

Abstract
Background
Innate immune and inflammatory responses mediated by Toll like receptors (TLRs) have been implicated in myocardial ischemia/reperfusion (I/R) injury. This study examined the role of TLR3 in myocardial injury induced by two models, namely, myocardial infarction (MI) and I/R.
Methods
First, we examined the role of TLR3 in MI. TLR3 deficient (TLR3−/−) and wild type (WT) mice were subjected to MI induced by permanent ligation of the left anterior descending coronary artery (LAD) for 21 days. Cardiac function was measured by echocardiography. Next, we examined whether TLR3 contributes to myocardial I/R injury. TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion for up to 3 days. Cardiac function and myocardial infarct size were examined. We also examined the effect of TLR3 deficiency on I/R-induced myocardial apoptosis and inflammatory cytokine production.
Results
TLR3−/− mice showed significant attenuation of cardiac dysfunction after MI or I/R. Myocardial infarct size and myocardial apoptosis induced by I/R injury were significantly attenuated in TLR3−/− mice. TLR3 deficiency increases Bcl2 levels and attenuates I/R-increased Fas, FasL, FADD, Bax and Bak levels in the myocardium. TLR3 deficiency also attenuates I/R-induced myocardial NF-κB binding activity, TNF-α and IL-1β production as well as I/R-induced infiltration of neutrophils and macrophages into the myocardium.
Conclusions
TLR3 plays an important role in myocardial injury induced by MI or I/R. The mechanisms involve activation of apoptotic signaling and NF-κB binding activity. Modulation of TLR3 may be an effective approach for ameliorating heart injury in heart attack patients.
Keywords: TLRs, myocardial I/R, apoptosis, NF-κB, inflammatory cytokines
Introduction
Cardiovascular disease is the number one killer in the United States[27]. Each year, an estimated 785,000 Americans will have a new coronary attack, 470,000 will have a recurrent attack and 195,000 Americans will have silent myocardial infarctions[27]. Despite extensive investigation, the cellular and molecular mechanisms that are involved in the initiation and progress of myocardial injury in response to ischemia/reperfusion (I/R) are still unclear.
Innate immune and inflammatory responses mediated by Toll-like receptors (TLRs) have been demonstrated to be involved in the pathophysiology of myocardial I/R injury[3,23]. TLRs are pattern recognition receptors that play an important role in the induction of innate immune and inflammatory responses[21,42]. TLR-mediated signaling predominately activates nuclear factor KappaB (NF-κB) which is an important transcription factor regulating the expression of genes associated with innate immunity and inflammatory responses as well as cell growth, cell survival, and cell death[21,42]. We and others have reported that TLR4 deficiency or modulation of TLR4 mediated signaling decreases myocardial injury following I/R[1,3,4,13,17,23].
TLR3 is located in intracellular endosomes and recognizes double-stranded RNA (dsRNA) and polyinosinic-polycytidylic acid (Poly I:C, a synthetic analog of dsRNA), resulting in induction of antiviral immune responses[20]. Recently, Cavassani et al reported that TLR3 deficient (TLR3−/−) mice showed an increased survival rate in cecal ligation and puncture induced sepsis[2]. We have shown that TLR3−/− mice exhibit protection against polymicrobial sepsis-induced cardiac dysfunction[10]. These data suggest that TLR3 plays an important role in cardiac function during sepsis. However, whether TLR3 contributes to myocardial injury induced by myocardial infarction or I/R has not been investigated. It is possible that TLR3 plays a role in myocardial ischemic injury by recognition of endogenous ligands, i.e. damage-associated molecular patterns (DAMPs) that are released during myocardial I/R injury.
We hypothesized that TLR3 contributes to myocardial injury by recognition of DAMPs during myocardial I/R. To evaluate our hypothesis, we examined the role of TLR3 in myocardial injury induced by either permanent ligation-induced myocardial infarction (MI) or ischemia/reperfusion (I/R) using TLR3 deficient (TLR3−/−) mice. We observed that TLR3 deficiency significantly attenuates myocardial dysfunction induced by both models, i.e MI and I/R. TLR3 deficiency also reduces infarct size and myocardial apoptosis after I/R injury. Our data indicate that TLR3 plays an important role in myocardial ischemic and I/R injury.
Materials and Methods
Animals
TLR3 knockout mice (TLR3−/−) and wild type (WT) genetic background control mice (C57BL/6) were obtained from Jackson Laboratory (Indianapolis, IN)[10]. The mice were maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU). The experiments outlined in this article conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication, 8th Edition, 2011). All aspects of the animal care and experimental protocols were approved by the ETSU Committee on Animal Care.
