

Abstract
The cytochrome P450 (CYP) 2C9 R150H (*8) allele occurs commonly in African Americans and is associated with lower warfarin dose requirements. We examined whether the CYP2C9*8 allele impacts warfarin clearance through a pharmacokinetic study in warfarin-treated African American patients and an in vitro kinetic study of S-warfarin 7-hydroxylation using cDNA-expressed CYP2C9 enzymes. We observed a 30% reduction in the unbound oral clearance of S-warfarin and 25% lower R- to S-warfarin plasma concentration in patients with the CYP2C9*8 allele (n=12) compared to CYP2C9*1 homozygotes (n=26). Consistent with these findings, the in-vitro intrinsic clearance of S-warfarin was 30% lower with the cDNA-expressed R150H protein compared to the wild-type protein. These data show that the R150H variant of the CYP2C9*8 allele reduces S-warfarin clearance, thus providing clinical and experimental evidence to explain lower warfarin dose requirements with the CYP2C9*8 allele.
Keywords: CYP2C9*8, warfarin, pharmacokinetics, polymorphism, metabolism
Introduction
Cytochrome P450 (CYP) 2C9 is responsible for the oxidative metabolism of approximately 15% of clinically used drugs, including warfarin.[1] Warfarin is the most commonly prescribed oral anticoagulant worldwide for the prevention of venous thromboembolism and stroke. Warfarin is characterized by a large inter-individual variability in the dose required for optimal anticoagulation and a narrow therapeutic index.[2] The CYP2C9 enzyme metabolizes the more potent S-warfarin enantiomer primarily to S-7-hydroxywarfarin, while CYP3A4 is the primary enzyme catalyzing R-warfarin hydroxylation.[3]
The highly polymorphic CYP2C9 gene has over 35 known variant alleles, many of which result in decreased enzyme activity.[4] In particular, it is well documented that the CYP2C9*2 (R144C) and *3 (I359L) alleles reduce enzyme activity, S-warfarin metabolism, and warfarin dose requirements.[3, 5-10] There is also evidence of increased bleeding risk with the CYP2C9*2 and *3 polymorphisms.[11, 12] The warfarin labeling was recently revised to include dosing recommendations based on CYP2C9 genotype, with recommendations limited to the CYP2C9*2 and *3 variants.[13]
While CYP2C9*2 and *3 are the predominant variants in Caucasians, with a prevalence of 30% to 35%, they occur in only about 6% of African Americans.[14, 15] Consequently, the CYP2C9*2 and *3 alleles explain less of the variance in warfarin dose among persons of African descent compared to European Caucasians.[16] The CYP2C9 R150H (rs7900194) polymorphism, which defines the CYP2C9*8 allele, is nearly twice as common as the CYP2C9*2 and *3 alleles combined in African Americans, with a frequency of 0.05 to 0.06.[17, 18] Thus, approximately 10% to 12% of African Americans carry the CYP2C9*8 allele. The CYP2C9 D360E (*5), 10601delA (*6), and R335W (*11) alleles also occur almost exclusively in African Americans, albeit at lower frequencies than the CYP2C9*8 allele, and are associated with reduced enzyme activity and clearance of CYP2C9 substrates.[19, 20]
We recently showed that African Americans with the CYP2C9*8 allele require significantly lower warfarin doses to achieve optimal anticoagulation compared to CYP2C9*1 allele homozygotes.[15] Similarly, Scott et al[17] described a single African American patient with the *8/*8 genotype who had a significantly lower warfarin maintenance dose than expected based on other genotypes and clinical factors. The CYP2C9*8 allele was also correlated with lower warfarin dose requirements in a South African population.[21] These data suggest that the CYP2C9*8 allele reduces warfarin clearance. However, there are limited data on the functional significance of the CYP2C9*8 allele, and existing data are conflicting. Specifically, the R150H variant was reported to increase metabolism of tolbutamide in vitro,[20] reduce metabolism of phenytoin in-vivo,[22] and have no effect on the in-vivo metabolism of losartan.[23] Importantly, there are no functional or pharmacokinetic studies of the CYP2C9*8 allele using warfarin as a phenotyping probe. Information about the extent to which the CYP2C9*8 allele impacts warfarin clearance is important to inform appropriate pharmacogenetic testing and warfarin dosing. In order to systematically investigate the impact of the CYP2C9*8 allele on warfarin clearance as an explanation for its association with lower warfarin dose requirements, we conducted an in vivo pharmacokinetic study in warfarin-treated patients and in vitro kinetic studies with cDNA-expressed CYP2C9.
