

Key Points
First prospective US cooperative trial group in preneoplastic gammopathies.
Prospective demonstration that genomic features of preneoplastic cells predict disease risk.
Abstract
All cases of clinical myeloma (CMM) are preceded by an asymptomatic monoclonal gammopathy (AMG), classified as either monoclonal gammopathy of undetermined significance (MGUS) or asymptomatic multiple myeloma (AMM). We analyzed data from AMG patients (n = 331) enrolled in a prospective, observational clinical trial (S0120). Baseline data from clinical variables, gene expression profiles (GEP) of purified tumor cells, and findings of magnetic resonance imaging (MRI) were correlated with the risk of progression to CMM requiring therapy. GEP of purified tumor cells revealed that all molecular subtypes of CMM are also represented in the AMG phase. An increased risk score (>-0.26) (based on a 70-gene signature, GEP70) was an independent predictor of the risk of progression to CMM. Combination of elevated serum free light chain, M-spike, and GEP70 risk score identified a subset with high risk (67% at 2 years) of progression to CMM requiring therapy. Importantly, absence of these factors in AMM patients predicted low risk similar to MGUS. Detection of multiple (>1) focal lesions by MRI also conferred an increased risk of progression. These data demonstrate that signatures associated with high-risk CMM impact disease risk and support inclusion of genomic analysis in the clinical management of AMGs. This trial was registered at www.clinicaltrials.gov as # NCT00900263.
Introduction
Multiple myeloma (MM) is a plasma cell (PC) malignancy characterized by lytic bone disease, anemia, hypercalcemia, renal failure, and infections.1 MM is preceded by a clinically asymptomatic precursor phase (asymptomatic monoclonal gammopathy [AMG]), which is more common than the malignancy.2,3 Current criteria for the diagnosis of clinical myeloma (CMM) and initiation of therapy are based on the degree of bone marrow PC infiltration, level of monoclonal immunoglobulin, and presence of myeloma-related end-organ/tissue injury.4 Patients with AMG lack myeloma-related end-organ/tissue injury and are classified as monoclonal gammopathy of undetermined significance (MGUS) or as asymptomatic MM (AMM), based on the level of monoclonal immunoglobulin (M spike; ≥3 g/dL for AMM) and/or bone marrow PC infiltration (≥10% for AMM). The estimated risk of progression from AMM to MM (approximately 10%/year) is higher than from MGUS (approximately 1%/year).4-8 Although current models to predict the risk of disease progression from AMGs have been useful in guiding clinical research,9,10 these models were based on retrospective analyses of cohorts tested for a limited set of clinical variables and did not incorporate data on genomic properties of tumor cells or modern imaging, and recent studies suggest poor concordance between the current models.11 This emphasizes the need for prospective studies that include a broader array of clinical and biological variables to identify risk factors for progression of AMG.
Advances in understanding the molecular biology and genetics of MM have demonstrated distinct genetic subtypes of the disease.12,13 Gene expression profiling (GEP) of purified CD138+ tumor cells has emerged as a powerful tool to dissect this biological heterogeneity and has been used to identify distinct molecular subgroups of MM. In addition to molecular classification, GEP has been used to develop validated signatures that identify patients with high-risk myeloma a subset of patients for whom current therapies resulted in extremely poor outcomes.14,15 It is not known whether GEP-defined molecular subtypes or risk groups of MM are also represented in the AMG precursor phase or whether molecular features impact the risk of progression to clinical MM. In addition, current imaging criteria for the diagnosis of bone disease use skeletal radiographs, which lack sensitivity. Magnetic resonance imaging (MRI) of the spine has emerged as a useful tool for evaluating the presence of marrow infiltration and focal lesions.16 MRI abnormalities were shown to correlate with increased risk in patients with AMM17 but need to be tested in the context of other variables in a prospective trial.
To address these issues, SWOG (formerly the Southwest Oncology Group) developed the first US cooperative trial group in AMGs in 2003 to prospectively examine a broad array of laboratory variables, genomic analyses, and, when possible, state-of-the-art imaging tools to evaluate the predictors of progression from AMG to MM that requires therapy.
Methods
Eligibility criteria and study design
Patients with PC proliferative diseases not requiring therapy were eligible for participation in a prospective, observational clinical trial (S0120). The objectives were to assess the feasibility of accruing patients with asymptomatic PC disorders in a national cooperative trial group and to identify biological correlates that may relate to progression to symptomatic disease. Other eligibility criteria included no prior therapy for the PC disorder and willingness to submit samples for research. At study entry, patients were classified into categories of MGUS, AMM, solitary plasmacytoma, or other PC proliferative disorders not requiring therapy. Diagnostic criteria for MGUS and AMM were based on the International Myeloma Working Group convention.4 All patients signed an informed consent, in keeping with the Declaration of Helsinki and federal and institutional guidelines. The protocol was approved by the National Cancer Institute and all participating centers’ internal review boards.
All patients underwent detailed clinical staging at initial registration. This included hemogram, multichemical scan, and MM-related measurements (included serum and urine electrophoresis to quantify serum-M concentration, daily urinary M-protein excretion, and quantification of 24-hour proteinuria). Nephelometric analysis was performed to determine serum immunoglobulin levels. Immunofixation analyses of serum and urine were performed to define the nature of the monoclonal protein present in serum or urine. Additional measurements included serum lactate dehydrogenase and β-2-microglobulin (B2M). Bone marrow aspirates and biopsies were obtained for cytological and histopathological evaluation of PC infiltration, including immunohistochemical clonality assessment. To evaluate bone marrow plasmacytosis (BMPC), data for both aspirate and biopsy were considered and the higher value was used. Metaphase karyotyping was performed on at least 20 Giemsa-stained metaphases. In most patients, serum free light chain (SFLC) assays were used to quantify κ and λ SFLCs. Imaging studies involved standard metastatic bone surveys by radiographic examination, and, when possible, MRI of the entire spine was used to identify focal lesions. For follow up, all patients were seen, at a minimum, for MM-related laboratory studies at 3, 6, and 12 months in the first year, and then every 6 to 12 months.
