

Abstract
In the past three years, major advances in understanding cytochrome P450 2B (CYP2B) structure-function relationships have been made through determination of multiple ligand-bound and one ligand-free X-ray crystal structure of CYP2B4 and one ligand-bound X-ray crystal structure of CYP2B6. These structures have provided insight into the features that provide the high degree of plasticity of the enzymes. A combination of a phenylalanine cluster that allows for concerted movement of helices F through G and a conserved set of electrostatic interactions involving Arg262 facilitates movement of this region to accommodate binding of ligands of various sizes without perturbing most of the P450 fold. Integrating solution based techniques such as NMR or deuterium exchange mass spectrometry (DXMS) with computational methods including molecular docking has provided further insight into enzyme behavior upon ligand binding. In addition, extended molecular dynamics simulations have provided a link between an open and a closed conformation of ligand-free CYP2B4 found in crystal structures. Other studies revealed the utility of rational engineering in improving stability of P450s to facilitate structural studies. The solution and computational results combined with the X-ray crystal structures yield a comprehensive picture of how these enzymes adopt different conformations to bind various ligands.
Keywords: cytochrome P450, CYP2B, crystallography, NMR, deuterium exchange mass spectrometry, molecular modeling, molecular dynamics simulation
INTRODUCTION
Cytochrome P450-dependent monooxygenases (P450) are involved in the biotransformation of a broad range of endogenous and exogenous chemicals including drugs, hormones, and environmental pollutants [1]. The P450 2B (CYP2B) subfamily of enzymes has served as a traditional model for investigation of the mechanism of gene expression mediated by drugs and environmental contaminants and for exploration of the structural plasticity of mammalian drug metabolizing P450s [2, 3]. CYP2B enzymes catalyze the oxidation of a broad range of substrates, with preference for angular, medium-sized neutral or basic compounds [4, 5]. In addition, CYP2B enzymes display a relatively low degree of catalytic conservation across mammalian species when compared with other P450 subfamilies [6, 7]. The identification of distinct allelic variants of a single rat CYP2B enzyme and of functionally distinct but highly structurally related CYP2B enzymes in rats and rabbits followed by sequence comparisons, site-directed mutagenesis, and heterologous expression systems allowed for the identification of many of the key residues that dictate ligand orientation in the active site and contribute to substrate and inhibitor specificity [8–10]. The structural basis of CYP2B enzyme specificity as investigated by mutagenesis was summarized previously [3, 11].
In order to elucidate in more depth the structural basis for CYP2B function, many studies have turned to techniques that can provide atomic level structural detail, which include X-ray crystallography and NMR. While the traditional application of NMR has been limited by the large size of P450s, which have in excess of 400 residues, X-ray crystallography has provided a wealth of information on bacterial and mammalian P450s [12–14]. Although the first structure of a bacterial P450 was determined in the early 1980s, the first structure of a mammalian P450 was not solved until 2000 [15, 16]. Later, the structure of CYP2B4 in the absence of added ligand was determined at 1.6 Å, which remains the highest resolution structure of a mammalian P450 reported to date [17] only equaled by the recently reported structure of CYP46A1 [18]. In this CYP2B4 structure, the protein formed a dimer in the crystal through coordination of His226 to the heme iron of the opposite monomer, with each protein chain displaying an “open” conformation of the enzyme that would allow ligand access to the active site. The subsequent structure of CYP2B4 complexed with 4-(4-chlorophenyl)imidazole (4-CPI) showed the enzyme in a more “closed” conformation than the previous structure, indicating how the enzyme is able to adjust to bind smaller ligands [19]. A third structure of CYP2B4 in complex with the antifungal agent bifonazole provided yet another conformation of the enzyme [20]. This structure suggests that CYP2B4 has the ability to widen the active site to accommodate very bulky ligands in a manner that differs from the “open” ligand-free structure. Major conclusions concerning these three structures were previously discussed elsewhere [3].
Concurrently, solution thermodynamic studies using isothermal titration calorimetry (ITC) revealed marked differences in thermodynamic binding parameters for CYP2B4 and imidazole inhibitors of different ring chemistry and side chains [21]. Continued crystallization efforts yielded another “closed” structure of CYP2B4 when complexed with an isomer of 4-CPI, 1-(4-chlorophenyl)imidazole (1-CPI). Furthermore, a complex of CYP2B4 with 1-biphenyl-4-methyl-1H-imidazole (1-PBI) revealed an “intermediate” conformation and indicated that the F-G cassette moves in concert around a pivot to accommodate ligand binding [22]. More recently, two structures of CYP2B4 were determined in the presence of the antiplatelet drugs ticlopidine and clopidogrel [23], which were the first complexes solved in the presence of a non-imidazole compound. Finally, the structural analysis of CYP2B4 has come full circle with another ligand-free structure of CYP2B4 in a “closed” conformation similar to the 4-CPI-complex, rather than the previously mentioned “open” structure [24]. In the meantime, the first crystal structure of human CYP2B6 was also determined in the presence of 4-CPI [25]. Interestingly, the CYP2B4 and CYP2B6 4-CPI structures were more similar to each other than the structures of the CYP2B4 complexes with 4-CPI or 1-CPI.
In addition to the wealth of information generated through crystallographic studies, structural knowledge of CYP2B enzymes has also benefitted from other approaches. Utilizing the heme iron as a paramagnetic center, NMR was employed for the first time with a CYP2B enzyme to determine the orientation of ticlopidine in the active site, since electron density maps from the crystal structure were ambiguous with regard to ligand orientation [23]. Ligand docking and simulated annealing also provided predictive information concerning the orientation of ticlopidine in the active site of CYP2B4. The results suggest that ticlopidine orients differently in the active sites of CYP2B4 and CYP2B6. In parallel, examination of the solution structural behavior of CYP2B4 was made possible through the first use of deuterium exchange coupled to mass spectrometry (DXMS) applied to a mammalian drug metabolizing P450 [24]. The results from these DXMS experiments indicated that CYP2B4 is in a predominantly open conformation in solution. Molecular dynamics (MD) simulation showed that the average open conformation of the enzyme in solution is energetically accessible starting with the “closed” ligand-free structure.
In addition to the approaches already discussed, rational mutagenesis has extended the knowledge of how CYP2B enzyme structure relates to function. Finally, rational mutagenesis has identified single point mutants that significantly increase expression levels of CYP2B6 [26] or CYP2B6 and CYP2B11 and provide increased structural fidelity [27]. This review will cover structural features of CYP2B enzymes revealed by crystal structures, major findings found through computational and solution structural investigations, and results of rational protein engineering of CYP2B enzymes.
