

Abstract
Background
General anesthetics induce apoptotic neurodegeneration in the developing mammalian brain. General anesthesia (GA) also causes significant disturbances in mitochondrial morphogenesis during intense synaptogenesis. Mitochondria are dynamic organelles that undergo remodeling via fusion and fission. The fine balance between these two opposing processes determines mitochondrial morphometric properties, allowing for their regeneration and enabling normal functioning. As mitochondria are exquisitely sensitive to anesthesia-induced damage, we examined how GA affects mitochondrial fusion/fission.
Methods
Seven-day-old rat pups received anesthesia containing a sedative dose of midazolam followed by a combined nitrous oxide and isoflurane anesthesia for 6 h.
Results
GA causes 30% upregulation of reactive oxygen species (n = 3–5 pups/group), accompanied by a 2-fold downregulation of an important scavenging enzyme, superoxide dismutase (n = 6 pups/group). Reactive oxygen species upregulation is associated with impaired mitochondrial fission/fusion balance, leading to excessive mitochondrial fission. The imbalance between fission and fusion is due to acute sequestration of the main fission protein, dynamin-related protein 1, from the cytoplasm to mitochondria, and its oligomerization on the outer mitochondrial membrane. These are necessary steps in the formation of the ring-like structures that are required for mitochondrial fission. The fission is further promoted by GA-induced 40% downregulation of cytosolic mitofusin-2, a protein necessary for maintaining the opposing process, mitochondrial fusion (n = 6 pups/group).
Conclusions
Early exposure to GA causes acute reactive oxygen species upregulation and disturbs the fine balance between mitochondrial fission and fusion, leading to excessive fission and disturbed mitochondrial morphogenesis. These effects may play a causal role in GA-induced developmental neuroapoptosis.
Recent animal and emerging human data suggest that general anesthetics commonly used in pediatric medicine could be damaging to the developing nervous system. The neurotoxic effects are described as apoptotic in nature1–4 and are accompanied by severe and long-lasting disturbances in synaptogenesis.5–8 It appears that the impairment of synaptic development involves not only deletion of the existing synapses, but also a disturbance in the formation of novel synapses.9
Proper morphogenesis, function, and regional distribution of mitochondria are crucially important in the development and function of immature synapses and, consequently, for the formation of functional brain circuitries. Our recent studies indicate that general anesthesia (GA) causes statistically significant decrease in synapses, and disturbances in mitochondrial morphogenesis in the vicinity of synaptic connections, thus pointing at mitochondria as organelles likely to be responsible for anesthesia-induced impairment of neuronal development and synaptic function.10 Furthermore, we previously reported that the general anesthetic isoflurane, when combined with midazolam and nitrous oxide, causes apoptotic neurodegeneration that is, in part, mitochondria dependant.4 These findings collectively suggest that mitochondria could be an important and early target for GA-induced impairment of neuronal development and synaptogenesis.
Mitochondria are highly dynamic. Their ability to provide adequate support to the developing neurons relies on constant remodeling via fusion and fission.11 A fine dynamic balance between these two opposing processes depends on the physiological and metabolic requirements of a neuron. Overactive fission leads to mitochondrial fragmentation, whereas overactive fusion leads to undue mitochondrial enlargement. Both phenomena may cause impaired mitochondrial function.
Fusion and fission in mammalian neurons are controlled by many proteins. A protein of particular interest in the control of fission is an important member of the dynamin superfamily of proteins, dynamin-related protein 1 (Drp-1), which mediates the remodeling of the inner and outer mitochondrial membranes.12,13 Drp-1 translocates to the mitochondrial outer membrane and polymerizes to form a ring-like structure that enables mitochondrial division. A protein of particular interest in the control of fusion is mitofusin-2 (Mfn-2), a member of the Mfn family of proteins.11 Mfn-2 stabilizes the interaction between two adjacent mitochondria. 14 Interestingly, Mfn-2 also controls mitochondrial oxidative metabolism and the redox state of a neuron,15 a function that was of interest in view of our recently published findings, suggesting that GA causes upregulation of reactive oxygen species (ROS).16
We examined the acute in vivo effects of GA on the dynamic balance between mitochondrial fission and fusion, two key processes in mitochondrial proliferation, regeneration, and function. We administered a routine anesthesia cocktail containing isoflurane, nitrous oxide, and midazolam to rats during the intense stage of their brain development (at postnatal day [P] 7). We confirmed that acute anesthesia exposure results in an imbalance of ROS homeostasis, caused, in part, by modulation of the function of scavenging enzymes. We also discovered that GA causes downregulation of the fusion protein Mfn-2 and translocation of a fission protein, Drp-1, from cytoplasm to mitochondria, followed by the enhancement of Drp-1 oligomerization, ultimately leading to excessive mitochondrial fission.
Materials and Methods
Animals
Sprague–Dawley rat pups (Harlan Laboratories, Indianapolis, IN) at P7 were used for all experiments. This postnatal day is when rat pups are most vulnerable to anesthesia-induced neuronal damage.4 Our routine anesthesia protocol was used as previously described.10 Briefly, experimental rat pups were exposed to 6 h of anesthesia and controls were exposed to 6 h of mock anesthesia (vehicle + air). After the administration of anesthesia, rats were randomly divided into three groups: one group for ultrastructural analysis of the subiculum using electron microscopy, one group for assessing expression of several proteins using the Western blotting technique, and one group for functional studies of superoxide dismutase (SOD) and catalase activity using ELISA. Rat pups assigned for histological studies were reunited with their mothers and killed 24 h postanesthesia (at P8). Rat pups assigned for Western blot and ELISA studies were killed immediately postanesthesia (at P7).
The experiments were approved by the Animal Use and Care Committee of the University of Virginia Health System, Charlottesville, Virginia, and were performed in accordance with the Public Health Service’s Policy on Human Care and Use of Laboratory Animals. Efforts were made to minimize the number of animals used.