Models of Myocardial infarction (MI) and ischemia/reperfusion (I/R) injury
Myocardial infarction was induced by permanent ligation of the left anterior descending (LAD) coronary artery as described previously[18]. Myocardial I/R injury was induced as described previously[11,13,17,38]. Briefly, TLR3−/− and age-matched WT male mice (26–28 gram body weight) were anaesthetized by 5.0% isoflurane inhalation, intubated and ventilated with room air using a rodent ventilator. Anesthesia was maintained by inhalation of 1.5% isoflurane driven by 100% oxygen flow. Body temperature was regulated at 37°C by surface water heating. Following the skin incision, the hearts were exposed through a left thoracotomy in the fourth intercostal space. For induction of MI, the LAD coronary was permanently ligated with 8-0 silk ligature[18]. For induction of I/R injury, the LAD coronary artery was ligated with 8-0 silk ligature that was tied using a ‘shoestring knot’ over a 1 mm polyethylene tube (PE-10). After completion of 45 min of occlusion, the coronary artery was reperfused by pulling on the exteriorized suture to release the knot. Cardiac function was measured by echocardiography[26,38]. After completion of the experiments, the mice were euthanized by CO2 inhalation and the hearts were harvested.
Evaluation of myocardial infarct size
Myocardial infarct size was evaluated by triphenyltetrazolium chloride (TTC, Sigma-Aldrich) staining as described previously[11,13,17,38]. Briefly, the hearts were perfused with saline on a Langendorff system to wash blood from the coronary vasculature. The LAD coronary artery was re-ligated at the previous site of ligation prior to staining with 1% Evans Blue in order to assess area at risk. Each heart was then sliced horizontally to yield five slices. The slices were incubated in 1% TTC for 15 min at 37°C, fixed by immersion in 10% neutral buffered formalin. The area of infarction on both sides of each slice was determined by an image analyzer, corrected for the weight of each slice, and summed for each heart. Ratios of risk area vs. left ventricle area (RA/LV) and infarct area vs. risk area (IA/RA) were calculated and expressed as a percentage.
Echocardiography
Transthoracic two-dimensional M-mode echocardiogram and pulsed wave Doppler spectral tracings were obtained using a Toshiba Aplio 80 Imaging System (Toshiba Medical Systems, Tochigi, Japan) equipped with a 12-MHz linear transducer as described previously[26,38]. Percent fractional shortening (%FS) and percent ejection fraction (%EF) were calculated as described previously [26,38]. All measurements were made by one observer who was blinded with respect to the identity of the tracings. All data were collected from 10 cardiac cycles.
Western blot
Western blots were performed as described previously[11,13,17,38]. Briefly, the cellular proteins were separated by SDS-polyacrylamide gel electrophoresis, transferred onto Hybond ECL membranes (Amersham Pharmacia, Piscataway, NJ). The membranes were incubated with appropriate primary antibody [anti-Fas (CD95), anti-FasL, anti-FADD, anti-vascular cell adhesion molecule-1 (VCAM-1), anti-intercellular adhesion molecule-1 (ICAM-1), anti-Bcl-2, anti-Bax, and anti-Bak, (Santa Cruz Biotech, Santa Cruz, CA), and anti-phospho-IκBα, (Cell Signaling Technology, Inc., Danvers, MA), respectively, followed by incubation with peroxidase-conjugated second antibodies (Cell Signaling Technology) and analysis by the ECL system (Amersham Pharmacia, Piscataway, NJ). To control for lane loading, the same membranes were probed with anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase, Biodesign, Saco, Maine) after being washed with stripping buffer. The signals were quantified using the Syngene G: Box gel imaging system (Syngene, USA, Fredrick, MD).
In situ apoptosis assay
Myocardial apoptosis was examined as described previously[11,13,17,38] using the In Situ Cell Death Detection Kit, Fluorescein (Roche, USA). Briefly, hearts were harvested and slices cut horizontally. One slice was immersion-fixed in 4% buffered paraformaldehyde, embedded in paraffin and cut at 5 μm thick. The sections were incubated with the commercially prepared labeling mixture supplied by the manufacturer at 37°C for one hr. Nuclei of living and apoptotic cells were counterstained with Hoechst 33342 (Invitrogen). Three slides from each block were evaluated for percentage of apoptotic cells and four fields on each slide were examined at the border areas using a defined rectangular field area with 20x magnification. The numbers of apoptotic cardiac myocytes are presented as a percentage of total cells counted.
Caspase-activity
Caspase-3/7 and -8 activities in heart tissues were measured as described previously[19] using a Caspase-Glo assay kit (Promega).
Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were isolated from heart samples as previously described[11,13,17,38] and NF-κB binding activity was measured using a LightShift Chemiluminescent EMSA kit (Thermo Fisher Scientific, Waltham, MA) according to the instructions of manufacturer.
ELISA quantification of cytokines
The levels of inflammatory cytokines (TNF-α and IL-1β) in the serum were assessed by ELISA (PeproTech, Rocky Hill, NJ) according to the instructions provided by the manufacturer[10,24,38].
