

Abstract
Context:
Short stature is a common reason for referral to pediatric endocrinology centers. Frequently, the underlying etiology of short stature is unknown, resulting in a diagnosis of idiopathic short stature. Rare genetic defects in the GH/IGF-1 axis have been found to cause short stature.
Objective:
The objective of this study was to identify the genetic etiology of short stature in a patient with Idiopathic Short Stature and to review the clinical presentation of patients with genetic defects in IGF1, and specifically IGF-1 haploinsufficiency.
Design/Setting/Participants:
The index patient was evaluated at an academic medical center, and DNA was obtained from the proband and both parents.
Intervention:
Genome-wide copy number analysis was performed in the proband with confirmatory quantitative PCR in the proband and his parents.
Main Outcome Measure:
We measured novel copy number variants (CNVs) thought to explain the patient's short stature.
Results:
CNV analysis revealed that the proband carried a paternally inherited heterozygous IGF1 gene deletion. His phenotypic features are consistent with those found in previous case reports of IGF-1 deficiency.
Conclusions:
This study, as the first case of a complete heterozygous 1GF1 deletion, provides insight into the effects of true IGF-1 haploinsufficiency. Given the similarities in phenotype between the present proband and those previously described, it is highly likely that his IGF1 deletion is the cause for his short stature. Broadly, this study emphasizes how CNV analysis and other genetic sequencing techniques are evolving as an important tool to identify genetic causes underlying human disease, allowing for improved diagnosis and targeted treatment.
Short stature is a common reason for referral to pediatric endocrinology centers. Despite the frequency of these referrals, standard endocrine evaluation in the clinical setting does not often lead to a diagnosis, and thus, many children are labeled with idiopathic short stature (ISS). It is estimated that the underlying cause for short stature remains unknown in approximately 80% of patients (1). Numerous genes have been identified in which rare mutations cause short stature, including genes in the GH/IGF-1 pathway as well as genes involved in skeletal development (2). With the advent of new, more readily available genomic investigative techniques, including copy number variant (CNV) analysis and whole exome sequencing, it is now possible to identify likely causative genetic variants in several of these children who previously had unknown reasons for their poor linear growth (3). Two recent studies identified a higher frequency of rare CNVs in patients with short stature (1, 4). To that end, in this case report, we demonstrate how genome-wide CNV analysis revealed a complete heterozygous IGF1 deletion as the likely cause for short stature in our proband, who previously had been diagnosed with ISS.
Although many mutations causing disruptions in the GH/IGF-1 system have been implicated in growth failure, mutations in the IGF1 gene itself are relatively rare (2, 5). To our knowledge, this is the first report identifying a patient with a complete heterozygous deletion on the IGF1 gene allowing us to better characterize and understand the implications of IGF-1 haploinsufficiency on growth and development.
Subject and Methods
Molecular analysis
DNA samples were obtained from the proband and his parents. Written informed consent was obtained, and the study was approved by the institutional review board of Boston Children's Hospital.
CNV analysis
The DNA sample from the proband was analyzed using an Agilent SurePrint G3 custom comparative genomic hybridization and SNP Microarray 4x180k (Agilent Technologies), following the manufacturer's instructions. This microarray platform contains 150 000 oligonucleotide probes for the detection of CNVs and 30 000 probes for the detection of single-nucleotide polymorphisms. CNV probes were densely populated in genes of interest potentially related to short stature (detection sensitivity is less than 10 kb) as previously described (3, 6). For the remaining regions of the genome, CNV probe coverage provided detection sensitivity of about 400 kb.
Quantitative PCR (qPCR) analysis
qPCR was performed in the proband and his parents to verify the CNV analysis results and to assess for segregation in the family. All qPCR assays were performed on the iCycler iQ Multi-Color Real Time PCR Detection System (Bio-Rad) and iQ5 Software (Bio-Rad). The assays were scaled to 20-μL reactions using the Roche FastStart Universal SYBR Green Master Mix (catalog no. 04913850001; Roche Diagnostics). Each reaction contained either 50 or 100 ng of total genomic DNA, and the final concentration for the IGF-1 qPCR primers were at 150 ng/μL (forward, 5′-GCTCTTCAGTTCGTGTGTGGA-3′; reverse, 5′-GCCTCCTTAGATCACAGCTCC-3′). A two-step thermal cycler program was used in the following order: one cycle at 95°C for 5 minutes, 45 cycles of 95°C for 15 seconds, then 60°C for 1 minute, one cycle at 95°C for 1 minute, and the final step held at 55°C. Fluorescence was normalized with the reference dye (Rox) in the SYBR Green Master Mix.