X-RAY CRYSTAL STRUCTURES OF 2B ENZYMES
With twelve entries deposited in the protein data bank (PDB), CYP2B enzymes show the most plasticity of any P450 studied by X-ray crystallography. The structures of these proteins are composed of structurally conserved regions and plastic regions (PR) [3]. The structurally conserved regions include most of the secondary structural elements involved in heme binding and those on the proximal side of the enzyme, consistent with their role in binding of electron transfer partners NADPH-cytochrome P450 reductase and cytochrome b5. Five plastic regions account for about one third of the protein: PR1 (residues 39–57), PR2 (residues 101–140), PR3 (residues 177–188), PR4 (residues 203–298), and PR5 (residues 474–480). Comparison of two of these regions, PR2 and PR4, of these structures allows one to group the structures in four distinct conformations: “open”, “closed”, “expanded”, and “intermediate” (Fig. 1). The first structure of CYP2B4 was in an “open” configuration with an apparent means of substrate access to the active site [17]. Binding of the smaller molecules 1-CPI, 4-CPI, ticlopidine, or clopidogrel to CYP2B4 and 4-CPI to CYP2B6 produces a “closed” conformation similar to other family 2 enzymes [28]. The large antifungal drug bifonazole produced an “expanded” conformation that was more spread out than the “open” conformation [20]. Finally, an “intermediate” conformation was observed in the structure of CYP2B4 complexed with 1-PBI [22]. Table 1 provides a list of crystal structures of CYP2B enzymes. Whereas the crystallization efforts for the “open” structure utilized an engineered enzyme with modifications limited to the N-terminal 30 amino acids of the wild-type enzyme sequence [29], subsequent crystallization efforts utilized protein modified to interrupt dimerization through a H226Y modification [19, 24].
Figure 1.

Conformations observed in CYP2B X-ray crystal structures. CYP2B4 forms a compact structure (A) when bound to the small inhibitors 4-CPI (1SUO), 1-CPI (2Q6N), ticlopidine (3KW4), or clopidogrel (3ME6). Rearrangement of the B′/C loop and F-G cassette accommodates the larger inhibitors (B) bifonazole (2BDM) and (C) 1-PBI (3G5N). Interestingly, the absence of ligand has yielded structures in two conformations: a closed structure similar to the CYP2B4-4-CPI complex (A) and an open conformation (D). Stick diagrams show the chemical structures of ligands.
Table 1.
Crystal Structures of Mammalian P450 2B Enzymes
| Enzyme | Ligand | PDB Code | Cavity volume† (Å3) | Reference |
|---|---|---|---|---|
| CYP2B4 | 4-CPI | 1SUO | 253 | [19] |
| CYP2B4 | 1-CPI | 2Q6N | 421 | [31] |
| CYP2B4 | Ticlopidine | 3KW4 | 276 | [23] |
| CYP2B4 | Clopidogrel | 3ME6 | 343 | [23] |
| CYP2B4 | 3MVR | 358 | [24] | |
| CYP2B6 | 4-CPI | 3IBD | 582 | [25] |
| CYP2B4 | 1-PBI | 3G5N | 391 | [22] |
| CYP2B4 | 1PO5 | NA§ | [17] | |
| CYP2B4 | Bifonazole | 2BDM | 457 | [20] |
Probe-occupied cavities were calculated using a 1.4 Å probe radius in Voidoo [53].
Not Applicable - Formation of dimer fills active site.
Open Conformation
Structurally, the majority of this CYP2B4 structure resembles previously determined P450 structures [15], but key areas of the enzyme show marked differences. The packing of the core region, helices E, J, K, and L, is highly conserved. However, an open cleft is seen consisting of the F-G cassette on one side and the B′/C loop and helix C on the other side [17]. Interestingly, the cleft is filled by portions of the F-G cassette of the symmetry-related molecule in the dimer, where H226 forms an intermolecular bond to the heme iron of the other monomer. These regions form a closed conformation in most P450 structures. For example, the structure of CYP2C5 bound with 4-methyl-N-methyl-N-(2-phenyl-2H-pyrazol-3-yl)benzene sulfonamide (DMZ) exhibits a closed conformation where helix B′ is stabilized by contacts with helix G residues [30]. The F-G cassette, which forms the ceiling of the active site in the CYP2C5-DMZ structure, flexes away from the protein core and helix B′ in the open CYP2B4 structure. This allows a means of direct transfer of substrates from bulk solvent or the lipid bilayer to the catalytic center of CYP2B4 [17].
Closed Conformation
The structures with a closed conformation are virtually superimposable, showing less than 1 Å root mean square deviation (RMSD) for the Cα overlays using the CYP2B4-4-CPI complex as a reference structure. Structures included in this group are CYP2B4 without ligand (3MVR) [24] or in complex with the small molecules 4-CPI (1SUO) [19], 1-CPI (2Q6N) [31], ticlopidine (3KW4) [23], or clopidogrel (3ME6) [23], as well as a CYP2B6 genetic variant complexed with 4-CPI (3IBD) [25]. In this conformation, the enzyme adopts a relatively compact structure with active site residues identified by site-directed mutagenesis and by analogy with CYP2C5 structures forming a closely packed pocket around the ligand [16, 19, 30]. There is no apparent access or egress channel for the tightly sequestered ligands. Helix B′ and the N-terminal portion of helix I move toward each other, bringing helix B′ closer to helix G (Fig. 2). The F-G cassette containing helices F through G moves toward the β1 sheet and interacts with the N-terminal structures, helix B′ and the B′/C loop, the N-terminus of helix I, and the β4 sheet, capping the active site. Contacts between helices G and I and helix B′ appear to close the active site, and the F-G cassette provides a roof for the active site. Differences between the original “open” conformation of the enzyme and subsequent “closed” structures provided insight into the structural flexibility of the CYP2B enzymes.
Figure 2.
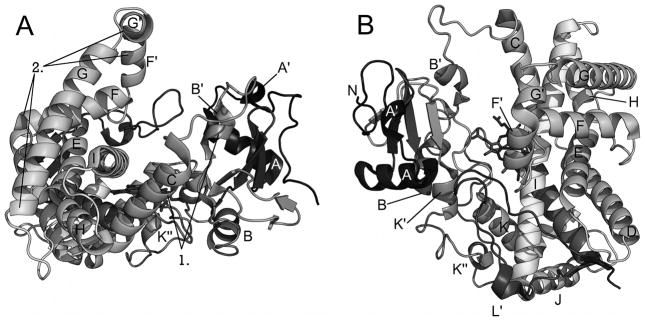
Structure of CYP2B4. (A) View of CYP2B4 along helix I showing large open cleft with elements of the B′-C loop (1) and F-G cassette (2) labeled. (B) Perspective looking down on heme and active site.
