

Abstract
During murine kidney development, canonical WNT signalling is highly active in the renal tubules until about embryonic day E16-E18 when β-catenin transcriptional activity becomes progressively restricted to the nephrogenic zone. Several in vitro studies have found a link between cilial signalling and β-catenin regulation. The cilial protein genes PKD1 and PKD2 are known to be mutated in autosomal dominant polycystic kidney disease and previous studies proposed that these mutations could lead to a failure to suppress canonical WNT signalling activity; aberrant activity might then contribute to the cystic phenotype. However, in this study, we show that suppression of canonical WNT activity, defined by a TCF/β-catenin-lacZ reporter, is normal in two independent models of polycystic kidney disease. We crossed a TCF/β-catenin-lacZ reporter mouse with mice Pkd1 or Pkd2 mutations and found that there was no β-galactosidase staining in cells lining the renal cysts. This suggests that excessive β-catenin transcriptional activity may not contribute to cystogenesis in these models of autosomal dominant polycystic kidney disease.
INTRODUCTION
WNTs are secreted signalling molecules critical to many developmental processes such as organogenesis and their aberrant activity is associated with a number of disorders including diabetes and Alzheimer’s disease.1–3 In mammals, 19 different WNTs and 10 WNT receptors (Frizzleds) have been discovered thus far. Dependent on the context of the cell receiving a WNT signal, WNT molecules can be categorized as canonical or non-canonical. When a canonical WNT binds its cognate frizzled receptor with LRP5/6 co-receptor, the effector molecule β-catenin is stabilized in the cytosol and translocates to the nucleus where it binds T-cell factor (TCF)/Lymphoid Enhancer-binding Factor 1 (LEF1) to activate transcription of proliferative genes, such as c-myc.4, 5 WNTs can also activate β-catenin-independent (non-canonical) pathways that direct the asymmetric localization of intracellular proteins involved in determining cell shape, movement, and cytokinesis.6 Such processes involve numerous effector molecules whose functions remain less well understood, particularly in kidney development.
In 2005, Simons et al. proposed that, during normal kidney development, the onset of urinary flow-dependent cilial signals effects a switch from predominantly canonical to non-canonical WNT signalling activity.7 That study suggested that loss of genes involved in cilial signalling may lead to failure of events initiated by non-canonical WNT pathways and promote aberrant canonical WNT activity; either scenario could contribute to a cystic kidney phenotype. In support of the proposal that there is a developmental switch between canonical and non-canonical WNT signalling pathways, Karner et al. showed that WNT9b is capable of delivering a canonical message required for nephron induction and a later non-canonical signal involved in tubule elongation.8
During kidney development, intense canonical WNT activity is normally associated with rapid proliferation, branching morphogenesis of the ureteric bud tip, and the twisting morphogenesis of the emerging nephron.9, 10 Thus, it is tempting to speculate that failure to suppress canonical WNT signalling during tubular maturation may account for the development of cysts in autosomal dominant polycystic kidney disease (ADPKD). Indeed, a mouse model with unregulated β-catenin activity develops a severe cystic phenotype11 and a number of studies have linked cilial signalling to the downregulation of β-catenin activity.12–14 Based on these studies the current belief is that β-catenin is not suppressed normally in ADPKD and is aberrantly active in the epithelial cells lining cysts. However, Simons’ hypothesis also predicts a failure of non-canonical WNT pathways activation. In order to develop new therapeutic strategies for ADPKD, it is important to understand whether aberrant canonical WNT signalling or faulty non-canonical WNT signaling (or both) drive cystogenesis in ADPKD.
ADPKD is one of the most common monogenic disorders, affecting 1 in 400 to 1 in 1000 individuals and is characterized by the progressive development of bilateral renal cysts which eventually reduce renal function.15 The disease is caused by loss-of-function mutations in the PKD1 or PKD2 genes, which encode the membrane proteins polycystin-1 (PC-1) and PC-2 respectively.16 During kidney development in individuals carrying a mutated allele of PKD1 or PKD2, it is thought that some cells acquire second “hits” to the wildtype allele resulting in an aberrant cell phenotype. If the aberrant cell phenotype is due to ectopic β-catenin activity, then inhibition of the canonical WNT pathway might provide an effective therapeutic strategy.
In this study, we analyze the regulation of canonical WNT activity in ADPKD. We begin by observing that in normal mice, the infrastructure required for the developmental switch is in place by E16. Specifically, primary cilia are present on renal epithelial cells, the ureters have established a patent connection with the bladder and distension of Bowman’s capsule is evidence indicating that glomerular ultrafiltration has begun. To determine if β-catenin is inappropriately active in ADPKD, we crossed a TCF/β-catenin-lacZ reporter mouse, validated previously,17 to mice with mutations in Pkd1 or Pkd2 and determined the ontogeny of cyst formation relative to the normal developmental downregulation of TCF/β-catenin transcriptional activity.18
In homozygous Pkd1 null mice, cysts arose at E16 but canonical WNT reporter activity was suppressed normally. Since Pkd1 null mice die in utero we studied the hypermutable Pkd2/WS25 mice, which are viable and develop cysts by 2 weeks of age.19 The TCF/β-catenin-lacZ reporter activity was suppressed normally in the perinatal period. By 8 weeks of age, renal cysts were widespread but no TCF/β-catenin-lacZ reporter activity was observed. This study provides direct evidence that β-catenin transcriptional activity, as detected by the TCF/β-catenin-lacZ reporter, is unlikely to contribute to cystogenesis in ADPKD.