Results
Pharmacokinetic study
A total of 38 African Americans on a stable dose of warfarin were enrolled, including 26 with the CYP2C9*1/*1, 10 with the CYP2C9 *1/*8, and two with the CYP2C9*8/*8 genotype. The majority of patients were female (Table 1) and taking warfarin for secondary prevention of venous thromboembolism (81%). Body surface area (BSA) was correlated with unbound oral clearance (CLpo,u) of S-warfarin in our cohort (P<0.05). Thus, BSA-adjusted clearance values are reported. No other clinical factor, demographic characteristic, or VKORC1 genotype was associated with S-warfarin clearance. With the exception of BSA-normalized CLpo,u (S), all data (including warfarin dose) were normally distributed. Warfarin dose requirements were lower in CYP2C9*8 allele carriers versus noncarriers, as we previously reported.[15] When excluding the two CYP2C9*8 homozygotes, doses remained lower in those with the *1/*8 genotype (5.2 ± 2.0 mg/day) compared to CYP2C9*1 allele homozygotes (P=0.02). All other characteristics and VKORC1 genotype distribution were similar between CYP2C9*8 allele carriers and noncarriers.
Table 1. Patient characteristics of CYP2C9*8 carriers and CYP2C9*1 homozygotes.
| Characteristic | *1/*8 or *8/*8 (n=12) |
*1/*1 (n=26) |
|---|---|---|
| Age (yrs) | 57 ± 12 | 54 ± 12 |
| Female | 9 (75) | 24 (92) |
| Body weight (kg) | 105 ± 37 | 99 ± 28 |
| Body surface area (m2) | 2.11 ± 0.33 | 2.02 ± 0.28 |
| Targ et INR | ||
| 2.0 | 9 (75) | 24 (92) |
| 2.5 | 3 (25) | 2 (8) |
| INR on enrollment | 2.7 ± 0.4 | 2.6 ± 0.4 |
| Estimated CrCl (ml/min) | 57 ± 27 | 62 ± 23 |
| Warfarin dose (mg/d) | 5.4 ± 1.9 | 7.4 ± 3.2* |
| Tobacco use | 2 (17) | 3 (12) |
| Alcohol use | 0 | 1 (4) |
| VKORC1 -1639 G>A Genotype | ||
| GG | 9 (75) | 23 (88) |
| AG | 3 (25) | 3 (12) |
Values are presented as No (%) or mean ± SD.
BSA, body surface area; INR, international normalized ratio; CrCl, creatinine clearance
P=0.02
Clinical pharmacokinetic data are shown in Table 2. As expected, CLpo (R) was similar between CYP2C9*8 carriers and *1 allele homozygotes. However, BSA-normalized CLpo,u(S) was 30% lower, and the Css ratio of R- to S-warfarin (i.e., Cp R:S) was 25% lower in CYP2C9*8 carriers. When excluding the two CYP2C9*8 homozygotes, BSA-normalized CLpo,u (S) was 28% lower in CYP2C9*8 heterozygotes compared to the CYP2C9*1 homozygotes, as shown in Fig. 1.
Table 2. Clinical pharmacokinetic parameters of R- and S-warfarin in CYP2C9*8 carriers and CYP2C9*1 homozygotes.
| Kinetic parameter | *1/*8 or *8/*8 (n=12) |
*1/*1 (n=26) |
P value |
|---|---|---|---|
| CLpo (R) (ml/min) | 1.94 ± 0.79 | 1.89 ± 0.58 | 0.850 |
| BSA- normalized CLpo (R) (ml/min/m2) | 0.90 ± 0.22 | 0.94 ± 0.27 | 0.626 |
| CLpo (S) (ml/min) | 2.39 ± 0.90 | 3.27 ± 1.70 | 0.044 |
| BSA-normalized CLpo (S) | 1.12 ± 0.36 | 1.63 ± 0.86 | 0.015 |
| CLpo,u (S) (ml/min) | 182 ± 69 | 248 ± 142 | 0.062 |
| BSA-normalized CLpo,u (S) (ml/min/m2) | 85 ± 27 | 122 ± 66 | 0.033* |
| Cp (R)/(S) | 1.29 ± 0.42 | 1.73 ± 0.69 | 0.021 |
Values are presented as mean ± SD.
P value for log-transformed data
Figure 1.