Anesthesia
We used our routine anesthesia protocol as previously described.10,16 Briefly, nitrous oxide and oxygen were delivered using a calibrated flowmeter. Isoflurane was administered using an agent-specific vaporizer that delivers a set percentage of anesthetic into the anesthesia chamber. Midazolam (Sigma–Aldrich Chemical, St. Louis, MO) was dissolved in 0.1% dimethyl sulfoxide just before administration. For control animals, 0.1% dimethyl sulfoxide was used alone. To administer a specific concentration of nitrous oxide/oxygen and isoflurane in a highly controlled environment, an anesthesia chamber was used. Rats were kept normothermic and normoxic while glucose homeostasis was maintained within normal limits throughout the experiment, as previously described.17,18 For control experiments, air was substituted for the gas mixture. After initial equilibration of the nitrous oxide/oxygen/isoflurane or air atmosphere inside the chamber, the composition of the chamber gas was analyzed by infrared analyzer (Datex Ohmeda, Madison, WI) to establish the concentrations of nitrous oxide, isoflurane, carbon dioxide, and oxygen. P7 rat pups received a single injection of midazolam (9 mg/kg, intraperitoneally) followed by 6 h of nitrous oxide (75%), isoflurane (0.75%), and oxygen (approximately 24%). Thus, the measured fraction of inspired oxygen in both control and experimental conditions was 0.21–0.24. Several studies have shown that this protocol causes significant developmental neuroapoptosis.1,4,10,16,19
Histopathologic Studies
On P8, each pup was deeply anesthetized with phenobarbital (65 mg/kg, intraperitoneally) (University of Virginia Pharmacy, Charlottesville, Virginia). Perfusion and fixation of brain tissue were performed as previously described.10,16 Briefly, the left ventricle was cannulated, the descending aorta was clamped, and an initial flush was carried out with Tyrodes solution (30–40 ml) (Sigma–Aldrich Chemical). For morphometric analyses of pyramidal neurons, we perfused with paraformaldehyde (2%) and glutaraldehyde (2%). After the perfusion, we removed the rats’ brains and stored them in the same fixative overnight. Both control and experimental pups were perfused by an experienced experimenter on the same day, using the same solution to assure uniform tissue fixation. Any brain considered to have been inadequately perfused was not processed for electron microscopy analysis. Our routine electron microscopy protocol has been described elsewhere.6,10 Briefly, fixed brains were coronally sectioned (50–75 μm thick) with a DTK-1000 microslicer (Ted Pella, Tools for Science and Industry, Redding, CA). The subiculum was localized as described in anatomical maps,20 fixed in 2% osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA), stained with 4% uranyl acetate (Electron Microscopy Sciences), and embedded in aclar sheets using epon–araldite resins. The subiculum was then dissected from the aclar sheets and embedded in BEEM capsules (Electron Microscopy Sciences). To prepare capsules for microtome cutting (Sorvall MT-2 microtome, Ivan Sorvall, Norwalk, CT), the tips were manually trimmed so that ultrathin slices (silver interference color, 600–900 Å) could be cut using a diamond knife (Diatome, Hatfield, PA). Ultrathin sections were placed on grids and examined using a 1230 TEM electron microscope (Carl Zeiss, Oberkochen, Germany).
Morphometric Analyses
Our protocol for morphometric analyses of mitochondria was previously described.10 Briefly, as the cytoplasmic soma of pyramidal neurons cannot be captured in their entirety with a single photo frame at such high magnification (×6,000–×12,000), we took multiple sequential pictures using a 16-megapixel digital camera (SIA-12C digital cameras, Scientific Instruments and Applications, Duluth, GA), then tiled them seamlessly together to make a mosaic of one whole cell body. We analyzed 5 neurons from each animal (n = 4 pups/group) for a total of 20 neurons in the control group and anesthesia-treated group each. For statistical analysis, we used n = 4 pups/group after we obtained the average from five neurons in each pup. From these mosaic pictures, the cytoplasmic and mitochondrial areas were measured using Image-Pro Plus 6.1 computer software (MediaCybernetics, Bethesda, MD). The number of animals necessary for these complex and time-consuming ultrastructural histological studies was determined based on our previously published studies (n = 4 pups in each group from four different litters. Equal numbers of male and female pups were used for each experimental condition. Control and experimental pups were equally represented from each litter).6,10 The investigator analyzing electron micrographs was blinded to the experimental conditions.
Catalase and SOD Activity Assays
Control and experimental groups of rats were killed immediately postanesthesia, and the subicular and thalamic brain tissues were removed quickly. The tissues were homogenized in 20 mm HEPES buffer at pH 7.2, containing 1 mm EDTA, mannitol, and sucrose per gram of brain tissue (for SOD) or cold phosphate-buffered saline (for catalase) with 1mm EDTA. Upon centrifugation at 1,500g, followed by centrifugation at 10,000g at 4°C, the supernatants were collected and the assays were carried out at 25°C. SOD activity was assayed using a commercially available kit (Superoxide Dismutase Assay kit, Cayman Chemical, Ann Arbor, MI) that can detect the activity of all three forms of SOD—Cu/Zn-, Mn-, and Fe-SOD—as absorbance at 440–460 nm. Catalase activity was assayed using a commercially available kit (OxiSelect Catalase Activity Assay kit, Cell Biolabs Inc., San Diego, CA) with absorbance detected at 520 nm. The assays were performed following the manufacturer’s instructions using a microplate reader (VersaMax, Molecular Devices, Chicago, IL). Total protein was measured for each sample on the day of the assay using a commercially available protein determination kit (Bradford method) (Cayman Chemical, Ann Arbor, MI). The activities of SOD and catalase were expressed in arbitrary units per milligram protein. The group sizes for each experimental condition are indicated in the figure legends.
