP, FLAG-HSD17B12, FLAG-MTHFD1, FLAG-XPO1, FLAG-HBxIP, and FLAG-FKBP5) were produced by inserting a PCR product amplified from a commercial fully sequenced cDNA (Thermo Scientific, Lafayette, CO, USA) using Pfl23II/NotI enzymes into pcDNA3.1_FLAG-MCS(MB) expression vector. MYC-tagged versions of HCV proteins (MYC-Core, MYC-NS3/4A, MYC-NS2, and MYC-NS5B) were cloned into pcDNA3.1_MYC-MCS expression vector using EcoRV/HindIII enzymes. STAT1 and viral ORFs were amplified via PCR from the human fetal brain cDNA library (OriGene, Lake Forest, CA) and from JFH-1 (Core, NS2) or HCV 1b genotype (Con1 isolate, GenBankTM accession number AJ238799) by using KOD polymerase (EMD Millipore, Billerica, MA). STAT1, NS3/4A, and NS3/4A S139A constructs were cloned using a recombinational cloning system (Gateway, Invitrogen) into pDONR207 or pDONR223. After validation by sequencing, these ORFs were transferred by means of Gateway recombinational cloning from pDONR207 or pDONR223 to the expression vector pCI-neo-3xFLAG (3xFLAG fusion, a gift from Y. Jacob). The mutant NS3/4A S139A was generated via site-directed mutagenesis using the Quickchange kit (Stratagene, La Jolla, CA, USA). The pISRE-Fluc cis-reporter plasmid (Stratagene) containing the interferon-stimulated response element (ISRE) upstream of the firefly luciferase (Fluc) gene was used in combination with the pRL-SV40 plasmid (Promega, Madison, WI). pRL-SV40 contains the SV40 enhancer upstream of the Renilla luciferase (Rluc) gene and was used as an internal control reporter.
Antibodies to FLAG and MTHFD1 were purchased from Sigma (St-Louis, MO, USA, F3165 and HPA001290); FKBP5, EXOC7, MYC, STAT1, and XPO1 were purchased from Santa Cruz Biotechnology (Dallas, TX sc-13983, sc-365825, sc-789, sc-346, and sc-74454); EGLN1, KPNB1, and NS3 were purchased from Abcam (Toronto, Ontario, Canada, ab4561, ab7811, and ab13830); DDX3X and ILF2 were purchased from Bethyl Laboratories (Montgomery, TX A-300–474A and A303–147A); Core was purchased from ABR-Affinity Bioreagents (Golden, CO, USA, MA1–080); HSP90 was purchased from Cell Signaling Technology, Inc, (Danvers, MA 4874); HSD17B12 was purchased from NovusBiologicals (Littleton, CO NBPI-81567); and ACTIN was purchased from Chemicon International (Billerica, MA MAB1501R). TARDBP antibody was a kind gift from C. Vande Velde.
Immunoprecipitation and Purification of HCV Interactors
HCV protein-expressing cells were washed twice with PBS and lysed in 10 mm Tris-HCl, 100 mm NaCl, 0.5% Triton X-100, pH 7.6, with protease inhibitor mix (Roche). Resulting cell extracts were adjusted to 1 mg/ml and subjected to IP as follows: preclearing of the lysates was done by incubating lysates with a 50:50 slurry of immunoglobulin G-Sepharose (GE Healthcare) prepared in the lysis buffer with IgG beads for 1 to 2 h. Supernatants were immunoprecipitated by adding 40 μl of anti-Flag affinity gel (Sigma-Aldrich) prepared in TBS buffer (50 mm Tris-HCl, 150 mm NaCl, pH 7.4) as described by the manufacturer. Immunoprecipitates were washed three times in lysis buffer and three times with TBS. For MS/MS sample preparation, immune complexes were eluted twice from the resin for 15 min at 4 °C using 100 μl of NH4OH 0.5 m (pH 11.7) and dried using a SpeedVac. For interaction validation analysis, elution was performed using the FLAG or 3xFLAG purified peptide for 45 min at 4 °C (Sigma-Aldrich) at a concentration of 250 ng/μl.
Mass Spectrometry
Affinity-purified protein complexes were resuspended in 50 μl of 50 mm ammonium bicarbonate. Tris(2-carboxyethyl)phosphine was added to the protein samples at a final concentration of 5 mm. Samples were incubated at 37 °C at 650 rpm for 30 min. 30 μl of chloroacetamide at 55 mm was added and the samples were incubated at 37 °C at 650 rpm for 30 min. Tryptic digestion (1 μg each; Promega) was performed overnight at 37 °C, and samples were dried down in a SpeedVac and resolubilized in 50 μl of aqueous 5% acetonitrile (formic acid, 0.2%). Samples (20 μl each) were injected onto a C18 precolumn (0.3 mm inner diameter × 5 mm), and peptides were separated on a C18 analytical column (150 μm inner diameter × 100 mm) using an Eksigent nanoLC-2D system. A 56-min gradient from 10% to 60% acetonitrile (0.2% formic acid) was used to elute peptides at a flow rate of 600 nl/min.
The LC system was coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher). MS analyses were performed using data-dependent acquisition in which each full MS spectrum was followed by six collision-induced dissociation MS/MS spectra in the linear ion trap for the most abundant multiply charged ions. The conventional MS spectra (survey scan) were acquired in the Orbitrap at a resolution of 60,000 for m/z 400 after the accumulation of 106 ions in the linear ion trap. Mass calibration used a lock mass from ambient air (protonated (Si(CH3)2O)6; m/z 445.120029) and provided mass accuracy within 15 ppm for precursor ion mass measurements. The dynamic exclusion of previously acquired precursor ions was enabled (repeat count 1, repeat duration of 15 s, exclusion duration of 15 s). MS/MS spectra were acquired in collision-induced dissociation mode using an isolation window of 2 Da, and precursor ions were sequentially isolated and accumulated to a target value of 10,000 with a maximum injection time of 100 ms.
Protein Identification and Quantitative Analysis
The centroided MS/MS data were merged into a single peak-list file (Distiller, V2.4.2.0) and searched with the Mascot search engine (version 2.2, Matrix Science, London, UK) against a concatenated forward and reversed human IPI database (IPI human v3.54) containing 150,858 forward protein sequences. Mascot was searched with a parent ion tolerance of 10 ppm, a fragment ion mass tolerance of 0.5 Da, and one miscleavage. Carbamidomethylation of cysteine; oxidation of methionine; deamidation; amd phosphorylation of serine, threonine, and tyrosine residues were specified as variable modifications. The false discovery rate was calculated as the percentage of positive hits in the decoy database versus the target database, and a false discovery rate of 1% was considered for both proteins and peptides. Protein identification is reported only for those assigned with a minimum of two peptides per protein. The data were processed using the Mascot 2.2 (Matrix Science) search engine.
Label-free quantitative proteomics was used to profile protein abundance across sample sets, as reported previously (22, 23). Briefly, Mascot peptide identifications were matched to ion intensity (MS peak intensity, minimum threshold: 10,000 counts) extracted from the aligned MS raw data files (tolerances set to m/z = 15 ppm and retention time = 1 min). For each LC-MS run, we normalized peptide ratios so that the median of their logarithms was zero, to account for unequal protein amounts across conditions and replicates. Intensities were summed across fractions for peptides of identical sequences, and protein ratios were calculated as the median of all peptide ratios, while minimizing the effect of outliers. Only proteins defined by two or more peptide quantification events and identified in more than two independent experiments were considered. For each identified host protein, the fold enrichment change was determined from the ratio of the proportion for one target viral protein to the average proportion for other viral baits. Host proteins with greater than 3-fold enrichment and in the upper 5% of enriched host proteins for one viral protein were considered as specific interactors of viral proteins.
HCV Production and HCV Replication/Infection Assay
J6/JFH(p7-Rluc2A) RNA was produced from lineated Xba1 DNA template (a kind gift from C. Rice). Following phenol/chloroform extraction and ethanol precipitation, RNA was generated using the MEGAscript T7 in vitro transcription kit (Ambion, Life Technologies, Burlington, ON, Canada) according to the manufacturer's instructions and purified with an RNeasy Mini Kit (Qiagen, Toronto, ON, Canada) using the RNA cleanup protocol. RNA electroporation was conducted in cytomix buffer with Huh7.5 cells resuspended at a concentration of 1.7 × 107 cells ml−1 as previously described by Kato et al. (24). Cells were plated in 150-mm cell culture dishes with complete media, and the media was changed after 24 h. At 72 hours post-electroporation, cells were trypsinized and re-plated in five 150-mm dishes. Virus-containing supernatants were collected four days later, cleared through a 0.45-μm filter, and stored at −80 °C. For infection assays, Huh7.5 cells were seeded at 4,000 cells per well in a 96-well white plate at day 0 and transduced with shRNA-expressing lentivirus at multiplicity of infection (MOI) 10 for gene silencing. At day 1, transduced or control cells were incubated with J6/JFH-1 virus preparations (75 μl of culture supernatant) for 8 h, and media was then replaced by fresh complete media. HCV replication was measured at day 4 as follows: cell media was replaced with 50 μl/well PBS, and 50 μl of 2 mm EDTA (pH 8)/5 μm coelanterazin (Nanolight, Pinetop, AZ) was added to measure Rluc activities. Gene silencing shRNA screens were performed with J6/JFH-1 electroporated cells as follow: two days after electroporation, cells were seeded in 96-well white plates at 4,000 cells per well containing complete media and polybrene and transduced with shRNA-expressing lentivirus for three days before HCV replication was measured based on Fluc activity using a MicroBeta JET luminescence counter (PerkinElmer Life Sciences).
Lentiviral shRNA Library Production
From the MISSION TRC lentiviral library (Sigma-Aldrich), 234 MS hits were selected and shRNA were produced as follows. Five different shRNA-expressing lentiviruses per gene were produced individually in HEK 293T cells (2 × 104) that were plated one day prior to transfection. Transfections were performed using a Biomek FX (Beckman Coulter) enclosed in a class II cabinet according to the MISSION® Lentiviral Packaging Mix protocol (SHP001). Viruses were collected at 24 and 48 h post-transfection and were pooled prior to freezing. A nontarget sequence (NT) shRNA-expressing control lentivirus and 4% of random samples of each plate were used to measure lentiviral titers for quality control purposes. Titers were determined by limiting dilution assays using HeLa cells. Briefly, samples were diluted in complete DMEM (1:400 or 1:10,000) and added to HeLa cells. Media was changed at days 3 and 5 with complete DMEM containing 1 μg/ml puromycin (Wisent). After four days of selections, cells were stained with 1.25% crystal violet and plaque-forming units were counted to determine the viral titer.