For evaluation, the residues within 5 Å of the ligand in the ligand-bound complex are listed in Table 2. Upon closer comparison, small differences among the closed structures begin to appear. Of the eleven residues within 5 Å of 4-CPI in the CYP2B4-4-CPI complex, six are within the same distance from the ligand in other structures with a “closed” conformation (I114, F297, A298, T302, I/L363, V367). In the structures of CYP2B4 complexed with 4-CPI, 1-CPI and ticlopidine and CYP2B6 complexed with 4-CPI, I101 is also within the 5 Å cutoff. Additionally, in the structure of CYP2B4 complexed with 1-CPI and CYP2B6 complexed with 4-CPI E301 swings out to interact with H172 and a water molecule, respectively. The Q172H substitution is a naturally occurring polymorphism of CYP2B6, and differences in how E301 interacts with glutamine versus histidine may provide a clue as to differences in stability and functionality of the wild-type CYP2B6 with Q172 compared to the CYP2B6 Q172H variant. Interestingly, E301 interacts with ticlopidine but not clopidogrel in the respective complexes with CYP2B4.
Table 2.
Residues Within 5Å of Ligand in Crystal Structures
| Structure Identified by PDB Code and ligand | |||||||
|---|---|---|---|---|---|---|---|
| Residue | 1SUO1 4-CPI | 3IBD2 4-CPI | 2Q6N 1-CPI | 3KW4 ticlopidine | 3ME6 clopidogrel | 3G5N 1-PBI | 2BDM bifonazole |
| I101 | X | X | X | X | |||
| V104 | X | X | X | ||||
| F108 | X | ||||||
| I114 | X | X | X | X | X | ||
| F115 | X | X | X | X | |||
| S128 | X | ||||||
| M132 | X | ||||||
| F206 | X | X4 | X | X | |||
| I209 | X | X | X | X | |||
| S210 | X | ||||||
| V292 | X | ||||||
| S294 | X | ||||||
| L295 | X | ||||||
| F296 | X | ||||||
| F297 | X | X | X | X | X | ||
| A298 | X | X | X | X | X | X | X |
| G299 | X4 | X | X | ||||
| E301 | X | X | X | ||||
| T302 | X | X | X | X | X | X | X |
| I/L3633 | X | X | X4 | X | X | X | X |
| G366 | X | ||||||
| V367 | X | X | X | X | X | X | |
| P368 | X | ||||||
| V477 | X | X | X | X | X | ||
| G478 | X | X | |||||
Determined using the PyMOL Molecular Graphics System, Version 1.2, Schrödinger, LLC. Residues that interact with the ligand in each of the structures are highlighted in gray.
Structure of CYP2B4 complexed with 4-CPI
Structure of CYP2B6 complexed with 4-CPI
Residue 363 is Isoleucine in CYP2B4 and Leucine in CYP2B6
These residues are at the 5Å cutoff.
Other differences in active site residues as defined by those residues within 5 Å of 4-CPI in the CYP2B4-4-CPI complex, show slight displacement of the following residues away from the ligand in the respective structures: V104 in the CYP2B6-4-CPI and the CYP2B4-ticlopidine complexes, F115 in the CYP2B6-4-CPI complex, and V477 in the CYP2B4-1-CPI and CYP2B4-clopidogrel complexes. The small difference in orientation of the isomers of CPI with respect to the heme in the complexes with CYP2B4 explains why V477 is not within 5 Å of 1-CPI. Additionally, F206 and I209 come into proximity of the ligand in the CYP2B6-4-CPI complex and in the CYP2B4-1-CPI, CYP2B4-ticlopidine, and CYP2B4-clopidogrel complexes, and G299 is at the cutoff distance in the CYP2B6-4-CPI complex. In the CYP2B4-ticlopidine, S210 is in close proximity to the ligand, and S294 is close to clopidogrel in the complex with CYP2B4; G478 is within the 5 Å distance in both these structures. Clopidogrel also interacts with F108. One may note that some residues included here are not discussed in previous work; residues with side chains not facing toward the active site (i.e. - L362) were discarded as not interacting with the ligand. Interestingly, a subsequent “closed” CYP2B4 ligand-free structure (3MVR) [24] shows that at least two conformations of CYP2B4 are accessible in solution, and rearrangement of the plastic regions of the protein can occur in the absence of ligand binding.
Expanded Conformation
Initial efforts to trap an “intermediate” conformation of CYP2B4 involved crystallization of the enzyme with the antifungal bifonazole [20]. The resulting complex displayed a further expanded active site when compared with the “open” conformation. Helices C, E, H, and G and the N-terminal end of helix I move to accommodate bifonazole binding and relative protein packing. There is no contact between helix G and residues on the B′/C loop region. The enzyme does not close around the bound inhibitor. The set of residues within 5 Å of the ligand consists of ten residues in this structure (Table 2), but only four of those residues are involved in binding both bifonazole and 4-CPI (A298, E301, T302, I363). Furthermore, CYP2B4-bifonazole forms a dimer in which the F-G cassette of one molecule inserts into the widely open active site cleft of the other monomer, and vice versa. Since this region of the enzyme is thought to be a membrane-binding motif in microsomal P450s, structural rearrangement likely occurs in this region upon extraction from the membrane. The greatly expanded conformation of this complex is likely a product of minimizing water interactions with the hydrophobic walls of the active site. Further influence on the conformation is contributed by dimerization of the enzyme in the crystal structure. However, the wide-open arrangement of the protein should be possible in solution or during interactions with the membrane.
Intermediate Conformation
The “intermediate” conformation is represented by the structure of CYP2B4 complexed with 1-PBI [22]. Similar to other conformations of ligand-bound enzyme, the CYP2B4-1-PBI complex retains the overall P450 fold, while molding to the inhibitor molecule. The core of the fold (the C-terminus of helix A through helix B, helices D and E, the C-terminus of helix I through helix K, and all the β-sheets) remains relatively unchanged when compared with other conformations of CYP2B4. Differences between this structure and structures defining the other conformations of the enzyme again occur in plastic regions of the enzyme [3]. The B′/C-loop makes a shift toward the F-G cassette and contains two small helices to compact the structure. In contrast, structures in the “closed” conformation of the enzyme have an elongated helix B′ that actually enters the active site to close around the small molecule, and the CYP2B4-bifonazole complex displays a fully elongated B′/C-loop that is extended toward helix F′ [22]. The active site lid formed by the F-G cassette adopts a conformation intermediate to those seen in the 4-CPI and bifonazole structures. The ligand interacts with six residues (A298, T302, I363, V367, P368, V477) and is in close proximity to five others (S128, M132, V292, L295, F296) in addition to coordinating to the heme iron. This represents a hybrid of the residues involved in bifonazole binding and those involved in 4-CPI binding, with three being common in each structure of CYP2B ligand complexes (A298, T302, I363). In addition to the active site interactions with 1-PBI, two other molecules of 1-PBI appear to fill residual surface pockets in the structure. However, these extra 1-PBI molecules do not appear to affect the structure, as it is virtually identical to that obtained at a lower concentration of ligand where only a single 1-PBI molecule is observed. The CYP2B4-1-PBI complex forms a crystallographic tetramer, with each dimer similar to the symmetric dimer found in the crystals of CYP2B4-bifonazole complex [20]. Furthermore, each monomer in the tetramer is in roughly the same conformation with an RMSD of less than 0.2 Å.