Subcellular Fractionation
Mainly subicular tissues (with some thalamic contamination) were collected immediately postanesthesia. Briefly, the tissues were cut in small pieces and gently homogenized in ice-chilled Dounce homogenizers (20 strokes) using isotonic extraction buffer A from a Mitochondrial Isolation Kit (Sigma-Aldrich Co.) with protease inhibitor cocktail (Roche, Indianapollis, IN). The homogenates were centrifuged at 1,000g for 5 min to remove unbroken cells and nuclei. Supernatants were transferred into new tubes and centrifuged at low speed (3,500g for 10 min) to yield mitochondria-enriched fractions without lysosome and peroxisome contamination. Supernatants were removed and centrifuged at 70,000g to obtain pure cytosol fractions, and the mitochondria-enriched pellets were carefully resuspended and washed again in 1× extraction buffer A. The mitochondrial fractions then were re-pelleted by centrifugation at 1,000 and 3,500g for 5 and 10 min, respectively. Proteins from the mitochondria-enriched pellets were extracted by vortexing for 1 min in lysis buffer containing 20 mm Tris-HCl at pH 8.0, 137 mm NaCl, 10% glycerol, 1% nonidet P-40, and 2 mm EDTA. Following centrifugation at 13,000g, the supernatants were removed and protein concentration in the pellets was determined using the bicinchoninic acid micro-protein assay (Micro BCA protein assay kit, Pierce Inc., Rockford, IL). Subcelullar fractionation was performed at 4°C. The pellets are considered to contain the “heavy” mitochondrial fraction—enriched with mitochondria with substantially diminished presence of lysosomes and peroxisomes, which are common contaminants of this fraction.
Western Blotting
The protein concentration of the lysates was determined with the Total Protein kit (Sigma–Aldrich Chemical Co.). For separation of Mfn-2 and Drp-1 monomers, protein samples (30 μg per lane) were heat-denaturized in 2× Laemmli sample buffer, electrophoresed on a 10% sodium dodecyl sulfate-polyacrylamide gel, and transferred to a nitrocellulose membrane (Hybond ECL, Amersham International, Buckinghamshire, United Kingdom). To investigate Drp-1 oligomerization, the boiling step was omitted and 60 μg of samples were subjected to nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (without β-merkaptoethanol or dithlothreltol in 2× loading buffer) on 8% acrylamide gel21 and transferred to a polyvinylidene difluoride membrane (Millipore, Danvers, MA).
The membranes subsequently were incubated and probed with the anti-Drp-1 primary antibody or anti-Mfn 2 antibody at a dilution of 1:1000 each (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) in Tris-buffered saline–Tween overnight, followed by the incubation with the appropriate secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Inc.). Immunoreactive bands were visualized using enhanced chemiluminescence (Pierce, Inc.). β-ACTIN (1:10,000, Sigma-Aldrich) and porin (1:2500, Invitrogen, Eugene, OR) were used as loading controls for cytosolic and mitochondrial fraction, respectively. The molecular size of the proteins of interest was determined by comparison to pre-stained protein markers (BioRad, Hercules, CA). All gels were densitometrically analyzed in GBOX-chemi (Syngene, Frederick, MD) using the computerized image analysis program ImageQuant 5.0 (GE Heathcare, Life Sciences, Piscataway, NJ). Data were analyzed first as a ratio between the protein of interest and β-actin (or porin) and expressed as a percent change from a control density. The group sizes for each experimental condition are indicated in the figure Legends.
Spectrophotometric Detection of ROS
Control and experimental groups of rats were killed immediately postanesthesia, and the subicular and thalamic brain tissues were quickly removed. ROS were measured as hydrogen peroxide using the horseradish peroxidase-linked spectrophotometric assay kit according to the manufacturer’s instructions (Amplex Red, Invitrogen). Briefly, extracted brain mitochondria samples (120 μg) were added to a 96-well plate containing 100 μl of reaction buffer consisting of 0.1 U/ml of the horseradish peroxidase, 50 μm Amplex UltraRed, and 1 μl of dimethyl sulfoxide. Reactions were incubated at room temperature for 30 min and protected from light. Resorufin absorptions were followed at 560 nm using a VersaMax tunable microplate reader (Molecular Devices, Chicago, IL). Hydrogen peroxide levels are expressed in arbitrary units (per milligram protein). The group sizes for each experimental condition are indicated in the figure legends.
Statistical Analysis
Single comparisons among groups were made using an unpaired two-tailed t test. When ANOVA with repeated measures was needed, the Bonferroni correction was used to help maintain prescribed alpha levels (e.g., 0.05). Histograms in cumulative frequency analysis were compared with chi-square-test. Using the standard version of GraphPad Prism 5.01 software (Media Cybernetics, Inc., Bethesda, MD), we considered P < 0.05 to be statistically significant. All the data are presented as mean + SEM. No experimental data were missing or lost to statistical analysis.
Results
GA Induces Excessive Mitochondrial Fission in Developing Neurons
Our ultrastructural analysis of mitochondrial morphology in subicular pyramidal neurons revealed that anesthesia-treated animals contain numerous small round mitochondria displaying globular morphology 24 h postanesthesia exposure (on P8). Compared to controls (fig. 1A), it appeared that anesthesia-treated subicular neurons contained significantly more mitochondria (fig. 1B). The mitochondrial matrix was pale and showed signs of swelling. Although the inner and outer membranes appeared somewhat intact, the cristae seemed distorted and difficult to discern (fig. 1C), suggesting ultrastructural damage to mitochondria undergoing excessive fission.
Fig. 1.
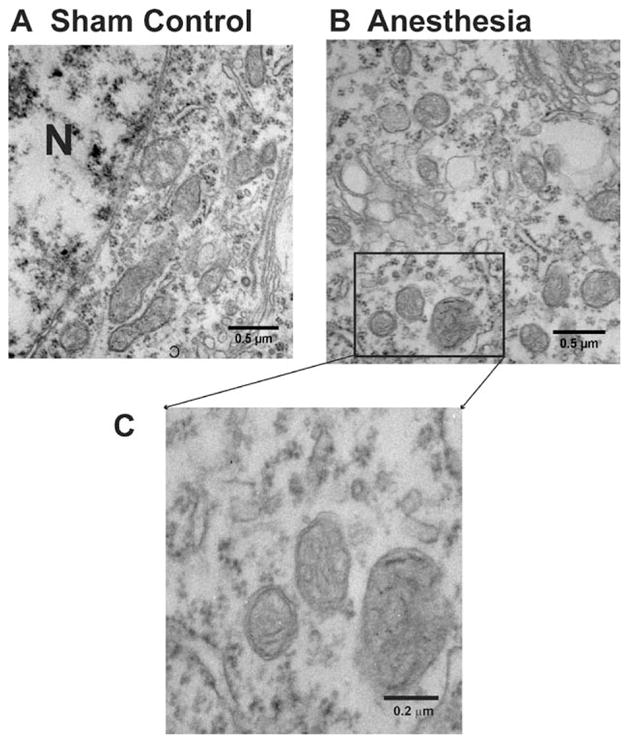
Anesthesia causes acute ultrastructural changes in mitochondria of pyramidal subicular neurons of 8-day-old rats. (A) Mitochondria in the cytoplasm of subicular pyramidal neurons from sham control animals resemble long tubules with intact inner and outer membranes and numerous cristae tightly packed inside healthy looking matrix. (B) Mitochondria in the cytoplasm of subicular pyramidal neurons from anesthesia- treated animals are numerous. The mitochondria are round, small, and display globular morphology 24 h postanesthesia exposure (on P8). Their matrix is pale and shows the signs of swelling. Although the inner and outer membranes appear somewhat intact, the cristae seem distorted and difficult to discern (C). N = nucleus.