Large-scale shRNA Production
293T cells were transfected with pRSV-REV, pMDLg/pRRE, pMD2-VSVg, and various shRNA-expressing pLKO.1-puro constructs (Sigma-Aldrich) using linear 25-kDa PEI (Polysciences, Inc.) at 3 μg PEI to 1 μg DNA. 48 h after transfection, cell media was collected, filtered (0.45-μm filter), and aliquoted. The MOI was determined using limiting dilution assays as described in a preceding section. Lentivirus infections were conducted overnight in media containing 4 μg/ml polybrene. For Western blot analysis, puromycin was added to the medium at a concentration of 3 μg/ml to select transduced cells.
Gene Silencing shRNA Screening
5,000 cells per well were seeded in 96-well plates in 90 μl of completed DMEM containing 4 μg/ml polybrene. After plating, infection with shRNA-expressing lentivirus at an MOI of 10 was performed using a Biomek FK (Beckman Coulter) in a class II cabinet. The readout was measured after three days. For subgenomic replicon assay, the Fluc fluorescence of cell lysates was determined using a buffer consisting of 100 mm Tris acetate, 20 mm Mg acetate, 2 mm EGTA, 3.6 mm ATP, 1% Brij 58, 0.7% β-mercaptoethanol, and 45 μg/ml luciferine pH 7.9. For AlamarBlue assays, cells were cultured in black 96-well plates. 10 μl AlamarBlue reagent (Invitrogen; diluted 1:4 in PBS) was added to the cells, and the cells were then incubated for 3 h at 37 °C prior to fluorescence measurement using an EnVision plate reader (PerkinElmer Life Sciences) at 595 nm (excitation wavelength = 531 nm). A control plate with media and AlamarBlue only (no cells) was used to determine the background that was subtracted from the fluorescence value. For Rluc assays following J6/JFH-1(p7-Rluc2A), 50 μl of PBS and 50 μl of 2 mm EDTA 237 (pH8)/5 μm coelenterazine E (Nanolight) were added to the washed cells. For the control, shRNA NT, shRNA YB-1 (TRCN0000007952), shRNA DDX3X (TRCN0000000003), and BILN2061 0.1 μm were included in each plate, and values were normalized with shRNA NT. Fluc and Rluc activities were measured using a MicroBeta JET luminescence counter (PerkinElmer Life Sciences).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Assays
Cells were cultured in transparent 96-well plates. 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide stock solution (5 mg/ml in PBS) was added to the cells, and the cells were then incubated for 1 h at 37 °C. After complete removal of the medium, cells were incubated at room temperature under mild shaking for 10 min with 150 μl of dimethyl sulfoxide containing 2 mm glycine pH 11 to dissolve crystals. The absorbance was read at 570 nm.
Detection of Tyrosine-phosphorylated STAT1
HeLa cells were grown on six-well plates and transfected with empty vector (pCI-neo3xFlagGW), NS3, NS3/4A, and STAT1 expression plasmids using JetPEI™ transfection reagent (Polyplus Transfection, New York, NY). Two days post-transfection, cells were serum starved for 4 h and were either untreated or treated with 1,000 IU/ml of human IFN-β (Calbiochem) for 30 min. 3xFLAG-tagged viral proteins, STAT1, and tyrosine-phosphorylated STAT1 at position Y701 were immunodetected via Western blot analysis using monoclonal anti-FLAG M2 (Sigma), polyclonal anti-STAT1 (Santa Cruz Biotechnology), and monoclonal anti-STAT1 (pY701) (BD Transduction Laboratories), respectively.
ISRE Luciferase Reporter Gene Assay
HeLa cells were grown on six-well plates and transfected with the indicated expression plasmids (1.5 μg/well) together with pISRE-Fluc reporter plasmid (1.5 μg/well) and pRL-SV40 reference plasmid (0.15 μg/well) using JetPEI™ transfection reagent (Polyplus Transfection). 24 h post-transfection, cells were seeded in 96-well plates in DMEM without FBS. 24 h post–serum starvation, cells were incubated with 800 IU/ml human IFN-α2b (Merck, France) for 6 h. After the addition of Dual-GloTM Luciferase Substrate (Promega), luciferase activities were counted with a Top Count NXTTM reader (Packard). The luciferase activity was calculated as the ratio of Fluc activity to reference Rluc activity.
Indirect Immunofluorescence and Confocal Microscopy
HeLa cells were plated on 12-mm-diameter glass coverslips and transfected with various expression plasmids. 48 h post-transfection, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Double stainings were performed via sequential incubations with FLAG® M2 monoclonal antibody-Cy3 conjugate (Sigma) and rabbit anti-myc antibody (Bethyl Laboratories) in combination with Alexa 488-labeled anti-rabbit F(ab)2′ fragment (Molecular Probes, Life Technologies, Burlington, ON, Canada). For STAT1 nuclear translocation experiments, cells were incubated with 1,000 IU/ml human IFN-β (Calbiochem, Merck Millipore, Billerica, MA, USA) for 45 min before paraformaldehyde fixation. For HCV infection, Huh7.5 cells were plated on coverslips in six-well plates and transfected with either JFH-1 Δcore 153–167 or NS3+ Q221L mutants. Six days post-transfection, cells were serum starved for 4 h and then stimulated with either 5 ng/ml IFN-γ or 1000 IU/ml IFN-α for 30 min. For gene knockdown, Huh7 cells were infected with a lentivirus encoding the shRNA NT or KPNB1 (MOI = 10). Three days post-infection, cells were serum starved for 4 h and then stimulated with 5 ng/ml IFN-γ for 30 min. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. Cells were blocked with 5% bovine serum albumin (BSA), 0.02% sodium azide, and 10% goat serum in PBS and incubated with the following primary antibodies diluted in 5% BSA–0.02% sodium azide: STAT1 (1:200), NS3 (1:100), and Core (1:200). Cells were then probed with Alexa Fluor-488 and -594 secondary antibodies (Invitrogen) diluted at 1:1,000 in 5% BSA–0.02% sodium azide, washed, and incubated with Hoechst dye (Invitrogen) at 1 μg/ml in PBS. Slides were mounted using DABCO (Sigma-Aldrich) as an antifading agent. Cells were examined via laser-scanning confocal microscopy (Axiovert 100 M LSM510, Zeiss, Toronto, ON, Canada), and images were processed using LSM Image Browser (Zeiss).
Functional Enrichment Analysis
The DAVID database was used for functional annotation (25, 26). The DAVID functional annotation chart tool was used to perform Gene Ontology biological process and InterPro protein domain analysis. Terms with a Benjamini–Hochberg corrected p value of less than 5 × 10−2 were considered as significantly overrepresented.
Overlap Size Significance
The significance of the overlap between cellular interactors identified in this screen and cellular interactors listed in the literature was assessed by means of random simulation using R (27). For each list, we randomly drew as many proteins as interactors from the human proteome. The procedure was repeated 10,000 times, and the overlap size was systematically measured. The p value was obtained by comparing the distribution of overlap sizes with the observed number of overlapping interactors (significant overlap size: p value of 5 × 102).
HCV–Human Protein–Protein Interactions from the Literature
Interactions between HCV proteins and human proteins were retrieved from an up-to-date and expert manual curation of the literature.
Human Interactome
Human–human protein–protein interactions were downloaded from iRefIndex 9.0 (28) and filtered in order to reconstruct a good-quality human interactome.
Network Representation
Protein interaction networks were visualized with Cytoscape (29).
Topological Analysis
The R statistical environment was used to perform statistical analysis, and the igraph R package was used to compute network topology measures (27, 30).
Network Interconnectivity Analysis
Statistical significance for the interconnectivity of targeted proteins was assessed via a random resampling testing procedure (n = 10,000 permutations) using R (27). For each permutation, we randomly extracted from the human interactome a number of proteins equivalent to the number of targeted proteins, and the number of shared interactions was determined. The randomization procedure was weighted and corrected according to the connectivity of proteins in order to prevent inspection bias on highly studied proteins. A theoretical distribution was computed for the 10,000 resampled values. From this distribution, an empirical p value was computed by counting the number of resampled values greater than the value observed for virus interactors.
RESULTS
Identification of HCV-associated Host Proteins
In an effort to decipher the HCV–host interactome, we used an IP-MS/MS approach and ectopic expression of individually tagged HCV proteins. Briefly, HEK 293T cells were transfected with vectors encoding Core, NS2, NS3/4A, NS4B, NS5A, or N5B viral proteins fused with 3xFLAG tag or the control empty 3xFLAG MCS vector (Fig. 1). Individual HCV bait proteins were tagged according to the predicted membrane topology of cleaved HCV proteins (31). The reciprocal protein interaction ability of HCV tagged proteins was confirmed by the positive strong bioleminescence resonance energy transfer signal obtained upon homodimer interaction (data not shown). All viral proteins were tagged at the N-terminal extremity except NS4B, which was tagged at the C terminus. Following individual protein expression, cellular lysates were subjected to IP using a resin coupled to anti-FLAG antibodies. As illustrated in a schematic representation in Fig. 1A, viral protein-containing complexes were isolated following several washing steps and elution with a high-pH buffer. The expression of viral tagged proteins was first confirmed in cell lysates using an anti-FLAG antibody at the expected molecular weight (Fig. 1B). Following elution with ammonium hydroxide, proteins were subjected to silver staining (Fig. 1C) to allow visualization of the immunopurification of viral and host protein complexes. The silver-staining analyses of the corresponding protein complexes showed protein bands at the expected molecular weights for 3XFLAG-Core, 1XFLAG-NS2, 3XFLAG-NS3, NS4B-3XFLAG, 3XFLAG-NS5A, and 3XFLAG-NS5B, which were confirmed by Western blot analysis (data not shown). In addition, the silver-staining analysis showed co-immunoprecipitated host partners specific for each HCV protein, whereas only weak bands in the FLAG-MCS control condition were detected corresponding to nonspecifically bound material.
Fig. 1.

Immunopurification of viral protein complexes. A, schematic representation of 3xFLAG-tagged viral protein immunoprecipitation. Briefly, 3xFLAG-tagged viral proteins were expressed in 293T cells and cell lysates were incubated with FLAG resin, extensively washed, and eluted with ammonium hydroxide to enrich viral protein and its attached host–protein complexes before being submitted for LC-MS/MS analysis. B, immunoblot detection of the viral-tag protein at expected weight in cell extracts. C, silver staining of SDS-PAGE gel for the different IP conditions with viral proteins (black dots) and other bands corresponding to co-IP host proteins.
Biological replicates of immunoprecipitated cell extracts were digested with trypsin, and the corresponding digests were subjected to LC-MS/MS analyses. Label-free quantitative proteomics was used to profile protein abundances across sample replicates and conditions. From three independent experiments, data were clustered to correlate the abundance of interacting proteins between immunoaffinity extracts. Triplicate LC-MS/MS analyses from all isolated protein complexes enabled the identification of 5,585 peptides from 1,254 proteins with a false discovery rate of less than 1% (supplemental Table S1). For each immunoprecipitation, 90% of the identified peptides showed a fold change of less than 3 relative to the mock sample. The 10% of peptides with greater than a 3-fold change were used for the identification of host proteins interacting with HCV proteins. Overall, 426 proteins were reproducibly detected in multiple immunoaffinity protein extracts with at least two peptides per protein (supplemental Table S2). By determining the proportion of individual host proteins for each immunoprecipitated viral bait condition (Σ proportions = 1 for all baits), we calculated the fold enrichment change, and its distribution was used in a threshold for the stringent selection of the upper 5% of statistically enriched host proteins for each viral protein (supplemental Fig. S1). A total of 98 host proteins interacting more specifically with one viral protein were identified that corresponded to those criteria, and these are illustrated in a heat map generated from the data reflecting the proportion of each host protein for the different viral baits (Fig. 2A and supplemental Table S3).