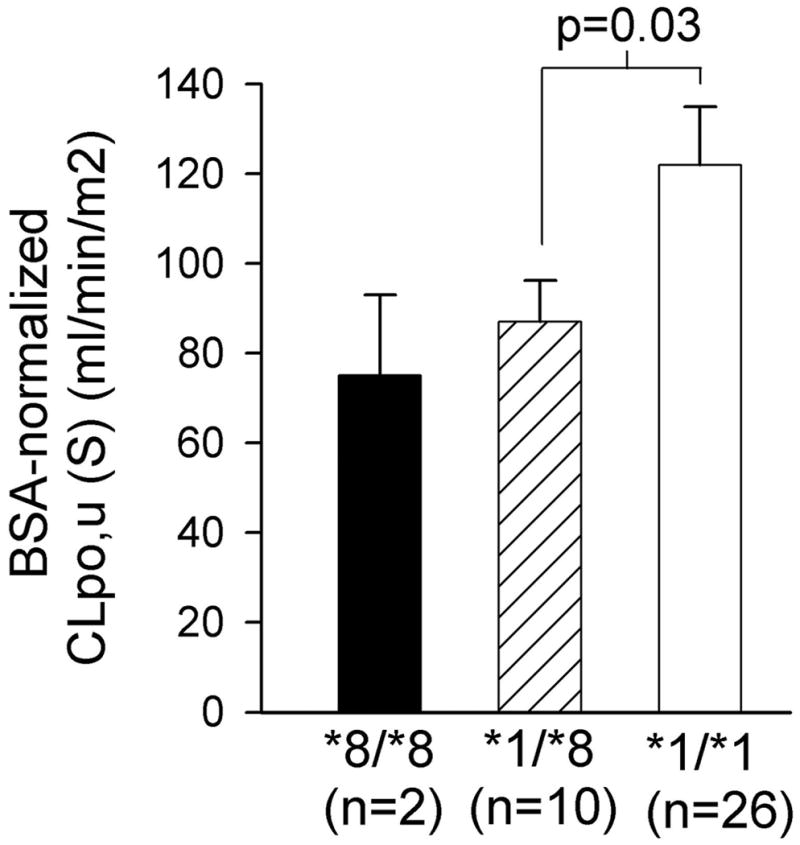
Mean ± SEM body-size adjusted unbound, oral clearance of S-warfarin according to CYP2C9 R150H (*8) genotype. P value by one-sided unpaired t-test of log-transformed data.
In vitro enzyme kinetic study
Enzyme kinetic profiles for S-warfarin 7-hydroxylation and diclofenac 4′-hydroxylation in cDNA-expressed wild-type CYP2C9 and its three variants are shown in Fig. 2, and the resulting kinetic parameter estimates are presented in Table 3. In all of the reactions studied, the CYP2C9 R144C (*2) and I359L (*3) proteins showed appreciable decreases in metabolic activity compared with wild-type CYP2C9. Intrinsic clearance (Vmax/Km) of both S-warfarin 7-hydroxylation and diclofenac 4′-hydroxylation were lower for R144C and I359L compared to the wild type protein, consistent with previous studies.[8, 9, 24] The apparent Km of S-warfarin 7-hydroxylation with the R150H variant was similar to that of the wild-type, but the apparent Vmax was significantly lower than that of wild type protein (p<0.01), resulting in an overall 30% reduction of the intrinsic clearance compared with the wild-type protein (p<0.05). In contrast, the R150H protein catalyzed diclofenac 4′-hydroxylation at rates comparable to, but slightly lower than, those of the wild-type CYP2C9.
Figure 2.
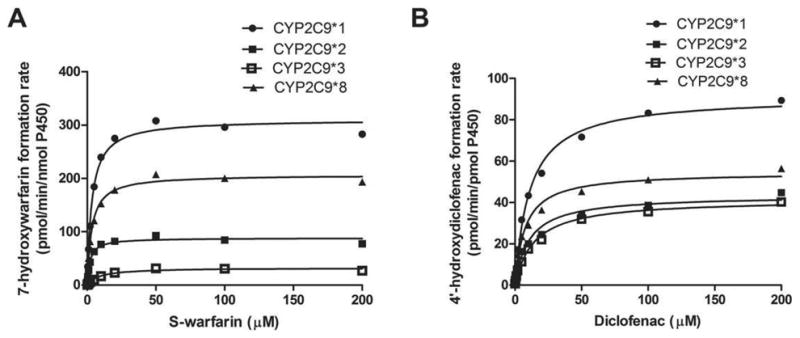
Kinetics of S-warfarin 7-hydroxylation (A) and diclofenac 4′-hydroxylation (B) in cDNA-expressed wild-type CYP2C9 and its three variants. All data points shown represent the mean of three independent experiments in duplicate measurements.
Table 3.
Kinetic parameters for S-warfarin 7-hydroxylation and diclofenac 4′-hydroxylation in cDNA-expressed wild-type CYP2C9 and its variants.
| CYP2C9*1 | CYP2C9*2 | CYP2C9*3 | CYP2C9*8 | |
|---|---|---|---|---|
| S-warfarin | ||||
| Vmax (pmol/min/nmol P450) | 310 ±10 | 88±16** | 32±4** | 207±30** |
| Km (μM) | 3.3±0.1 | 2.0±0.3** | 9.1±1.0** | 3.3±0.5 |
| Vmax/Km (μl/min/nmol P450) | 92.5±1.3 | 43.8±3.0** | 3.6±0.1** | 63.8±7.7* |
| Diclofenac | ||||
| Vmax (pmol/min/pmol P450) | 91±12 | 44±3** | 41±3** | 55±6** |
| Km (μM) | 11.1±0.4 | 11.0±2.4 | 14.4±1.4* | 7.4±0.4** |
| Vmax/Km (μl/min/pmol P450) | 8.2±1.1 | 4.1±0.8** | 2.9±0.4** | 7.3±0.7 |
Values are presented as mean ± SD of three independent experiments in duplicate.