GEP of purified CD138+ tumor cells
When possible, an aliquot of bone marrow aspirate was collected to isolate CD138+ PCs with immunomagnetic bead selection (autoMACS; Miltenyi Biotec), as described.13 Purity of PC was monitored by flow cytometry and was >85%. Total RNA from these PCs was used to measure GEP with Affymetrix U133 Plus 2.0 microarrays. Resultant GEP data were used to determine whether molecular subtypes of clinical MM could be identified in this cohort to generate a polyclonal PCs score for assessing the contribution of polyclonal PCs18 and to generate risk scores based on a validated 70-gene model (GEP-70)14 for high-risk MM.
Statistical analysis
Baseline features of patients with MGUS and AMM were compared using χ2 and Fisher's exact tests. Cumulative incidence analysis was used to model timing and onset of overt MM that required therapy in the presence of death as a competing risk.19 Cox proportional hazards regression was used to model univariate and multivariate (MV) associations of baseline features with progression to MM.20 Running log rank tests and recursive partitioning were used to identify statistically optimal binary splits for continuous baseline predictors.21,22 In several instances, these cut-points were rounded to simplify clinical implementation. In the absence of clear optimality, conventional cut-points were implemented. The R2 statistic was used to evaluate the predictive power of different models in this dataset.23
Results
Clinical characteristics
Between June 1, 2003 and January 3, 2011, there were 375 patients enrolled in the S0120 clinical trial, of whom 361 patients were eligible. Analysis is restricted to 331 patients with non-immunoglobulin M (IgM) monoclonal gammopathies who met the IMWG criteria for MGUS (n = 152) or AMM (n = 179) and for whom follow-up data are available (supplemental Figure 1, available on the Blood Web site). Clinical characteristics of the cohort are shown in Table 1. Patients with AMM had greater proportions of patients with elevated serum levels of involved SFLC and B2M, and lower serum levels of uninvolved immunoglobulins. MRI imaging of the spine revealed focal lesions in 25 of 156 patients tested. The cohort with available MRI data had a lower proportion of patients with hypoalbuminemia and abnormal free light chains (FLC) than those without, but did not differ in terms of other features such as marrow plasmacytosis or M spike (data not shown).
Table 1.
Patient characteristics
| Factor | Overall n/N (%) | MGUS | AMM | P value |
|---|---|---|---|---|
| Age ≥65 y | 143/331 (43%) | 63/152 (41%) | 80/179 (45%) | .553 |
| Female | 153/331 (46%) | 77/152 (51%) | 76/179 (42%) | .136 |
| SWOG performance status 0 | 228/327 (70%) | 101/151 (67%) | 127/176 (72%) | .301 |
| Hemoglobin <12 g/dL | 84/331 (25%) | 32/152 (21%) | 52/179 (29%) | .096 |
| Platelets <240 × 103/μL* | 206/331 (62%) | 88/152 (58%) | 118/179 (66%) | .133 |
| Albumin <4 g/dL* | 154/329 (47%) | 64/151 (42%) | 90/178 (51%) | .139 |
| Serum B2M >3 mg/L* | 86/322 (27%) | 31/147 (21%) | 55/175 (31%) | .037 |
| Serum B2M >5.5 mg/L | 17/322 (5%) | 8/147 (5%) | 9/175 (5%) | 1.000† |
| Bone marrow PCs ≥10% | 176/330 (53%) | 0/152 (0%) | 176/178 (99%) | <.001 |
| Bone marrow PCs ≥20%* | 84/330 (25%) | 0/152 (0%) | 84/178 (47%) | <.001 |
| Bone marrow PCs ≥60% | 2/330 (1%) | 0/152 (0%) | 2/178 (1%) | .502† |
| Serum M-protein ≥3 g/dL* | 34/330 (10%) | 0/152 (0%) | 34/178 (19%) | <.001 |
| Urine M-protein >0 | 48/227 (21%) | 21/110 (19%) | 27/117 (23%) | .462 |
| IgA isotype M-protein | 45/313 (14%) | 14/138 (10%) | 31/175 (18%) | .058 |
| IgG isotype M-protein | 264/313 (84%) | 121/138 (88%) | 143/175 (82%) | .149 |
| Light chain only | 4/313 (1%) | 3/138 (2%) | 1/175 (1%) | .324† |
| Uninvolved immunoglobulins low‡ | 212/309 (69%) | 66/135 (49%) | 146/174 (84%) | <.001 |
| κ light chain clonal isotype | 190/303 (63%) | 86/137 (63%) | 104/166 (63%) | .982 |
| Invovlved/uninvolved SFLC ratio >10* | 79/228 (35%) | 13/87 (15%) | 66/141 (47%) | <.001 |
| Involved SFLC >25 mg/dL | 42/228 (18%) | 10/87 (11%) | 32/141 (23%) | .034 |
| Abnormal metaphase cytogenetics | 24/250 (10%) | 5/95 (5%) | 19/155 (12%) | .068 |
| CD4 <920/μL | 121/178 (68%) | 42/66 (64%) | 79/112 (71%) | .341 |
| GEP 70-gene risk >−0.26* | 37/126 (29%) | 6/39 (15%) | 31/87 (36%) | .021 |
| GEP poly-PC >11.60* | 62/126 (49%) | 32/39 (82%) | 30/87 (34%) | <.001 |
| GEP PI >−2.73* | 50/126 (40%) | 10/39 (26%) | 40/87 (46%) | .031 |
| GEP CD-1 subgroup | 6/126 (5%) | 3/39 (8%) | 3/87 (3%) | .373† |
| GEP CD-2 subgroup | 28/126 (22%) | 14/39 (36%) | 14/87 (16%) | .013 |
| GEP HY subgroup | 31/126 (25%) | 4/39 (10%) | 27/87 (31%) | .012 |
| GEP LB subgroup | 28/126 (22%) | 10/39 (26%) | 18/87 (21%) | .537 |
| GEP MF subgroup | 17/126 (13%) | 6/39 (15%) | 11/87 (13%) | .677 |
| GEP MS subgroup | 11/126 (9%) | 1/39 (3%) | 10/87 (11%) | .170† |
| GEP PR subgroup | 5/126 (4%) | 1/39 (3%) | 4/87 (5%) | 1.000† |
| MRI focal lesions ≥1 | 25/156 (16%) | 7/64 (11%) | 18/92 (20%) | .148 |
| MRI focal lesions >1 | 9/156 (6%) | 3/64 (5%) | 6/92 (7%) | .738† |
CD-2, cyclin D-2; HY, hyperdiploid; LB, low bone; n, number with factor; N, number with valid data for factor; PI, proliferation index; poly-PC, polyclonal PCs.