Fig. 2.

Heat map representation of enriched HCV-associated host proteins. A, proportions for each of the seven experimental conditions are represented for host protein hits (Σ 7 conditions = 1 for each host protein). The darker color correlates with the absence of the host protein in the condition, and brighter green indicates a high prevalence of the host protein in the condition. Orders of hit representation are according to function of viral protein specificity and are sorted by ascending proportion. B, Venn diagram of the overlap between HCV interactors identified in our study and those reported in the literature with the list of the 24 common interactors. C, statistical analysis of the overlap. The 10,000 overlap sizes obtained via random simulation are plotted. The observed value of x is indicated by the vertical arrow, and its significance is given.
From such analysis, the Core protein was found to interact specifically with a set of 22 host proteins out of the 98 proteins. The two most enriched Core-associated proteins were nucleosome assembly protein 1-like 4 and nucleosome assembly protein 1-like 1, which were recently reported as Core interactors (32). The known interactors ATP-dependent RNA helicase DDX3X and chromosome 14 open reading frame 166 (33) were also highly enriched in the 3xFLAG-Core by more than 8-fold and 4-fold, respectively, further validating the use of the approach for the identification of relevant HCV-associated host proteins. All other proteins, including highly enriched 2-hydroxyacylsphingosine 1-beta-galactosyltransferase (UGT8), monocarboxylate transporter 1 (SLC16A1), and protein tyrosine phosphatase-like protein (PTPLAD1), were newly identified HCV partners. Notably, the characterized Core interactant complement component 1 Q subcomponent-binding protein (34, 35) was identified in our study, although with a weak enrichment factor of 2 resulting from detection in many other viral bait conditions (for others, see Fig. 2A and supplemental Table S3). In the IP 3xFLAG-NS2 condition, eight interactants were identified that included hepatitis B virus X-interacting protein (HBxIP) as the most enriched host protein (supplemental Table S3). Most of the identified NS2 partners were novel, including the protein calnexin, which was identified as an HCV cofactor in an RNAi screen (18) and characterized as a cell apoptosis inhibitor induced in HCV-positive hepatocellular carcinoma (36). The IP-NS3/4A showed the greatest number of interacting partners on silver staining gel, and this was reflected in the MS/MS analysis with a total of 33 statistically enriched hits. The IP 3xFLAG-NS3/4A retrieved previously identified TAR DNA binding protein (TARDBP), tubulin, and importin subunit β-1 (KPNB1 or NTF97) (37, 38). All other proteins, including the most enriched ataxin-10 and trifunctional purine biosynthetic protein adenosine-3 (GART), isoform 1 of voltage-dependent anion-selective channel protein 3, and isoform 1 of exocyst complex component 7 (EXOC7), were newly identified HCV partners. Interestingly, NS3/4A strongly interacted with proteins that function in nucleocytoplasmic transport: KPNB1, Exportin-5 (XPO5), RAN binding protein 1, Transportin 1 isoform 1 (TNPO1 or KPNB2), and RAN, a member of the RAS oncogene family. In the IP 3xFLAG-NS4B condition, the fold-enrichment Gaussian curve showed a shift to the left, corresponding to a low level of co-immunoprecipitated proteins with NS4B (supplemental Fig. S1). Despite this irregular distribution, four proteins having fold enrichments of greater than 3-fold were identified: lysine deficient protein kinase 1 (WNK1) with the highest degree of stringency criteria, interleukin enhancer-binding factor 2 (ILF2 or NF45), calumenin, and Fanconi anemia group I protein (supplemental Table S3). Unexpectedly, the NS4B interactor ILF2 was recently reported as a Core partner (33), whereas in our study ILF2 showed a weak 0.7-fold enrichment in the 3xFLAG-Core condition. From the data for the IP-NS5A condition, 8 interacting proteins out of 17 confirmed in our study were previously identified via either yeast two-hybrid or IP-MS/MS analysis: VAPB, VAPA, BIN1, FKBP8, MOBKL1A, HSPA5, SMYD3, and USP19 (16, 32, 39–45). Novel NS5A partners included girdin (CCDC88A), egl nine homolog 1 (EGLN1), F-actin-capping protein subunit α-1, and E3 ubiquitin-protein ligase HUWE1 among the most enriched proteins (Fig. 2A and supplemental Table S3). For NS5B, 15 proteins were immunoprecipitated that matched selection criteria, including the most enriched partners heat shock protein 90kDa α class A member 1 (HSP90AA1), FK506-binding protein 5 (FKBP5), creatine kinase B-type, and proteasome activator complex subunit 3. In this proteomics analysis, only MORF4 family-associated protein 1 was enriched in two conditions simultaneously, and immunoprecipitated with both NS2 and NS3/4A proteins.
The comparison of the 98 HCV-associated human proteins found in this study to the 543 host proteins identified from an extensive manual curation of all reported HCV–human interactions and PPIs showed 24 common interactors (Fig. 2B). The observed number of interactions was significantly greater than expected (p value < 10−4) for a random simulation of overlapping human interactors (Fig. 2C). Overall, this approach identified 98 putative HCV partners (99 PPIs) with a high degree of stringency, including 74 novel host proteins interacting specifically with Core, NS2, NS3/4A, NS4B, NS5A, or NS5B proteins.
Validation of the HCV–Host Protein-mapping Approach
In order to confirm the specificity of selected virus–host protein partners identified in the MS analysis, we performed a first set of IPs of 3xFLAG-tag viral proteins with a competitive 3xFLAG peptide elution procedure followed by Western blot analysis of the eluant to detect endogenous host interactor proteins. The peptide elution procedure could not be used in the MS/MS analysis because the large amount of 3xFLAG peptide interfered with peptide detection. Fig. 3 (left-hand panel) shows similar levels of selected host interactor proteins in cell extracts upon the expression of each viral protein or control FLAG-MCS. The anti-FLAG immunoblots (bottom) confirmed the presence of viral FLAG-tagged HCV proteins in cell extracts (left-hand panel) and after immunopurification (right-hand panel). In accordance with previous reports, DDX3X was specifically immunopurified with Core protein and detected only in the IP-Core condition (34, 35, 46). TARDBP, EXOC7, and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) were confirmed as specific NS3/4A interactors. TARDBP was the only NS3/4A partner previously retrieved in an MS/MS analysis, by Lai et al. (37), although no validation was available for this interaction. Similarly, EGLN1 was confirmed as a specific NS5A interacting protein (known as FKBP8 binding partner (47)), and immunophilins FKBP5 and HSP90 were specifically detected as NS5B binding partners. We also performed two reciprocal co-immunoprecipitation studies. Fig. 4A (left-hand panel) shows the expression in cell extracts of the FLAG-tagged host proteins HSD17B12, MTHFD1, XPO1, HBxIP, and FKBP5, as well as FLAG-eYFP, used as negative control, and (right-hand panel) specific co-immunoprecipitation of the expected viral partners as revealed by the Western blot detection of Myc-tagged viral proteins in the FLAG peptide eluants. Fig. 4B shows the immunoprecipitation of endogenous host proteins DDX3X, ILF2, EXOC7, XPO1, MTHFD1, KPNB1, EGLN1, and FKBP5, and again revealed the specific interactions with the expected viral partners via the Western blot detection of 3xFLAG-tagged viral proteins in the IP. These confirmation results further strengthen the degree of confidence in our IP-MS/MS analysis for the identification of specific HCV partners.
Fig. 3.

Validation by immunoprecipitation–Western blotting of selected virus–host protein partners identified by MS analysis. Selected hits retrieved by IP-MS/MS were validated by FLAG IP coupled to FLAG peptide elution and Western blot detection. DDX3X was present and specific to the Core condition; TARDBP, MTHFD1, and EXOC7 were NS3/4A partners; EGLN1 was a specific NS5A interactor; and FKBP5 and HSP90 interacted specifically with NS5B. Flag immunoblots show the expression of viral protein in the lysates and IP conditions.
Fig. 4.

Immunoprecipitation of host interactor proteins validates their interactions with viral HCV proteins. A, immunoprecipitation of FLAG-tagged host interactor proteins HSD17B12, MTHFD1, XPO1, HBxIP, and FKBP5 coupled to FLAG peptide elution and Western blotting detection of Myc-tagged HCV proteins (reciprocal co-IP). Immunoprecipitation of FLAG-eYFP was used as a negative control. B, immunoprecipitation of endogenous host interactor proteins DDX3X, ILF2, EXOC7, XPO1, MTHFD1, KPNB1, EGLN1, and FKBP5 followed by Western blotting detection of 3xFLAG-tagged HCV proteins (reciprocal co-IP). Immunoprecipitation of Human influenza hemagglutinin (HA) was used as a negative control. Ab, antibody.
Gene Ontology Term Enrichment
Each list of HCV-associated proteins was submitted to a Gene Ontology term analysis using the DAVID Internet resource (Table I). The identified set of host proteins from IP of HCV proteins showed an enriched Gene Ontology term associated with the endoplasmic reticulum category, which is consistent with a putative role of most of these proteins in viral replication functions. HCV-associated proteins also showed enrichment of the ontology term “transport proteins” with a major contribution of NS3/4A interactors (50%). This is reflected by the significant tendency of NS3/4A to interact with cytoplasmic proteins having HEAT repeat structures, which are involved in intracellular transport and mRNA maturation processes as a scaffold for protein–protein interactions (Interpro, Benjamini p value = 2.4 × 10−4). Another striking observation was that Core interactors were highly enriched in transmembrane proteins (Protein Information Resource, Benjamini p value = 2.9 × 10−3, 15/22 interactors). Although not statistically significant, six out of eight NS2 interactors were also transmembrane proteins. All these interactors were novel, illustrating the necessity of diversifying technologies in order to provide a comprehensive protein interactome. Indeed, transmembrane proteins are often problematic and hardly detectable in the transcription-based yeast two-hybrid technologies, in contrast to the IP-MS/MS approach used in this study.