Structural Plasticity of CYP2B Enzymes
Previous examination of the “open”, “closed”, and “expanded” conformations of CYP2B4 defined plastic regions (PR) of the protein and examined relative reshaping of the molecule in response to ligand binding [3]. However, further insight into the plasticity of the enzyme has emerged with subsequent structures. Where PR2 (residues 101–140), PR3 (residues 177–188), and PR4 (residues 203–298) show concerted movements to convert among the various conformations of the enzyme, PR2 (containing helix B′) and PR4 (containing the F-G cassette) appear to be the most structurally dynamic regions of the protein. PR2 appears to adjust position to cap the active site in response to ligand size. A portion of PR2 including the C-terminal end of helix C has been identified in cytochrome b5 and NADPH-cytochrome P450 reductase binding [32, 33], so plasticity of this area of the protein likely is central to the mechanism by which P450s bind redox partners [30, 32, 34, 35]. Interestingly, heme binding is affected by rearrangements in this region, and R98, W121, and R125 make varying contributions to heme propionate hydrogen bonding in each CYP2B structure.
On the other hand, alignment of the F-G cassette from the CYP2B4-4-CPI, bifonazole, and 1-PBI complexes shows rearrangement of helix F′ in response to extension of that region into the active site in the bifonazole and 1-PBI complexes. However, the orientation of these helices relative to each other does not appear to change [22]. This concerted movement appears to be facilitated by a series of complex rearrangements of ten phenylalanine residues (F184, F188, F195, F202, F203, F206, F244, F264, F296, F297). Examination of an alignment of CYP2B enzymes shows that four of these residues are absolutely conserved (184, 188, 195, 296). Residues 203 and 244 are bulky aromatic amino acids (phenylalanine or tyrosine) across the entire subfamily. Residues 264 and 297 are phenylalanine across the majority of the subfamily; however, at position 264, CYP2B6 (leucine), CYP2B9 (tyrosine), and CYP2B22 (leucine) are different, and at position 297, CYP2B3 has a leucine. Residues 202 and 206 are long chain aliphatic (isoleucine, leucine, methionine) or bulky aromatic residues. CYP2B12 is the only outlier at these two residues, having valine and serine at these locations, respectively. Furthermore, examination of the plastic regions in the CYP2B6-4-CPI complex revealed a highly conserved environment surrounding residue 262 [25]. In both CYP2B4 and the genetic variant of CYP2B6 (K262R), R262 is part of a hydrogen-bonding network including H252, T255, D263, and D266. Alignment of CYP2B enzymes indicates that the only divergence in this cluster of residues is K262 in CYP2B6. Despite the large shifts in orientation of PR4, this network of interactions is maintained in every CYP2B structure to date [17, 19, 20, 22–25, 31].
The remaining three plastic regions show varying degrees of differences among the crystal structures. PR1 shows only slight differences in the N-terminal end of the A-helix, but the A′-helix shifts away from the heme pocket in the “open” ligand-free structure and is disordered in the bifonazole complex. PR3 exhibits shifts in the C-terminal end of the E-helix with the intermediate and expanded conformations showing the greatest difference from the remaining structures. PR5 shows small differences among the conformations with the region unchanged in each of the structures with a “closed” conformation; this region shifts toward the A-helix in the “intermediate” conformation and toward the F-G cassette in the “expanded” conformation. PR5 shifts slightly away from the heme pocket in the “open” conformation. The spatial shifts seen in these regions are much smaller than those seen in PR2 and PR4.
SOLUTION AND COMPUTATIONAL METHODS
While X-ray crystal structures provide a wealth of information about structural features of P450s, they are effectively snapshots of single conformational states. Initial efforts to decipher solution structural behavior of CYP2B enzymes utilized ITC to monitor energetic changes in CYP2B4 upon binding of ligands of varying size [3, 21, 31, 36]. To complement the knowledge gained from ITC and X-ray crystal structures, solution (NMR and DXMS) and computational (molecular docking and MD simulation) methods were employed to deepen our understanding of CYP2B solution structural behavior and enzyme plasticity [23, 24].
Molecular Docking
Clopidogrel and ticlopidine are mechanism-based inactivators of CYP2B6 [37]. After showing that these drugs bind to CYP2B4, crystallization efforts were undertaken. The crystal structure of CYP2B4-clopidogrel complex shows that clopidogrel clearly binds with the chlorophenyl moiety closest to the heme iron [23]. In contrast, ticlopidine could be modeled into the active site with either the chlorophenyl moiety or the thiophene portion of the molecule, where most of the oxidation by CYP2B6 occurs [38], toward the heme iron (Fig. 3A–B) [23].
Figure 3.

Electron density maps and docking results of CYP2B4 and ticlopidine. The Fo-Fc simulated annealing omit map for ticlopidine contoured at 3σ shows two lobes of electron density above the plane of the heme. Ticlopidine fits this density well either in the thiophene down (A) or the chlorophenyl down (B) orientation. Molecular docking studies using Autodock4 or simulated annealing in GROMACS4 show that the preferred orientation for CYP2B4 binding of ticlopidine is with the chlorophenyl group toward the heme as represented by the results of the simulated annealing experiment (C). Modified from originally published figures in Gay et al. [23] and reproduced with permission from the American Chemical Society. © the American Chemical Society.
Initial docking experiments were undertaken to evaluate the ticlopidine orientations modeled in the electron density maps of the CYP2B4-ticlopidine complex. Using Autodock4 [39], an experiment was performed with 100 docking runs where protein side chains were fixed in place; two clusters of poses were observed. The most populous cluster contained 95 poses with a mean binding energy of −7.79 kcal/mol. These poses had the chlorophenyl ring closest to the heme, consistent with the chlorophenyl down orientation observed in the X-ray crystal structure. The second cluster consisted of the other five poses (binding energies ranging from −7.77 to −7.76 kcal/mol), where the thiophene ring was closest to the heme with the chlorophenyl ring above it. Due to bond rotations, this cluster does not correlate with the thiophene down orientation from the crystal structure. Similar experiments with CYP2B6 indicated that the thiophene group is likely to be closer to the heme consistent with the mechanism-based inactivation of CYP2B6 [38].