RESULTS
Primary cilia on renal tubular cells appeared by E15 and were present at the ureteric bud tips where β-catenin transcriptional activity is high in wildtype mice
In order to determine appearance of cilial structures in the developing kidney, frozen sections of wildtype kidneys were stained for anti-α-tubulin and analyzed by immunofluorescent histochemistry to visualize the primary cilia on renal tubular cells. Cilia were first observed at E15 (Figure 1A–C). To confirm the presence of cilia, ultrathin sections of wildtype kidneys from E15.5 embryos were analyzed with transmission electron microscopy (TEM). Primary cilia on renal epithelial cells including the basal bodies were clearly visible at E15.5 (Figure 1D). At E18.5, scanning electron microscopy (SEM) shows numerous primary cilia on the luminal side of the renal tubular cells (Figure 1E).
Figure 1. Ontogeny of primary cilia on renal epithelial cells.
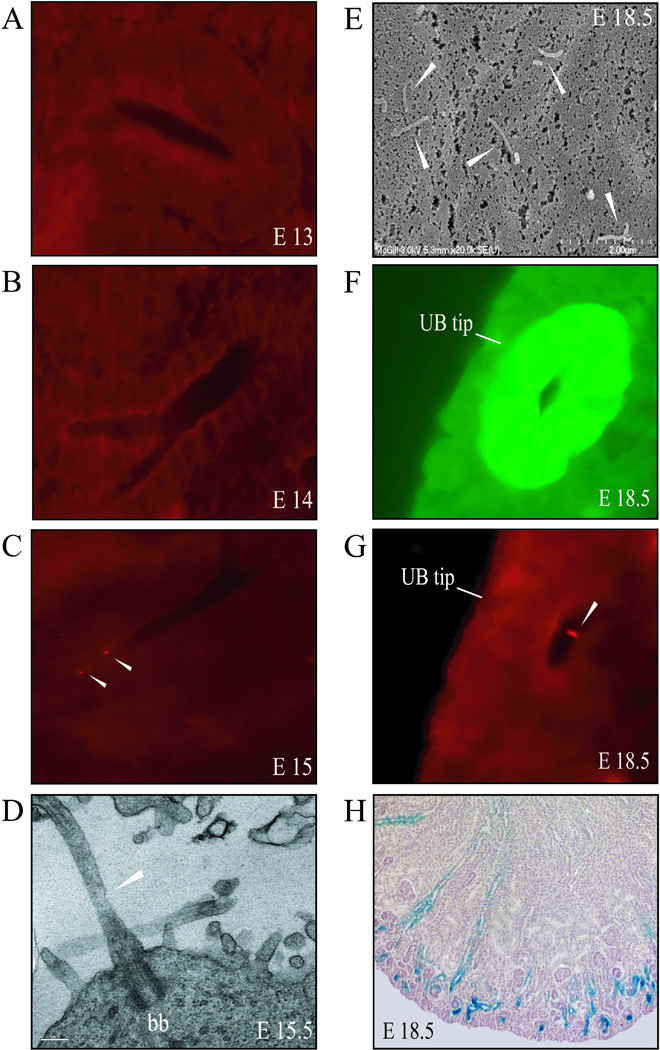
(A–C) Representative frozen sections of embryonic kidneys stained with anti-α-tubulin (red) show that cilia were absent at E13 and E14 but were easily detected at E15. (D) TEM confirmed the presence of primary cilia in E15.5 kidney samples. Basal body is indicated (bb). Scale bar represents 0.2µm. (E) At E18.5, SEM demonstrated that the primary cilia project up to 12µm into the lumen of the renal tubules. (F) A ureteric bud tip at the perimeter of the nephrogenic zone is identified by expression of a Hoxb7-GFP transgene. (G) The same E18.5 tissue section was stained with anti-α-tubulin antibody to identify cilia in the UB tip. (H) Characteristic β-catenin-lacZ staining (blue) at E18.5; the most intense reporter activity is seen in renal tubules in the nephrogenic zone. White arrowheads indicate cilia.
The ureteric bud (UB) tips were clearly distinguishable in frozen sections of E18.5 kidneys from a Hoxb7-GFP embryo (Figure 1F). Staining for anti-α-tubulin on the same E18.5 sample showed the presence of primary cilia at the UB tips (Figure 1G), which is an area of the developing kidney that exhibits strong TCF/β-catenin-lacZ reporter activity (Figure 1H).
By E16 the ureters have established a patent connection with the bladder and glomerular distension is evident
Simons’ work in vitro showed that renal tubular flow could reduce levels of active β-catenin.7 We used Hoxb7-GFP embryos and analyzed the time of contact between ureters and the bladder, which is a requirement for the initiation of renal tubular flow. The Hoxb7 promoter is active in the UB lineage and its derivatives, including the ureters,20 making the Hoxb7-GFP embryos ideal for studying the migratory path of the ureters. Embryos were micro-dissected and the GFP-containing ureters were visualized by fluorescent microscopy. By E15.5 the ureters had made a physical connection with the bladder, providing the outlet for urinary flow (Figure 2A). Tubular flow also requires an input provided by glomerular ultrafiltration, which is associated with distension of Bowman’s capsule. By E15.5 embryos had numerous distended glomerular structures (Figure 2B). To demonstrate patency at the uretero-vesicular junction, we micro-injected E16 kidneys with Lissamine Green. The dye entered the embryonic bladder and refluxed up the contralateral ureter (Figure 2C and D).
Figure 2. Conditions for urinary flow are present by E16.