Table I. Gene Ontology enrichment.
| Category | Count | Genes | Fold enrichment | Benjamini p value |
|---|---|---|---|---|
| GOTERM_BP_FAT | ||||
| GO:0015031 | 16 | CANX, KPNB1, TNPO1, EXOC7, HSPA9, IPO7, SEC61B, TRAM1, XPO1, SSR3, RAN, XPO5, CLTC, PCNA, SEC63, COPB1 | 3.6 | 0.0017 |
| Protein Transport | ||||
| GO:0006461 | 12 | TUBB, CAPZA1, HSP90AA1, IPO7, KPNB1, TNPO1, HSPD1, XPO1, TUBA1B, TUBB2A, CCDC88C, TUBB4B | 4.0 | 0.0067 |
| Protein Complex Assembly | ||||
| GO:0006611 | 4 | XPO1, XPO5, RAN, HSPA9 | 37.6 | 0.0068 |
| Protein Export from Nucleus | ||||
| GO:0006606 | 6 | IPO7, KPNB1, TNPO1, SEC61B, XPO1, RAN | 11.8 | 0.0071 |
| Protein Import into Nucleus | ||||
| GO:0006986 | 5 | DNAJA1, HSP90AA1, VAPB, HSP90AB1, HSPD1 | 11.9 | 0.0282 |
| Response to Unfolded Protein | ||||
| GOTERM_CC_FAT | ||||
| GO:0005635 | 9 | IPO7, KPNB1, TNPO1, XPO1, TMEM109, LBR, CACYBP, RAN, PCNA | 6.8 | 0.0011 |
| Nuclear Envelope | ||||
| GO:0005783 | 16 | VAPA, CANX, SET, IKBIP, VAPB, FKBP8, SEC61B, TRAM1, PGRMC1, TMEM109, SSR3, CCDC88A, HSPA5, CALU, SEC63, HSD17B12 | 2.6 | 0.0137 |
| Endoplasmic Reticulum | ||||
| GO:0031974 | 24 | ILF2, LYAR, KPNB1, SET, FANCI, TUFM, DDX20, DDX3X, HUWE1, PRKDC, HSPA9, QKI, HSPD1, SMN1, PGRMC1, XPO1, TMEM109, MYBBP1A, CACYBP, HSPA5, RAN, TARDBP, CALU, PCNA | 2.0 | 0.0147 |
| Membrane-enclosed Lumen | ||||
| GOTERM_MF_FAT | ||||
| GO:0008565 | 6 | IPO7, KPNB1, TNPO1, SEC61B, XPO1, XPO5 | 11.5 | 0.0208 |
| Protein Transporter Activity | ||||
| GO:0000166 | 27 | TUBB, ILF2, DDX3X, PHGDH, TUBB4B, PRKDC, HSP90AA1, WNK1, GART, RAN, CCT4, HSPA5, ATP1A1, GNAI3, CKB, TUFM, DDX20, TUBA1B, ABCD3, HSPA9, DDX1, HSP90AB1, HSPD1, TBRG4, TUBB2A, TARDBP, VDAC3 | 2.0 | 0.0219 |
| Nucleotide Binding |
Effect of Silencing HCV-associated Proteins on HCV Replication
We next determined whether host proteins identified via the IP-MS/MS approach were involved in vRNA replication by means of a lentiviral-mediated gene knockdown approach. The targeted functional RNAi screen was extended to a total of 234 proteins. These included 60 proteins out of the 98 statistically enriched proteins and 174 out of the 426 proteins pulled down in the different viral IP conditions that met our stringent selection threshold (see supplemental Table S4 for the complete screening data and the extended gene list). Three functional assays were performed in cells transduced with shRNA-expressing lentivirus to knock down targeted genes in a medium-throughput screening approach. Huh7-derived cells stably expressing the bi-cistronic Con1b subgenomic replicon reporter system (Huh7-Con1-Fluc) were first exploited to investigate the gene-silencing effect on vRNA replication (Fig. 5A). In a second assay, knockdown cells were electroporated with the full-length infectious HCV RNA genome J6/JFH-1(p7Rluc2A), which carries out a complete virus life cycle (Fig. 5B). As a third assay, an AlamarBlue counterscreen was performed to measure the viability of cells after gene knockdown. In the screening, five shRNA per gene were tested individually in each assay and performed in triplicate per experiment for the 234 genes (supplemental Table S3). The average of three experiments was determined for the identification of HCV co-factors, and data illustrated a low standard deviation of biological replicates for the three assays (supplemental Table S4). For quality purposes, the following controls were included in each plate in duplicate: protease inhibitor BILN 2061-treated cells, YB-1 knockdown cells (shRNA YB-1), nontarget shRNA transduced cells (shRNA NT), and untransduced cells. The specific HCV inhibitor BILN2061 was used as a control of HCV replication (48). YB-1 knockdown cells were included because we previously reported a significant inhibition effect on viral replication in these cells (38, 49). Nontransduced and shRNA NT transduced control cells were included to confirm the absence of effects resulting solely from infection with lentivirus and were used to standardize results arbitrarily set to 1.
Fig. 5.

Effect of silencing potential HCV-associated host proteins by shRNA on HCV viral replication. A, B, schematic representation of the HCV reporter systems used in the shRNA screen. A, Huh7.5-Con1-Fluc subgenomic replicon contains the 5′UTR and 3′UTR of the HCV RNA genome that are required for its replication and expresses Fluc under the control of the HCV internal ribosome entry site and the viral replicative unit (protein NS3 to NS5B of genotype 1b) under an encephalomyocarditis virus internal ribosome entry site. B, J6/JFH-1/p7Rluc2a chimeric construct is composed of the HCV 5′UTR region extending to NS2 from a J6 sequence, genetically engineered to express Rluc, and the sequence from NS3 through the 3′UTR from the JFH-1 isolate (genotype 2a). The reporter protein is inserted between p7 and NS2 with a foot-and-mouth-disease virus peptide cleavage site resulting in self-cleavage from the polyprotein. C, TIGR MultiExperiment Viewer (TMEV) representation of the shRNA screen results for 32 gene hits whose silencing significantly inhibited HCV viral replication with no observed changes in cell viability. Results are presented as an inhibition heat map and were normalized according to cells treated with shRNA NT (negative control set to 1 (black)) based on an average of three independent experiments. The following criteria were applied to select hits: >25% decrease of viral replication on J6/JFH-1 model; AlamarBlue effect of <25%; and >50% decrease in Con1b system with two shRNAs or >70% if only one shRNA was efficient. Hits are clustered by their corresponding viral binding partners, and the last two digits of the hit name correspond to the shRNA TRC number from the Sigma bank. D, comparison of host factors identified in this study and those from a genome-wide screen by Tai et al. (7).
We found that silencing the expression of all HCV interactors tested did not have a significant positive effect on viral RNA replication, and thus no antiviral restriction factors were identified in this study using the Huh7-derived cells permissive to HCV replication. We identified 32 gene hits out of 234 that significantly inhibited viral replication with no observed changes in cell viability based on an average of three independent gene-silencing experiments (Fig. 5C). The threshold criteria for the selection of gene hits were >50% decrease in Fluc activities in Huh7-Con1-Fluc assays for two or more shRNAs (or >70% decrease if only one shRNA was efficient), >25% decrease in J6/JFH-1(p7Rluc2A) activities, and <25% effect in AlamarBlue viability assay. We observed a significant effect with knockdown of identified HCV partners for each viral bait and for hits previously described in the genome-wide RNAi screen (Fig. 5D) (7). In our study, all hits inhibiting J6/JFH-1(p7Rluc2A) infection also affected vRNA replication in the HCV replicon assay.
In order to confirm the functional effects of gene hits in the RNAi silencing screen, shRNA-expressing lentiviruses for selected genes were produced at a large scale to transduce (MOI of 10) Con1-Fluc cells or J6/JFH-1(p7Rluc2A)-infected cells (a different infectious model than the HCV RNA-electroporated cells used in the shRNA screening assay). A viability 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay was carried out in parallel, and experiments were performed in quintuplicate. Controls were included (shRNA targeting YB-1, shRNA NT, and untransduced cells) similar to those in screening assays. We first demonstrated the knockdown for the selected enriched HCV partners EGLN1, TARDBP, HSD17B12, and FKBP5 in 239T cells following three days of transduction and in the presence of puromycin selection pressure for the last two days (Fig. 6A). In both HCV reporter assays, the fold change in Fluc or Rluc activity for each gene knockdown was determined as a ratio of the shRNA NT, arbitrarily set to 1. We observed a significant inhibitory effect on HCV replication upon silencing of those genes that was not attributable to cell mortality, as demonstrated by the absence of an effect in the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay conducted under similar cell conditions (Figs. 6B and 6C). In these experiments, gene knockdown significantly reduced HCV replication in Huh7.5 cells infected with cell-culture-grown virus J6/JFH-1(p7Rluc2A) at three days post-infection. MTHFD1 and WNK1 (not included in the shRNA screen) also showed a major decrease in HCV RNA replication when tested in similar gene-silencing conditions (Fig. 6A and supplemental Fig. S2). This infection model provides additional evidence of the involvement of host proteins in the viral life cycle and supports the relevance of our functional RNAi screening.
Fig. 6.

Validation of the silencing efficiency of selected shRNA targeting HCV cofactors and their effect on viral replication. A, the gene silencing effect was evaluated for each cofactor via Western blot detection. A decrease of the target protein was observed with the gene-specific shRNA, illustrating the efficacy of gene silencing. Actin immunoblot was performed as a loading control. B, C, the gene silencing effects of shRNA-expressing lentiviruses for each cofactor were determined on HCV replication-dependent reporter activities (MOI of 10) at day 3 post-transduction with HCV replicon-containing cells (B) and in the infection assay (C). The cellular viability was assessed with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cytotoxicity assay performed in parallel.
Among our statistically enriched host interactors of HCV, 60 genes were silenced in this study (available in our shRNA collection). Of these, 13 HCV cofactors were identified according to our stringent criteria (supplemental Table S4) and protein knockdown validation (Fig. 6), for an identification rate of 21.7%. Interestingly, the rates of virus replication modulators identified from genome-wide siRNA screens are 0.45% (7) and 1.36% (21). These cofactors are HSD17B12 (Core); COPB1, EXO7, KPNB1, MTHFD1, RAN, TARDBP, and TNPO1 (NS3/4A); WNK1 (NS4B); BIN1, EGLN1, and SMYD3 (NS5A); and FKBP5 (NS5B). The two cofactors RAN and COPB1 were previously identified in a genomic screen of HCV replication by Tai et al. (7). In addition, PTCD1, identified as an enriched Core partner but not tested in our RNAi screen, was previously reported as a cofactor for HCV replication by Li et al. (21). Therefore, combining interactomic screens with genetic screens enhances the rate of functional validation, providing lists of cellular proteins enriched in modulators of viral replication (exact Fisher test, p value < 0.01). Overall, the RNAi screening and validation data provide associations between host proteins pulled down with viral baits and functional contributions to the viral life cycle.