Ticlopidine binding was further explored by distance-restrained simulated annealing utilizing the GROMACS software package [40]. The final ten preferred orientations deduced from the annealing simulation (Fig. 3C) position the chlorophenyl ring closest to the heme in an orientation consistent with both the X-ray crystal structure chlorophenyl down orientation and the predominant cluster from Autodock experiments [23]. Interestingly, the conformation of ticlopidine obtained by simulated annealing would allow for oxidation of the tetrahydropyridine nitrogen by CYP2B4; ongoing experiments indicate this is the preferred site of oxidation of ticlopidine by CYP2B4 [41].
NMR
Traditional protein NMR assigns resonances to each amino acid in a protein, generates restraints for distances, angles, and orientations, and calculates the structure of the entire protein. However, utilizing longitudinal (T1) relaxation measurements and existing crystal structures, NMR may be utilized to monitor ligand interaction with P450s [42–44]. In the case of CYP2B enzymes, T1 relaxation measurements were used to complement results obtained from X-ray crystallography [23].
In these NMR experiments, T1 paramagnetic relaxation rates for protons on the chlorophenyl ring were 71% higher than those on the thiophene group, suggesting that the molecule is preferentially oriented with chlorophenyl moiety closer to the heme than the thiophene group. Due to lower binding affinity of CYP2B4 for ticlopidine compared with previously crystallized imidazole inhibitors, it is “fast exchange” in solution and likely adopts multiple conformations within the enzyme active site. Recent studies indicate that both CYP2B4 and CYP2B6 can produce multiple metabolites from ticlopidine, with time courses that deviate rapidly from linearity and are consistent with mechanism-based inactivation [41]. Compared with CYP2B6, CYP2B4 catalyzes more N-oxidation and less oxidation of the thiophene ring. These results emphasize how ligand dynamics affect P450-ligand complexes.
Deuterium Exchange Mass Spectrometry
Deuterium exchange mass spectrometry (DXMS) is a solution method used to observe protein structural dynamics, protein-protein interactions, and protein-ligand interactions [45–48]. Available crystal structures of CYP2B enzymes indicate a wide range of conformations in response to ligand binding [3, 22–25, 31], and ligand-free CYP2B4 has been crystallized in two conformations: “open” [17] and “closed” [24]. However, these results do not indicate which structure is closer to the solution behavior of the enzyme. DXMS experiments indicate that CYP2B4 adopts a predominantly open conformation in solution [24]. Furthermore, amide hydrogens are provided differing solvent protection depending upon the absence or presence of ligand and the identity of that ligand (Fig. 4). Peptides covering large portions of PR2 and PR4 showed significant slowing of the H-D exchange rate in the presence of 4-CPI when compared with the exchange rate of ligand-free CYP2B4, consistent with “closed” and “open” structures, respectively. Conversely, CYP2B4 with and without 1-PBI shows little difference in H-D exchange rate across the entire protein. This finding indicates that binding of 1-PBI does not increase protection from amide hydrogen exchange for the plastic regions of the protein in the B′/C loop and the F-G cassette but does not contradict the differences in conformation between the crystal structures of “open” ligand-free and 1-PBI-bound CYP2B4. While DXMS demonstrates that the large majority of the protein adopts a more open conformation in the absence of ligand, averaging across the entire population of protein means that closed conformers representing minor populations would not be observed with this method.
Figure 4.

CYP2B4 behavior in solution upon ligand binding. Time course of deuterium exchange in the B′-C loop region and the F-G cassette, respectively, of peptides 1 (residues 95–115) and 2 (residues 225–243), which show differences in DXMS exchange. Peptides in the D-E helices and I-J helices, respectively are illustrated as 3 (residues 155–178) and 4 (residues 313–340). The scale of the axis is the maximum number of exchangeable amides. Modified from originally published figure in Wilderman et al. [24] and reproduced with permission from the American Society for Biochemistry and Molecular Biology. © the American Society for Biochemistry and Molecular Biology.
Molecular Dynamics Simulation
With the demonstration by DXMS that CYP2B4 is predominantly open in solution and ligand-free crystal structures in two distinct conformations, the question of ease of transition from across the energy landscape arose. To address this, a molecular dynamics (MD) simulation was performed for 15 ns to investigate the energetically accessible and preferred conformations of CYP2B4 [24] using the GROMACS software package [40]. Comparing the RMSD of the Cα backbone during the MD simulation versus the starting “closed” ligand-free structure (3MVR) showed rapid equilibration in ~100 ps to a structure differing from the starting model by an RMSD of 2 Å, as reported previously [36]. A slower equalization representing larger molecular motions followed that steadied in ~ 10 ns with an RMSD of ~ 4 Å and persisted for at least 5 ns. Conformational differences among structures in the MD simulation became apparent when the starting structure was aligned to the structure of the enzyme at 15 ns. Notably, the B′/C loop and the F-G cassette were shifted away from the heme when compared with their initial positions, as seen in comparing the final MD structure (MDEND) with the initial MD structure, the closed ligand-free crystal structure (3MVR, closed conformation), the crystal structure of 2B4dH in complex with bifonazole (2BDM, expanded conformation), the crystal structure of 2B4dH in complex with 1-PBI (3G5N, intermediate conformation), and the “open” ligand-free crystal structure (1PO5, open conformation) (Fig. 5A–D). Furthermore, comparison of the structures in the MD simulation to available crystal structures of CYP2B4 provided a quantitative comparison of position differences (Fig. 5E). The 4-CPI (1SUO) and 1-CPI (2Q6N) were most similar to the starting structure (3MVR) with differences in RMSD of less than 1 Å. The average RMSD of the equilibrated ligand-free CYP2B4 structure during the MD simulation (MDEND) was similar to the RMSD of the CYP2B4 complex with 1-PBI (2.7 Å, 3G5N and 3G93). Larger RMSD values were observed for the “open” ligand-free enzyme (3.16 Å, 1PO5) and the bifonazole-bound structure (3.97 Å, 2BDM). The MD simulation shows that a wide range of conformations are energetically accessible to CYP2B4.
Figure 5.

Comparison of results from MD simulation to existing crystal structures of CYP2B4. Overlay of the ending structure of the MD simulation (MDEND, dark gray) with A) the closed ligand-free 2B4dH crystal structure (3MVR, closed conformation, cyan), B) the crystal structure of 2B4dH with bifonazole (2BDM, expanded conformation, red), C) the crystal structure of 2B4dH with three molecules of 1-PBI (3G5N, intermediate conformation, green), or D) the crystal structure of “open” ligand-free 2B4dH (1PO5, open conformation, yellow). E) RMSD of the Cα backbone of the P450 2B4dH crystal structures and MD simulation from the closed ligand-free P450 2B4dH structure (3MVR). The category axis is labeled with the PDB ID of the crystal structure or with MDEND for the average RMSD between 12 ns and 15 ns of the MD simulation (1SUO, P450 2B4dH with 4-CPI; 2Q6N, 2B4dH with 1-CPI; 3G5N, P450 2B4dH with three 1-PBI molecules; 3G93, P450 2B4dH with one 1-PBI molecule; 1PO5, P450 2B4 in the open conformation; 2BDM, P450 2B4dH with bifonazole). Modified from originally published figure in Wilderman et al. [24] and reproduced with permission from the American Society for Biochemistry and Molecular Biology. © the American Society for Biochemistry and Molecular Biology.