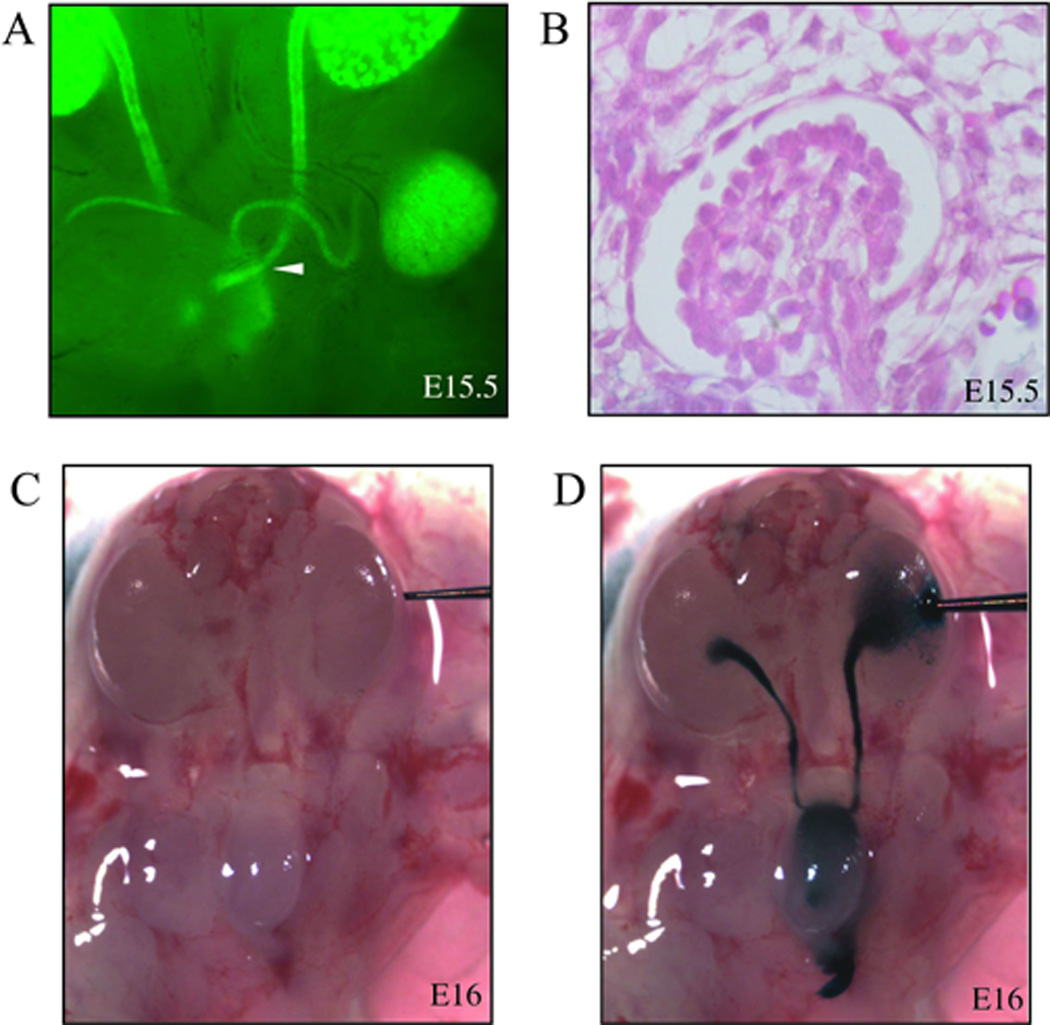
(A) Side-view of an E15.5 Hoxb7-GFP embryo showing a physical connection between ureter and bladder. White arrowhead indicates where ureter and bladder make contact. (B) A distended glomerulus in the kidney of an E15.5 embryo (hematoxylin/eosin). (C) Before and (D) after Lissamine Green micro-injection of an E16 kidney shows the dye in the renal pelvis, ureter, embryonic bladder, and contralateral ureter; demonstrating patency of uretero-vesicle junctions.
TCF/β-catenin transcriptional activity was restricted normally in homozygous Pkd1 null mutants
The TCF/β-catenin-lacZ reporter mouse demonstrates restriction of canonical WNT activity in the medulla while maintaining intense β-catenin transcriptional activity in the renal tubules of the nephrogenic zone beginning around E16 and continuing until the end of kidney development.18 In order to characterize the pattern of canonical WNT signalling activity in a cystic context, we crossed the TCF/β-catenin-lacZ reporter mouse with a previously described Pkd1 cystic model.21 In these mutant mice, exons 2–4 were removed resulting in a functionally null allele. Homozygous mice do not survive past E18 and develop kidney cysts.
Homozygous Pkd1 mutants bearing the TCF/β-catenin-lacZ reporter lacked visible renal cysts at E15 but, by E16, cysts were easily observed and by E17 cysts were widespread throughout the renal cortex and medulla (Figure 3). As previously reported, these mutant embryos do not survive to E18; the ontogeny of cyst formation closely duplicated that of previous studies.21 However, in the cystic kidneys, TCF/β-catenin-lacZ reporter activity was not seen in the cells lining the cysts suggesting that no aberrant canonical WNT signaling was present.
Figure 3. Cyst ontogeny and β-catenin reporter activity in Pkd1 homozygous mutant embryos.