Results
Case report
Labor was induced at 41 weeks gestational age, given concern for intrauterine growth restriction (IUGR). The patient's birth weight was 2.7 kg (SD score [SDS], −1.5), with a birth length of 47.6 cm (SDS, −1.2). At initial presentation to the endocrine clinic at age 2 years and 3 months, his weight was 8.8 kg (SDS, −3.8) and length was 77.5 cm (SDS, −3.1), indicating postnatal growth failure with further decrease in height SDS from birth (Table 1). Ancillary testing to evaluate for other causes for his poor growth was normal, including negative celiac testing, a negative sweat test, normal thyroid function testing, normal complete blood count, alanine aminotransferase, albumin, and erythrocyte sedimentation rate. The IGF-1 level was on the low end of the normal range with a normal IGF binding protein-3 (IGFBP-3) (Table 1).
Table 1.
Clinical, Laboratory, and Radiographic Data
| Age | Bone Age | Height, cm (SDS)a | Weight, kg (SDS)a | HC, cm (SDS)b | IGF-1, ng/mLc | IGFBP-3, mg/Ld | ALS, mg/Le |
|---|---|---|---|---|---|---|---|
| Birth | 47.6 (−1.2) | 2.7 (−1.5) | |||||
| 2.3 y | 26 months (normal) | 77.5 (−3.1) | 8.8 (−3.8) | 43.7 (low/normal) | 2.3 (normal) | ||
| 4.3 y | 4 y (normal) | 91.9 (−2.8) | 12.2 (−3.1) | ||||
| 5.3 y | 6 y (normal) | 96.9 (−2.9) | 14.4 (−2.5) | 46.5 (−3.3) | 58.5 (low/normal) | 4.3 (normal) | 10 (normal) |
| 8.4 y | 8 y (normal) | 114.9 (−2.7) | 21.9 (−1.5) | 47.8 (−3.4) | 100 (normal) | 5.4 (normal) |
Height and weight SDSs based on CDC growth curves.
Head circumference SDSs.
IGF-1 normal ranges per age in Tanner 1 males (ng/mL): age 1–2 years, 30–122; age 5 years, 30–174; and age 8 years, 52–231.
IGFBP-3 normal ranges per age in Tanner 1 males (mg/L): age 2 years, 0.8–3.9; age 5 years, 1.1–5.2; age 8 years, 1.6–6.5.
ALS, normal range for a male at 5.3 years, 2.3–11 mg/L.
At the age of 5 years and 3 months, he underwent repeat evaluation; his height was 96.9 cm (SDS, −2.9), and head circumference was 46.5 cm (SDS, −3.3) (Figure 1). Repeat IGF-1 was again low-normal, with normal IGFBP-3 (Table 1). GH binding protein and acid labile subunit (ALS) values were also in the normal range. GH stimulation testing was deferred at the family's request. SHOX gene mutation testing was performed, with no mutation identified.
Figure 1.

Growth chart of proband. Centers for Disease Control and Prevention (CDC) growth chart depicting age-adjusted percentiles (top, 95%; bottom, 5%). Black dots represent proband's height over a span of 6 years. Arrow depicts midparental height (MPH) target.
At his most recent visit at age 8 years and 5 months, height was 114.9 cm (SDS, −2.7), weight was 21.9 kg (SDS, −1.5), and head circumference was 47.8 cm (SDS, −3.4). On physical examination, he had two café-au-lait macules, mild micrognathia, mild epicanthus, and bilateral fifth finger clinodactyly. IGF-1 and IGFBP-3 were both in the normal range. His bone age was consistent with chronological age throughout his evaluations (Table 1).
Additional history includes long-standing developmental delay. He struggles primarily with speech and reading and was held back a grade in school. He currently has an individualized education plan in place for learning difficulties in school.