To quantify the observed effect, which suggested that GA may increase mitochondrial density, we performed detailed morphometric analysis of each mitochondrion and determined mitochondrial density in the soma of pyramidal subicular neurons. We calculated mitochondrial density by counting the number of mitochondrial profiles per unit area (μm2) of cytoplasmic soma in each pyramidal neuron. We found that there were approximately 30% more mitochondrial profiles in experimental neurons compared to controls (* P = 0.0179) (fig. 2A). However, when the sum of mitochondrial areas was presented as a percent of the cytoplasmic area of pyramidal neurons, we found that mitochondria in the control and experimental neurons occupied approximately the same percent of the cytoplasmic soma (P = 0.8067) (fig. 2B), suggesting that the higher density of mitochondrial profiles could be due to enhanced fission, resulting in a shift of mitochondrial pool from a larger to a smaller category (n = 4 control and four experimental pups). To examine this notion, we performed frequency distribution analysis by grouping mitochondria by their area and counting the number of mitochondria in each bin as shown in figure 2C. We found that there were significantly more mitochondria smaller than 0.16 μm2 in experimental animals when compared to controls (P < 0.001, horizontal bar), suggesting a leftward shift in mitochondrial size due to GA treatment. Indeed, when we performed cumulative frequency analysis (in percentage), designed to take into account the differences in overall mitochondrial number in control versus experimental neurons, we found a leftward shift toward the smaller category (fig. 2D). For example, although mitochondria smaller than 0.012 μm2 were detected in GA-treated pyramidal neurons, none that small could be detected in control neurons. In addition, more than 50% of mitochondria in GA-treated neurons were smaller than 0.1 μm2, whereas about 30% were found in that size category in control animals. This finding suggests that GA may enhance mitochondrial fission.
Fig. 2.

Anesthesia induces excessive fission of mitochondria in the soma of pyramidal subicular neurons of 8-day-old rats. (A) Mitochondrial density was assessed by counting the number of mitochondrial profiles per unit area (μm2) of cytoplasmic soma in each pyramidal neuron. There are approximately 30% more mitochondrial profiles in anesthesia-treated neurons compared with controls (*P = 0.0179). (B) The summation of mitochondrial areas, presented as a percent of the cytoplasmic area of pyramidal neurons, reveals that mitochondria in the control and experimental neurons occupy approximately the same area of the cytoplasmic soma (P = 0.8067). (C) Frequency distribution analysis by grouping of mitochondrial area indicates that there are significantly more mitochondria smaller than 0.16 μm2 (indicated with horizontal bar) in experimental animals compared with controls (P < 0.001). (D) Cumulative frequency analysis (in percentage), designed to take into account the differences in overall mitochondrial number in control vs. experimental neurons, indicates a leftward shift toward smaller mitochondria after anesthesia treatment, with over 50% of mitochondria in the category of lesser than 0.1 μm2. In addition, mitochondria smaller than 0.012 μm2 were detected in the anesthesia-treated pyramidal neurons, whereas none that small could be detected in the control neurons (n = 4 control and four experimental pups, five neurons from each pup).
GA Causes Excessive Accumulation of ROS
To begin to understand the mechanism(s) by which anesthesia may cause excessive mitochondrial fission, we examined whether anesthesia exposure causes undue accumulation of ROS. This idea stems from the fact that oxidative stress, implicated in promoting fission and inhibiting fusion,21 may disturb the fine balance between these two processes crucial for proper mitochondrial remodeling.11 We measured ROS with a kit that detects hydrogen peroxide in fresh brain homogenate obtained from P7 rats immediately after 6 h of anesthesia. As shown in figure 3, the level of ROS in experimental animals was significantly increased (about 30%) compared to that in sham controls (*P = 0.0357) (n = 3 rat pups in the control group; n = 5 rat pups in the experimental group), suggesting that anesthesia promotes significant ROS accumulation.
Fig. 3.
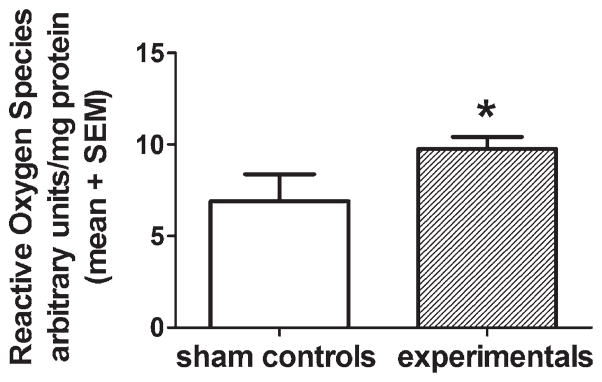
Anesthesia causes acute reactive oxygen species upregulation. Reactive oxygen species were measured in fresh homogenates of subicular and thalamic tissues obtained from P7 rats immediately after 6 h of anesthesia or sham treatment using a kit that detects hydrogen peroxide as described in Methods. We found that the level of reactive oxygen species in anesthesia-treated animals was increased significantly (about 30%) compared to that in sham controls (*P < 0.0357) (n = 3 rat pups in control group; n = 5 rat pups in the experimental group). P7 = postnatal day 7.