P < 0.05;
P < 0.01 vs. CYP2C9*1
Discussion
Our group and others have reported lower warfarin dose requirements among persons of African descent who have a CYP2C9*8 allele, as defined by the R150H amino acid substitution.[15, 17, 21] These data suggest that, similar to CYP2C9*2 and *3 alleles, the CYP2C9*8 variant reduces enzyme activity. However, data on the clinical and functional significance of the CYP2C9*8 allele are inconsistent and even conflicting. Specifically, in-vitro data with the CYP2C9*8 allele show greater activity toward tolbutamine.[20] This is in contrast to clinical pharmacokinetic studies reporting a reduction in phenytoin metabolism and no effect on losartan metabolism with the CYP2C9*8 allele.[22, 23] To our knowledge, our study is the first to examine the effects on the CYP2C9*8 variant on warfarin kinetics.
We found that warfarin-treated patients with the CYP2C9*8 allele have significantly lower body size-adjusted unbound clearance of S-warfarin, a good indicator of the hepatic metabolizing activity, compared to CYP2C9*1 homozygotes, indicating that the CYP2C9*8 variant results in a reduction of S-warfarin elimination. In contrast, as expected, there was no significant difference in the clearance of R-warfarin between genotype groups. Because S-warfarin possess more potent anticoagulant activity than the R-enantiomer, [2] and S-warfarin 7-hydroxylation dominates its oral, unbound clearance, [25] our data support and explain our previous observation of lower warfarin dose requirements with the CYP2C9*8 allele.[15]
Only two patients had the *8/*8 genotype, limiting comparisons between the 3 possible genotypes at the R150H position. However, body size-adjusted unbounded clearance of S-warfarin was significantly lower with the *1/*8 compared to the *1/*1 genotype, suggesting that pharmacokinetic differences by the R150H genotype were not driven by inclusion of the two homozygotes for the variant allele.
Our in vitro kinetic data are consistent with our in-vivo data and offer experimental evidence that the reduction of S-warfarin clearance results from decreased enzyme activity due to the R150H substitution. The mechanism underlying the effects of the R150H variant on S-warfarin 7-hydroxylation remains unclear and requires further investigation. The R150H variant is located in exon 3 distal to the R144C substitution that defines the CYP2C9*2 allele. The R144C substitution is located outside the active site of CYP2C9. Nonetheless, it is well documented that the CYP2C9*2 variant moderately reduces enzyme activity and dose requirements of warfarin.[5, 6, 8, 10] The reduced catalytic activity of R144C may be related to an altered interaction between CYP2C9 and cytochrome P450 reductase[9] or differential uncoupling to shunt products.[26] Given the fairly close proximity of the R144C and R150H substitutions, R150H may reduce enzyme activity through a mechanism similar to that of the R144C allele; although this has yet to be determined.
Our clinical and in-vitro findings with warfarin are consistent with previous findings of reductions in phenytoin metabolism with the CYP2C9*8 allele.[22] However, our in-vitro data indicate that R150H catalyzes diclofenac 4′-hydroxylation at rates comparable to those of the wild-type, a result different from that with warfarin. The magnitude of reduction in enzyme activity with different CYP2C9 variants is reported to be highly substrate-dependent.[27] For example, similar to our findings, Dickmann et al.[19] showed that the CYP2C9*3 and *5 variants have a greater impact on S-warfarin 7-hydroxylation than on diclofenac 4′-hydroxylation. Our disparate in-vitro findings with warfarin and diclofenac, together with conflicting data on CYP2C9*8 effects on the metabolism of other substrates, support substrate-dependent activity of the R150H allele. Kumar et al.[28] suggest that substrates bind to different regions (or orientation) within the large active site of CYP2C9, and this may provide a potential explanation for substrate-dependent effects of CYP2C9 variants on enzyme activity. In the absence of confirmatory data on the substrate specificity of the CYP2C9*8 variant, our disparate findings with warfarin and diclofenac should serve as a caution against generalizing our findings with warfarin and the CYP2C9*8 allele to other substrates.