Immunohistochemistry staining
Immunohistochemistry was performed as described previously[10,38]. Briefly, heart tissues were immersion-fixed in 4% buffered paraformaldehyde, embedded in paraffin, and cut at 5 um sections. The sections were stained with specific goat anti-ICAM-1 (1:50 dilution, Santa Cruz Biotechnology) and rabbit anti-VCAM-1 (1:50 dilution, Santa Cruz Biotechnology), respectively, and treated with the ABC staining system (Santa Cruz Biotechnology) according to the instructions of the manufacturer. Three slides from each block were evaluated, counterstained with hematoxylin, and examined with brightfield microscopy. Four different areas of each section were evaluated.
Accumulation of neutrophils and macrophages
Neutrophil accumulation in the heart tissues was examined by staining with naphtol AS-D Chloroacetate Esterase (Sigma-Aldrich, St. Louis, MO) as described previously[10,38]. Macrophages in the myocardium were examined with the macrophage specific antibody F4/80 (1:50 dilution, Santa Cruz, CA). Three slides from each block were evaluated, counterstained with hematoxylin, and examined with brightfield microscopy. Four different areas of each section were evaluated. The results are expressed as the numbers of macrophages/field (40x).
Statistical analysis
All data were expressed as mean ± SEM. Comparisons of data between groups were made using one-way analysis of variance (ANOVA) and Tukey’s procedure for multiple range tests was performed. P< 0.05 was considered to be significant.
Results
TLR3 deficiency attenuates cardiac dysfunction induced by myocardial infarction
To investigate whether TLR3 plays a role in cardiac dysfunction following myocardial infarction, we performed permanent ligation of LAD artery in TLR3−/− and WT mice and measured cardiac function before and 3, 7 14 and 21 days after induction of myocardial infarction. As shown in Figure 1A, ejection fraction (EF%) and fractional shortening (%FS) values were significantly reduced, compared with baseline, in WT and TLR3−/− mice following permanent ligation. The EF% and %FS in TLR3−/− and WT mice decreased as a function of time. However, TLR3 deficiency resulted in significantly greater attenuation of cardiac dysfunction after myocardial infarction. There was no significant difference in EF% and %FS of baseline between TLR3−/− and WT mice. The data indicates that TLR3 contributes to cardiac dysfunction following induction of myocardial infarction.
Figure 1. TLR3 deficiency attenuates cardiac dysfunction following myocardial infarction or I/R injury and decreases myocardial infarct size following myocardial I/R injury.

TLR3−/− and age-matched WT mice were subjected to myocardial infarction (MI) by permanent ligation of LAD artery (A) or myocardial ischemia (45 min) followed by reperfusion for up to 3 days (B). Cardiac function was measured by echocardiography before (baseline) and after ischemia/reperfusion. (C) TLR3 deficiency decreases myocardial infarct size. TLR3−/− and age-matched WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion (4 hrs). Infarct size was determined by TTC staining. Blue color shows non-ischemic areas, red color indicates ischemic areas. Pale (white) indicates necrotic tissues. Ratios of risk area vs. left ventricle area (RA/LV) and infarct area vs. risk area (IA/RA) were calculated and are presented in the graphs. Photographs of representative heart sections are shown above. There were 8 mice in each group. *p < 0.05 compared with indicated groups. #p < 0.05 compared with respective baseline.
TLR3 deficiency attenuates cardiac dysfunction following transient myocardial ischemia followed by reperfusion
Next, we examined the role of TLR3 in cardiac function following myocardial I/R injury. TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion for up to 3 days. Cardiac function was assessed by echocardiography before and after reperfusion for 1 and 3 days. Figure 1B showed that EF% and %FS in WT and TLR3−/− mice were significantly decreased following myocardial I/R injury compared with the baseline. However, TLR3 deficiency markedly attenuated I/R-induced cardiac dysfunction. The EF% and %FS values in TLR3−/− mice were significantly greater on day 1 (45.1% and 52.7%) and on day 3 (39.7% and 42.3%) after reperfusion than in WT I/R mice, respectively. The data further confirm that TLR3 contributes to cardiac dysfunction in myocardial I/R injury.
TLR3 deficiency reduces myocardial infarct size following myocardial I/R injury
We also examined the role of TLR3 in myocardial infarct size following I/R. TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) following by reperfusion (4 hrs). Hearts were harvested for the evaluation of infarct size. As shown in Figure 1C, ischemia followed by reperfusion induced significant myocardial injury as denoted by the infarct size in WT mice. In contrast, infarct size was significantly reduced (42.9%) in TLR3−/− mice following I/R, when compared with WT mice. There was no significant difference in the ratio of risk area/left ventricle (RA/LV), which reflects the position of the coronary artery ligation, between TLR3−/− and WT mice.