GA Acutely Impairs SOD but not Catalase Activity
As excessive ROS accumulation could be the result of ineffective scavenging machinery (responsible for maintaining ROS levels within normal limits), we set out to examine whether anesthesia has an acute effect on two important scavenging enzymes, SOD and catalase. We measured their activities in fresh brain homogenate obtained from P7 rat pups immediately after 6 h of anesthesia or sham treatment. The activity of SOD and catalase was expressed in units per milligram of protein. As shown in figure 4, there was a significant, 2-fold decrease in SOD activity immediately after anesthesia treatment compared to that in sham controls (fig. 4A) (**P = 0.0011; n = 6 pups in control group; n = 6 pups in experimental group). When we measured the activity of catalase, we found no change in the experimental groups compared to that in the sham controls (fig. 4B) (P = 0.6631; n = 6 pups in control group; n = 6 pups in experimental group).
Fig. 4.
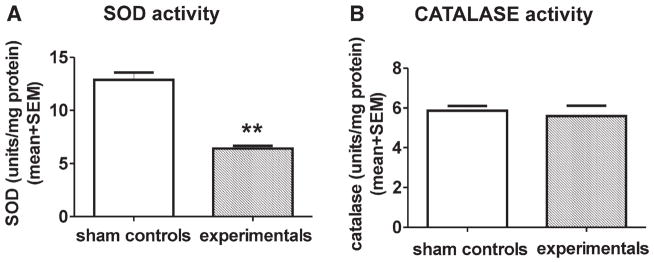
Anesthesia acutely impairs superoxide dismutase (SOD) but not catalase activity. The activities of SOD and catalase were measured in fresh homogenates of subicular and thalamic tissues obtained from P7 rat pups immediately after 6 h of anesthesia or sham treatment and are expressed in units per milligram of protein. (A) We found a significant 2-fold decrease in SOD activity immediately after anesthesia treatment compared to that in sham controls (**P = 0.0011) (n = 6 pups in the control group; n = 6 pups in experimental group). (B) There was no difference in catalase activity between the sham control and experimental groups (P = 0.6631) (n = 6 pups in the control group; n = 6 pups in the experimental group). P7 = postnatal day 7.
GA Modulates Expressions of Mfn-2 and Drp-1 Proteins, Two Important Regulators of Mitochondrial Fusion and Fission, Respectively
In view of our findings that anesthesia may cause excessive mitochondrial fission and inappropriate ROS accumulation, and the fact that oxidative stress can lead to disturbances of fine balance between mitochondrial fusion and fission, we assessed whether anesthesia modulates the expression of two key proteins responsible for maintaining mitochondrial dynamics, Mfn-2 and Drp-1. As Mfn-2 and Drp-1 proteins are involved in active remodeling of the outer and inner mitochondrial membranes, and could be localized in cytoplasm or be sequestered in the mitochondrial membrane, we measured their expression in both cytosolic and mitochondrial compartments. As shown in figure 5, there was about a 40% decrease in cytosolic Mfn-2 protein in GA-treated animals compared to that in sham controls (*P = 0.026; n = 6 pups in control group; n = 6 pups in experimental group) (fig. 5A). However, we found a slight but nonsignificant difference (P = 0.075) between Mfn-2 expression in the mitochondrial fractions of GA-treated versus sham controls (fig. 5B) (n = 9 pups in the control group; n = 9 pups in the experimental group).
Fig. 5.

Anesthesia decreases expression of mitofusin-2 (Mfn-2) in the cytosolic fraction. The expression of Mfn-2 protein was estimated from Western blotting in fresh cytosolic and mitochondrial fractions of subicular and thalamic tissues obtained from P7 rats immediately postanesthesia or sham treatment. The protein levels are estimated from Western blotting as percent change from sham controls after normalization to β-actin (cytosolic fraction) or porin (mitochondrial fraction). (A) In the anesthesia- treated group (Treat), Mfn-2 protein expression in the cytosolic fraction was decreased by about 40% compared to that in the sham controls (Cont) (*P = 0.026) (n = 6 pups in the control group; n = 6 pups in the experimental group). (B) In the anesthesia-treated group (Treat), Mfn-2 protein expression in the mitochondrial fraction was approximately the same as that in the experimental group compared to that in the sham controls (Cont) (P = 0.0745) (n = 9 pups in control group; n = 9 pups in experimental group). The molecular mass standards (in kDa) are shown at the right of the representative Western blots (C: cytosolic Mfn-2; D: mitochondrial Mfn-2). (*) Indicates a nonspecific band detected by anti-Mfn2 antibody. P7 = postnatal day 7.
As shown in figure 6, we detected a significant decrease (about 40%) (***P < 0.0001) in Drp-1 protein expression in cytosol from GA-treated versus sham-treated animals (fig. 6A) (n = 11 pups in the control group; n = 11 pups in the experimental group), and a significant increase (about 50%) (***P = 0.0002) in Drp-1 protein expression in the mitochondrial fraction of GA-treated versus sham-treated animals (fig. 6B), suggesting substantial translocation of Drp-1 to the mitochondrial membranes (n = 10 pups in the control group; n = 10 pups in the experimental group).
Fig. 6.

Anesthesia decreases expression of Drp-1 in the cytosolic fraction and increases Drp-1 in the mitochondrial fraction. The expression of Drp-1 protein was estimated by Western blotting in fresh cytosolic and mitochondrial fractions of subicular and thalamic tissues obtained from P7 rat pups immediately postanesthesia or sham treatment. The protein levels were expressed as percent change from sham controls after normalization to β-actin (cytosolic fraction) or porin (mitochondrial fraction). (A) In the anesthesia-treated group (Treat), Drp-1 protein expression in the cytosolic fraction was significantly decreased compared to sham controls (Cont) (***P < 0.0001) (n = 11 pups in control group; n = 11 pups in experimental group). (B) In the anesthesia-treated group (Treat), Drp-1 protein expression in the mitochondrial fraction was significantly increased compared to that in sham controls (Cont) (***P = 0.0002) (n = 10 pups in the control group; n = 10 pups in the experimental group). The molecular mass standards (in kDa) are shown at the right of the representative Western blots (C = cytosolic Drp-1; D = mitochondrial Drp-1). (*) Indicates alternate splice variant typically recognized by antibody against Drp-1. Drp-1 = dynamin-related protein 1; P7 = postnatal day 7.