In order to more clearly elucidate the effects of the CYP2C9*8 allele on warfarin clearance, we excluded patients taking potent CYP2C9 inducers or inhibitors or with significant liver disease. As such, we cannot draw any conclusions about the effect of the CYP2C9*8 allele on warfarin clearance in these types of patients. We also excluded patients with the CYP2C9*8 allele and another CYP2C9 variant (e.g. *2/*8). The CYP2C9*1/*2 and *2/*2 genotypes are reported to reduce S-warfarin clearance by approximately 40% and 70%, respectively, whereas we observed a 28% reduction in clearance with the CYP2C9*1/*8 genotype. Thus, one might expect a reduction in clearance within the intermediate range of 40% to 70% with the *2/*8 genotype. Another limitation to our study is that we limited our analysis to the CYP2C9*8 allele. The less common CYP2C9*6 and *11 alleles also occur almost exclusively in African Americans, and their effects on warfarin clearance remain to be determined.
In conclusion, our pharmacokinetic and in vitro data indicate that the R150H substitution defining the CYP2C9*8 allele results in an intermediate reduction of S-warfarin metabolism. The present findings shed light on the mechanism underlying reduced warfarin dose requirements with the CYP2C9*8 allele. Ultimately, consideration of CYP2C9*8 genotype, in addition to CYP2C9*2, CYP2C9*3, and other variants that result in reduced warfarin clearance may improve the accuracy of warfarin dosing and potentially reduce the risk for supratherapeutic anticoagulation and bleeding across racial groups.
Methods
Clinical pharmacokinetic study
Patients
Warfarin-treated African American patients with the CYP2C9 R150H variant were identified among patients genotyped as part of previous warfarin pharmacogenetic studies.[15, 18] Inclusion criteria for the current study were treatment with a fixed maintenance dose of warfarin for at least 2 weeks and an international normalized ratio (INR) within 0.1 units of the target range by point-of-care testing using the ProTime® monitor (ITC, Edison NJ, USA). Exclusion criteria were concomitant treatment with moderate to potent inducers or inhibitors of CYP2C9-mediated warfarin metabolism (e.g. phenytoin, carbamazepine, rifampin, amiodarone, metronidazole, sulfonamindes), documented history of hepatic disease or recent (within the previous 6 months) serum transaminase levels greater than 2 times the upper limit of normal, and nonadherence to warfarin within the previous 2 weeks. Of 26 patients from our clinic who were previously genotyped for the R150H allele (6 homozygotes and 20 heterozygotes),[15, 18] 10 were no longer followed in the clinic, 4 had another CYP2C9 variant (e.g. *5 or *11) or were taking a CYP2C9 inducer (e.g. carbamazepine) and were excluded, and the remaining 12 were enrolled. Additional patients with the CYP2C9*1/*1 genotype, based on absence of the CYP2C9*2, *3, *5, *6, *8, or *11 allele, who met the same eligibility criteria and were of similar age and gender as R150H carriers were consecutively enrolled as they presented to the Antithrombosis Clinic for their regularly scheduled visit.
Procedures
Patients were approached about study participation during a regularly scheduled visit to the pharmacist-managed anticoagulation clinic at the University of Illinois at Chicago. After obtaining written informed consent, a venous blood sample was collected 12 to 16 hours after the last warfarin dose for determination of warfarin enantiomer concentrations and confirmation of CYP2C9 genotype. Clinical data were collected via subject interview and review of the medical record. Creatinine clearance was estimated using the Cockcroft-Gault equation with ideal body weight.[29] The study protocol was approved by the University of Illinois at Chicago Institutional Review Board and conducted according to the Declaration of Helsinki.
CYP2C9 genotyping
Genomic DNA was isolated from whole blood using a Puregene kit (Qiagen, Valencia, CA). The CYP2C9 R144C (*2), I359L (*3), D360E (*5), 10601delA (*6), and R335W (*11) polymorphisms and the VKORC1 -1639G>A (rs9923231) genotype were determined by PCR and pyrosequencing, and R150H (*8) was determined by PCR and capillary sequencing, as previously described.[30-32]
Pharmacokinetic data analysis
Total and free concentrations of warfarin enantiomers were determined by a chiral HPLC-based method as previously described.[33, 34] The oral clearance (CLpo) of both enantiomers and unbound oral clearance (CLpo,u) of S-warfa rin were calculated according to equations 1 and 2:[34]
| (1) |
| (2) |
in which D is the daily dose of racemic warfarin, τ is the dosing interval, Css is the average total plasma concentration of S- or R-warfarin at steady state, and Cuss is the average unbound plasma concentration of S-warfarin at steady state.. This formula assumes, based on experimental data, that the oral bioavailability of racemic warfarin is complete, and that warfarin plasma concentration measured 12 to 16 hours after dosing at steady state is very close to the Css.[34] Pharmacokinetic parameters were corrected for BSA based on a correlation between BSA and CLpo,u in our cohort and previous evidence that physiologic parameters relevant to drug metabolism and elimination (e.g., renal and hepatic function) are proportional to body surface area.[35]
In-vitro kinetic studies
Chemicals
S-warfarin, 7-hydroxywarfarin, diclofenac, 4′-hydroxydiclofenac, mebendazole, indomethacin, isocitric acid, isocitric acid dehydrogenase, and NADP+ were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents were of HPLC grade or of the highest grade commercially available.