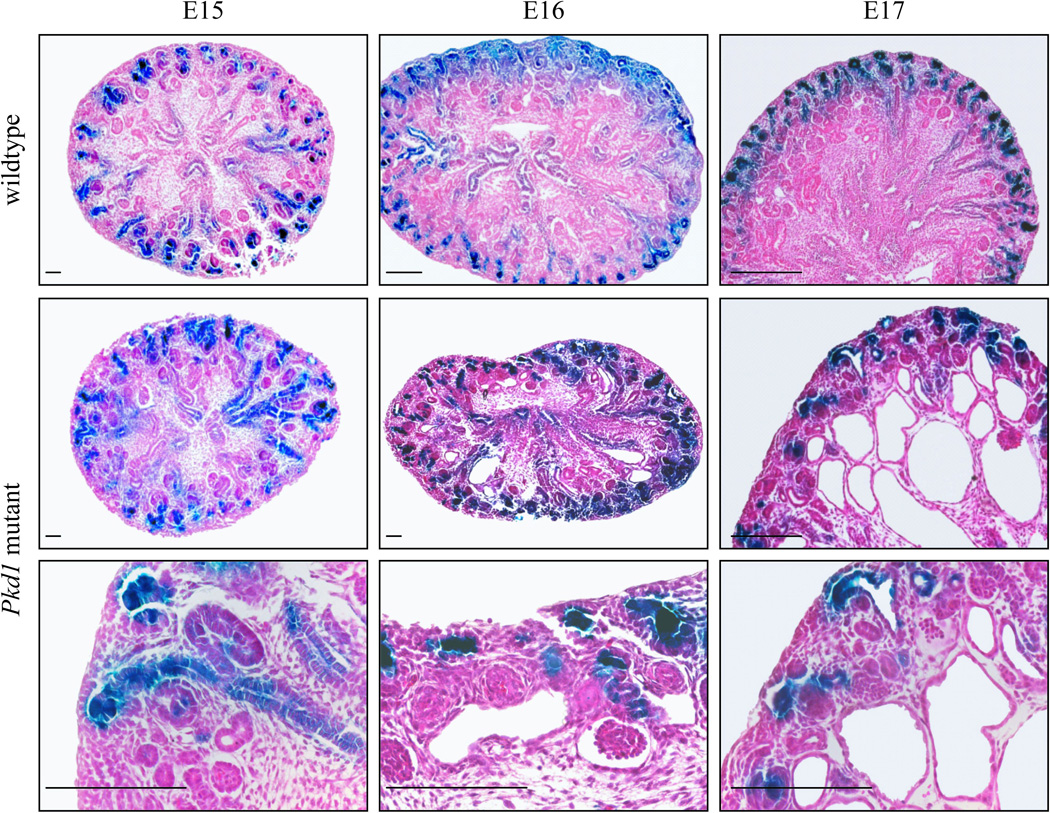
Kidney sections from wildtype littermates at E15, E16, and E17 (top panel) illustrate the normal restriction of β-catenin activity to the renal tubules of the cortical zone. Cysts were absent in embryos with homozygous mutations in Pkd1 at E15, but began to appear by E16 and were widespread at E17 (middle panel). Despite the development of renal cysts in Pkd1 null embryos, β-catenin activity was progressively restricted to the nephrogenic zone between E15 and E17, and no lacZ reporter activity was observed in cells lining ADPKD cysts (bottom panel). Scale bars represent 20µm.
TCF/β-catenin transcriptional activity was not present in postnatal cysts in Pkd2/WS25 mutants
The embryonic lethality of the homozygous Pkd1 mutants did not allow the examination of β-catenin activity in postnatal cysts. This led us to study the hypermutable Pkd2/WS25 model of ADPKD19 in combination with the TCF/β-catenin-lacZ reporter. The Pkd2 allele in these mice contains a neomycin cassette inserted into the first exon of the Pkd2 gene, which generates a premature stop codon and a null allele.19 The WS25 allele carries a large duplication of the 5’ region of the Pkd2 gene, which creates a highly unstable gene structure capable of frequent somatic recombination to produce a null allele.19 The Pkd2/WS25 mice bearing the lacZ reporter developed one to two cysts around 2 weeks of age and by 8 weeks of age the cysts were widespread (Figure 4).
Figure 4. No aberrant β-catenin activity is observed in Pkd2/WS25 mice.
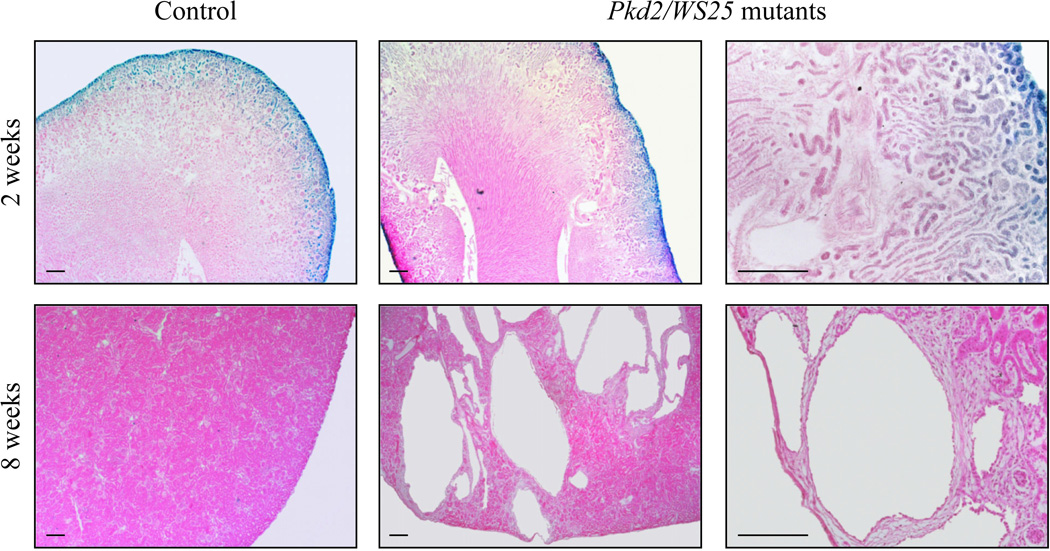
Paraffin-embedded kidney sections from 2 week old Pkd2/WS25 embryos carrying the TCF/β-catenin-lacZ reporter showed normal restriction of canonical WNT activity and a few cysts (top panel). At 8 weeks of age, cysts are clearly visible in the Pkd2/WS25 kidneys but no β-catenin activity was observed (bottom panel). Scale bars represent 20µm.