TLR3 deficiency attenuates I/R-induced myocardial apoptosis
Cardiac myocyte apoptosis contributes to myocardial I/R injury[36]. We examined whether TLR3 deficiency will attenuate myocardial apoptosis following I/R injury. Figure 2A shows that I/R significantly increased the TUNEL positive apoptotic cells by 9.1 fold in the WT myocardium compared with WT sham control. I/R also induced myocardial apoptosis in TLR3−/− mice by 5.2 fold, when compared with TLR3−/− sham control mice. However, myocardial apoptotic cells in TLR3−/− I/R mice were markedly reduced by 41.1% compared with WT I/R mice.
Figure 2. TLR3 deficiency attenuates I/R-induced myocardial apoptosis.

TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion (24 h). Sham surgical operation served as sham control. Myocardial apoptosis was examined by the TUNEL assay in the heart sections. (A) DAPI stained nuclei are blue color and TUNEL positive cells show green fluorescence. The bar graph shows the percent of apoptotic cells. n=3 in each group. (B) TLR3 deficiency prevents I/R-induced activity of caspase-3/7 and caspase-8 in the myocardium. There were 6 mice in each group. *p < 0.05 compared with indicated groups.
Caspase-3 and -8 activities are the specific markers for apoptosis. As shown in Figure 2B, I/R significantly induced caspase-3/7 activity by 51.1% and caspase-8 by 45% respectively, in the WT I/R myocardium, when compared with sham control. In contrast, TLR3−/− mice showed a significant attenuation of I/R-induced caspase-3/7 and caspase-8 activities following myocardial I/R injury. The levels of caspase-3/7 and caspase-8 activity in TLR3−/− I/R mice were reduced by16.4% and 16.6% (p<0.05) when compared with WT I/R mice.
TLR3 deficiency attenuates up regulation of pro-apoptotic factors in the myocardium
Activation of Fas/FasL mediated apoptotic signaling plays a role in I/R-induced myocardial apoptosis[36]. Bax and Bak1 are pro-apoptotic mediators[36]. We examined the effect of TLR3 deficiency on I/R-induced activation of Fas/FasL-mediated extrinsic apoptotic signaling in the myocardium. Figure 3A–C show that I/R significantly increased the levels of Fas, FasL and FADD, respectively in the WT myocardium when compared with WT sham control. In contrast, TLR3 deficiency markedly attenuated I/R-induced increases in the levels of Fas by 27.2% and prevented I/R-increased FasL and FADD, respectively, compared with WT I/R mice.
Figure 3. TLR3 deficiency attenuates I/R-increased FasL, Fas, FADD, Bak, and Bax expression, and prevents I/R-induced decreases in myocardial Bcl-2 levels.

TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion (24 h). Sham surgical operation serve as sham control. TLR3 deficiency attenuates I/R-increased the levels of Fas (A), FasL (B), FADD (C), Bak (D), and Bax (E) in the myocardium. (F) TLR3 deficiency increases the levels of Bcl2 in the myocardium following myocardial I/R. There were 6 mice in each group. *p < 0.05 compared with indicated groups.
We also examined the effect of TLR3 deficiency on I/R-induced activation of intrinsic apoptotic signaling in the myocardium. In intrinsic apoptotic signaling, Bcl-2 is an important anti-apoptotic factor while Bak and Bax are pro-apoptotic mediators[36]. Figure 3D–F shows that I/R markedly increased the levels of Bax (↑52.6%) and Bak (↑52.9%) and decreased the levels of Bcl2 in the myocardium compared with sham control. In contrast, TLR3 deficiency prevented I/R increases in the levels of Bax and Bak, when compared with WT I/R mice. Importantly, TLR3−/− completely prevented the deleterious effect of I/R on myocardial Bcl2 levels. In addition, the levels of Bcl2 in TLR3−/− sham mice were significantly greater than in WT sham mice.
TLR3 deficiency prevents I/R-induced myocardial NF-κB binding activity and systemic TNF-α and IL-1β production
We have previously reported that I/R significantly induced myocardial NF-κB binding activity[12,13,16,17]. Inhibition of NF-κB activity has been shown to attenuate myocardial injury following I/R[22,29]. We determine the whether TLR3 deficiency will attenuate I/R-induced myocardial NF-κB activity. Figure 4A shows that I/R significantly induced NF-κB binding activity by 1.76 fold in WT mice compared with sham control. TLR3 deficiency prevented I/R-induced NF-κB binding activity in the myocardium. Activation of NF-κB stimulates the expression of numerous genes, including pro-inflammatory cytokines[21,42]. Figure 4B shows that circulating levels of TNFα and IL-1β were markedly increased by 1.2 fold and 2.6 fold, respectively, following myocardial I/R compared with sham control. However, TLR3 deficiency attenuated I/R-induced increases in the production of TNFα and IL-1β in the serum.
Figure 4. TLR3-deficiency prevents I/R-induced myocardial NF-κB binding activity and pro-inflammatory cytokine production in the circulation.