Optimal cut-points when applicable are based on approximate running log-rank test statistic.
P values computed using Fisher's exact test.
Uninvolved immunoglobulins low: <600 mg/dL if IgG, <50 mg/dL if IgM, <100 mg/dL if IgA.
GEP data were available for purified tumor cells from 126 patients. The cohort with available GEP data had a greater proportion of patients with marrow plasmacytosis >20% and elevated FLC >25 mg/dL than those without such data (not shown). All major molecular subtypes of MM13 were detected in both MGUS and AMM cohorts. Patients with AMM had a higher proportion of hyperdiploid subtype and a lower proportion of cyclin D-2 subtype than MGUS patients (Table 1). In comparison with MGUS, the AMM cohort had a higher proportion of patients with GEP signatures of high risk, according to the GEP-70 model, as well as proliferation index,14 and a lower proportion of patients with a signature of polyclonal PC13 (Table 1).
Univariate analyses
With a median follow up of 43 months, 54 patients experienced disease progression that required initiation of anti-myeloma therapy (Figure 1A). Risk of disease progression at 2 years was 1.6% in the MGUS subgroup and 23.3% in the AMM subgroup, consistent with current estimates (Figure 1B). The most common mode of progression was the development of bone disease accounting for approximately 50% of patients. Concurrent with myeloma, AL amyloidosis was diagnosed in 4 patients and polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes (POEMS) syndrome suspected in 2 patients. Several factors assessed at initial registration were analyzed to determine whether they were associated with the time-to-progression to CMM requiring therapy. Optimal cutoffs for continuous variables were used to assess prognostic impact in univariate analysis and were further evaluated in MV Cox proportional hazards models (Table 2). Univariate analysis showed that increased risk of progression to CMM requiring therapy was associated with the following clinical variables: age ≥65 years, hemoglobin <12 g/dL, serum albumin <4 g/dL, serum B2M >3 mg/L, elevated serum M (≥3 g/dL), and urine M-protein (>0 g/dL), and low levels of uninvolved immunoglobulins, a level of involved SFLC (>25 mg/dL), elevated ratio of involved/uninvolved SFLC (>10), and increased bone marrow PCs (≥20%) (Table 2).
Figure 1.

Analysis of time to progression to CMM requiring therapy. (A) Time-to-progression for the entire cohort. (B) Time to progression by AMG disease type (MGUS/AMM) by International Myeloma Working Group criteria. N, number.
Table 2.