In the mouse, nephrogenesis is complete around 2 weeks after birth. Shortly after the kidney is fully developed, TCF/β-catenin-lacZ reporter activity disappears, making this an excellent reporter to study canonical WNT activity in postnatal cystic kidneys. In 8 week old Pkd2/WS25 mice carrying the TCF/β-catenin reporter, kidneys were removed and stained for β-galactosidase activity. No TCF/β-catenin-lacZ reporter activity was observed in the cystic kidneys (Figure 4) suggesting that canonical WNT signaling was not active in the cysts of Pkd2/WS25 mice. We also performed immunohistochemistry with an antibody to β-galactosidase on sections of E16 and 2 week cystic kidneys. Strong expression of the enzyme was evident at E16 in tips of the arborizing UB, S-shaped body, and emerging tubules, conforming to the pattern of TCF/β-catenin-lacZ reporter activity (Figure 5A and B). No β-galactosidase immunostaining was evident in renal tissue at 2 weeks (Figure 5C and D) nor in tissue from a mouse lacking the TCF/β-catenin-lacZ reporter (not shown).
Figure 5. The pattern of immunohistochemical staining for β-galactosidase conforms to the pattern of TCF/β-catenin-lacZ reporter activity.

Wildtype embryos bearing the TCF/β-catenin-lacZ reporter were subjected to immunohistochemistry for β-galactosidase without (A) and with (B) the addition of the primary antibody at embryonic day E16. Arrowheads show location of β-galactosidase in UB and comma-shaped body. At 2 weeks of age, no immunostaining for β-galactosidase was evident in the normal or cystic compartments of Pkd2/WS25 kidneys. Representative images at 10X magnification (A–C) and 40X magnification (B–D) are shown.
To further confirm our findings and corroborate the sensitivity of the TCF/β-catenin-lacZ reporter mouse to detect canonical WNT signalling activity, quantitative Western blots for unphosphorylated (active) β-catenin were performed on nuclear lysates of E16 and 2 week kidneys (Figure 6). No significant differences were observed in the amount of unphosphorylated β-catenin found in wildtype kidneys compared to Pkd2/WS25 kidneys.
Figure 6. Quantitative western blotting for active β-catenin shows no difference between wildtype and Pkd2/WS25 kidneys.

Western blots for active β-catenin were performed on nuclear lysates from E16 (A) and 2 week old (B) kidneys. Quantification relative to actin demonstrates no change in the levels of active β-catenin among the different genotype groups. M, marker.
DISCUSSION
At the onset of metanephric kidney development in the mouse, robust canonical WNT signalling is evident throughout the arborizing UB.18 Activation of TCF/LEF1/β-catenin gene targets appears to be critical for cell proliferation and motility that support branching at the UB tip; conditional knockout of β-catenin in the UB leads to premature terminal differentiation and cessation of branching.10, 22 Canonical WNT signalling is also seen in the early renal vesicle and S-shaped body, where it is crucial for nephrogenesis23; targeted inactivation of β-catenin in the induced mesenchyme prevents the normal emergence of nephrons.9, 24 As kidney development proceeds, the activity of our TCF/β-catenin transcription reporter becomes highly restricted. The signal fades from cells of the maturing collecting duct trunk but is sustained in outer UB tip cells where arborization continues. In the emerging S-shaped body it is downregulated in tubules as differentiation proceeds. By the early postnatal period, canonical WNT signalling activity is confined to a narrow rim where the final generations of nephrons are being formed; residual pathway activity is not readily detectable in adult kidney.
While canonical WNT signalling is crucial for the early events that drive nephrogenesis, the pathway must be downregulated for normal development to proceed. Transgenic mice with inappropriately sustained canonical WNT signalling develop cystic renal dysplasia.11 In 2005, Simons et al. proposed that canonical β-catenin signalling is downregulated by the cilial protein, inversin.7 They showed that inversin targets the signalling pathway molecule, dishevelled (DVL), for degradation. They also showed that inversin activates separate non-canonical WNT signalling pathways that are required for cell movement during embryonic animal cap elongation and gastrulation. Finally, they demonstrated that inversin expression is increased by fluid movement over MDCK cells in monolayer culture. Based on these observations, they proposed that inversin might act as “a molecular switch between different WNT signalling pathways” and that onset of tubular flow along the ciliated tubular epithelium might provide the signal for normal downregulation of WNT signalling during kidney development.7 Conversely, failure of this mechanism might underlie the renal cystic phenotype.