The family is primarily Irish in origin. Maternal height is 154.9 cm (SDS, −1.3), and paternal height is estimated to be 162 cm (SDS, −2.0). Paternal head circumference was reported to be small, and as an adult, the proband's father wears a youth-size baseball cap. The father was unavailable for examination but did provide a sample for genetic evaluation.
Mutational analysis
Chromosomal microarray revealed a heterozygous 262-kb deletion of chromosome 12:102613657–102875825 (hg19 reference genome), which includes the entire IGF1 gene (Figure 2). There are no other genes in the deleted region. qPCR confirmed the heterozygous deletion in the proband and his father. The deletion was not present in the mother or controls of normal height (Supplemental Figure 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org).
Figure 2.
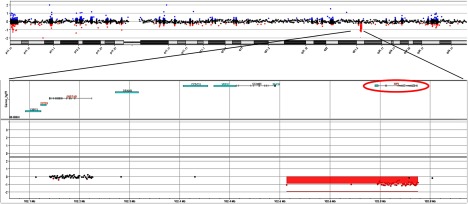
CNV analysis of proband reveals heterozygous deletion of IGF-1. Top, Schematic of chromosome 12 segment. Each dot represents a single copy number probe. Dot color indicates areas of deletion (red) and duplication (blue). Bottom, Magnified schematic segment containing the deletion of interest (red box). The location of the IGF-1 gene (circled above) is noted to be included in this region.
Discussion
In this study we describe a patient with IUGR, short stature, microcephaly, and cognitive delay who was diagnosed with ISS. He was found, using genome-wide CNV analysis, to have a paternally inherited heterozygous IGF1 deletion, which is likely the underlying cause for his short stature. As the first case reporting an isolated deletion of the entire IGF1 gene, this patient provides important insight into the effects of IGF-1 haploinsufficiency.
To our knowledge, there are only six previous case studies describing the effects of IGF1 gene mutations on both physical and neurodevelopmental growth in humans (5, 7–11). From these case reports, consisting of patients and relatives with both homozygous and heterozygous IGF1 mutations, there is a range in phenotypic severity with features including IUGR, postnatal growth failure, microcephaly, varying degrees of developmental delay (from none to severe), sensorineural hearing loss, and hyperinsulinism. In general, patients with heterozygous mutations have short stature, but milder associated phenotypes than those seen in the patients with homozygous IGF1 mutations. Relatives of probands, who themselves have heterozygous mutations, have also been noted to have lower than average recorded heights, with some meeting criteria for short stature (SDS ≤ −2) (2, 5, 7–12).
Table 2 and Supplemental Figure 2 provide a review of the previously reported cases of patients with IGF1 gene mutations, comparing them with the phenotypic, biochemical, and genetic findings of our current study patient. Although the present subject's phenotype is milder than some of the previously reported patients with homozygous IGF1 mutations, he has several overlapping features including IUGR, postnatal growth failure, microcephaly, micrognathia, cognitive delay, and fifth finger clinodactyly. He does not have sensorineural hearing loss or severe developmental delay as described in most of the cases with homozygous mutations (Table 2).
Table 2.