RATIONAL MUTAGENESIS
CYP2B6 and CYP2B11 were subjected to rational engineering efforts to enhance soluble protein expression and protein stability and to support solution biophysical and X-ray crystallographic efforts [26, 27]. First, utilization of the GroEL/ES chaperone system for coexpression of CYP2B6 was employed, which increased yields of CYP2B6 significantly [26]. Furthermore, a comparison of expression and stability of CYP2B enzymes revealed eleven locations where the residues were the same in CYP2B1, CYP2B4, and CYP2B11 but different from those in CYP2B6. The residues in CYP2B6 were mutated to those in the other three enzymes (F58L, M103V, V129L, I154V, M198L, L264F, S284H, E350D, Y354H, L390P, T394S). Steady state kinetic analysis with 7-EFC as a substrate revealed similar kcat/Km values to wild-type CYP2B6 for each mutant. L264F showed an ~4°C increase in melting temperature (Tm) compared with wild-type enzyme with a slightly higher catalytic tolerance to temperature (kinact). This mutant also showed minimal changes in functional properties of CYP2B6 in the presence of the imidazole inhibitor 4-CPI and the substrate benzphetamine. The improved stability of CYP2B6 L264F allowed the performance of ITC experiments using 1-benzylimidazole as the ligand that were not possible with the wild-type [26].
Subsequently, comparison of the relatively more stable CYP2B1 and CYP2B4 with the relatively less stable CYP2B6 and CYP2B11 revealed seven locations of interest. At these positions, the residues were similar/identical in CYP2B1 and CYP2B4 and different from the residue in CYP2B6 and CYP2B11, which were also similar/identical to each other. These residues in CYP2B6 and CYP2B11 were mutated to the residue identity found in CYP2B1 and CYP2B4 (V/I81T, V234I, E254A, Y352Q, P334S, I427M, Q473K); the L295H mutant, which was beneficial in CYP2B1 [49], was also included. For both CYP2B6 and CYP2B11, P334S showed increased expression levels by ~1.5-fold, and Y325Q and I427M expressed at approximately the same level as wild-type enzyme. P334S was the only mutant to exhibit an increased Tm in either CYP2B6 or CYP2B11; this mutant also decreased the thermal kinact of CYP2B6 and CYP2B11 and provided increased protection from pressure inactivation for both enzymes. Interestingly, introducing the S334P mutant into CYP2B1 and CYP2B4 decreased the Tm, increased thermal inactivation, and made the enzymes more susceptible to pressure inactivation.
PERSPECTIVES
In conclusion, additional high-resolution crystal structures, including two very recent CYP2B4 structures with a covalent adduct [50], have provided further insight into the remarkable plasticity of the CYP2B enzymes. The use of solution methods (NMR and DXMS) and computational methods (molecular docking and MD simulations) extends the knowledge of how these enzymes bind ligands of different sizes and shapes, providing a dynamic glimpse of enzyme behavior in addition to the snapshots provided by crystallography.
Accurate prediction of the metabolism of drug candidates is one of the long-term goals in the studies of P450s. This is challenging due to the conformational diversity and plasticity of drug metabolizing P450s. Crystal structures of CYP2B4 have been determined with the clinically relevant drugs clopidogrel and ticlopidine that are also metabolized by CYP2B6. In addition, a CYP2B6 structure in complex with 4-CPI has been solved; however, crystal structures of CYP2B6 substrate complexes have yet to be solved. With the addition of enzyme-substrate complexes, the current set of crystal structures and solution structural information from NMR and DXMS, and accelerated molecular dynamic simulation methods [51, 52], a good sampling of CYP2B “conformational space” should be available to provide a base of knowledge for modeling studies of P450-ligand interactions. Computational methods utilizing docking and high throughput screening of compound libraries, especially natural products, would provide further insight into possible clinically relevant substrates and inhibitors of these enzymes.
Rational mutagenesis guided by sequence comparison of CYP2B enzymes will allow the study of how non-active site residues modulate substrate specificity and catalytic efficiency. The highly polymorphic CYP2B6 is an excellent model system for studying the roles of non-active site residues in structure-function of P450s. Analysis of CYP2B6 polymorphisms via homology modeling and docking studies with CYP2B substrates and inhibitors will provide a basic understanding of pharmacological consequences of these amino acid differences. Additionally, future questions involving structure-function of CYP2B enzymes likely will require a blend of crystallography, solution, computational, and mutagenesis techniques.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grant ES003619 (to J.R.H.). Dr. P. Ross Wilderman is supported by National Institutes of Health Grant T32-DK007233. We appreciate the critical review of this manuscript by Dr. Sean Gay.