Interaction Network and Analysis
In order to visualize the interactome on a broader level, we produced an interaction network of the 98 statistically enriched host proteins interacting with HCV (Fig. 7A). In agreement with observations from previous virus–host interactome studies, HCV proteins tended to interact with highly central proteins in the human interactome (16, 50). Indeed, the degree distribution of targeted human proteins was significantly higher than the degree distribution in the human interactome (U test, p value < 2.2 × 10−16) (Fig. 7B). Similarly, the betweenness distribution of targeted human proteins was significantly higher than the betweenness distribution in the human interactome (U test, p value < 7.8 × 10−16) (Fig. 7C). This suggests that HCV proteins preferentially target pleiotropic cellular proteins. Finally, HCV interactors were significantly interconnected as assessed by random simulation (Fig. 7D), indicating that they are involved in similar biological processes. Recomputation of these metrics including literature data showed the same centrality features, with a high degree distribution, a high betweenness distribution, and high interconnectivity (supplemental Fig. S3). Overall, our data are consistent with the complete picture of the HCV–host protein interactome.
Fig. 7.

Interaction network of host interactors with viral proteins and degree, betweenness, and interconnectivity of 98 interactors. A, HCV–human protein–protein interaction network from this work. Viral proteins were manually connected to their hit host proteins and host proteins were connected together automatically using Cytoscape. Red nodes: HCV protein. Blue nodes: human protein. Red lines: HCV–human protein–protein interactions identified in this work. Blue lines: human–human protein–protein interactions. Arrowhead nodes: cofactors identified in this work. Nodes are disposed according to a force-directed layout. B, degree distributions of human proteins and human proteins targeted by HCV proteins in the human interactome. P(k) is the probability that a node will connect to k other nodes in the network. Solid lines represent linear regression fits. Vertical dashed lines indicate the mean degree of each distribution. C, betweenness distributions of human proteins and human proteins targeted by HCV proteins in the human interactome. P(b) is the probability that a node will have a betweenness value of b in the network. Solid lines represent linear regression fits. Vertical dashed lines indicate the mean betweenness value for each distribution. D, statistical analysis of the overlap size. The 10,000 overlap sizes obtained via random simulation are plotted. The observed value of x is indicated by the vertical arrow, and its significance is given.
NS3/4A Protein Blocks STAT1 Nuclear Accumulation
Given that a majority of the statistically enriched host proteins selectively interacting with HCV proteins neither affect HCV replication as host cofactors nor enhance replication as restriction factors upon gene silencing, we postulate that some enriched host proteins are targeted by viral proteins to subvert the antiviral innate immune responses. NS3/4A showed a significant tendency to interact with transport proteins such as karyopherins (KPNB1, TNPO1, XPO1) and with the GTP-binding/RAN GTPase-activating protein complex involved in nucleocytoplasmic transport (RAN, RANBP1, XPO5). As one of the essential cargoes of karyopherins is STAT1 (51), we investigated both the response to type I IFN and STAT1 subcellular localization in the presence of NS3/4A (Fig. 8). First, NS2, NS3, NS3/4A, and NS3/4A inactive protease mutant (NS3/4A S139A) were expressed in HeLa cells containing an ISRE-driven reporter Fluc (ISRE-Fluc) gene. Cell-expressing HCV proteins were then treated with type I IFN for 6 h, and cells extracts were prepared to determine the luciferase activities (ratio of Fluc to Rluc) as a readout of antiviral response. The NS3/4A showed a reduced response to type I IFN, whereas NS3 and inactive NS3/4A S139A had no significant effect, indicating a correlation between the functional inhibition of ISRE-dependent activities and expression of the protease-dependent mature NS3/4A protein with proper subcellular localization (Fig. 8A).
Fig. 8.

NS3/4A protein disrupts the type I IFN response. A, an ISRE luciferase reporter assay was performed to examine the effect of viral proteins on the response to type I IFN. HeLa cells were transfected with plasmids encoding viral proteins (NS2, NS3, NS3/4A, or serine protease inactive NS3/4A S139A mutant) and reporter plasmids (pISRE-Fluc and pRL-SV40 Rluc, Stratagene). 48 h post-transfection, cells were treated with 800 IU/ml IFN-β for 6 h, and then luciferase activity was determined using Dual-Glo Luciferase (Promega). Data are expressed as the percentage of luciferase activity relative to the signal obtained with HCV NS2. B, most representative subcellular localizations of STAT1, NS3, and NS3/4A. The effect of HCV NS3/4A on STAT1 subcellular localization was examined via immunofluorescence assay. HeLa cells were co-transfected with plasmids encoding 3xFLAG-NS3 or -NS3/4A and myc-STAT1. 48 h post-transfection, cells were treated with 1,000 IU/ml IFN-β for 45 min. Cells were fixed, permeabilized, stained with anti-FLAG (red) or anti-myc (green) antibody, and examined with a laser-scanning confocal microscope. C, relative localization of STAT1 in nucleus, cytoplasm, or both was analyzed in 100 cells expressing NS3 or NS3/4A. D, effect of NS3 protease activity on STAT1 phosphorylation. HeLa cells were transfected with plasmids encoding indicated viral proteins with or without myc-STAT1 plasmid. 48 h post-transfection, cells were either untreated or treated with 1,000 IU/ml IFN-β for 30 min and lysed. STAT1 tyrosine phosphorylation was assessed by immunoblotting of total lysates with anti-pY701 STAT1 and anti-STAT1 antibodies. Viral protein expression was detected with anti-FLAG antibody.
To investigate whether NS3/4A protein affects the nuclear transport of STAT1, HeLa cells transfected with plasmids encoding myc-STAT1 and Flag-tagged NS3 or NS3/4A and treated with type I IFN were analyzed for the subcellular localization of STAT1 via immunofluorescence microscopy (Figs. 8B and 8C). In about 50% of cells expressing NS3 or NS3/4A, STAT1 could be detected in both cytoplasmic and nuclear compartments. In 47% of cells expressing NS3, STAT1 displayed strict nuclear localization, whereas a minority of cells showed a strict cytoplasmic pattern (5%). This distribution was drastically modified in cells expressing NS3/4A, in which STAT1 nuclear localization was reduced (from 47% in NS3 expressing cells to 23% in NS3/4A expressing cells) in favor of cytoplasmic localization (from 5% in NS3 expressing cells to 27% in NS3/4A expressing cells). Fig. 8B shows the most representative subcellular localization of STAT1, NS3, and NS3/4A; STAT1 was prevented from being transported to the nucleus only when NS3/4A was present. The overall data suggest that the nuclear translocation of the karyopherin cargo STAT1 is affected by the NS3/4A protein. To verify that the impaired nuclear accumulation of STAT1 in the presence of NS3/4A does not result from a defect in upstream STAT1 activation, we examined the STAT1 tyrosine phosphorylation status. Cells overexpressing myc-STAT1 and the different viral proteins were treated with human type I IFN before analysis of the tyrosine 701 phosphorylation level of STAT1 via immunoblotting (Fig. 8D). In the absence of IFN treatment, no phosphorylation of endogenous or transfected STAT1 could be detected. Upon IFN treatment, both endogenous and transfected STAT1 were equally phosphorylated independently of the presence of viral proteins. The data indicate that NS3/4A prevents the nuclear translocation of STAT1 without interfering with the early steps of the Tyr701-phophorylated STAT1 activation process.
HCV core protein was previously reported to block STAT1 tyrosine phosphorylation and inhibit IFN signaling (52). To validate the phenotype of STAT1 localization seen in NS3/4A-expressing HeLa cells in the context of HCV infection, Huh7.5 cells were transfected with the JFH-1 mutant Δcore 153–167, which encodes a defective core protein, and the wild-type JFH1 Q221L variant, which produces 20 times more viral particles than JFH-1. Fig. 9 shows that when both Δcore mutant-transfected cells and JFH-1-infected cells were treated with IFN-γ, STAT1 did not accumulate in the nucleus of infected cells. Similar phenotypes were obtained upon the treatment of cells with IFN-α (supplemental Fig. S4). This suggests that NS3/4A independently affects the localization of STAT1 and does not require the cooperation of Core.
Fig. 9.
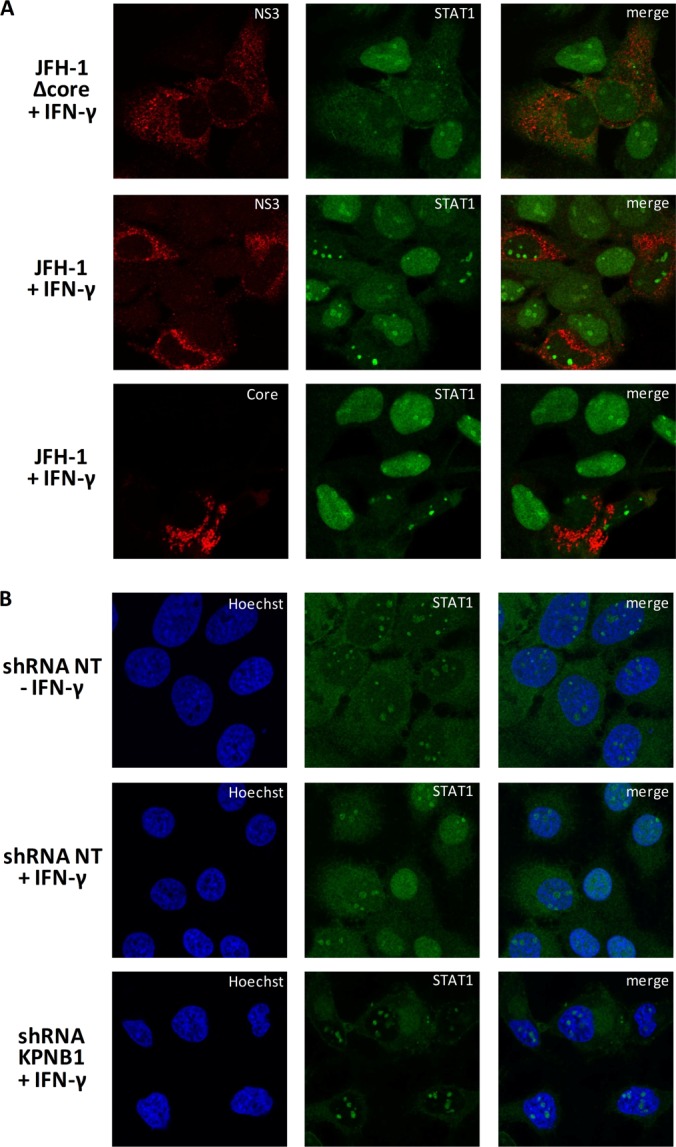
NS3/4A and its interactor KPNB1 prevent STAT1 nuclear localization. A, Huh7.5 cells were transfected with JFH-1 mutant Δcore 153–167 or NS3+ Q221L. Six days post-transfection, cells were serum starved for 4 h and then stimulated with 5 ng/ml of IFN-γ for 30 min. Cells were fixed, permeabilized, and stained with anti-NS3 or anti-core (red) and anti-STAT1 (green) and then examined via laser-scanning microscopy. B, Huh7 cells were infected with lentiviruses encoding shRNA NT or KPNB1 for three days (MOI = 10). Three days post-infection, cells were serum starved for 4 h and then stimulated with 5 ng/ml of IFN-γ for 30 min. Cells were fixed, permeabilized, and stained with anti-STAT1 (green). Nuclei were stained with Hoechst. Cells were examined via laser-scanning confocal microscopy.