Once Drp-1 is translocated from the cytoplasm to mitochondrial membranes, the Drp-1 monomer undergoes self-assembly (i.e., oligomerization) on the mitochondrial outer membrane, a step that allows the formation of the ring-like structure necessary for mitochondrial fission.12,13 As our findings suggest that anesthesia causes oxidative stress and may promote Drp-1 translocation to mitochondria, and in view of the fact that the oxidative stress can cause Drp-1 oligomerization, we set out to examine whether anesthesia promotes excessive formation of Drp-1 oligomers, which could, in part, explain enhanced mitochondrial fission. Indeed, we found that anesthesia increases the amount of the oligomerized form of Drp-1 protein by about 45% compared to that in controls (fig. 7; **P = 0.0037; n = 7 pups in the control group; n = 7 pups in the experimental group).
Fig. 7.

Anesthesia enhances Drp-1 oligomerization in the mitochondria. The samples for Drp-1 oligomer analysis were subjected to a nonreducing gel sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anesthesia (Treat) increases the protein content of the oligomerized form of Drp-1 in the mitochondrial fraction by about 45% compared to sham controls (Cont) (**P = 0.0037; n = 7 pups in the control group; n = 7 pups in the experimental group). The molecular mass standards (in kDa) are shown at the right of the representative Western blots. Drp-1 = dynamin-related protein 1.
Discussion
Early exposure to GA causes acute upregulation of ROS that is, in part, due to downregulation of SOD activity and lack of compensatory modulation of catalase activity. ROS upregulation is associated with impaired mitochondrial fission and fusion. This could be due to differential modulation of mitochondrial fission/fusion proteins. On the one hand, GA causes a decrease in Drp-1 protein in the cytoplasm due to an apparent translocation to mitochondria, with subsequent Drp-1 oligomerization on the outer mitochondrial membrane, a necessary step in the formation of the ring-like structures and fission. On the other hand, a lack of compensatory modulation of Mfn-2, a protein necessary for mitochondrial fusion, tips the fine equilibrium toward excessive mitochondrial fission (fig. 8). Based on previous reports suggesting that the GA causes massive developmental neuroapoptosis,1,4,18,19 and on others suggesting that excessive mitochondrial fission is associated with apoptotic cell death,21–23 we propose that excessive mitochondrial fission may be important for GA-induced developmental neurotoxicity.
Fig. 8.

Proposed pathways that may be responsible for the excessive mitochondrial fission caused by an early exposure to anesthesia. Anesthesia causes downregulation of superoxide dismutase activity accompanied by a lack of compensatory modulation of catalase activity, and these effects are associated with reactive oxygen species upregulation. Elevated reactive oxygen species differentially modulate mitochondrial fission and fusion. They are suggested to induce acute downregulation of Drp-1 protein in the cytoplasm due to its translocation to mitochondria, followed by its oligomerization on the outer mitochondrial membrane, a necessary step in the formation of the ring-like structures required for mitochondrial fission. General anesthesia also causes acute downregulation of mitofusin-2 (Mfn-2), a protein necessary for mitochondrial fusion, thus tipping the fine equilibrium between fission and fusion toward excessive mitochondrial fission. Mitochondria that undergo excessive fission are less functional and more likely to generate excessive amounts of reactive oxygen species, thus further promoting reactive oxygen species upregulation in the setting of downregulated superoxide dismutase activity. In addition, down-regulation of Mfn-2 in the cytoplasm disturbs the redox balance in the neuron, leading to additional reactive oxygen species accumulation. Drp-1 = dynamin-related protein 1.
Here we confirm that GA causes acute ROS upregulation.16 Moreover, we propose that a GA-induced disturbance in the neuronal redox state is likely caused by an imbalance between ROS production and ROS scavenging. Because mitochondria exposed to GA undergo excessive fission, and because unbalanced fission may lead to mitochondrial dysfunction11,21 resulting in excessive ROS generation, it is possible that overproduction of ROS is the likely cause of ROS upregulation as dysfunctional mitochondria could be the greatest intracellular source of ROS. Indeed, Barsoum et al.24 have shown that persistent mitochondrial fission leads to mitochondrial dysfunction and excessive production of ROS, one of the earliest signs of disturbed homeostasis leading to neuronal cell death.
Nevertheless, a fully functional scavenging system is also very important. For example, in order to manage a substantial increase in production of superoxide ions (normally generated with a rate constant that is already 3–8 times that of superoxide decomposition by SOD), the activity of SOD has to increase substantially.25 However, we show that GA induces statistically significant decrease in SOD activity, thus placing ROS homeostasis in double jeopardy: impaired scavenging in the setting of enhanced ROS production. The GA effect on scavenging enzymes seems to be selective. Despite a decrease in SOD activity, the activity of catalase is spared. Although the reason for this selective effect remains unclear, the lack of a compensatory increase in catalase activity in the setting of upregulated levels of its substrate, hydrogen peroxide, may worsen the acute oxidative stress in developing neurons. Although GA induces differential effects on the activity of these scavenging enzymes, it remains to be determined whether GA has an effect on their protein content.
It remains unclear whether GA-induced mitochondrial fission is the main cause of acute oxidative stress (especially as impaired mitochondria could be a powerful source of ROS) or its outcome, as mitochondria are also an important target of ROS. Disturbed redox state of the neuron regulates the translocation of Drp-1 from the cytosol to mitochondria and its oligomerization in vitro, thus directing mitochondrial dynamics toward excessive fission.21 Here we show that in vivo upregulation of ROS is associated with similar changes in Drp-1. Although the mechanism of Drp-1 oligomerization in vivo remains unsettled, previous in vitro studies have suggested that oxidative stress promotes the formation of thiol cross-links from cysteine residues in Drp-1, which could, in turn, promote the formation of Drp-1 oligomers.26
In some neurodegenerative diseases27 and some forms of acute brain injury,28 there is a shift toward excessive mitochondrial fission, leading to neuronal death. Consequently, Drp-1 inhibitors such as the Mdivi family of compounds were shown to prevent mitochondrial fission, loss of mitochondrial membrane potential, and neuronal death both in vitro and in vivo.28 In fact, Drp1 translocation to mitochondrial membrane and subsequent mitochondrial fission were the key features that preceded neuronal death. Although the safety profile of presently available Drp-1 antagonists in very young animals remains to be established, it is possible that these agents may offer some benefit in protecting against GA-induced disturbance in mitochondrial fission as Drp-1 may be an important cellular target of GA-induced developmental neurotoxicity.