Univariate Cox proportional hazards analysis of risk factors of progression to clinical MM requiring therapy
| Variable | n/N (%) | Time to MM requiring therapy | |
|---|---|---|---|
| HR (95% CI) | P value | ||
| Age ≥65 y | 143/331 (43%) | 2.21 (1.28, 3.81) | .004 |
| Female | 153/331 (46%) | 0.62 (0.35, 1.08) | .093 |
| SWOG performance status 0 | 228/327 (70%) | 0.80 (0.45, 1.43) | .452 |
| Hemoglobin <12 g/dL | 84/331 (25%) | 2.01 (1.15, 3.49) | .014 |
| Platelets <240 × 103/μL* | 206/331 (62%) | 1.47 (0.83, 2.62) | .189 |
| Albumin <4 g/dL* | 154/329 (47%) | 1.90 (1.10, 3.28) | .021 |
| Serum B2M >3 mg/L* | 86/322 (27%) | 2.61 (1.51, 4.49) | <.001 |
| Serum B2M >5.5 mg/L | 17/322 (5%) | 1.23 (0.38, 3.93) | .732 |
| Bone marrow PCs ≥10% | 176/330 (53%) | 7.78 (3.32, 18.20) | <.001 |
| Bone marrow PCs ≥20%* | 84/330 (25%) | 4.94 (2.86, 8.54) | <.001 |
| Bone marrow PCs ≥60% | 2/330 (1%) | 6.54 (0.90, 47.55) | .063 |
| Serum M-protein ≥3 g/dL* | 34/330 (10%) | 5.48 (3.08, 9.78) | <.001 |
| IgA isotype M-protein | 45/313 (14%) | 1.44 (0.72, 2.86) | .299 |
| IgG isotype M-protein | 264/313 (80%) | 0.93 (0.48, 1.81) | .840 |
| Uninvolved immunoglobulins low† | 212/309 (69%) | 4.05 (1.73, 9.48) | .001 |
| κ light chain clonal isotype | 190/303 (63%) | 1.08 (0.61, 1.91) | .787 |
| Involved SFLC >25 mg/dL* | 42/228 (18%) | 3.71 (2.01, 6.83) | <.001 |
| Involved/uninvolved SFLC ratio >10* | 79/228 (35%) | 3.54 (1.92, 6.52) | <.001 |
| Abnormal metaphase cytogenetics | 24/250 (10%) | 1.67 (0.75, 3.73) | .210 |
| GEP 70-gene risk >−0.26* | 37/126 (29%) | 5.85 (2.56, 13.34) | <.001 |
| GEP poly-PC >11.60* | 62/126 (49%) | 0.17 (0.06, 0.48) | .001 |
| GEP PI >−2.73* | 50/126 (40%) | 4.41 (1.84, 10.58) | <.001 |
| GEP CD-1 subgroup | 6/126 (5%) | 0.00 (0.00, NE) | .989 |
| GEP CD-2 subgroup | 28/126 (22%) | 0.64 (0.22, 1.86) | .413 |
| GEP HY subgroup | 31/126 (25%) | 1.48 (0.64, 3.43) | .361 |
| GEP LB subgroup | 28/126 (22%) | 0.62 (0.21, 1.80) | .374 |
| GEP MF subgroup | 17/126 (13%) | 0.54 (0.13, 2.31) | .407 |
| GEP MS subgroup | 11/126 (9%) | 2.36 (0.81, 6.88) | .116 |
| GEP PR subgroup | 5/126 (4%) | 4.56 (1.35, 15.35) | .014 |
| MRI focal lesions ≥1 | 25/156 (16%) | 2.81 (1.19, 6.65) | .018 |
| MRI focal lesions >1 | 9/156 (6%) | 4.71 (1.57, 14.11) | .006 |
| Center-University of Arkansas | 246/331 (74%) | 1.30 (0.65, 2.59) | .452 |
P value from Wald χ2 test in Cox regression.
CD-1, cyclin D-1; CD-2, cyclin D-2; CI, confidence interval; HR, hazard ratio; HY, hyperdiploid; MS, multiple myeloma-SET; NE, not estimable; PI, proliferation index.
Optimal cut-points when applicable are based on approximate running log-rank test statistic.
Uninvolved immunoglobulins low: <600 mg/dL if IgG, <50 mg/dL if IgM, <100 mg/dL if IgA.
Several GEP-based variables were analyzed for correlation with increased risk of disease progression. GEP-70 risk score >−0.26, and GEP-proliferation index >−2.73 predicted increased risk, whereas the GEP-polytypic-PC score >11 correlated with reduced risk (Table 2). The GEP70 risk score correlated moderately with the proliferation index, but not with the polytypic-PC score (supplemental Figure 2). Of the GEP-based molecular subtypes, only the proliferation (PR) subtype had a significantly increased risk. In particular, musculoaponeurotic fibrosarcoma (MF) subtype associated with poor outcome in CMM did not portend increased risk. No other molecular subtypes had altered risk of progression to CMM (Table 2). There was no difference in the risk of disease progression based on availability of GEP data in either MGUS or AMM cohorts (not shown).
In addition to the clinical and genomic variables, the presence of focal lesions on MRI of the spine (using cutoffs of >1 focal lesion, as well as >1 focal lesion) was also significantly associated with increased risk of progression to CMM (Table 2). The factors associated with increased risk of progression were similar to those in the entire cohort when the analysis was restricted to patients with AMM (supplemental Table 1). Changes in M spike over time may also be a marker of disease risk.10,24 To study the effect of a progressive phenotype during a 4-month landmark, we classified patients as those with a high M spike (≥3 g/dL) at baseline (Hi), those who retained an M spike (<3 g/dL) throughout the 3-month period (Lo-Lo), and those with an increase in M spike to ≥3 g/dL (Lo-Hi subset). Risk of progression for patients in the Lo-Hi subset was similar to that for patients who had high M spikes at baseline (supplemental Figure 3), indicating that increase in M spike may also be an important risk factor for progression to CMM.
MV models
In the MV Cox proportional hazards model utilizing clinical variables alone, serum M-protein ≥3g/dL, BMPC ≥20%, and age ≥65 years were independent predictors of the risk of progression to CMM requiring therapy (Table 3). The same variables emerged when the analysis was restricted to patients with available SFLC data (data not shown). Because several GEP-related variables were highly significant predictors of outcome in univariate analysis, we developed an MV model analyzing the importance of GEP variables in the context of other clinical variables. In this model, GEP70 risk score >−0.26, serum M spike ≥3g/dL and involved SFLC >25 mg/dL emerged as the significant prognostic variables (Table 3). The model with both genomic and clinical data was superior to the model with clinical variables alone and yielded higher cumulative R2 values. To directly evaluate the additional impact of GEP as a variable, we examined the effect of directly adding GEP to the clinical variable-based model. Addition of GEP improved cumulative R2 by approximately 11% (supplemental Table 2). Serum M and GEP70 were the only significant variables in the MV model, wherein serial measurement of M spike was considered using a 4-month landmark analysis (supplemental Table 3). When the MV model was restricted to AMM, GEP70 risk score >−0.26, serum M spike ≥3g/dL, and involved SFLC >25 mg/dL again emerged as the significant prognostic variables (supplemental Table 4). When GEP70 was specifically excluded from the model, GEP-based proliferation index entered the model, which is consistent with the correlation between these variables (supplemental Table 5 and supplemental Figure 2). In an MV analysis restricted to patients with available MRI data, the presence of >1 MRI-detected focal lesions did not emerge as an independent variable (data not shown). The presence of multiple (>1) focal lesions was detected by MRI in only 9 (6%) patients. However, the presence of multiple (>1) MRI-detected focal lesions was an independent predictor of increased risk of disease progression (supplemental Table 6).