KPNA1 (also known as importin subunit α-5) is the adaptor protein that binds to STAT1 (51), and the carrier to import this particular adaptor and cargo is KPNB1. To elucidate a possible mechanism connecting NS3/4A with its interactors, we performed the knockdown of one of its interactors, KPNB1. We observed a phenotype (Fig. 9B) similar to the one seen in HCV infection, in which STAT1 is unable to accumulate to the nucleus. Therefore, the silencing of KPNB1 phenocopied HCV infection in preventing STAT1 from accumulating in the nucleus upon IFN-γ or IFN-α stimulation. Altogether, the data provide evidence that the interaction between NS3/4A and KPNB1 mediates the blocking of STAT1 nuclear accumulation and highlights a viral subversion strategy for evading the antiviral immune response mediated by type I IFN.
DISCUSSION
To uncover novel HCV–host PPIs and their role in the vRNA replication process, we used an IP-MS/MS proteomic approach and functional RNAi screening of genes encoding newly identified host proteins in HCV replicon-containing cells and J6/JFH-1-infected cells. The IP-MS/MS approach identified 98 distinct human proteins as putative interactors of HCV proteins, including 24 previously reported, which were identified using very stringent selection criteria. An important concern with proteomic studies is the purity of the starting material for LC-MS/MS and the specificity of host interactions either directly or indirectly with binding partners. This issue was addressed in part by the reproducible detection of interactors in three independent experiments and, more important, by parallel purification for the six viral baits in each experiment. This experimental procedure allowed the identification of high-ranking enriched HCV interactors from the global analysis of LC-MS/MS data that correspond to a specific set of cellular proteins interacting predominantly with one virus-specific protein. Indeed, the overlap between this set of statistically enriched HCV interactors and those from previous large-scale yeast two-hybrid or proteomics studies reported in the literature is significant, with 24.5% of host interactors shared overall (24/98), demonstrating the reliability, robustness, and value of our approach. It also indicates that the interactome dataset is now close to completion. In fact, there is significant overlap between our data and data from a previously reported MS/MS analysis of Core and NS5A by Pichlmair et al. (32). Three hits with Core (DDX3X, NAP1L1, and NAP1L4) and eight hits with NS5A (USP19, HSPA5, SMYD3, BIN1, VAPA, MOBKL1A, VAPB, and FKBP8) were recovered in both studies. In addition, many HCV partners were also found to be enriched in detergent-resistant proteomics studies (53) (HSPD1, creatine kinase B-type, and HSP90AB1), involved in lipid metabolism with localization to vesicles containing the HCV replication complex (54) (estradiol 17-β-dehydrogenase family member HSD17B11), or as hit proteins retrieved in genome-wide RNAi studies (7, 18, 21) (DDX3X, PTCD1, calnexin, COPB1, RAN, VAPA, and VAPB; see supplemental Table S5). This important overlapping of identified host binding partners from our work and previous studies reinforces the hits' representation of true HCV-associated host proteins with a role in the HCV life cycle. Additionally, the data identify common host proteins that likely play a role in the life cycle of divergent viruses. When we queried the set of 98 HCV partners for cellular proteins that were previously identified as binding partners of others viruses, remarkably, we identified a total of 624 distinct virus–host interactions involving 52 different virus species (supplemental Table S6). Indeed, 80 host proteins out of the 98 interacting with HCV (82%) also correspond to known targets of at least one other virus. This emphasizes previous observations that a restricted number of host factors are critical to the virus life cycle and the targets of multiple viruses often with a different role.
To more directly address the biological significance of host proteins presumably associated with HCV proteins and increase our confidence that the hits represented true HCV-associated host proteins, we employed an average of five shRNA-expressing lentiviruses per gene that ensured efficient targeting of the transcript of putative virus-associated proteins. In total, 37 genes significantly affected HCV replication upon gene silencing according to rigorous selection criteria and reproducible inhibition in the two cell models. Even more reassuring was the identification of 13 modulators of HCV replication among our statistically enriched HCV partners (Core: HSD17B12; NS3/4A: COPB1, EXO7, KPNB1, RAN, TARDBP, TNPO1, MTHFD1; NS4B: WNK1; NS5A: BIN1, EGLN1, SMYD3; NS5B: FKBP5), among which a role for 11 novel cofactors has never been reported in HCV replication. Two cofactors, RAN and COPB1, were previously identified as high-ranking hits in a genomic screen of HCV replication by Tai et al. (7). Those authors unraveled a role of the COPI coat complex in HCV replication that is involved in retrograde traffic from the cis-Golgi to the rough endoplasmic reticulum. In the study, six coatomer subunits demonstrated a substantial effect on vRNA replication upon gene silencing, namely, COPA, COPB1, COPB2, ARCN1, COPG, and COPZ1. Our proteomics data show an interaction between NS3/4A and COPB1 (8-fold enrichment) and, to a lesser extent, with COPA (1.7-fold enrichment). COPA was also identified in the IP-NS5A condition but did not meet our stringent selection threshold (data not shown). Indeed, COPB1 tested in our screen had a substantial effect on HCV replication in both model systems. It is interesting to note that many viral proteins interact with COPB1, including Nef HIV1 (55–57), NS1 RSV (58), V measle virus (59), NSs Bunyavirus La Crosse (32), BBRF2, BNLF2a EBV (60), ST-Ag (SV40) (60), and E5A HPV6b (60). Nevertheless, to our knowledge, this is the first indication of a physical interaction between an HCV protein and a subunit of the COPI protein complex. Additional studies are required in order to define the role of a COPI-containing protein complex in the HCV infection life cycle that relies on its involvement in vesicle trafficking. Similarly, the cofactor EXOC7 as an NS3/4A selective partner is a component of the exocyst protein complex that ensures the targeting of exocytotic vesicles to the plasma membrane via interaction with PIP2 (61). Interestingly, EXOC7 is selective to PI(4,5)P2 but can also bind PI(4)P. As a critical role of the host PI4KA was reported in HCV replication and for the local enrichment of PI(4)P at the HCV membranous web, one can speculate on the recruitment of such a viral–host protein complex at the site of replication. Future studies will characterize the hijacking of EXOC7 and its function in vesicle trafficking at the site of replication from Golgi to plasma membrane to regulate HCV secretion.
It has long been known that TARDBP (TDP-43) modulates HIV expression (62). TARDBP is an hnRNP protein that can bind both DNA and RNA and is involved in RNA processing. Previous studies conducted in our lab (data not shown) demonstrated that TARDBP binds YB-1, which is a host factor that is co-opted by assembling capsids to regulate the equilibrium between vRNA replication and HCV production (38, 63). Here, knockdown of TARDBP was associated with a significant decrease in replication of the infectious HCV genome. Further investigations will be required in order to determine the existence and dynamics of a protein complex involving YB-1, TARDBP, and NS3/4A along the viral replication and assembly steps.
Immunophilins, like cyclophilins, possess prolyl isomerase activity and are involved in protein folding. Previous reports revealed the importance of this family of proteins in HCV replication and cell survival with the involvement of FKBP8 (44, 45). This interaction was also retrieved in our IP-NS5A condition. Moreover, the FKBP8 interacting protein EGLN1 (propylyl-4-hydroxylase domain-containing 2) (47), an enzyme involved in the cellular response to hypoxia, was identified as an NS5A partner and validated as a cofactor for HCV RNA replication. Additionally, FKBP5 was retrieved in the IP-MS/MS analysis and confirmed as an NS5B partner and HCV cofactor. Likewise, HSP90AA1 and HSP90AB1 strongly interacted with NS5B, as reflected by the fold enrichment in our IP-MS/MS analysis. However, their individual silencing did not affect HCV replication, in contrast to previous reports describing HSP90 as part of a complex with NS5A and FKBP8 and with an important role in HCV RNA replication (44, 64). It is possible that this discrepancy is due to some redundancy in virus–host PPIs or insufficient protein silencing. Taking all these data together, it is tempting to speculate that these immunophilins in complex with HSP provide a link between NS5A and NS5B within the HCV RNA replication machinery. Nevertheless, these data add strong evidence that members of this protein family are highly important for HCV replication, as reported for other viruses (see the review in Ref. 65).
Interestingly, a role of the very stringent NS4B partner and cofactor WNK1 identified in our study was demonstrated in vaccinia virus replication, but with increased growth of vaccinia virus upon WNK1 silencing, in contrast to the inhibition of HCV replication (32). A novel role of members of the WNK kinase family in the antiviral innate response was also reported. It is thus interesting to see that viruses have evolved in different ways while exploiting the same cellular protein. Noteworthy, the poor number of interacting proteins retrieved in the NS4B condition is consistent with what can be found in the literature (16).
WNK1 host protein is an example of an increasing number of targeted host proteins that regulate, either positively or negatively, multiple replicative virus life cycles through interaction with viral proteins and simultaneously affect immunologically antiviral responses. Indeed, only a fraction of the HCV partners identified via IP-MS/MS in this study scored as hits in the RNAi screen, and one possible explanation is that host proteins targeted by virus are more critical to the antiviral innate immune response than essential cofactors to vRNA replication. This delicate balance between the role of the host protein in innate immunity and vRNA replication is often difficult to demonstrate experimentally, and clearly depends on the immune competence of the cell types used in the study. Another example is NS3/4A, which strongly interacts with members of nuclear protein transport. These proteins (XPO1, IPO7, KPNB1, RAN, TNPO1, and XPO5) are also highly targeted by many viruses and in different functions (supplemental Table S6). Noteworthy, three of these proteins (KPNB1, XPO1, and RAN) have a potential NS3/4A consensus cleavage motif (D/E)X4(C/T)↓(S/A) (66, 67), although we could not demonstrate any cleavage of these proteins in cells expressing NS3/4A or in the context of the replicon (data not shown). As all three are major actors in nucleocytoplasmic transport, by interacting with these proteins NS3/4A might modify the subcellular localization of cargoes. Indeed, IFN-induced nuclear import of STAT1 is mediated by members of the importin NPI-1 (51), and the nuclear export of STAT1 is regulated by Ran/XPO1 (68). In this study, we demonstrated that HCV infection, expression of an HCV Δcore mutant, or expression of HCV NS3/4A partially blocks the response to IFN and STAT1 nuclear translocation, in contrast to expression of the NS3/4A precursor with an inactive protease (S139A). In addition, knockdown of the carrier KPNB1 also prevented STAT1 nuclear accumulation in the nucleus, suggesting that this interaction between NS3/4A and KPNB1 interferes with the nuclear translocation of STAT1. Finally, the silencing of two out of the three nucleocytoplasmic transport genes (KPNB1 and RAN) predominantly inhibited HCV replication in Huh7-Con1 or Huh7.5 cells, which are characterized by an abrogated virus-induced IFN response. For this reason, we were not expecting these gene products to act as restriction factors for HCV replication in these cells. Nevertheless, it is intriguing to conceive of specific virus–host PPIs that contribute to the viral replication and interfere with the antiviral innate response, possibly by taking place at different stages of the virus life cycle in order to confer a growth advantage upon infection (similar to a role of hte NS4B partner WNK1 in viral replication and innate immunity). The ability of NS3/4A to interact with several proteins with a HEAT domain also illustrates the need shared by viral proteins from viruses with small proteomes to develop strategies to hit multiple cellular targets (50). Although the HEAT recognition by NS3/4A is not directly established, this interactome suggests that this viral protein has evolved molecular structures to interact with this domain and thus with the diversity of proteins having this domain.