Although the focus of this study was not on following the neuronal fate during excessive mitochondrial fission, we and other researchers previously have reported that early exposure to GA enhances developmental neuroapoptosis by massive activation of caspase-3.1,3,4,9,18,19 As excessive fission is considered to be an early occurrence during apoptosis, 22,23 the question remains whether apoptosis is caused by fission or whether fission is its consequence. Based on our previously published findings, one would suggest the former conclusion. We know that our GA protocol causes early cytochrome c leak, resulting in caspase-3 and -9 activation and apoptotic neurodegeneration that is considered to be intrinsic (mitochondria)-dependent.4 Although the exact timing of these processes vis-à-vis mitochondrial fission remains to be deciphered, we suggest that GA-induced mitochondrial fission promotes acute cytochrome c leak, leading to caspase activation. Some earlier reports support this view. For example, dominant negative mutants of Drp-1, which antagonize mitochondrial fission, have been shown to block cytochrome c release, apoptotic activation, and cell death.22,24,29,30 In addition, overexpression of the Mfn family of proteins (that promote mitochondrial fusion) has been known to curtail cytochrome c release and inhibit apoptosis. 31 However, other reports suggest that mitochondrial fission is an epiphenomenon that accompanies apoptosis. For instance, overexpression of the anti-apoptotic protein Bcl-xL (known to protect mitochondrial membrane) was demonstrated to block cytochrome c release in vitro, but did not block mitochondrial fission. This suggests that mitochondrial fission may not be the causal event in apoptosis-associated cytochrome c release.32 Regardless of what view is accepted, GA-induced ROS upregulation disturbs fusion/fission balance and promotes excessive mitochondrial fission, as we have shown here, while promoting excessive cytochrome c release and caspases 9 and -3 activation, as we have shown previously.4
We know that the long-term effects of early exposure to GA are manifested as mitochondrial enlargement.10 Here we report that the acute effects of GA are manifested as a decrease, not an increase, in mitochondrial size. Although it remains to be examined whether GA has long-term effects on fission/fusion machinery, possibly causing excessive fusion long after the initial exposure occurs, it is more likely that the mitochondria undergoing undue fission become vulnerable to fragmentation of cristae and inner mitochondrial membranes. This could, in turn, cause significant impairment of mitochondrial membrane integrity, leading to “leakiness” and swelling. Supporting this notion is our earlier report describing that mitochondrial swelling in addition to derangement and fragmentation of cristae indeed occurs when mitochondria are examined two weeks postanesthesia.10 In addition, a report by Barsoum et al.24 showed that excessive fission can impair the integrity of mitochondrial cristae often referred to as “cristae remodeling.”30,33
Although our study was not designed for live imaging of mitochondrial dynamics in terms of mitochondrial localization, antero versus retrograde transport along the neuronal axon, or speed of movement, it is possible that the disturbed fission and fusion could cause impaired mitochondrial agility and dynamics. Well-designed in vitro studies in which neurons can be examined during anesthesia exposure using time lapse confocal imaging will be needed to confirm this notion.
The concern regarding the maintenance of physiological homeostasis during anesthesia brings into focus the potential role of hypoxia in anesthesia-induced ROS upregulation. Indeed hypoxia may cause a significant rise in ROS in ischemic neurons, which could in turn initiate mitochondrial injury and neuronal death.34 As our earlier studies using the same GA protocol have ruled out the occurrence of hypoxia,1,4,18 it is unlikely that the GA-induced ROS upregulation we report herein is due to inadequate oxygenation.
The role of Mfn-2 protein extends beyond controlling fusion. Mfn-2 modulates metabolism via electron transport chain complexes I, IV, and V35,36 and controls oxygen consumption and electrochemical potential.15 Hence, Mfn-2 plays an important role in controlling the redox and metabolic state of the cell. Due to its role in curtailing oxidative stress and cytochrome c release, while interacting with anti-apoptotic bcl family of proteins,37 Mfn-2 may be crucial for maintaining mitochondrial integrity. Consequently, Mfn-2 is regarded as an important therapeutic target for the treatment of diseases caused by disturbed mitochondrial homeostasis and morphogenesis.38 Indeed some studies have shown that compensatory upregulation of Mfn-2 is the key to blocking the upregulation of ROS and mitochondrial fragmentation. 39,40 Because of the vital role of Mfn-2 in mitochondrial and neuronal function and its apparent downregulation by GA, we suggest that Mfn-2 could be an important target for preventive strategies aimed at curtailing GA-induced developmental neuroapoptosis.
Although our anesthesia protocol is a reliable model for studying developmental neurodegeneration, it is based on the use of anesthetics in combination. Therefore, despite the fact that clinical anesthesia commonly relies on the use of more than one anesthetic to achieve the desired effect, we were unable to examine the relative contribution of each agent. Further studies with individual anesthetics could decipher their relative importance.
In conclusion, early exposure to GA impairs mitochondrial morphogenesis and disturbs fission/fusion. This is accompanied by the disturbance of neuronal scavenging machinery and excessive ROS accumulation.