ABBREVIATIONS
- 7-BR
7-benzyloxyresorufin
- 1-CPI
1-(4-chlorophenyl)imdazole
- 4-CPI
4-(4-chlorophenyl)imidazole
- CYP
cytochrome P450-dependent monooxygenase
- CYP2B
cytochrome P450 2B subfamily of enzymes
- DXMS
hydrogen-deuterium exchange mass spectrometry
- 7-EFC
7-ethoxy-4-trifluoromethylcoumarin
- ITC
isothermal titration calorimetry
- kcat
enzyme turnover number
- kinact
thermal inactivation rate
- Km
Michaelis-Menten constant
- MD
molecular dynamics
- P450
cytochrome P450-dependent monooxygenase
- 1-PBI
1-biphenyl-4-methyl-1H-imidazole
- PDB
protein data bank
- PR
plastic region
- T1
longitudinal NMR relaxation
- Tm
thermal melting point of protein
References
- 1.Johnson EF, Stout CD. Structural diversity of human xenobiotic-metabolizing cytochrome P450 monooxygenases. Biochem Bioph Res Co. 2005;338(1):331–336. doi: 10.1016/j.bbrc.2005.08.190. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, Negishi M. Transcriptional regulation of cytochrome P450 2B genes by nuclear receptors. Curr Drug Metab. 2003;4(6):515–525. doi: 10.2174/1389200033489262. [DOI] [PubMed] [Google Scholar]
- 3.Zhao Y, Halpert JR. Structure-function analysis of cytochromes P450 2B. BBA-Gen Subjects. 2007;1770(3):402–412. doi: 10.1016/j.bbagen.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Lewis DFV, Lake BG, Ito Y, Anzenbacher P. Quantitative structure-activity relationships (QSARS) within cytochromes P450 2B (CYP2B) subfamily enzymes: The importance of lipophilicity for binding and metabolism. Drug Metabol Drug Interact. 2006;21(3–4):213–31. doi: 10.1515/dmdi.2006.21.3-4.213. [DOI] [PubMed] [Google Scholar]
- 5.Lewis DFV, Ito Y, Lake BG. Quantitative structure-activity relationships (QSARS) for inhibitors and substrates of CYP2B enzymes: Importance of compound lipophilicity in explanation of potency differences. J Enzym Inhib Med Ch. 2010;25(5):679–684. doi: 10.3109/14756360903514149. [DOI] [PubMed] [Google Scholar]
- 6.Kedzie KM, Philpot RM, Halpert JR. Functional expression of mammalian cytochromes P450IIB in the yeast Saccharomyces cerevisiae. Arch Biochem Biophys. 1991;291(1):176–186. doi: 10.1016/0003-9861(91)90121-x. [DOI] [PubMed] [Google Scholar]
- 7.Kedzie KM, Grimm SW, Chen F, Halpert JR. Hybrid enzymes for structure-function analysis of cytochrome P-450 2B11. Biochim Biophys Acta. 1993;1164(2):124–132. doi: 10.1016/0167-4838(93)90238-m. [DOI] [PubMed] [Google Scholar]
- 8.Kedzie KM, Balfour CA, Escobar GY, Grimm SW, He YA, Pepperl DJ, Regan JW, Stevens JC, Halpert JR. Molecular basis for a functionally unique cytochrome P450IIB1 variant. J Biol Chem. 1991;266(33):22515–21. [PubMed] [Google Scholar]
- 9.Mizukami Y, Sogawa K, Suwa Y, Muramatsu M, Fujiikuriyama Y. Gene structure of a phenobarbital-inducible cytochrome P-450 in rat liver. P Natl Acad Sci USA. 1983;80(13):3958–3962. doi: 10.1073/pnas.80.13.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gasser R, Negishi M, Philpot RM. Primary structures of multiple forms of cytochrome P-450 isozyme 2 derived from rabbit pulmonary and hepatic cdnas. Mol Pharmacol. 1988;33(1):22–30. [PubMed] [Google Scholar]
- 11.Domanski TL, Halpert JR. Analysis of mammalian cytochrome P450 structure and function by site-directed mutagenesis. Curr Drug Metab. 2001;2(2):117–137. doi: 10.2174/1389200013338612. [DOI] [PubMed] [Google Scholar]
- 12.Poulos TL. Cytochrome P450 flexibility. P Natl Acad Sci USA. 2003;100(23):13121–13122. doi: 10.1073/pnas.2336095100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis DFV, Ito Y. Human CYPs involved in drug metabolism: Structures, substrates and binding affinities. Expert Opin Drug Met. 2010;6(6):661–674. doi: 10.1517/17425251003674380. [DOI] [PubMed] [Google Scholar]
- 14.Poulos TL. Structural and functional diversity in heme monooxygenases. Drug Metab Dispos. 2005;33(1):10–18. doi: 10.1124/dmd.104.002071. [DOI] [PubMed] [Google Scholar]
- 15.Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. The 2.6-Å crystal structure of Pseudomonas putida cytochrome P-450. J Biol Chem. 1985;260(30):16122–16130. [PubMed] [Google Scholar]
- 16.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: Structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5(1):121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 17.Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. An open conformation of mammalian cytochrome P450 2B4 at 1.6-Å resolution. P Natl Acad Sci USA. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mast N, Charvet C, Pikuleva IA, Stout CD. Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem. 2010;285(41):31783–31795. doi: 10.1074/jbc.M110.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott EE, White MA, He YA, Johnson EF, Stout CD, Halpert JR. Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9-Å resolution: Insight into the range of P450 conformations and the coordination of redox partner binding. J Biol Chem. 2004;279(26):27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, White MA, Muralidhara BK, Sun L, Halpert JR, Stout CD. Structure of microsomal cytochrome P450 2B4 complexed with the antifungal drug bifonazole: Insight into P450 conformational plasticity and membrane interaction. J Biol Chem. 2006;281(9):5973–5981. doi: 10.1074/jbc.M511464200. [DOI] [PubMed] [Google Scholar]
- 21.Muralidhara BK, Negi S, Chin CC, Braun W, Halpert JR. Conformational flexibility of mammalian cytochrome P450 2B4 in binding imidazole inhibitors with different ring chemistry and side chains: Solution thermodynamics and molecular modeling. J Biol Chem. 2006;281(12):8051–8061. doi: 10.1074/jbc.M509696200. [DOI] [PubMed] [Google Scholar]
- 22.Gay SC, Sun L, Maekawa K, Halpert JR, Stout CD. Crystal structures of cytochrome P450 2B4 in complex with the inhibitor 1-biphenyl-4-methyl-1h-imidazole: Ligand-induced structural response through α-helical repositioning. Biochemistry. 2009;48(22):4762–4771. doi: 10.1021/bi9003765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gay SC, Roberts AG, Maekawa K, Talakad JC, Hong W-X, Zhang Q, Stout CD, Halpert JR. Structures of cytochrome P450 2B4 complexed with the antiplatelet drugs ticlopidine and clopidogrel. Biochemistry. 2010;49(40):8709–8720. doi: 10.1021/bi100914z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilderman PR, Shah MB, Liu T, Li S, Hsu S, Roberts AG, Goodlett DR, Zhang Q, Woods VL, Jr, Stout CD, Halpert JR. Plasticity of cytochrome P450 2B4 as investigated by hydrogen-deuterium exchange mass spectrometry and X-ray crystallography. J Biol Chem. 2010;285(49):38602–38611. doi: 10.1074/jbc.M110.180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gay SC, Shah MB, Talakad JC, Maekawa K, Roberts AG, Wilderman PR, Sun L, Yang JY, Huelga SC, Hong W-X, Zhang Q, Stout CD, Halpert JR. Crystal structure of a cytochrome P450 2B6 genetic variant in complex with the inhibitor 4-(4-chlorophenyl)imidazole at 2.0-Å resolution. Mol Pharmacol. 2010;77(4):529–538. doi: 10.1124/mol.109.062570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, Zhao Y, Sun L, Negi SS, Halpert JR, Muralidhara BK. Rational engineering of human cytochrome P450 2B6 for enhanced expression and stability: Importance of a Leu264->Phe substitution. Mol Pharmacol. 2007;72(5):1191–1199. doi: 10.1124/mol.107.039693. [DOI] [PubMed] [Google Scholar]