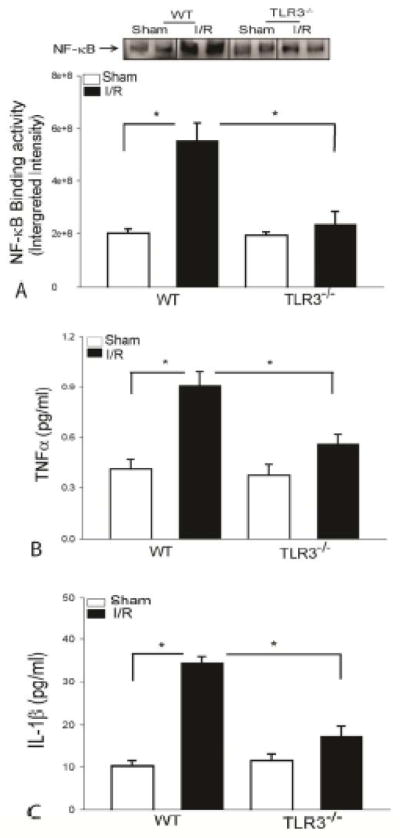
TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion (24 h). Sham surgical operation serve as sham control. Hears were harvested for the preparation of the nuclear and cytoplasmic proteins. TLR3 deficiency prevents I/R-induced NF-κB binding activity (A), TNF-α (B), and IL-1β (C) production. There were 6 mice in each group. *p < 0.05 compared with indicated groups.
TLR3 deficiency attenuates I/R-induced infiltration of neutrophils and macrophages into the myocardium
Infiltration of neutrophils and macrophages into the myocardium plays an important role in cardiac dysfunction and myocardial I/R injury[7,14]. We examined the effect of TLR3 deficiency on the infiltration of neutrophils and macrophages into the myocardium following myocardial I/R injury. Figure 5A shows that I/R increased the number of neutrophils in the WT myocardium by 20 fold on day 1 and by 30 fold on day 3 after reperfusion, respectively, when compared with sham control. I/R also induced the infiltration of neutrophils into the myocardium in TLR3−/− mice when compared with sham control. However, the numbers of neutrophils in the myocardium of TLR3−/− I/R mice were markedly reduced by 53.3% on day 1 and by 38.8% on day 3 after reperfusion, respectively compared with WT I/R mice. Figure 5B shows that the numbers of positively staining macrophages were markedly increased in the WT myocardium by 8.6 fold on day 1 and by 15 fold on day 3 after reperfusion compared with sham control. In contrast, I/R induced infiltration of macrophages into the myocardium in TLR3−/− mice was significantly reduced, when compared with WT I/R mice.
Figure 5. TLR3 deficiency attenuates I/R-induced infiltration of neutrophils and macrophages into the myocardium.


TLR3−/− and WT mice were subjected to myocardial ischemia (45 min) followed by reperfusion for indicated time. Sham surgical operation served as sham control. (A) TLR3 deficiency decreases the infiltration of neutrophils (A) and macrophages (B) into the myocardium. The pink color indicates positive neutrophils in the myocardium (A). The dark brown color indicates positive macrophages (B). There were 3 mice in each group. TLR3 deficiency prevents I/R-induced increases in the expression of VCAM-1 (C) and ICAM-1 (D) in the myocardium. The dark brown color indicates positive staining of VCAM-1 or ICAM-1 in the myocardium. There were 6 mice in each group. *p < 0.05 compared with indicated groups.
TLR3 deficiency attenuates I/R-induced expression of adhesion molecules in the myocardium
Increased expression of adhesion molecules, such as VCAM-1 and ICAM-1, promotes neutrophil and macrophage infiltration into the myocardium during myocardial I/R injury[8,9]. We examined the role of TLR3 deficiency in I/R-induced the expression of adhesion molecules in the myocardium. As shown in Figure 5C, I/R significantly increased the levels of VCAM-1 by 57.8% and ICAM-1 by 118.7% in the myocardium of WT mice compared with sham control. Immunohistochemistry also showed more positive staining of VCAM-1 and ICAM-1 in the heart tissues of WT I/R mice (Figure 5D). In contrast, TLR3 deficiency attenuated I/R-increased the levels of VCAM-1 by 40.7% and ICAM-1 by 31.4%, respectively compared with WT I/R mice. Immunohistochemistry showed that there was less staining of VACAM and ICAM-1 in the heart tissues of TLR3−/− I/R mice.
Discussion
The present study demonstrates that TLR3 contributes to myocardial injury induced by either permanent ligation-induced myocardial infarction (MI) or myocardial I/R. Specifically, we observed that TLR3−/− mice showed significantly attenuated cardiac dysfunction following either permanent ligation of the LAD artery or transient ischemia followed by reperfusion. Myocardial I/R-induced infarct size observed in WT mice was also significantly reduced in TLR3−/− mice. In addition, TLR3 deficiency attenuated I/R-induced myocardial apoptosis, prevented I/R-induced NF-κB binding activity, and sequestration of inflammatory cells into the myocardium. The data suggest that TLR3 plays an important role in myocardial I/R injury through activation of apoptotic signaling, stimulation of inflammatory responses, and promotion of inflammatory cell infiltration into the myocardium. Thus, TLR3 may be an important target for the management and treatment of myocardial I/R injury.