Table 3.
MV Cox proportional hazards analysis of risk factors of progression to clinical MM requiring therapy
| Variable | n/N (%) | HR (95% CI) | P value | Cumulative R2 | |
|---|---|---|---|---|---|
| Clinical Variables | |||||
| Serum M-protein ≥3 g/dL* | 32/297 (11%) | 3.52 (1.86, 6.65) | <.001 | 22.23 | |
| Bone marrow PCs ≥20%* | 78/297 (26%) | 3.22 (1.77, 5.84) | <.001 | 40.65 | |
| Age ≥65 y | 127/297 (43%) | 2.10 (1.19, 3.69) | .010 | 46.14 | |
| Clincal + GEP variables | |||||
| GEP 70-gene risk >−0.26* | 32/117 (27%) | 6.81 (2.90, 15.97) | <.001 | 39.94 | |
| Serum M-protein ≥3 g/dL* | 17/117 (15%) | 6.49 (2.78, 15.18) | .006 | 63.12 | |
| Involved SFLC >25 mg/dL* | 27/117 (23%) | 3.15 (1.40, 7.08) | <.001 | 70.01 |
P value from Wald χ2 test in Cox regression. MV model uses stepwise selection with entry level 0.1 and variable remains if meet the 0.05 level.
Variables considered for clinical model: age ≥65, female, hemoglobin <12 g/dL, albumin <4 g/dL, serum B2M >3 mg/L, bone marrow PCs ≥20%, M-protein ≥3 g/dL, and uninvolved immunoglobulins low (<600 mg/dL if IgG, <50 mg/dL if IgM, <100 mg/dL if IgA).
Variables considered for clinical + GEP model: age ≥65, female, hemoglobin <12 g/dL, albumin <4 g/dL, serum B2M >3 mg/L, bone marrow PCs ≥20%, M-protein ≥3 g/dL, uninvolved immunoglobulins low (<600 mg/dL if IgG, <50 mg/dL if IgM, <100 mg/dL if IgA), and involved SFLC >25 mg/dL, involved/uninvolved (SFLC ratio >10, GEP 70-gene risk >−0.26, GEP poly-PC >11.60, GEP PR subgroup).
Optimal cut-points based on approximate running log-rank test statistic.
Risk models
Combination of serum M spike, involved SFLC and GEP-70 identified 3 distinct risk groups. Patients with 2 to 3 risk factors had the highest risk with 66.7% risk of progression to CMM requiring therapy in 2 years (Figure 2A). The risk of progression at 2 years in patients with 0 or 1 risk factor was 3.4% and 21.9%, respectively. Risk model based on the MV analysis with only clinical variables identified 3 groups with 3.2%, 13.8%, and 39.8% risk of progression at 2 years (Figure 2B). GEP-70 associated risk was more evident in the AMM cohort than in MGUS (Figure 2C). The combination of serum M spike, GEP-70 risk, and involved SFLC also identified 3 distinct risk groups in AMM (Figure 2D). Notably, AMM patients lacking these risk factors have a low risk of progression to CMM, similar to that in MGUS (Figure 2D).
Figure 2.
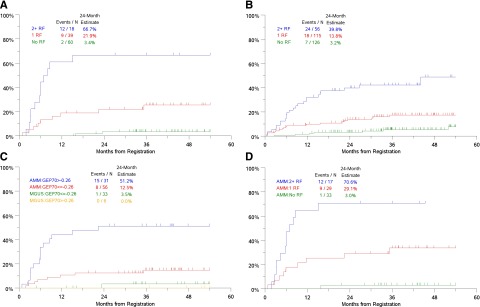
Risk groups in AMGs based on clinical/genomic risk factors. (A) Risk groups based on independent variables in the MV model including GEP-SFLC >25 mg/dL, serum M spike ≥3 g/dL, and GEP70 risk score >−0.26. (B) Risk groups based on independent clinical variables in the MV model: serum M-protein ≥3 g/dL, bone marrow PCs ≥20%, and age ≥65 years. (C) Time-to-progression to CMM requiring therapy in cohorts based on GEP70 score and disease subtype (AMM or MGUS). (D) Time-to-progression to CMM requiring therapy in AMM patients based on risk factors 0, 1, or 2+ (risk factors: SFLC >25 mg/dL, serum M spike ≥ 3g/dL, and GEP70 risk score >−0.26). N, number; RF, risk factors.
Current models for risk stratification in AMGs initially classify patients into MGUS and AMM based on BMPC and M spike, and further identify high-risk subgroups in AMM based on FLC ratio. In contrast, the SWOG model, which takes GEP into account, does not include data on percentage of BMPC. We used cumulative R2 to directly compare these models. The R2 score in the SWOG model was 70.1, whereas the R2 values with the BMPC-based models with FLC ratio cutoffs at 8 (model 1) and 100 (model 2) in this patient population were 57.4 and 61.1, respectively (Table 4).