In this study, we report that primary cilia are evident in the renal tubular cells of wildtype mice by embryonic day E15.5. At this point, the caudally migrating nephric duct has penetrated the bladder wall, ultrafiltration has begun in some maturing glomeruli, as evidenced by distension of Bowman’s capsule, and patency at the ureteric orifice of the bladder was confirmed at E16. These events are rapidly followed by progressive loss of TCF/β-catenin-lacZ reporter activity between E16-E17. Thus, our observations define a developmental window between E15-E17 during which flow-mediated cilial function may begin, in temporal association with the suppression of canonical WNT signalling downstream of functioning glomeruli. Interestingly, renal development appears to progress normally in Pkd1 null knockout mice until this point and renal cysts first appear at about E16.
The molecular mechanisms which link cilial function to downregulation of the canonical WNT pathway are complex, but appear to converge on stabilization of cytoplasmic β-catenin by reduced phosphorlyated DVL. Recently, Kisimoto et al. discovered that seahorse, a cilial protein which binds to inversin, directly suppresses phosphorylation of DVL by casein II kinase-1; in the absence of phosphorylated DVL, β-catenin undergoes proteasome-dependent degradation.25 Corbit et al. reported a similar effect of the cilial protein, KIF3A, on DVL phosphorylation.26 Gerdes et al. reported that the basal body protein BBS4 promotes proteasome-dependent degradation of β-catenin itself.27 Taken together, these observations have lent support to the speculation that all human ciliopathies, including ADPKD, might reflect a failure to suppress canonical WNT signalling. Surprisingly, our observations show that TCF/β-catenin-lacZ reporter activity is downregulated normally in the embryonic kidneys of mice with homozygous inactivation of the Pkd1 gene. At E17, Pkd1 null cyst cells are devoid of TCF/β-catenin-lacZ reporter activity. Similarly, activity of the reporter transgene was fully suppressed when tracked into the postnatal period in Pkd2/WS25 mice. Canonical WNT signalling pathway activity detected by the transgene was restricted to the thin nephrogenic rim by two weeks of age and no ectopic canonical pathway signal was seen at 8 weeks postnatal age. Although flow-dependent cilial signals may suppress the β-catenin pathway, it appears that polycystins are not required.
There are several lines of indirect evidence that prompted the hypothesis that aberrant canonical WNT signalling might drive cystogenesis in human polycystic kidney disease. Lal et al. performed expression microarray analysis on cystic tissue from ADPKD patients at the time of nephrectomy and found an increase in β-catenin gene (CTNNB1) expression.12 However, changes in β-catenin transcript level may easily be secondary to chronic renal disease and do not necessarily reflect nuclear signalling activity of the pathway. Kim et al. reported a modest increase in cellular β-catenin protein in Pkd2 knockout mice, but immunohistochemistry for β-catenin and c-MYC did not establish clear evidence for increased nuclear levels of these proteins.13
Our observations support those of Lancaster et al., who examined canonical WNT signalling with a TCF/β-catenin reporter transgene in adult mice with knockout of the Ahi1 gene, mutated in Joubert Syndrome.28 Using a similar TCF/β-catenin-lacZ reporter transgene, they demonstrated a low level of residual pathway activity in adult kidney by increasing the sensitivity of the standard β-galactosidase assay. However, in Ahi1 null homozygotes, adult canonical WNT signalling was unexpectedly reduced in renal cysts following cisplatin administration.
In conclusion, this study supports the hypothesis by Simons et al. that primary cilia may preside over a developmental switch in WNT signalling pathways during renal development. Our observations suggest a window between E15-E17 during which primary cilia appear on renal epithelial cells, tubular flow begins, and canonical WNT signalling is progressively restricted in maturing segments of the nephron downstream of functional glomeruli. In mice with homozygous inactivation of Pkd1 or Pkd2 genes, renal development appears to proceed normally until the developmental transition at E15-E16, but tubular cysts arise thereafter. Surprisingly, however, developmental downregulation of canonical WNT signalling appears to be normal and we could find no evidence of aberrant TCF/β-catenin transcriptional activity that could contribute to the cystic phenotype. On the other hand, if ADPKD involves a failure of the developmental switch in WNT signaling pathways, as proposed by Simons et al., then the possibility remains that cystogenesis may be driven primarily by failure of polycystin-dependent activation of non-canonical WNT signalling.
METHODS
Immunofluorescent histochemistry
C3H embryonic kidneys from stages E13 to E18.5 were fixed overnight at 4°C in 4% paraformaldehyde and embedded in OCT (Sakura Finetek U.S.A Inc, Torrance, CA) and frozen. Twelve micron-sections were washed in 1X PBS, fixed in cold acetone for 10 minutes at room temperature, air-dried for 30 minutes and washed with 1X PBS. Sections were blocked with MOM mouse IgG blocking reagent and universal blocking serum (Vector Laboratories, Burlingame, CA) for 1 hour at room temperature followed by incubation with monoclonal anti-acetylated tubulin antibody (Sigma, Oakville, Canada) at 1:800 overnight at 4°C. After three washes with 1X PBS, sections were incubated with anti-mouse IgG (1:50) antibody (Sigma) for 1 hour at room temperature and washed three times in 1X PBS.
Transmission and scanning electron microscopy
For transmission electron microscopy (TEM), C3H embryonic kidneys were fixed overnight in 2.5% glutaraldehyde/0.1 M sodium cacodylate buffer at 4°C and washed 3 times in 0.1M cacodylate buffer for 10 minutes at 4°C. After, tissues were osmicated in 1% OsO4/1.5% KfeCN for 2 hours at 4°C. Tissue samples were then dehydrated with increasing percentages of acetone and infiltrated with solutions of varying ratios of acetone:epon. The samples were embedded in epon at 58°C for 48 hours. Ultrathin sections (80nm) were visualized with a JEOL JEM 2000 FXII transmission electron microscope (JEOL Ltd, Tokyo, Japan).