TLR3 recognizes viral double-stranded RNA (dsRNA) and induces antiviral immune responses[20]. Our data show that TLR3 deficiency attenuates myocardial injury in response to I/R, suggesting that TLR3 may recognize endogenous ligands during myocardial I/R. At present we do not fully understand the nature of the endogenous ligands or DAMPs that activate TLR3 during myocardial I/R injury. Recently, Cavassani et al [2] have demonstrated that TLR3 serves as an endogenous sensor that recognizes RNA released from necrotic cells, resulting in amplification of inflammation in experimental polymicrobial peritonitis and ischemic gut injury. Interestingly, recent studies have shown that TLRs serve as microRNA receptors[5,6]. MicroRNAs (miRs) are 21 to 23 nucleotide non-protein-coding RNA molecules, which have been identified as novel regulators of gene expression at the post-transcriptional level by binding to target messenger RNAs (mRNAs)[30,32]. Recent data indicates that several miRs are involved in ischemic heart disease[25,28,39]. Whether TLR3 is involved in the recognition of the increased miRs, resulting in amplification of inflammation during myocardial I/R has not been established.
We have observed that TLR3 deficiency significantly attenuated cardiac dysfunction following either myocardial infarction induced by permanent ligation of LAD artery or transient ischemia followed by reperfusion. Since transient ischemia followed by reperfusion injury is the most common clinical presentation among patients with heart attack, we employed the I/R model of myocardial injury for the subsequent mechanistic studies.
TLR3 mediates signaling via TIR/TRIF which interacts with either 1) RIP1/Peli1 to activate TRAF6, leading to NF-κB activation and nuclear translocation; 2) RIP1/RIP3 to stimulate FADD-dependent apoptotic signaling; and 3) TRAF3/TBK1 to activate IRF3/7, resulting in stimulating interferon production[41]. At the present, the role of IRF3-mediated signaling in myocardial I/R injury is unclear. Yang et al reported that HMGB1 mediates brain I/R injury via TRIF adaptor independent TLR4 signaling[40]. Since TRIF is an important component of TLR3-mediated signaling[41], the data indicates that TLR3/TRIF signaling may contribute to brain ischemic injury. On the other hand, recent studies have shown that IRF3-mediated signaling may contribute to preconditioning induced protection against cerebral I/R injury[34]. For example, Stevens et al reported that multiple preconditioning paradigms converge on IRF3/7 dependent signaling to promote tolerance to ischemic brain injury[33].
TLR3 also mediates activation of NF-κB which is an important transcription factor controlling innate immune and inflammatory cytokine gene expression[21,42]. Activation of NF-κB contributes to myocardial I/R injury[22,29]. We have previously reported that I/R significantly increased the levels of TLR4-mediated MyD88-dependent NF-κB activation in the myocardium[12,13,16,17]. In the present study, we observed that I/R markedly induced NF-κB activation in WT mice but not in TLR3 deficient mice, suggesting that TLR3 plays a role in the induction of myocardial NF-κB nuclear translocation and binding activity during myocardial I/R injury. TLR3 is located in intracellular endosomes and recognizes double stranded RNA and poly (I:C), a synthetic analog of dsRNA and byproducts from apoptotic and necrotic cells[2,15,31]. The TLR3-mediated signaling pathway predominately activates IRF3 and NF-κB through TRIF-dependent pathways[21,42]. Activation of these pathways results in the expression of various inflammatory cytokines including TNFα, IL-1β and IL-6 as well as IFNs[21,42]. It is possible that I/R results in the production of endogenous ligands, i.e. disease associated molecular patterns which are recognized by TLR3, leading to NF-κB and subsequent inflammatory cytokine production. Indeed, we have observed that TLR3 deficiency markedly attenuated I/R-increased inflammatory cytokine production in the circulation.
The infiltration of neutrophils and macrophages into the myocardium plays an important role in mediating cardiac dysfunction following myocardial I/R injury[7,14]. Recent studies have shown that ischemia induces rapid recruitment of circulating macrophages into the myocardium[7,14]. These recruited inflammatory cells release inflammatory cytokines and chemokines which further attract neutrophil infiltration and promote inflammatory responses[8,9]. We have observed in the present study that TLR3 deficiency significantly attenuated I/R-induced infiltration of macrophages and neutrophils into the myocardium which were observed in the myocardium of WT I/R mice. These results positively correlated with I/R-induced increases in the expression of adhesion molecules, such as ICAM-1 and VCAM-1. However, adhesion molecule expression in the myocardium was significantly attenuated by TLR3 deficiency. It has been well documented that activation of NF-κB regulates the expression of inflammatory cytokines and chemokines during myocardial I/R. Therefore, it is possible that TLR3 deficiency attenuated I/R-induced infiltration of inflammatory cells into the myocardium by preventing I/R-induced NF-κB binding activity.