Table 4.
Comparison of risk models
| Model | Variables | Cumulative r2 |
|---|---|---|
| Model 1 | Marrow PCs >10% | 57.47 |
| Serum M spike >3 g/dL | ||
| FLC ratio >8 | ||
| Model 2 | Marrow PCs >10% | 61.1 |
| Serum M spike >3 g/dL | ||
| FLC ratio >100 | ||
| SWOG | Serum M spike >3 g/dL | 70.01 |
| Involved FLC >25 mg/dL | ||
| GEP risk score >−0.26 |
Discussion
These data represent the first prospective evaluation of clinical, genomic, and imaging features of AMGs in the context of a US cooperative trial group. AMGs are the most common form of PC dyscrasias, and nearly all cases of CMM are preceded by an AMG precursor state.2,3,25 Understanding the factors predicting risk of progression from AMG to CMM will allow patients with the highest risk to be considered for innovative therapies aimed at preventing CMM. Findings from this study demonstrate the importance of integrating data from genomics of tumor cells in the clinical management of AMGs.
It is now well documented that MM consists of several GEP-defined molecular subtypes based on properties such as the nature of IgH translocation, expression of d-type cyclins, and proliferation signature in tumor cells.12,13 The data presented here demonstrate that all major GEP-defined molecular subtypes of MM are already present in the precursor stages, indicating that the molecular heterogeneity of MM is established early in the course of the disease, and is consistent with prior studies that show all the major cytogenetic abnormalities in MM are also observed in MGUS.26 Of the GEP-defined molecular subtypes, only the PR subtype was associated with an altered risk of progression to CMM, indicating that the genetic features that define several of these subtypes are likely not the key determinants of the transition to clinical malignancy. This was most evident for the MF subtype, which portends an aggressive course in CMM, but did not predict increased risk of progression to CMM. A recent retrospective analysis described an increased risk of disease progression in AMM patients with fluorescence in situ hybridization (FISH)-detected t(4:14) translocation, but did not include patients with MGUS who also carry this genetic lesion.27 Further prospective studies are needed to confirm this observation, as the prognostic impact of FISH-detected t(4:14) in CMM can be variable.28 Differences in prognostic risk with t(4:14) in this study vs that by Rajkumar et al27 may relate to the inclusion of MGUS patients in this study and the use of FISH vs GEP to detect this subset.
GEP analysis of purified CD138+ PCs has also emerged as a powerful tool to predict high-risk disease in clinical MM.14,15 Shaughnessy et al14 proposed a 70-gene signature for high-risk MM (GEP-70), which has been validated in several datasets. In the current study, the GEP-70 risk score was an independent predictor of the risk of progression to clinical MM from AMM. Nearly 30% of genes in GEP-70 are derived from chromosome 1; therefore, these data are consistent with prior cytogenetic studies implicating amplification of chromosome 1q21 in MM pathogenesis.29 A recent study by Neben et al30 also identified an increased risk of disease progression in patients with abnormalities of chromosome 1q21, which is consistent with this analysis. The GEP-70 score correlated with the proliferative index and may also, in part, reflect the proliferative capacity of tumor cells. Consistent with this, the PI entered the MV model when GEP70 was excluded. Proliferative capacity of tumor cells has previously emerged as an important prognostic variable in MM across several genetic subtypes.31 Notably, in the MV models that considered the genomics of tumor cells, the degree of bone marrow plasmacytosis was no longer a significant independent variable. Thus, the analysis of genomic properties of tumor cells may potentially substitute for marrow plasmacytosis in the evaluation of clinical MM risk. Assessing BMPC can be challenging in some patients due to sampling bias related to focal lesions and the potential for blood contamination in marrow aspirates.32 The finding that a higher polytypic-PC risk score predicts reduced risk of progression is consistent with data from studies correlating disease risk with the proportion of phenotypically abnormal PCs.10,24 The polytypic-PC score and the level of uninvolved immunoglobulins were significant variables in univariate analysis, but did not emerge as independent variables in the MV models when other GEP variables were included. These data suggest that in spite of the potential caveat of contaminating normal PCs, GEP of purified CD138+ PCs can provide powerful information regarding risk of disease progression in AMGs. Further analyses that integrate genetic analyses of tumor cells with the biology of the tumor microenvironment may further refine our ability to predict risk of AMG progression to clinical MM.33,34
MRI imaging of the spine is a valuable tool for clinically managing MM,16 and it is particularly useful for detecting tumor foci in this typically multifocal disease. In prior retrospective studies, the presence of MRI-detected focal lesions has been associated with increased disease risk in AMM. In this study, the presence of multiple (>1) MRI-detected focal lesions was an independent risk factor, which is consistent with findings of Hillengass et al.17 However, this correlation is based on only 9 patients with multiple lesions on MRI. Therefore, further prospective studies are needed to validate this finding.
An important strength of this study is its prospective multicenter nature and the inclusion of genomic analyses. However, some limitations of the current analysis should also be considered. With limitation of current follow up, the present analysis is biased toward factors predicting early disease progression primarily in AMM and further follow up of this cohort is needed to better identify predictors of risk in lower risk patients. GEP analysis depends on the ability to isolate adequate CD138+ PCs, which may be a challenge in MGUS. Indeed, GEP data were available only on a subset of patients in this study. In the present study, most of the predictive utility with GEP was in patients with AMM and further studies are needed to understand its utility in patients with MGUS. Additional genomic analyses including genome sequencing/mutational analysis and epigenetic changes may further improve the detection of genetic features predicting increased risk.