For scanning electron microscopy (SEM), samples were fixed in 3% glutaraldehyde. The samples were dehydrated and examined with a Hitachi field emission gun SEM (FE-SEM S-4700, Hitachi High Technologies America, Pleasanton, CA).
Visualization of ureter-bladder connection
C3H mice bearing a GFP transgene under the control of the Hoxb7 promoter have been described elsewhere.20 Timed crossings of the Hoxb7-GFP mice were done to generate embryos which underwent micro-dissection to visualize the location of the ureters relative to the bladder as performed previously.29
Micro-injection of Lissamine Green into E16 kidneys
Kidneys of C3H E16 embryos were micro-injected with 3% Lissamine Green (Sigma-Aldrich Ltd., St. Louis, MO) as described elsewhere.29
Generation of cystic mouse models
CD1 mice bearing a β-catenin-responsive lacZ reporter gene have been previously described.17 Briefly, this transgene contains six TCF/LEF response elements cloned upstream of a minimal Hsp68 promoter driving the lacZ reporter gene. The β-catenin reporter mice were crossed to Sv129 mice heterozygous for a deletion of exons 2 through 4 in the Pkd1 gene, which results in a functionally null allele as described elsewhere.21 An F1 intercross of hybrid mice carrying both the β-catenin reporter and Pkd1 deletion was performed to generate reporter bearing embryos homozygous for the Pkd1 mutation.
In parallel, the β-catenin reporter CD1 mice were also crossed to C57BL6 mice harboring the WS25 hypomorphic allele of the Pkd2 gene. Hybrid mice heterozygous for both the lacZ reporter and WS25 allele were backcrossed to C57BL6 mice bearing a Pkd2 null allele to generate animals with the β-catenin reporter and both mutant alleles of Pkd2. The Pkd2/WS25 model has been described previously.19 Animal procedures followed the guidelines established by the Canadian Council of Animal Care and were approved by the Animal Care Committee from McGill University.
β-galactosidase activity in kidney sections from embryonic and postnatal mice with autosomal dominant polycystic kidney disease
The protocol for β-galactosidase staining has been described elsewhere.30 Following the staining procedure, kidneys were embedded in paraffin, sectioned (6µm), and counterstained with haematoxylin/eosin.
Immunohistochemistry for β-Galactosidase on sections of E16 and P14 kidneys
Kidneys from P15 Pkd2/WS25 animals and control littermates bearing the TCF/β-catenin-lacZ reporter were fixed in 4% paraformaldehyde overnight at 4°C, embedded in paraffin, and cut into 6µm sections. Tissue sections were de-paraffinized in xylene and rehydrated by ethanol series. Sections were quenched with 3% hydrogen peroxide in methanol for 30 minutes and washed in 1X PBS for 5 minutes. Blocking was performed with horse serum from the Vectastain Universal Elite ABC Kit (Vector Laboratories) at room temperature for 30 minutes. The tissues were incubated in anti-β-galactosidase antibody (Promega Corp., Madison, WI) at 1:25 overnight at 4°C in a humidified chamber. The biotinylated secondary antibody solution was applied at 1:500 followed by incubation in the ABC Mix for 30 minutes each, at room temperature. The tissues were exposed to peroxidase substrate solution for a maximum of 2 minutes, or until color development was evident. Sections were counterstained with Gill’s Haemotoxylin (Electron Microscopy Sciences, Hatfield, PA) for 20 seconds followed by vigorous washing with tap water until stain was completely removed, and tissues were dehydrated by ethanol series.
Western blot of E16 and P14 Pkd2/WS25 kidney lysates for unphosphorylated β-catenin
Kidneys from E16 and P15 Pkd2/WS25 animals and control littermates were homogenized in cold hypotonic swelling buffer (10mM Tris-HCl (pH=7.4), 10mM NaCl, 3mM MgCl2, protease inhibitor cocktail (F. Hoffmann-La Roche Ltd., Quebec, Canada)) and left for 20 minutes on ice. NP40 (Roche Diagnostics, Quebec, Canada) was added to a final proportion of 10% and the lysates were centrifuged at 13,000rpm for 1 minute at 4°C. The supernatant (cytoplasmic fraction) was removed and the pellet was resuspended in cold lysis buffer (20mM HEPES, 600mM KCl, 0.2mM EDTA, protease inhibitor cocktail) and incubated on ice for 20 minutes. Suspensions were centrifuged at 13,000 rpm for 10 minutes at 4°C. Western blots were performed with 30µg protein from nuclear extracts from the Pkd2/WS25 and wildtype kidneys using the anti-active β-catenin (anti-ABC), clone 8E7 antibody (Millipore, Billerica, MA) at 1:500 with the secondary anti-mouse IgG HRP-linked antibody (Cell Signaling Technology Inc., Danvers, MA) at 1:2000. The blots were stripped to allow quantification of unphosphorylated β-catenin relative to the house-keeping protein actin with the anti-actin (Ab-1) mouse mAb (JLA20) antibody (EMD Chemicals Inc., Darmstadt, Germany) at 1:3000 with the secondary anti-mouse IgM:HRP conjugate antibody (Stressgen Bioreagents Corp., British Columbia, Canada) at 1:2000. Band intensity was measured using the ImageQuant TL v2003.02 software and ratios of β-catenin to actin were determined.