IGF-1 Mutation Case Report Summary
| Measures | Proband (present study) | First Author, Year (Ref.) |
||||||
|---|---|---|---|---|---|---|---|---|
| Fuqua, 2012 (5) | Van Duyvenvoorde, 2010 (10), Sibling 2 | Van Duyvenvoorde, 2010 (10), Sibling 1 | Netchine, 2009 (9) | Walenkamp, 2005 (11) | Bonapace, 2003 (8) | Woods, 1996 (7) | ||
| Birth weight SDS | −1.5 | −1.5 | −1.2 | −2.9 | −2.4 | −3.9 | −4.0 | −3.9 |
| Birth length SDS | −1.2 | −0.6 | −1.0 | −3.8 | −3.7 | −4.3 | −6.5 | −5.4 |
| HC SDS | −3.4 | No microcephaly | −1.6 | −2.4 | −2.5 | −8.0 | −7.5 | −4.9 |
| Height SDS | −2.7 | −4.2 | −4.6 | −4.1 | −4.9 | −8.5 | −6.2 | −6.9 |
| Phenotype | ||||||||
| IUGR | Yes | No | NR | NR | Yes | Yes | Yes | Yes |
| Hearing impaired | No | No | NR | NR | No | Yes | Yes | Yes |
| Cognitive delay | Yes | No | NR | Yes (details NR) | Yes (IQ 70–75) | Yes (IQ < 40) | Yes (details NR) | Yes (details NR) |
| Micrognathia | Yes | NR | NR | NR | NR | Yes | NR | Yes |
| Clinodactyly | Yes | NR | NR | NR | Yes | NR | NR | Yes |
| IGF-1 | Low-normal | Low-normal | Low | Low | Low | Very high | Low | Undetectable |
| IGFBP-3 | Normal | Normal | Normal | Normal | High (after GH) | Normal | Normal | Normal |
| ALS | High-normal | High-normal | Normal | Normal | High (after GH) | High | NR | Normal |
| GH | NR | Basal, 0.82 ng/mL (nl, <10); stim, 15 ng/mL (nl, >10) | Borderline GH during stim (actual value NR) | GH max during stim, 10.7 μg/L (nl, >6.7) | Basal, 0.7 ng/mL; stim, 26 ng/mL (nl, >7) | Basal,13 ng/mL (high); stim, 127 ng/mL (high) | Basal, 10 ng/mL; stim, 18 ng/mL (high) (nl, 10–12 ng/mL) | Basal, 18 ng/mL (high); stim, 94 ng/mL (high) |
| Bone age | Normal | Normal | Delayed | Delayed | Delayed | Delayed | Delayed | Delayed |
| IGF-1 abnormality | Heterozygous; IGF-1 gene deletion | Heterozygous; exon 4 splicing excision → frameshift mutation and early stop codon (c.402 + 1G>C, p.N74Rfs*8) | Heterozygous; 4 bp duplication → frameshift mutation and early stop codon (c.243–246dupCAGC, p.S83Qfs*13) | Heterozygous; 4 bp duplication → frameshift mutation and early stop codon (c.243–246dupCAGC, p.S83Qfs*13) | Homozygous; missense mutation with 4 × lower IGF1R affinity (p.R36Q) | Homozygous; missense mutation with 90 × lower IGF1R affinity (p.V44M) | Homozygous; T to A transversion of poly-A tail leading to truncated exon 6 | Homozygous; deletion of exons 4 and 5 |
| Parental height SDS | ||||||||
| Maternal | −1.3 | −4.6a | −3.5a | −3.5a | −2.9a | −2.4a | −1.6a | −1.4a |
| Paternal | −2.0a | +1.2 | −1.3 | −1.3 | −1.0a | +0.3a | −1.9a | −1.8a |
| Treatment response (rhGH vs rhIGF-1) | NR | After 9 mo rhGH (44 μg/kg/d): GV, 4.1 cm/y; height, SDS − 3.8 (from − 4.19). Followed by: 8 mo rhIGF-1 (120 mg/kg twice a day), GV, 6 cm/y; height, SDS − 3.5 | After 2 y rhGH (1.4 mg/m2/d): height SDS increased by +1.5; plasma IGF-1 SDS increased from −2.4 to +0.3 | After 2 y rhGH (1.4 mg/m2/d): height SDS increased by +1.0; plasma IGF-1 SDS increased from −2.6 to +0.6 | On rhGH 0.4 mg/kg/wk, GV increased to max 9–10 cm/y. On rhGH 0.2 mg/kg/wk, GV decreased to 5 cm/y | NR | After 6 mo rhIGF-1 (40 μg/kg/d): GV, 4.4 cm/y. Followed by rhIGF-1 (80 μg/kg/d): GV max 7.9 cm/y. On rhIGF-1, HC SDS improved (−7.5 to −4.3) | On rhGH × 1.7 y, no GV improvement. On rhIGF-1 (40 μg/kg/d): GV 4.2 cm/y. On rhIGF-1 (80 μg/kg/d), GV 7.3 cm/y. On rhIGF-1, insulin resistance improved |
Abbreviations: HC, head circumference; stim, stimulation; nl, normal; NR, not reported; GV, growth velocity.
Heterozygous carrier parent.