These data are consistent with prior studies showing that the level of monoclonal immunoglobulin, percentage of marrow plasmacytosis, and increased SFLC ratio are important risk factors in AMG.6,9,35-38 Integration of GEP data with the clinical variables (SFLC and M spike) led to an improved risk model with higher cumulative R2 than the risk model based on clinical variables alone. Patients with 2 to 3 risk factors had an extremely high risk (67% at 2 years) of progression to CMM requiring therapy. Other studies have recently identified similarly high-risk patients based on extreme plasmacytosis (>60%) and SFLC ratio >100.39,40 GEP may be particularly important to identify a low-risk subset in AMM, as AMM patients lacking both GEP and FLC risk factors had a low risk similar to that in MGUS. The 2-year risk of disease progression for AMM and MGUS cohorts in this dataset is comparable to that reported earlier. However, it is possible that the AMM patients enrolled in this study may include fewer ultra-high risk patients than in other retrospective cohorts. This is objectively manifested as a low proportion of patients with multiple MRI-detected focal lesions, and extreme (>60%) marrow plasmacytosis in this study. Recent studies have begun to explore early initiation of antimyeloma therapy in AMM in an attempt to preserve organ function.41-43 Improved assessment of disease risk will be essential, when such approaches are considered.
In summary, this first prospective trial in AMGs demonstrated that the genomic heterogeneity of CMM as measured by GEP is established early during the precursor stage. Incorporation of genomic properties of tumor cells led to improved assessment of disease risk in AMGs and identified distinct cohorts with high- or low-risk disease. Importantly, when genomic features were considered, percentage of marrow plamacytosis, subject to potential sampling bias related to focal lesions, did not enter the risk model, suggesting the importance of tumor biology in addition to tumor bulk. Genomic features of tumor cells may be particularly important considerations in patients with a clinical diagnosis of AMM based on mild plasmacytosis. The surprising finding that the very genomic signatures (such as GEP-70) that predict high-risk disease in CMM also signal a higher risk of malignant transformation, suggests that understanding the biology of high-risk MM may be critical for not just therapy, but also for prevention of MM. Integration of genomics into routine management of these patients may improve the application of risk-adapted approaches to prevent clinical malignancy.
Supplementary Material
Acknowledgments
The authors thank Margaret E. Brenner for help with editing this manuscript.
This investigation was supported in part by the following Pooled Human Serum Cooperative Agreement grants awarded by the National Institutes of Health, National Cancer Institute, Department of Health and Human Services (CA32102, CA38926, CA37981, CA58416, CA12644, CA76447, CA46282, CA76462, CA35176, CA35119, CA58882, CA68183, CA20319, CA46441, CA27057, CA04919, and CA11083). M.V.D. is supported in part by funds from the National Institutes of Health, National Cancer Institute (CA135110 and CA106802).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.V.D. served as study chair, designed and coordinated research, and wrote the manuscript; B.B. and J.D.S. served as study co-chairs; B.B. served as chair of the SWOG Myeloma Committee; R.S., A.H., and J.C. carried out statistical analyses; S.W., S.U., X.P., B.N., and N.P. performed clinical research; and R.Z.O. serves as current chair of the SWOG Myeloma Committee.
Conflict-of-interest disclosure: J.D.S. is employed by and has equity shares in Myeloma Health, LLC, a clinical diagnostics company that uses J.D.S. patents in gene expression profiles service and analyses provided to clinicians treating patients with multiple myeloma. The remaining authors declare no competing financial interests.
Correspondence: Madhav Dhodapkar, Yale University, 333 Cedar St, New Haven, CT 06520; e-mail: madhav.dhodapkar@yale.edu.
References
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–5417. doi: 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749–757. [PubMed] [Google Scholar]
- 5.Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–2590. doi: 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 6.Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–569. doi: 10.1056/NEJMoa01133202. [DOI] [PubMed] [Google Scholar]
- 7.Kyle RA, Durie BG, Rajkumar SV, et al. International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121–1127. doi: 10.1038/leu.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexanian R. Localized and indolent myeloma. Blood. 1980;56(3):521–525. [PubMed] [Google Scholar]
- 9.Dispenzieri A, Kyle RA, Katzmann JA, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood. 2008;111(2):785–789. doi: 10.1182/blood-2007-08-108357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pérez-Persona E, Vidriales MB, Mateo G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–2592. doi: 10.1182/blood-2007-05-088443. [DOI] [PubMed] [Google Scholar]
- 11.Cherry BM, Korde N, Kwok M, et al. Modeling progression risk for smoldering multiple myeloma: results from a prospective clinical study. Leuk Lymphoma. 2013;54(10):2215–2218. doi: 10.3109/10428194.2013.764419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesi M, Bergsagel PL. Many multiple myelomas: making more of the molecular mayhem. Hematology Am Soc Hematol Educ Program. 2011;2011:344-353. [DOI] [PMC free article] [PubMed]
- 13.Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaughnessy JD, Jr, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 15.Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210–2221. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker R, Barlogie B, Haessler J, et al. Magnetic resonance imaging in multiple myeloma: diagnostic and clinical implications. J Clin Oncol. 2007;25(9):1121–1128. doi: 10.1200/JCO.2006.08.5803. [DOI] [PubMed] [Google Scholar]