It has been well demonstrated that cardiac myocyte apoptosis contributes to myocardial I/R injury[36]. We have observed that TLR3 deficiency significantly attenuated I/R-induced myocardial apoptosis. The mechanisms by which TLR3 deficiency attenuated I/R-induced myocardial apoptosis involve attenuation of Fas/FasL-mediated apoptotic signaling in the myocardium following I/R. In addition, TLR3 deficiency also prevented I/R-induced Bax and Bak expression and increased Bcl2 levels in the myocardium. Bax acts as an antagonist against anti-apoptotic Bcl2, while Bak-1 is the pro-apoptotic mitochondrial membrane protein. When Bcl2 is decreased and Bak-1 is oligomerized, it increases mitochondrial membrane permeability and promotes the release of cytochrome c[37,43]. Our data suggest that TLR3 contributes to I/R-induced myocardial apoptosis via activation of both extrinsic and intrinsic apoptotic signaling pathways. At present, we do not fully understand the mechanisms by which TLR3 activates apoptotic signaling during myocardial I/R injury. However, recent studies have shown that TLR3-mediated cell death is involved in the engagement of both extrinsic and intrinsic apoptotic pathways[35]. Sun et al reported that treatment of endothelial cells with the TLR3 agonist, poly (I:C) up-regulated the p53 family member,TAp63α, and initiated both intrinsic and extrinsic apoptotic pathways in a caspase-dependent manner leading to cell death[35]. The authors proposed that activation of TLR3 by either endogenous dsRNA or exogenous poly (I:C) induced the up-regulation of TAp63α, which translocated into the nucleus and bound to p53- or p63-responsive elements to up-regulate the expression of Noxa, the pro-apoptotic Bcl-2 family member and down-regulate anti-apoptotic Bcl-2[35]. TAp63α also up-regulated the expression of TRAIL and its receptors, DR4 and DR5. The interaction of TRAIL with DR4/5 activates caspase-8, resulting in the initiation of the extrinsic apoptotic signaling pathway[35]. Collectively, TLR3 may be a target for preventing I/R-induced myocardial apoptosis.
In summary, the present study demonstrated that TLR3 deficiency attenuates cardiac dysfunction induced by MI or I/R. TLR3−/− mice showed a significantly reduced myocardial infarct size following I/R injury. The mechanisms involve inhibition of I/R-activated apoptotic signaling and prevention of I/R-induced NF-κB binding activity, resulting in attenuation of I/R-induced infiltration of inflammatory cells into the myocardium. TLR3 may be an attractive target for the treatment and management of ischemic heart disease.
Highlights.
TLR3 deficiency attenuates cardiac dysfunction induced by permanent ligation of LAD or by I/R
TLLR3 deficiency decreases myocardial infarct size following I/R
TLR3 deficiency attenuates I/T-induced myocardial apoptosis
TLR3 deficiency prevents myocardial NF-κB binding activity and inflammatory cytokine production.
Acknowledgments
Funding:
This work was supported by NIH HL071837 to C.L., GM083016 to C.L. and D.L.W., GM53522 to D.L.W.