Cell Culture
HepG2 cells from ATCC (Manassas, VA) were cultured in complete DMEM supplemented with 10% fetal bovine serum (Gemini, Woodland, CA, USA), 2 mM L-glutamine, 100 U penicillin/ml, 100 μg streptomycin/ml, and 1% MEM nonessential amino acids.
Enzyme kinetics assays
To examine the effect of the R150H allele on CYP2C9 activity, a kinetic study of S-warfarin 7-hydroxylation was conducted using cDNA-expressed wild-type or variant CYP2C9 proteins. The CYP2C9 R144C (*2) and I359L (*3) proteins were included as positive controls. Diclofenac, a well-established substrate of CYP2C9, was included as a control substrate. cDNA-expressed CYP2C9*1 (wild type), *2 (R144C), *3 (I359L), and *8 (R150H) coexpressing human NADPH-P450 reductase (CPR) and human cytochrome b5 (b5) were purchased from BD Biosciences Corp. (Woburn, MA, USA). The P450 contents were provided by the manufacturer in the datasheets. A typical incubation mixture (200 μl total volume) contained recombinant CYP2C9*1, *2, *3, or *8 (final concentration: 8 pmol/incubation for S-warfarin and 4 pmol/incubation for diclofenac), 100 mM Tris-HCl buffer (pH 7.4), NADPH-generating system (5 mM isocitric acid, 0.2 unit/ml isocitric acid dehydrogenase, 5 mM magnesium chloride, 1 mM NADP+), and a range of concentrations of substrates of CYP2C9, including S-warfarin (0.2-200 μM), or diclofenac (0.2-200 μM). After pre-incubation at 37°C for 5 min, the reactions were started by addition of NADP+ and incubated at 37°C for 20 min for S-warfarin, or 10 min for diclofenac. The reactions were terminated by addition of 200 μl acetonitrile containing internal standard, followed by centrifugation at 16,100 ×g for 10 min to obtain the supernatant. Aliquots were then analyzed by LC/MS/MS (Applied Biosystems, 3200 Qtrap) equipped with an electrospray ion source.
Chromatographic separation was achieved with a Waters XterraTM MS C18 column (2.1 × 50 mm, 3.5 μm; Agilent Technologies, Santa Clara CA, USA). The mobile phase for S-warfarin consisted of 5 mM ammonium acetate buffer, pH 4.6 (A) and acetonitrile (B). For diclofenac, the mobile phase was 0.1% formic acid in water (A) and acetonitrile (B). Initial mobile-phase composition was 20% mobile phase B. The proportion of mobile phase B was increased to 90% over 2 min, then held constant for 1 min before returning to the starting composition. The system was operated in negative ion mode. 7-Hydroxywarfarin was detected by examining an ion pair of 323.1/176.9, and mebendazole was used as an internal standard (ion pair of 294.0/262.0). 4′-hydroxydiclofenac was detected by examining an ion pair of 310.1/266.1, and indomethacin was used as an internal standard (ion pair of 356.2/312.3). The metabolites were quantified by comparing the ratio of ion currents obtained for the metabolites and internal standards calibration curve. The apparent enzyme kinetic parameters Michaelis-Menten constant (Km) and maximum velocity of substrate conversion (Vmax) were determined by using nonlinear regression (GraphPad Prism 5 software, La Jolla, CA, USA).
Statistical analysis
For the clinical study, continuous data were tested for normality using the Kolmogorov-Smirnov normality test and log-transformed to obtain normality if necessary prior to analysis. Clinical characteristics and pharmacokinetic parameters were compared between CYP2C9*8 allele carriers and CYP2C9*1 allele homozygotes by the two-sided Student's unpaired t-test. Including at least 10 patients in each genotype group was estimated to provide 80% power to detect a 0.20 difference in R:S-warfarin plasma concentration between groups, assuming a SD of 0.15.[7] Comparisons among the in vitro data from different variants of CYP2C9 were made using one-way ANOVA followed by the Student's t-test. The threshold for statistical significance was set at p<0.05.
Acknowledgments
This work was supported an American Heart Association Midwest Affiliate Spring 2010 Grant-In-Aid (10GRNT3750024).
Footnotes
Conflict of interest: Dr. Cavallari is co-inventor for U.S. Utility Patent Application No. 12/572,908 entitled, CYP2C9*8 alleles correlate with decreased warfarin metabolism and increased warfarin sensitivity. Published: May 27, 2010; Pub. No. US 2010/0130599. The authors declared no other conflict of interest.
References
- 1.Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45:525–538. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) Chest. 2008;133:160S–198S. doi: 10.1378/chest.08-0670. [DOI] [PubMed] [Google Scholar]
- 3.Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics. 1994;4:39–42. doi: 10.1097/00008571-199402000-00005. [DOI] [PubMed] [Google Scholar]
- 4.CYP2C9 allele nomenclature. Updated 2, 5 2011 [cited 2011 September 5] Available from: http://www.cypalleles.ki.se/cyp2c9.htm.