- 17.Hillengass J, Fechtner K, Weber MA, et al. Prognostic significance of focal lesions in whole-body magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 2010;28(9):1606–1610. doi: 10.1200/JCO.2009.25.5356. [DOI] [PubMed] [Google Scholar]
- 18.Zhan F, Tian E, Bumm K, Smith R, Barlogie B, Shaughnessy J., Jr Gene expression profiling of human plasma cell differentiation and classification of multiple myeloma based on similarities to distinct stages of late-stage B-cell development. Blood. 2003;101(3):1128–1140. doi: 10.1182/blood-2002-06-1737. [DOI] [PubMed] [Google Scholar]
- 19.Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18(6):695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 20.Cox DR. Regression and life tables. Stat Soc. 1972;34:187–202. [Google Scholar]
- 21.Crowley J, LeBlanc M, Jacobson J, Salmon S. Some exploratory tools for survival analysis. In: Proceedings of the First Seattle Symposium in Biostatistics, Lecture Notes in Statistics. 1997;1:199-229. [Google Scholar]
- 22.LeBlanc M, Crowley J. Survival by goodness of split. J Am Stat Assoc. 1993;88:457–467. [Google Scholar]
- 23.Xu R, O'Quigley JA. R2 type measure of dependence for proportional hazards models. J Nonparametr Statist. 1999;12:83–107. [Google Scholar]
- 24.Rosiñol L, Bladé J, Esteve J, et al. Smoldering multiple myeloma: natural history and recognition of an evolving type. Br J Haematol. 2003;123(4):631–636. doi: 10.1046/j.1365-2141.2003.04654.x. [DOI] [PubMed] [Google Scholar]
- 25.Bladé J, Dimopoulos M, Rosiñol L, Rajkumar SV, Kyle RA. Smoldering (asymptomatic) multiple myeloma: current diagnostic criteria, new predictors of outcome, and follow-up recommendations. J Clin Oncol. 2010;28(4):690–697. doi: 10.1200/JCO.2009.22.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2(3):175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 27.Rajkumar SV, Gupta V, Fonseca R, et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia. 2013;27(8):1738–1744. doi: 10.1038/leu.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreau P, Attal M, Garban F, et al. SAKK; IFM Group. Heterogeneity of t(4;14) in multiple myeloma. Long-term follow-up of 100 cases treated with tandem transplantation in IFM99 trials. Leukemia. 2007;21(9):2020–2024. doi: 10.1038/sj.leu.2404832. [DOI] [PubMed] [Google Scholar]
- 29.Hanamura I, Stewart JP, Huang Y, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108(5):1724–1732. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neben K, Jauch A, Hielscher T, et al. The chromosome abnormalities Del(17p), t(4;14), and +1q21 predict progression from smoldering to symptomatic multiple myeloma [abstract]. Blood. 2012 120. Abstract 1806. [Google Scholar]
- 31.Hose D, Rème T, Hielscher T, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011;96(1):87–95. doi: 10.3324/haematol.2010.030296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terpstra WE, Lokhorst HM, Blomjous F, Meuwissen OJ, Dekker AW. Comparison of plasma cell infiltration in bone marrow biopsies and aspirates in patients with multiple myeloma. Br J Haematol. 1992;82(1):46–49. doi: 10.1111/j.1365-2141.1992.tb04592.x. [DOI] [PubMed] [Google Scholar]
- 33.Spisek R, Kukreja A, Chen LC, et al. Frequent and specific immunity to the embryonal stem cell-associated antigen SOX2 in patients with monoclonal gammopathy. J Exp Med. 2007;204(4):831–840. doi: 10.1084/jem.20062387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mitsiades CS, McMillin DW, Klippel S, et al. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am. 2007;21(6):1007–1034, vii-viii. doi: 10.1016/j.hoc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Melton LJ., III Long-term follow-up of 241 patients with monoclonal gammopathy of undetermined significance: the original Mayo Clinic series 25 years later. Mayo Clin Proc. 2004;79(7):859–866. doi: 10.4065/79.7.859. [DOI] [PubMed] [Google Scholar]
- 36.Cesana C, Klersy C, Barbarano L, et al. Prognostic factors for malignant transformation in monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. J Clin Oncol. 2002;20(6):1625–1634. doi: 10.1200/JCO.2002.20.6.1625. [DOI] [PubMed] [Google Scholar]
- 37.Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106(3):812–817. doi: 10.1182/blood-2005-03-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215–224. doi: 10.1038/leu.2008.307. [DOI] [PubMed] [Google Scholar]
- 39.Larsen JT, Kumar SK, Dispenzieri A, Kyle RA, Katzmann JA, Rajkumar SV. Serum free light chain ratio as a biomarker for high-risk smoldering multiple myeloma. Leukemia. 2013;27(4):941–946. doi: 10.1038/leu.2012.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kastritis E, Terpos E, Moulopoulos L, et al. Extensive bone marrow infiltration and abnormal free light chain ratio identifies patients with asymptomatic myeloma at high risk for progression to symptomatic disease. Leukemia. 2013;27(4):947–953. doi: 10.1038/leu.2012.309. [DOI] [PubMed] [Google Scholar]
- 41.Barlogie B, van Rhee F, Shaughnessy JD, Jr, et al. Seven-year median time to progression with thalidomide for smoldering myeloma: partial response identifies subset requiring earlier salvage therapy for symptomatic disease. Blood. 2008;112(8):3122–3125. doi: 10.1182/blood-2008-06-164228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajkumar SV, Gertz MA, Lacy MQ, et al. Thalidomide as initial therapy for early-stage myeloma. Leukemia. 2003;17(4):775–779. doi: 10.1038/sj.leu.2402866. [DOI] [PubMed] [Google Scholar]
- 43.Mateos MV, Hernández MT, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438–447. doi: 10.1056/NEJMoa1300439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.