What We Already Know about This Topic
Common general anesthetics induce apoptotic neurodegeneration in the developing mammalian brain and disturb mitochondrial morphogenesis during synaptogenesis and fission
What This Article Tells Us That Is New
Early exposure to general anesthetics causes acute reactive oxygen species upregulation and disturbs the fine balance between mitochondrial fission and fusion, implicating yet another causal role for general anesthetics-induced developmental neuroapoptosis
Acknowledgments
This study was supported by the National Institute of Health/The Eunice Kennedy Shriver National Institute of Child and Human Development 44517 (to Vesna Jevtovic-Todorovic) and 44517-S (to Vesna Jevtovic-Todorovic), Bethesda, Maryland; Harold Carron endowment (to Vesna Jevtovic-Todorovic), University of Virginia, Charlottesville, Virginia; John E. Fogarty Award 007423-128322 (to Vesna Jevtovic-Todorovic), National Institute of Health, Bethesda, Maryland; and March of Dimes National Award (to Vesna Jevtovic-Todorovic). Vesna Jevtovic-Todorovic was an Established Investigator of the American Heart Association (Dallas, Texas), National Award.
The authors thank Jan Redick, B.S., Laboratory Director of the Advanced Microscopy Facility at the University of Virginia Health System, Charlottesville, Virginia, for technical assistance with electron microscopy and data analyses. The authors thank Shawn D. Feinstein, B.S. (Medical Student, Virginia Commonwealth University School of Medicine, Richmond, Virginia), and Kirsten Rose, Ph.D. (Research Associate, Department of Anesthesiology, University of Virginia Health System), for technical assistance with mitochondrial analysis and Western blot studies, respectively.
Footnotes
Information on purchasing reprints may be found at www.anesthesiology.org or on the masthead page at the beginning of this issue. Anesthesiology’s articles are made freely accessible to all readers, for personal use only, 6 months from the cover date of the issue.
References
- 1.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizzi S, Carter LB, Ori C, Jevtovic-Todorovic V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008;18:198–210. doi: 10.1111/j.1750-3639.2007.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slikker W, Jr, Zou X, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC, Doerge DR, Scallet AC, Patterson TA, Hanig JP, Paule MG, Wang C. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–58. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 4.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–27. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 5.Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–25. doi: 10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lunardi N, Ori C, Erisir A, Jevtovic-Todorovic V. General anesthesia causes long-lasting disturbances in the ultrastructural properties of developing synapses in young rats. Neurotox Res. 2010;17:179–88. doi: 10.1007/s12640-009-9088-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briner A, De Roo M, Dayer A, Muller D, Habre W, Vutskits L. Volatile anesthetics rapidly increase dendritic spine density in the rat medial prefrontal cortex during synaptogenesis. Anesthesiology. 2010;112:546–56. doi: 10.1097/ALN.0b013e3181cd7942. [DOI] [PubMed] [Google Scholar]
- 8.Briner A, Nikonenko I, De Roo M, Dayer A, Muller D, Vutskits L. Developmental stage-dependent persistent impact of propofol anesthesia on dendritic spines in the rat medial prefrontal cortex. Anesthesiology. 2011;115:282–93. doi: 10.1097/ALN.0b013e318221fbbd. [DOI] [PubMed] [Google Scholar]
- 9.Lemkuil BP, Head BP, Pearn ML, Patel HH, Drummond JC, Patel PM. Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology. 2011;114:49–57. doi: 10.1097/ALN.0b013e318201dcb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez V, Feinstein SD, Lunardi N, Joksovic PM, Boscolo A, Todorovic SM, Jevtovic-Todorovic V. General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology. 2011;115:992–1002. doi: 10.1097/ALN.0b013e3182303a63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 12.Yoon Y, Pitts KR, McNiven MA. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell. 2001;12:2894–905. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–56. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–62. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–92. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 16.Boscolo A, Starr JA, Sanchez V, Lunardi N, DiGruccio MR, Ori C, Erisir A, Trimmer P, Bennett J, Jevtovic-Todorovic V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: The importance of free oxygen radicals and mitochondrial integrity. Neurobiol Dis. 2012;45:1031–41. doi: 10.1016/j.nbd.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jevtovic-Todorovic V, Benshoff N, Olney JW. Ketamine potentiates cerebrocortical damage induced by the common anaesthetic agent nitrous oxide in adult rats. Br J Pharmacol. 2000;130:1692–8. doi: 10.1038/sj.bjp.0703479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu LX, Yon JH, Carter LB, Jevtovic-Todorovic V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–15. doi: 10.1007/s10495-006-8762-3. [DOI] [PubMed] [Google Scholar]
- 19.Yon JH, Carter LB, Reiter RJ, Jevtovic-Todorovic V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis. 2006;21:522–30. doi: 10.1016/j.nbd.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Australia: Academic Press; 1944. [Google Scholar]
- 21.Tian C, Murrin LC, Zheng JC. Mitochondrial fragmentation is involved in methamphetamine-induced cell death in rat hippocampal neural progenitor cells. PLoS ONE. 2009;4:e5546. doi: 10.1371/journal.pone.0005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 23.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–63. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 24.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–11. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crichton RR, Ward RJ. Oxidative Stress and Redox Active Metal Ions. In: Crichton RR, Ward RJ, editors. Metal-based Neurodegeneration: From Molecular Mechanisms to Therapeutic Strategies. Chapter 2. John Wiley & Sons; 2006. pp. 21–51. [Google Scholar]
- 26.Costantini P, Belzacq AS, Vieira HL, Larochette N, de Pablo MA, Zamzami N, Susin SA, Brenner C, Kroemer G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 2000;19:307–14. doi: 10.1038/sj.onc.1203299. [DOI] [PubMed] [Google Scholar]
- 27.Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev. 2011;67:103–18. doi: 10.1016/j.brainresrev.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–58. doi: 10.1038/cdd.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–27. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–56. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011;278:941–54. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 32.Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–85. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Scorrano L. Opening the doors to cytochrome c: Changes in mitochondrial shape and apoptosis. Int J Biochem Cell Biol. 2009;41:1875–83. doi: 10.1016/j.biocel.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–38. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacín M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–7. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 36.Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacín M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–15. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 37.Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–73. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 38.Jahani-Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–98. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- 39.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–8. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Liu X, Wang H, Zhang W, Chan DC, Shi Y. Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc Natl Acad Sci USA. 2012;109:6975–80. doi: 10.1073/pnas.1120043109. [DOI] [PMC free article] [PubMed] [Google Scholar]