- 5.Rokitta D, Fuhr U. Comparison of enzyme kinetic parameters obtained in vitro for reactions mediated by human CYP2C enzymes including major CYP2C9 variants. Curr Drug Metab. 2010;11:153–161. doi: 10.2174/138920010791110872. [DOI] [PubMed] [Google Scholar]
- 6.Takanashi K, Tainaka H, Kobayashi K, Yasumori T, Hosakawa M, Chiba K. CYP2C9 Ile359 and Leu359 variants: enzyme kinetic study with seven substrates. Pharmacogenetics. 2000;10:95–104. doi: 10.1097/00008571-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Scordo MG, Pengo V, Spina E, Dahl ML, Gusella M, Padrini R. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on warfarin maintenance dose and metabolic clearance. Clin Pharmacol Ther. 2002;72:702–710. doi: 10.1067/mcp.2002.129321. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki H, Inoue K, Shimada T. Roles of two allelic variants (Arg144Cys and Ile359Leu) of cytochrome P4502C9 in the oxidation of tolbutamide and warfarin by human liver microsomes. Xenobiotica. 1998;28:103–115. doi: 10.1080/004982598239614. [DOI] [PubMed] [Google Scholar]
- 9.Crespi CL, Miller VP. The R144C change in the CYP2C9*2 allele alters interaction of the cytochrome P450 with NADPH:cytochrome P450 oxidoreductase. Pharmacogenetics. 1997;7:203–210. doi: 10.1097/00008571-199706000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Lindh JD, Holm L, Andersson ML, Rane A. Influence of CYP2C9 genotype on warfarin dose requirements--a systematic review and meta-analysis. Eur J Clin Pharmacol. 2009;65:365–375. doi: 10.1007/s00228-008-0584-5. [DOI] [PubMed] [Google Scholar]
- 11.Limdi NA, McGwin G, Goldstein JA, Beasley TM, Arnett DK, Adler BK, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther. 2008;83:312–321. doi: 10.1038/sj.clpt.6100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higashi MK, Veenstra DL, Kondo LM, Wittkowsky AK, Srinouanprachanh SL, Farin FM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002;287:1690–1698. doi: 10.1001/jama.287.13.1690. [DOI] [PubMed] [Google Scholar]
- 13.Coumadin (warfarin sodium) package insert. Princeton, NJ: Bristol-Myers Squibb; 2010. Jan, [Google Scholar]
- 14.Limdi NA, Arnett DK, Goldstein JA, Beasley TM, McGwin G, Adler BK, et al. Influence of CYP2C9 and VKORC1 on warfarin dose, anticoagulation attainment and maintenance among European-Americans and African-Americans. Pharmacogenomics. 2008;9:511–526. doi: 10.2217/14622416.9.5.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavallari LH, Langaee TY, Momary KM, Shapiro NL, Nutescu EA, Coty WA, et al. Genetic and clinical predictors of warfarin dose requirements in African Americans. Clin Pharmacol Ther. 2010;87:459–464. doi: 10.1038/clpt.2009.223. [DOI] [PubMed] [Google Scholar]
- 16.Schelleman H, Chen J, Chen Z, Christie J, Newcomb CW, Brensinger CM, et al. Dosing algorithms to predict warfarin maintenance dose in Caucasians and African Americans. Clin Pharmacol Ther. 2008;84:332–339. doi: 10.1038/clpt.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott SA, Jaremko M, Lubitz SA, Kornreich R, Halperin JL, Desnick RJ. CYP2C9*8 is prevalent among African-Americans: implications for pharmacogenetic dosing. Pharmacogenomics. 2009;10:1243–1255. doi: 10.2217/pgs.09.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perera MA, Gamazon E, Cavallari LH, Patel SR, Poindexter S, Kittles RA, et al. The missing association: sequencing-based discovery of novel SNPs in VKORC1 and CYP2C9 that affect warfarin dose in African Americans. Clin Pharmacol Ther. 2011;89:408–415. doi: 10.1038/clpt.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickmann LJ, Rettie AE, Kneller MB, Kim RB, Wood AJ, Stein CM, et al. Identification and functional characterization of a new CYP2C9 variant (CYP2C9*5) expressed among African Americans. Mol Pharmacol. 2001;60:382–387. doi: 10.1124/mol.60.2.382. [DOI] [PubMed] [Google Scholar]