In conclusion, a set of 98 statistically enriched HCV partners was identified, including 74 novel and 24 previously described partners, as a result of an IP-MS/MS approach and global data analysis for the Core, NS2, NS3/4A, NS4B, NS5A, and NS5B proteins. Eleven novel virus–host PPIs were further confirmed by IP-Western blotting. The investigation of the HCV-associated proteins' roles in the viral life cycle through gene silencing emphasized 37 HCV cofactors in total, of which 11 among the enriched set of HCV partners were novel modulators of HCV replication. This study contributes to the HCV interactome through the robust identification of HCV partners and host factors that may enhance the selection of novel host therapeutic targets with fewer side effects, along with the knowledge of the human interactome.
Supplementary Material
Acknowledgments
We thank Takaji Wakita (National Institute of Infectious Diseases, Tokyo), Charles Rice (Rockefeller University), and Apath LCC for the use of JFH-1, Huh7.5 cells, and J6-JFH(p7-Rluc2A) reporter virus; Ralf Bartenschlager (University of Heidelberg) for luciferase-encoding Con1b subgenomic replicon; Didier Trono (École Polytechnique Fédérale de Lausanne) for the lentiviral packaging plasmids; Amélie Cessieux (Inserm U1111) for her technical assistance in bioinformatics; Christine Vande Velde for providing the anti-TARDBP antibody; and Marie-Eve Racine for cloning the Myc-tagged HCV protein constructs. We also acknowledge IRIC's High Throughput Screening Core Facility; Jean Duchaine, Julie Lafontaine, and Karine Audette for lentivirus production and RNAi screening; IRIC's Genomics Core Facility; and Raphaëlle Lambert, Caroline Paradis, and Marianne Arteau for sequencing. P.T. holds the Canada Research Chair in Proteomics and Bioanalytical Spectrometry.
Footnotes
Author contribution: M.-A.G., L.C.-C., P.T., V.L., and D.L. were involved in the design of experiments and analysis of data; M.-A.G. performed experiments related to Figs. 1–3 and 5–7, supplemental Figs. S1 and S2, Table I, and supplemental Tables S2–S5; B.G. performed experiments related to Fig. 9 and supplemental Fig. S4; E.B. performed MS experiments and analysis related to supplemental Tables S1 and S2; F.P., B.d.C., and V.L. designed and performed experiments related to Fig. 8; L.M., B.d.C., and V.L were involved in bioinformatics analysis related to Figs. 2 and 7, supplemental Fig. S3, Table I, and supplemental Table S6; M.B. performed experiments related to Fig. 4; M.-A.G. and D.L. wrote the manuscript.
* This work was supported by an operating grant from the Canadian Institutes of Health Research (MOP-115058) awarded to D.L. and L.C.C. and by the Novartis/Canadian Liver Foundation Hepatology Research Chair awarded to D.L. L.M., B.d.C., F.P., and V.L. are supported by ANR, ANRS, Inserm, and the European Union's 7th program (FP7/2007–2013) under Grant Agreement No. 267429 (SysPatho). IRIC is supported in part by the Canadian Center of Excellence in Commercialization and Research, the Canada Foundation for Innovation (CFI), and the Fonds de Recherche du Québec en Santé (FRQS).
 This article contains supplemental material.
This article contains supplemental material.
Conflict of interest: D.L. is a shareholder in ViroCura Therapeutics Inc.
1 The abbreviations used are:
- HCV
- hepatitis C virus
- IFN
- interferon
- ILF2
- interleukin enhancer-binding factor 2
- IP
- immunoprecipitation
- IP-MS/MS
- immunoprecipitation coupled to tandem mass spectrometry identification
- ISRE
- interferon-stimulated response element
- EGLN1
- egl nine homolog 1
- EXOC7
- exocyst complex component 7
- FKBP
- FK506-binding protein
- Fluc
- firefly luciferase
- HSP
- heat shock protein
- Rluc
- Renilla luciferase
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MOI
- multiplicity of infection
- MS/MS
- tandem mass spectrometry
- MTHFD1
- methylenetetrahydrofolate dehydrogenase 1
- NS
- nonstructural
- NT
- nontarget sequence
- PPI
- protein–protein interaction
- RNAi
- RNA interference
- shRNA
- short hairpin RNA
- STAT1
- signal transducer and activator of transcription 1
- TARDBP
- TAR DNA binding protein
- TNPO1
- transportin 1 isoform 1
- vRNA
- viral RNA
- XPO
- exportin.
REFERENCES
- 1. Poynard T., Yuen M. F., Ratziu V., Lai C. L. (2003) Viral hepatitis C. Lancet 362, 2095–2100 [DOI] [PubMed] [Google Scholar]
- 2. Welsch C., Jesudian A., Zeuzem S., Jacobson I. (2012) New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut 61 Suppl 1, i36–i46 [DOI] [PubMed] [Google Scholar]
- 3. Blanchard E., Belouzard S., Goueslain L., Wakita T., Dubuisson J., Wychowski C., Rouille Y. (2006) Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80, 6964–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsukiyama-Kohara K., Iizuka N., Kohara M., Nomoto A. (1992) Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66, 1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Romero-Brey I., Merz A., Chiramel A., Lee J. Y., Chlanda P., Haselman U., Santarella-Mellwig R., Habermann A., Hoppe S., Kallis S., Walther P., Antony C., Krijnse-Locker J., Bartenschlager R. (2012) Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8, e1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moradpour D., Penin F., Rice C. M. (2007) Replication of hepatitis C virus. Nat. Rev. Microbiol. 5, 453–463 [DOI] [PubMed] [Google Scholar]
- 7. Tai A. W., Benita Y., Peng L. F., Kim S. S., Sakamoto N., Xavier R. J., Chung R. T. (2009) A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5, 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vaillancourt F. H., Pilote L., Cartier M., Lippens J., Liuzzi M., Bethell R. C., Cordingley M. G., Kukolj G. (2009) Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387, 5–10 [DOI] [PubMed] [Google Scholar]
- 9. Borawski J., Troke P., Puyang X., Gibaja V., Zhao S., Mickanin C., Leighton-Davies J., Wilson C. J., Myer V., Cornellataracido I., Baryza J., Tallarico J., Joberty G., Bantscheff M., Schirle M., Bouwmeester T., Mathy J. E., Lin K., Compton T., Labow M., Wiedmann B., Gaither L. A. (2009) Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83, 10058–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berger K. L., Kelly S. M., Jordan T. X., Tartell M. A., Randall G. (2011) Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85, 8870–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bianco A., Reghellin V., Donnici L., Fenu S., Alvarez R., Baruffa C., Peri F., Pagani M., Abrignani S., Neddermann P., De Francesco R. (2012) Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P Is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 8, e1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang F., Robotham J. M., Nelson H. B., Irsigler A., Kenworthy R., Tang H. (2008) Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 82, 5269–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coelmont L., Hanoulle X., Chatterji U., Berger C., Snoeck J., Bobardt M., Lim P., Vliegen I., Paeshuyse J., Vuagniaux G., Vandamme A. M., Bartenschlager R., Gallay P., Lippens G., Neyts J. (2010) DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5, e13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Flisiak R., Feinman S. V., Jablkowski M., Horban A., Kryczka W., Pawlowska M., Heathcote J. E., Mazzella G., Vandelli C., Nicolas-Metral V., Grosgurin P., Liz J. S., Scalfaro P., Porchet H., Crabbe R. (2009) The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 49, 1460–1468 [DOI] [PubMed] [Google Scholar]
- 15. Liang Y., Ishida H., Lenz O., Lin T. I., Nyanguile O., Simmen K., Pyles R. B., Bourne N., Yi M., Li K., Lemon S. M. (2008) Antiviral suppression vs restoration of RIG-I signaling by hepatitis C protease and polymerase inhibitors. Gastroenterology 135, 1710–1718, e1712 [DOI] [PubMed] [Google Scholar]
- 16. de Chassey B., Navratil V., Tafforeau L., Hiet M. S., Aublin-Gex A., Agaugue S., Meiffren G., Pradezynski F., Faria B. F., Chantier T., Le Breton M., Pellet J., Davoust N., Mangeot P. E., Chaboud A., Penin F., Jacob Y., Vidalain P. O., Vidal M., Andre P., Rabourdin-Combe C., Lotteau V. (2008) Hepatitis C virus infection protein network. Mol. Syst. Biol. 4, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tripathi L. P., Kataoka C., Taguwa S., Moriishi K., Mori Y., Matsuura Y., Mizuguchi K. (2010) Network based analysis of hepatitis C virus core and NS4B protein interactions. Mol. Biosyst. 6, 2539–2553 [DOI] [PubMed] [Google Scholar]
- 18. Randall G., Panis M., Cooper J. D., Tellinghuisen T. L., Sukhodolets K. E., Pfeffer S., Landthaler M., Landgraf P., Kan S., Lindenbach B. D., Chien M., Weir D. B., Russo J. J., Ju J., Brownstein M. J., Sheridan R., Sander C., Zavolan M., Tuschl T., Rice C. M. (2007) Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 104, 12884–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ng T. I., Mo H., Pilot-Matias T., He Y., Koev G., Krishnan P., Mondal R., Pithawalla R., He W., Dekhtyar T., Packer J., Schurdak M., Molla A. (2007) Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology 45, 1413–1421 [DOI] [PubMed] [Google Scholar]
- 20. Supekova L., Supek F., Lee J., Chen S., Gray N., Pezacki J. P., Schlapbach A., Schultz P. G. (2008) Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J. Biol. Chem. 283, 29–36 [DOI] [PubMed] [Google Scholar]
- 21. Li Q., Brass A. L., Ng A., Hu Z., Xavier R. J., Liang T. J., Elledge S. J. (2009) A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U.S.A. 106, 16410–16415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trost M., English L., Lemieux S., Courcelles M., Desjardins M., Thibault P. (2009) The phagosomal proteome in interferon-gamma-activated macrophages. Immunity 30, 143–154 [DOI] [PubMed] [Google Scholar]
- 23. Trost M., Sauvageau M., Herault O., Deleris P., Pomies C., Chagraoui J., Mayotte N., Meloche S., Sauvageau G., Thibault P. (2012) Posttranslational regulation of self-renewal capacity: insights from proteome and phosphoproteome analyses of stem cell leukemia. Blood 120, e17–e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kato T., Date T., Murayama A., Morikawa K., Akazawa D., Wakita T. (2006) Cell culture and infection system for hepatitis C virus. Nat. Protoc. 1, 2334–2339 [DOI] [PubMed] [Google Scholar]