The present subject's milder phenotype due to haploinsufficiency of IGF1 strengthens the previous data, which suggested that IGF-1 might act in a dose-dependent manner to affect phenotype. Previously reported height SDSs are lower for probands with homozygous mutations (−8.5 to −4.9), as compared to those with heterozygous mutations (−4.6 to −2.7) (Table 2). Similarly, some of the prior studies have evaluated the height of the probands' parents and other heterozygous family members to further comment on IGF-1 insufficient states. For example, heterozygous carriers of the IGF-1 p.R36Q mutation had height SDS ranging from −1 to −3.5 as opposed to −5 in the homozygous proband (9). Additionally, the heterozygous parents carrying the IGF1 exon 4 and 5 deletion had height SDSs of −1.4 and −1.8 consistent with mild short stature, compared with an SDS of −6.9 in the proband (7).
Another example supporting the concept of a dose-dependent effect of IGF-1 signaling is illustrated by comparison of the patients described by Walenkamp et al (11), with the p.V44M mutation, and Netchine et al (9), with the p.R36Q mutation. Although both of these patients were found to have homozygous recessive mutations of IGF1 leading to an IGF-1 protein product with decreased affinity for the IGF-1 receptor (IGF1R), the patient with the p.V44M variant produced an IGF-1 protein product with drastically lower IGF1R affinity (90 times less than normal) and a more severe phenotype. On the other hand, the patient with the p.R36Q variant produced an IGF-1 protein product that had an IGF1R affinity of only 3.9 times less than normal, with a milder phenotype that was, in fact, very similar to the phenotype observed in our patient with a heterozygous IGF-1 deletion (Table 2) (9, 11).
In terms of treatment recommendations, homozygous complete loss of function variants in IGF1 are not expected to respond to GH therapy. For this reason, two of the patients with homozygous IGF1 mutations were treated with recombinant human IGF-1 (rhIGF-1) therapy (rather than recombinant human GH [rhGH]) with moderate improvement in growth velocity (Table 2) (13, 14). However, patients with heterozygous deletions in IGF1 or those with partial impairment in IGF-1 signaling may respond to GH therapy. The responses in these cases have been variable, ranging from robust to more modest responses (Table 2) (5, 9, 10). This suggests that the current proband with a heterozygous IGF1 deletion may respond to GH therapy, although it is unclear to what degree. At this time, the subject and his family have elected not to pursue therapy.
Interestingly, not all of the patients with confirmed abnormalities in IGF1 demonstrated low IGF-1 levels by standard laboratory analysis. The proband with severely decreased IGF1R affinity had very elevated IGF-1 levels (11), and the patient with the heterozygous IGF-1 exon 4 splicing mutation (c.402+1G>C, p.N74Rfs*8) (5) had low-normal levels of IGF-1. Because the proband of this case carries an isolated IGF-1 deletion, we did not expect that IGFBP-3 values would be directly affected. If anything, because IGFBP-3 is a GH-dependent factor, it is possible that IGFBP-3 levels could increase in response to a compensatory increase in GH secretion; however, in this case, his IGFBP-3 values were normal. Like the patient with the exon 4 splicing mutation, our patient had circulating IGF-1 levels in the low-normal range. Given the normal IGFBP-3 levels, it is possible that free IGF-1 levels would be frankly low. It is also possible that circulating serum values may not accurately reflect local IGF-1 activity at the level of the growth plate. These case examples further support the argument that standard clinical and laboratory evaluation is not sufficient to detect disruptions in the GH/IGF-1 axis, and further genetic testing is often necessary to make the appropriate diagnosis and guide appropriate therapeutic strategies for these patients.
The present study, in the context of the prior case reports of patients with IGF-1 deficiency, highlights the fact that even in rare disorders caused by single gene mutations, there can still be a wide range of phenotypic variability. Using ever more precise genetic tools to identify the specific mutations at both the DNA and protein levels, we can better understand the basis for the range of observed phenotypes. This understanding also allows for better prediction of clinical outcomes for future patients with similar underlying mutations. It is possible that additional genetic defects in the GH/IGF-1 pathway or in other growth-related pathways may modify the effects of these individual variants. Additional research is needed in this area.