- 20.Blaisdell J, Jorge-Nebert LF, Coulter S, Ferguson SS, Lee SJ, Chanas B, et al. Discovery of new potentially defective alleles of human CYP2C9. Pharmacogenetics. 2004;14:527–537. doi: 10.1097/01.fpc.0000114759.08559.51. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell C, Gregersen N, Krause A. Novel CYP2C9 and VKORC1 gene variants associated with warfarin dosage variability in the South African black population. Pharmacogenomics. 2011;12:953–963. doi: 10.2217/pgs.11.36. [DOI] [PubMed] [Google Scholar]
- 22.Allabi AC, Gala JL, Horsmans Y. CYP2C9, CYP2C19, ABCB1 (MDR1) genetic polymorphisms and phenytoin metabolism in a Black Beninese population. Pharmacogenet Genomics. 2005;15:779–786. doi: 10.1097/01.fpc.0000174787.92861.91. [DOI] [PubMed] [Google Scholar]
- 23.Allabi AC, Gala JL, Horsmans Y, Babaoglu MO, Bozkurt A, Heusterspreute M, et al. Functional impact of CYP2C95, CYP2C96, CYP2C98, and CYP2C911 in vivo among black Africans. Clin Pharmacol Ther. 2004;76:113–118. doi: 10.1016/j.clpt.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Haining RL, Hunter AP, Veronese ME, Trager WF, Rettie AE. Allelic variants of human cytochrome P450 2C9: baculovirus-mediated expression, purification, structural characterization, substrate stereoselectivity, and prochiral selectivity of the wild-type and I359L mutant forms. Arch Biochem Biophys. 1996;333:447–458. doi: 10.1006/abbi.1996.0414. [DOI] [PubMed] [Google Scholar]
- 25.Porter RS, Sawyer WT. Warfarin. In: Evans WE, Schentag JJ, Jusko WJ, editors. Applied pharmacokinetics: principles of therapeutic drug monitoring. Applied Therapeutics Inc.; Vancouver: 1992. pp. 31–46. [Google Scholar]
- 26.Wei L, Locuson CW, Tracy TS. Polymorphic variants of CYP2C9: mechanisms involved in reduced catalytic activity. Mol Pharmacol. 2007;72:1280–1288. doi: 10.1124/mol.107.036178. [DOI] [PubMed] [Google Scholar]
- 27.Maekawa K, Harakawa N, Sugiyama E, Tohkin M, Kim SR, Kaniwa N, et al. Substrate-dependent functional alterations of seven CYP2C9 variants found in Japanese subjects. Drug Metab Dispos. 2009;37:1895–1903. doi: 10.1124/dmd.109.027003. [DOI] [PubMed] [Google Scholar]
- 28.Kumar V, Wahlstrom JL, Rock DA, Warren CJ, Gorman LA, Tracy TS. CYP2C9 inhibition: impact of probe selection and pharmacogenetics on in vitro inhibition profiles. Drug Metab Dispos. 2006;34:1966–1975. doi: 10.1124/dmd.106.010926. [DOI] [PubMed] [Google Scholar]
- 29.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 30.Hruska MW, Frye RF, Langaee TY. Pyrosequencing method for genotyping cytochrome P450 CYP2C8 and CYP2C9 enzymes. Clin Chem. 2004;50:2392–2395. doi: 10.1373/clinchem.2004.040071. [DOI] [PubMed] [Google Scholar]
- 31.Aquilante CL, Langaee TY, Lopez LM, Yarandi HN, Tromberg JS, Mohuczy D, et al. Influence of coagulation factor, vitamin K epoxide reductase complex subunit 1, and cytochrome P450 2C9 gene polymorphisms on warfarin dose requirements. Clin Pharmacol Ther. 2006;79:291–302. doi: 10.1016/j.clpt.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 32.Cavallari LH, Butler C, Langaee TY, Wardak N, Patel SR, Viana MAG, et al. Association of apolipoprotein E genotype with duration of time to achieve a stable warfarin dose in African-American patients. Pharmacotherapy. 2011;31:785–792. doi: 10.1592/phco.31.8.785. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi H, Kashima T, Kimura S, Muramoto N, Nakahata H, Kubo S, et al. Determination of unbound warfarin enantiomers in human plasma and 7-hydroxywarfarin in human urine by chiral stationary-phase liquid chromatography with ultraviolet or fluorescence and on-line circular dichroism detection. J Chromatogr B Biomed Sci Appl. 1997;701:71–80. doi: 10.1016/s0378-4347(97)00346-0. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi H, Kashima T, Nomizo Y, Muramoto N, Shimizu T, Nasu K, et al. Metabolism of warfarin enantiomers in Japanese patients with heart disease having different CYP2C9 and CYP2C19 genotypes. Clin Pharmacol Ther. 1998;63:519–528. doi: 10.1016/S0009-9236(98)90103-5. [DOI] [PubMed] [Google Scholar]
- 35.Crawford JD, Terry ME, Rourke GM. Simplification of drug dosage calculation by application of the surface area principle. Pediatrics. 1950;5:783–790. [PubMed] [Google Scholar]