- 25. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 26. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 27. R Development Core Team (2008) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 28. Razick S., Magklaras G., Donaldson I. M. (2008) iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinformatics 9, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Csardi G., Nepusz T. (2006) The igraph software package for complex network research. InterJournal Complex Systems 1695, 38 [Google Scholar]
- 31. Bartenschlager R., Lohmann V., Penin F. (2013) The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 11, 482–496 [DOI] [PubMed] [Google Scholar]
- 32. Pichlmair A., Kandasamy K., Alvisi G., Mulhern O., Sacco R., Habjan M., Binder M., Stefanovic A., Eberle C. A., Goncalves A., Burckstummer T., Muller A. C., Fauster A., Holze C., Lindsten K., Goodbourn S., Kochs G., Weber F., Bartenschlager R., Bowie A. G., Bennett K. L., Colinge J., Superti-Furga G. (2012) Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487, 486–490 [DOI] [PubMed] [Google Scholar]
- 33. Lee J. W., Liao P. C., Young K. C., Chang C. L., Chen S. S., Chang T. T., Lai M. D., Wang S. W. (2011) Identification of hnRNPH1, NF45, and C14orf166 as novel host interacting partners of the mature hepatitis C virus core protein. J. Proteome Res. 10, 4522–4534 [DOI] [PubMed] [Google Scholar]
- 34. Kittlesen D. J., Chianese-Bullock K. A., Yao Z. Q., Braciale T. J., Hahn Y. S. (2000) Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J. Clin. Invest. 106, 1239–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang S. M., Shin M. J., Kim J. H., Oh J. W. (2005) Proteomic profiling of cellular proteins interacting with the hepatitis C virus core protein. Proteomics 5, 2227–2237 [DOI] [PubMed] [Google Scholar]
- 36. Kurokawa Y., Honma K., Takemasa I., Nakamori S., Kita-Matsuo H., Motoori M., Nagano H., Dono K., Ochiya T., Monden M., Kato K. (2006) Central genetic alterations common to all HCV-positive, HBV-positive and non-B, non-C hepatocellular carcinoma: a new approach to identify novel tumor markers. Int. J. Oncol. 28, 383–391 [PubMed] [Google Scholar]
- 37. Lai C. K., Jeng K. S., Machida K., Lai M. M. (2008) Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A. J. Virol. 82, 8838–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chatel-Chaix L., Melancon P., Racine M. E., Baril M., Lamarre D. (2011) Y-box-binding protein 1 interacts with hepatitis C virus NS3/4A and influences the equilibrium between viral RNA replication and infectious particle production. J. Virol. 85, 11022–11037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tu H., Gao L., Shi S. T., Taylor D. R., Yang T., Mircheff A. K., Wen Y., Gorbalenya A. E., Hwang S. B., Lai M. M. (1999) Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 263, 30–41 [DOI] [PubMed] [Google Scholar]
- 40. Hamamoto I., Nishimura Y., Okamoto T., Aizaki H., Liu M., Mori Y., Abe T., Suzuki T., Lai M. M., Miyamura T., Moriishi K., Matsuura Y. (2005) Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 79, 13473–13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zech B., Kurtenbach A., Krieger N., Strand D., Blencke S., Morbitzer M., Salassidis K., Cotten M., Wissing J., Obert S., Bartenschlager R., Herget T., Daub H. (2003) Identification and characterization of amphiphysin II as a novel cellular interaction partner of the hepatitis C virus NS5A protein. J. Gen. Virol. 84, 555–560 [DOI] [PubMed] [Google Scholar]
- 42. Nanda S. K., Herion D., Liang T. J. (2006) The SH3 binding motif of HCV [corrected] NS5A protein interacts with Bin1 and is important for apoptosis and infectivity. Gastroenterology 130, 794–809 [DOI] [PubMed] [Google Scholar]
- 43. Taguwa S., Okamoto T., Abe T., Mori Y., Suzuki T., Moriishi K., Matsuura Y. (2008) Human butyrate-induced transcript 1 interacts with hepatitis C virus NS5A and regulates viral replication. J. Virol. 82, 2631–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okamoto T., Nishimura Y., Ichimura T., Suzuki K., Miyamura T., Suzuki T., Moriishi K., Matsuura Y. (2006) Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 25, 5015–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J., Tong W., Zhang X., Chen L., Yi Z., Pan T., Hu Y., Xiang L., Yuan Z. (2006) Hepatitis C virus non-structural protein NS5A interacts with FKBP38 and inhibits apoptosis in Huh7 hepatoma cells. FEBS Lett. 580, 4392–4400 [DOI] [PubMed] [Google Scholar]
- 46. Owsianka A. M., Patel A. H. (1999) Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 257, 330–340 [DOI] [PubMed] [Google Scholar]
- 47. Barth S., Nesper J., Hasgall P. A., Wirthner R., Nytko K. J., Edlich F., Katschinski D. M., Stiehl D. P., Wenger R. H., Camenisch G. (2007) The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol. Cell. Biol. 27, 3758–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lamarre D., Anderson P. C., Bailey M., Beaulieu P., Bolger G., Bonneau P., Bos M., Cameron D. R., Cartier M., Cordingley M. G., Faucher A. M., Goudreau N., Kawai S. H., Kukolj G., Lagace L., LaPlante S. R., Narjes H., Poupart M. A., Rancourt J., Sentjens R. E., St George R., Simoneau B., Steinmann G., Thibeault D., Tsantrizos Y. S., Weldon S. M., Yong C. L., Llinas-Brunet M. (2003) An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426, 186–189 [DOI] [PubMed] [Google Scholar]
- 49. Chatel-Chaix L., Germain M. A., Motorina A., Bonneil E., Thibault P., Baril M., Lamarre D. (2013) A host Yb-1 ribonucleoprotein complex is hijacked by hepatitis C virus for the control of Ns3-dependent particle production. J. Virol. 87, 11704–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meyniel-Schicklin L., de Chassey B., Andre P., Lotteau V. (2012) Viruses and interactomes in translation. Mol. Cell. Proteomics 11, M111.014738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sekimoto T., Imamoto N., Nakajima K., Hirano T., Yoneda Y. (1997) Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 16, 7067–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bode J. G., Ludwig S., Ehrhardt C., Albrecht U., Erhardt A., Schaper F., Heinrich P. C., Haussinger D. (2003) IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 17, 488–490 [DOI] [PubMed] [Google Scholar]
- 53. Hara H., Aizaki H., Matsuda M., Shinkai-Ouchi F., Inoue Y., Murakami K., Shoji I., Kawakami H., Matsuura Y., Lai M. M., Miyamura T., Wakita T., Suzuki T. (2009) Involvement of creatine kinase B in hepatitis C virus genome replication through interaction with the viral NS4A protein. J. Virol. 83, 5137–5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang H., Sun F., Owen D. M., Li W., Chen Y., Gale M., Jr., Ye J. (2007) Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 104, 5848–5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Benichou S., Bomsel M., Bodeus M., Durand H., Doute M., Letourneur F., Camonis J., Benarous R. (1994) Physical interaction of the HIV-1 Nef protein with beta-COP, a component of non-clathrin-coated vesicles essential for membrane traffic. J. Biol. Chem. 269, 30073–30076 [PubMed] [Google Scholar]
- 56. Janvier K., Craig H., Le Gall S., Benarous R., Guatelli J., Schwartz O., Benichou S. (2001) Nef-induced CD4 downregulation: a diacidic sequence in human immunodeficiency virus type 1 Nef does not function as a protein sorting motif through direct binding to beta-COP. J. Virol. 75, 3971–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Piguet V., Gu F., Foti M., Demaurex N., Gruenberg J., Carpentier J. L., Trono D. (1999) Nef-induced CD4 degradation: a diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 97, 63–73 [DOI] [PubMed] [Google Scholar]
- 58. Wu W., Tran K. C., Teng M. N., Heesom K. J., Matthews D. A., Barr J. N., Hiscox J. A. (2012) The interactome of the human respiratory syncytial virus NS1 protein highlights multiple effects on host cell biology. J. Virol. 86, 7777–7789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Komarova A. V., Combredet C., Meyniel-Schicklin L., Chapelle M., Caignard G., Camadro J. M., Lotteau V., Vidalain P. O., Tangy F. (2011) Proteomic analysis of virus-host interactions in an infectious context using recombinant viruses. Mol. Cell. Proteomics 10, M110.007443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rozenblatt-Rosen O., Deo R. C., Padi M., Adelmant G., Calderwood M. A., Rolland T., Grace M., Dricot A., Askenazi M., Tavares M., Pevzner S. J., Abderazzaq F., Byrdsong D., Carvunis A. R., Chen A. A., Cheng J., Correll M., Duarte M., Fan C., Feltkamp M. C., Ficarro S. B., Franchi R., Garg B. K., Gulbahce N., Hao T., Holthaus A. M., James R., Korkhin A., Litovchick L., Mar J. C., Pak T. R., Rabello S., Rubio R., Shen Y., Singh S., Spangle J. M., Tasan M., Wanamaker S., Webber J. T., Roecklein-Canfield J., Johannsen E., Barabasi A. L., Beroukhim R., Kieff E., Cusick M. E., Hill D. E., Munger K., Marto J. A., Quackenbush J., Roth F. P., DeCaprio J. A., Vidal M. (2012) Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487, 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu J., Zuo X., Yue P., Guo W. (2007) Phosphatidylinositol 4,5-bisphosphate mediates the targeting of the exocyst to the plasma membrane for exocytosis in mammalian cells. Mol. Biol. Cell 18, 4483–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ou S. H., Wu F., Harrich D., Garcia-Martinez L. F., Gaynor R. B. (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ling S. C., Albuquerque C. P., Han J. S., Lagier-Tourenne C., Tokunaga S., Zhou H., Cleveland D. W. (2010) ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U.S.A. 107, 13318–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gallo L. I., Lagadari M., Piwien-Pilipuk G., Galigniana M. D. (2011) The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress. J. Biol. Chem. 286, 30152–30160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma-Lauer Y., Lei J., Hilgenfeld R., von Brunn A. (2012) Virus-host interactomes—antiviral drug discovery. Curr. Opin. Virol. 2, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grakoui A., McCourt D. W., Wychowski C., Feinstone S. M., Rice C. M. (1993) Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 67, 2832–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang R., Durkin J., Windsor W. T., McNemar C., Ramanathan L., Le H. V. (1997) Probing the substrate specificity of hepatitis C virus NS3 serine protease by using synthetic peptides. J. Virol. 71, 6208–6213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Begitt A., Meyer T., van Rossum M., Vinkemeier U. (2000) Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc. Natl. Acad. Sci. U.S.A. 97, 10418–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.