ACKNOWLEDGEMENTS
This work was supported by an operating grant from the Canadian Institutes of Health Research (MOP 82904) and an infrastructure support grant to the McGill University Health Centre Research Institute from the Fonds de Recherches en Santé du Québec. Michelle Miller was a recipient of a National Science and Engineering Research Council postgraduate fellowship. Dr. Iglesias was a recipient of a Kidney Foundation of Canada postdoctoral fellowship. Dr. Germino was funded by NIH grant DK48006 and in part by the Intramural Research Program of the NIH (NIDDK). Dr. Goodyer was the recipient of a CIHR James McGill Research Chair.
Footnotes
DISCLOSURE
All the authors declare no competing interests.
REFERENCES
- 1.Pulkkinen K, Murugan S, Vainio S. Wnt signaling in kidney development and disease. Organogenesis. 2008;4:55–59. doi: 10.4161/org.4.2.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin T. The WNT signalling pathway and diabetes mellitus. Diabetologia. 2008;51:1771–1780. doi: 10.1007/s00125-008-1084-y. [DOI] [PubMed] [Google Scholar]
- 3.Behrens MI, Lendon C, Roe CM. A common biological mechanism in cancer and Alzheimer’s disease? Curr Alzheimer Res. 2009;6:196–204. doi: 10.2174/156720509788486608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He X, Semenov M, Tamai K, et al. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663–1677. doi: 10.1242/dev.01117. [DOI] [PubMed] [Google Scholar]
- 5.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 6.Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4:68–75. doi: 10.4161/org.4.2.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simons M, Gloy J, Ganner A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karner CM, Chirumamilla R, Aoki S, et al. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet. 2009;41:793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JS, Valerius MT, McMahon AP. Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development. 2007;134:2533–2539. doi: 10.1242/dev.006155. [DOI] [PubMed] [Google Scholar]
- 10.Bridgewater D, Cox B, Cain J, et al. Canonical WNT/beta-catenin signaling is required for ureteric branching. Dev Biol. 2008;317:83–94. doi: 10.1016/j.ydbio.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Saadi-Kheddouci S, Berrebi D, Romagnolo B, et al. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- 12.Lal M, Song X, Pluznick JL, et al. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim I, Ding T, Fu Y, et al. Conditional mutation of Pkd2 causes cystogenesis and upregulates beta-catenin. J Am Soc Nephrol. 2009;20:2556–2569. doi: 10.1681/ASN.2009030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X. Cilia put a brake on Wnt signalling. Nat Cell Biol. 2008;10:11–13. doi: 10.1038/ncb0108-11. [DOI] [PubMed] [Google Scholar]
- 15.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 17.Mohamed OA, Clarke HJ, Dufort D. Beta-catenin signaling marks the prospective site of primitive streak formation in the mouse embryo. Dev Dyn. 2004;231:416–424. doi: 10.1002/dvdy.20135. [DOI] [PubMed] [Google Scholar]
- 18.Iglesias DM, Hueber PA, Chu L, et al. Canonical WNT signaling during kidney development. Am J Physiol Renal Physiol. 2007;293:F494–F500. doi: 10.1152/ajprenal.00416.2006. [DOI] [PubMed] [Google Scholar]
- 19.Wu G, D’Agati V, Cai Y, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 20.Srinivas S, Goldberg MR, Watanabe T, et al. Expression of green fluorescent protein in the ureteric bud of transgenic mice: a new tool for the analysis of ureteric bud morphogenesis. Dev Genet. 1999;24:241–251. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<241::AID-DVG7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 21.Piontek KB, Huso DL, Grinberg A, et al. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- 22.Marose TD, Merkel CE, McMahon AP, et al. Beta-catenin is necessary to keep cells of ureteric bud/Wolffian duct epithelium in a precursor state. Dev Biol. 2008;314:112–126. doi: 10.1016/j.ydbio.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuure S, Popsueva A, Jakobson M, et al. Glycogen synthase kinase-3 inactivation and stabilization of beta-catenin induce nephron differentiation in isolated mouse and rat kidney mesenchymes. J Am Soc Nephrol. 2007;18:1130–1139. doi: 10.1681/ASN.2006111206. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt-Ott KM, Masckauchan TN, Chen X, et al. beta-catenin/TCF/Lef controls a differentiation-associated transcriptional program in renal epithelial progenitors. Development. 2007;134:3177–3190. doi: 10.1242/dev.006544. [DOI] [PubMed] [Google Scholar]
- 25.Kishimoto N, Cao Y, Park A, et al. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Dev Cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Corbit KC, Shyer AE, Dowdle WE, et al. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- 27.Gerdes JM, Liu Y, Zaghloul NA, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 28.Lancaster MA, Louie CM, Silhavy JL, et al. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med. 2009;15:1046–1054. doi: 10.1038/nm.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murawski IJ, Myburgh DB, Favor J, et al. Vesico-ureteric reflux and urinary tract development in the Pax2 1Neu+/− mouse. Am J Physiol Renal Physiol. 2007;293:F1736–F1745. doi: 10.1152/ajprenal.00221.2007. [DOI] [PubMed] [Google Scholar]
- 30.Mohamed OA, Jonnaert M, Labelle-Dumais C, et al. Uterine Wnt/beta-catenin signaling is required for implantation. Proc Natl Acad Sci U S A. 2005;102:8579–8584. doi: 10.1073/pnas.0500612102. [DOI] [PMC free article] [PubMed] [Google Scholar]