- 27.Talakad JC, Wilderman PR, Davydov DR, Kumar S, Halpert JR. Rational engineering of cytochromes P450 2B6 and 2B11 for enhanced stability: Insights into structural importance of residue 334. Arch Biochem Biophys. 2009;494(2):151–8. doi: 10.1016/j.abb.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gay SC, Roberts AG, Halpert JR. Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR. Future Med Chem. 2010;2(9):1451–1468. doi: 10.4155/fmc.10.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott EE, Spatzenegger M, Halpert JR. A truncation of 2B subfamily cytochromes P450 yields increased expression levels, increased solubility, and decreased aggregation while retaining function. Arch Biochem Biophys. 2001;395(1):57–68. doi: 10.1006/abbi.2001.2574. [DOI] [PubMed] [Google Scholar]
- 30.Wester MR, Johnson EF, Marques-Soares C, Dijols S, Dansette PM, Mansuy D, Stout CD. Structure of mammalian cytochrome P4502C5 complexed with diclofenac at 2.1 Å resolution: Evidence for an induced fit model of substrate binding. Biochemistry. 2003;42(31):9335–9345. doi: 10.1021/bi034556l. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Sun L, Muralidhara BK, Kumar S, White MA, Stout CD, Halpert JR. Structural and thermodynamic consequences of 1-(4-chlorophenyl)imidazole binding to cytochrome P450 2B4. Biochemistry. 2007;46(41):11559–11567. doi: 10.1021/bi7011614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bridges A, Gruenke L, Chang Y-T, Vakser IA, Loew G, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998;273(27):17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 33.Bumpus NN, Hollenberg PF. Cross-linking of human cytochrome P450 2B6 to nadph-cytochrome P450 reductase: Identification of a potential site of interaction. J Inorg Biochem. 2010;104(4):485–488. doi: 10.1016/j.jinorgbio.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams PA, Cosme J, Ward A, Angova HC, Vinkovic DM, Jhoti H. Crystal structure of human cytochrome P4502C9 with bound warfarin. Nature. 2003;424(6947):464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 35.Haines DC, Tomchick DR, Machius M, Peterson JA. Pivotal role of water in the mechanism of P450BM-3. Biochemistry. 2001;40(45):13456–13465. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- 36.Muralidhara BK, Halpert JR. Thermodynamics of ligand binding to P450 2B4 and P450EryF studied by isothermal titration calorimetry. Drug Metab Rev. 2007;39(2):539–556. doi: 10.1080/03602530701498182. [DOI] [PubMed] [Google Scholar]
- 37.Nishiya Y, Hagihara K, Ito T, Tajima M, Miura S, Kurihara A, Farid NA, Ikeda T. Mechanism-based inhibition of human cytochrome P450 2B6 by ticlopidine, clopidogrel, and the thiolactone metabolite of prasugrel. Drug Metab Dispos. 2009;37(3):589–593. doi: 10.1124/dmd.108.022988. [DOI] [PubMed] [Google Scholar]
- 38.Dalvie DK, O’connell TN. Characterization of novel dihydrothienopyridinium and thienopyridinium metabolites of ticlopidine in vitro: Role of peroxidases, cytochromes P450, and monoamine oxidases. Drug Metab Dispos. 2004;32(1):49–57. doi: 10.1124/dmd.32.1.49. [DOI] [PubMed] [Google Scholar]
- 39.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28(6):1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 40.Hess B, Kutzner C, van der Spoel D, Lindahl E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 41.Talakad JC, Shah MB, Walker GS, Xiang C, Halpert JR, Dalvie D. Comparison of in vitro metabolism of ticlopidine by human cytochrome P450 2B6 and rabbit cytochrome P450 2B4. 2011;39(3):539–550. doi: 10.1124/dmd.110.037101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cameron MD, Wen B, Allen KE, Roberts AG, Schuman JT, Campbell AP, Kunze KL, Nelson SD. Cooperative binding of midazolam with testosterone and α-naphthoflavone within the CYP3A4 active site: A NMR T1 paramagnetic relaxation study. Biochemistry. 2005;44(43):14143–14151. doi: 10.1021/bi051689t. [DOI] [PubMed] [Google Scholar]
- 43.Cameron MD, Wen B, Roberts AG, Atkins WM, Campbell AP, Nelson SD. Cooperative binding of acetaminophen and caffeine within the P450 3A4 active site. Chem Res Toxicol. 2007;20(10):1434–1441. doi: 10.1021/tx7000702. [DOI] [PubMed] [Google Scholar]
- 44.Regal KA, Nelson SD. Orientation of caffeine within the active site of human cytochrome P450 1A2 based on NMR longitudinal (T-1) relaxation measurements. Arch Biochem Biophys. 2000;384(1):47–58. doi: 10.1006/abbi.2000.2073. [DOI] [PubMed] [Google Scholar]
- 45.Eyles SJ, Kaltashov IA. Methods to study protein dynamics and folding by mass spectrometry. Methods. 2004;34(1):88–99. doi: 10.1016/j.ymeth.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Ferguson PL, Kuprowski MC, Boys BL, Wilson DJ, Pan JX, Konermann L. Protein folding and protein-ligand interactions monitored by electrospray mass spectrometry. Curr Anal Chem. 2009;5(2):186–204. [Google Scholar]
- 47.Liao WL, Dodder NG, Mast N, Pikuleva IA, Turko IV. Steroid and protein ligand binding to cytochrome P450 46A1 as assessed by hydrogen-deuterium exchange and mass spectrometry. Biochemistry. 2009;48(19):4150–4158. doi: 10.1021/bi900168m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frimpong AK, Abzatimov RR, Uversky VN, Kaltashov IA. Characterization of intrinsically disordered proteins with electrospray ionization mass spectrometry: Conformational heterogeneity of alpha-synuclein. Proteins. 2010;78(3):714–722. doi: 10.1002/prot.22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S, Sun L, Liu H, Muralidhara BK, Halpert JR. Engineering mammalian cytochrome P450 2B1 by directed evolution for enhanced catalytic tolerance to temperature and dimethyl sulfoxide. Protein Eng Des Sel. 2006;19(12):547–554. doi: 10.1093/protein/gzl042. [DOI] [PubMed] [Google Scholar]
- 50.Gay SC, Zhang H, Wilderman PR, Roberts AG, Liu T, Li S, Lin HL, Zhang Q, Woods VL, Stout CD, Hollenberg PF, Halpert JR. Structural analysis of mammalian cytochrome P450 2B4 covalently bound to the mechanism-based inactivator tert-butylphenylacetylene: Insight into partial enzymatic activity. Biochemistry. 2011;50(22):4903–11. doi: 10.1021/bi200482g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J Chem Phys. 2004;120(24):11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 52.Wereszczynski J, McCammon JA. Using selectively applied accelerated molecular dynamics to enhance free energy calculations. J Chem Theory Comput. 2010;6(11):3285–3292. doi: 10.1021/ct100322t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr Sect D-Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]