In summary, the present report highlights how the implementation of genome-wide evaluation can elucidate answers to clinical questions that would not have been otherwise uncovered using standard clinical and laboratory tools. As this genetic technology continues to become both more available and affordable, one can expect to significantly increase the ability to identify rare genetic variants underlying as yet undiagnosed conditions. Applying this technology will have a tremendous impact in improving the accuracy of diagnosis, predicting clinical outcomes, and targeting therapeutic approaches both on an individual and population-based scale.
Acknowledgments
The authors thank the family for participating in this research study and Timothy Miller for his support of this work.
This work was conducted with support from Harvard Catalyst, The Harvard Clinical and Translational Science Center (National Institutes of Health Award UL1 RR 025758 and financial contributions from Harvard University and its affiliated academic health care centers) as well as award number K23HD073351 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health, support from the Translational Research Program at Boston Children's Hospital, and a Clinical Scholar Award from the Pediatric Endocrine Society. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Center for Research Resources, or the National Institutes of Health.
Disclosure Summary: A.D. has previously consulted for Ipsen Pharma and Pfizer Inc. J.N.H. received grant support from Pfizer Inc. (2011 to the present). The remaining authors have no conflicts to report.
Footnotes
- ALS
- acid labile subunit
- CNV
- copy number variant
- IGFBP-3
- IGF binding protein-3
- IGF1R
- IGF-1 receptor
- IUGR
- intrauterine growth restriction
- ISS
- idiopathic short stature
- qPCR
- quantitative PCR
- rhGH
- recombinant human GH
- rhIGF-1
- recombinant human IGF-1
- SDS
- SD score.
References
- 1. Zahnleiter D, Uebe S, Ekici AB, et al. Rare copy number variants are a common cause of short stature. PLoS Genet. 2013;9:e1003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. David A, Hwa V, Metherell LA, et al. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. 2011;32:472–497 [DOI] [PubMed] [Google Scholar]
- 3. Wang SR, Carmichael H, Andrew SF, et al. Large-scale pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. J Clin Endocrinol Metab. 2013;98;E1428–E1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dauber A, Yu Y, Turchin MC, et al. Genome-wide association of copy-number variation reveals an association between short stature and the presence of low-frequency genomic deletions. Am J Hum Genet. 2011;89:751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fuqua JS, Derr M, Rosenfeld RG, Hwa V. Identification of a novel heterozygous IGF1 splicing mutation in a large kindred with familial short stature. Horm Res Paediatr. 2012;78:59–66 [DOI] [PubMed] [Google Scholar]
- 6. Carmichael H, Shen Y, Nguyen TT, Hirschhorn NJ, Dauber A. Whole exome sequencing in a patient with uniparental disomy of chromosome 2 and a complex phenotype. Clin Genet. 2013;84:213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363–1367 [DOI] [PubMed] [Google Scholar]
- 8. Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet. 2003;40:913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Netchine I, Azzi S, Houang M, et al. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab. 2009;94:3913–3921 [DOI] [PubMed] [Google Scholar]
- 10. van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, et al. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J Clin Endocrinol Metab. 2010;95:E363–E367 [DOI] [PubMed] [Google Scholar]
- 11. Walenkamp MJ, Karperien M, Pereira AM, et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab. 2005;90:2855–2864 [DOI] [PubMed] [Google Scholar]
- 12. Camacho-Hübner C, Woods KA, Miraki-Moud F, et al. Effects of recombinant human insulin-like growth factor I (IGF-I) therapy on the growth hormone-IGF system of a patient with a partial IGF-I gene deletion. J Clin Endocrinol Metab. 1999;84:1611–1616 [DOI] [PubMed] [Google Scholar]
- 13. Woods KA, Camacho-Hübner C, Bergman RN, Barter D, Clark AJ, Savage MO. Effects of insulin-like growth factor I (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J Clin Endocrinol Metab. 2000;85:1407–1411 [DOI] [PubMed] [Google Scholar]
- 14. Concolino D, Muzzi G, Sestito S, Vega G, Bonapace G, Strisciuglio P. Long-term treatment with recombinant insulin-like growth factor 1 (IGF-1) in a child with IGF-1 gene mutation. Eur J Pediatr. 2010;169:245–247 [DOI] [PubMed] [Google Scholar]