Footnotes
Conflict of Interest:
There was no conflict of interest for the authors in the present study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ao L, Zou N, Cleveland JC, Jr, Fullerton DA, Meng X. Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: mediating KC and MCP-1 expression induced by extracellular HSC70. Am J Physiol Heart Circ Physiol. 2009;297:H21–H28. doi: 10.1152/ajpheart.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cha J, Wang Z, Ao L, Zou N, Dinarello CA, Banerjee A, Fullerton DA, Meng X. Cytokines link Toll-like receptor 4 signaling to cardiac dysfunction after global myocardial ischemia. Ann Thorac Surg. 2008;85:1678–1685. doi: 10.1016/j.athoracsur.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–179. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 5.Fabbri M, Paone A, Calore F, Galli R, Croce CM. A new role for microRNAs, as ligands of Toll-like receptors. RNA Biol. 2013;10:169–74. doi: 10.4161/rna.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, Zanesi N, Crawford M, Ozer GH, Wernicke D, Alder H, Caligiuri MA, Nana-SInkam P, Perotti D, Groce CM. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. PNAS. 2012;109:E2110–E2116. doi: 10.1073/pnas.1209414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Formigli L, Manneschi LI, Nediani C, Marcelli E, Fratini G, Orlandini SZ, Perna AM. Are Macrophages Involved in Early Myocardial Reperfusion Injury? Ann Thorac Surg. 2001;71:1596–1602. doi: 10.1016/s0003-4975(01)02400-6. [DOI] [PubMed] [Google Scholar]
- 8.Frangogiannis NG, Entman ML. Chemokines in Myocardial Ischemia. Trends Cardiovasc Med. 2005;15:163–169. doi: 10.1016/j.tcm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 10.Gao M, Ha T, Zhang X, Liu L, Wang X, Kelley J, Singh K, Kao R, Gao X, Williams D, Li C. Toll-like receptor 3 plays a central role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2012;40:2390–2399. doi: 10.1097/CCM.0b013e3182535aeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ha T, Hu Y, Liu L, Lu C, McMullen JR, Shioi T, Isumo S, Kelley J, Kao RL, Williams DL, Gao X, Li C. TLR2 ligands induce cardioprotection against ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovascular Research. 2010;87:694–703. doi: 10.1093/cvr/cvq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua F, Ha T, Ma J, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. Blocking the MyD88-dependent pathway protects the myocardium from ischemia/reperfusion injury in rat hearts. Biochem Biophys Res Comm. 2005;338:1118–1125. doi: 10.1016/j.bbrc.2005.10.068. [DOI] [PubMed] [Google Scholar]
- 13.Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li C. Protection against Myocardial Ischemia/Reperfusion Injury in TLR4 Deficient Mice is Mediated through a Phosphoinositide 3-Kinase Dependent Mechanism. J Immunol. 2007;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- 14.Kakio T, Matsumori A, Ono K, Ito H, Matsushima K, Sasayama S. Roles and Relationship of Macrophages and Monocyte Chemotactic and Activating Factor/Monocyte Chemoattractant Protein-1 in the Ischemic and Reperfused Rat Heart. Lab Invest. 2000;80:1127–1136. doi: 10.1038/labinvest.3780119. [DOI] [PubMed] [Google Scholar]
- 15.Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 16.Li C, Browder W, Kao RL. Early activation of transcription factor NF-kB during ischemia in perfused rat heart. Am J Physiol. 1999;276:H543–H552. doi: 10.1152/ajpheart.1999.276.2.H543. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Ha T, Kelley J, Gao X, Qiu Y, Kao RL, Browder W, Williams DL. Modulating Toll-like receptor mediated signaling by (1-->3)-b-D-glucan rapidly induces cardioprotection. Cardiovascular Research. 2003;61:538–547. doi: 10.1016/j.cardiores.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Zhang Y, Li C, Xie J, Liu Y, Zhu W, Zhang X, Jiang S, Liu L, Ding Z. HSPA12B attenuates cardiac dysfunction and remodelling after myocardial infarction through an eNOS-dependent mechanism. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt139. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.Lu C, Hua F, Liu L, Ha T, Kalbfleisch J, Schweitzer J, Kelley J, Kao R, Williams D, Li C. Scavenger receptor class-A has a central role in cerebral ischemia/reperfusion injury. J Cereb Blood Flow Metab. 2010;30:1972–1981. doi: 10.1038/jcbfm.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60:805–812. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Medzhitov R, Preston-Hurlburt P, Jr, Janeway CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 22.Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- 23.Oyama J, Jr, Blais C, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 24.Ren D, Wang X, Ha T, Liu L, Kalbfleisch J, Gao X, Williams D, Li C. SR-A deficiency reduces myocardial ischemia/reperfusion injury; involvement of increased microRNA-125b expression in macrophages. Biochimica Et Biophysica Acta - Molecular Basis of Disease. 2012;1832:336–46. doi: 10.1016/j.bbadis.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren XP, Wu J, Wang X, Sartor MA, Qian J, Jones K, Nicolaou P, Pritchard TJ, Fan GC. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119:2357–2366. doi: 10.1161/CIRCULATIONAHA.108.814145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ripa RS, Wang Y, Jorgensen E, Johnsen HE, Hesse B, Kastrup J. Intramyocardial injection of vascular endothelial growth factor-A15 plasmid followed by granulocyte-colony stimulating factor to induce angiogenesis in patients with severe chronic ischaemic heart disease. Eur Heart J. 2006;27:1785–1792. doi: 10.1093/eurheartj/ehl117. [DOI] [PubMed] [Google Scholar]
- 27.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, De Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart Disease and Stroke Statistics -- 2011 Update. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salic K, De Windt LJ. MicroRNAs as Biomarkers for Myocardial Infarction. Curr Atheroscler Rep. 2012;14:193–200. doi: 10.1007/s11883-012-0238-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawa Y, Morishita R, Suzuki K, Kagisaki K, Kaneda Y, Maeda K, Kadoba K, Matsuda H. A novel strategy for myocardial protection using in vivo transfection of cis element ‘decoy’ against NFkB binding site. Evidence for a role of NFkB in ischemia-reperfusion injury. Circulation. 1997;96:II-280–II-285. [PubMed] [Google Scholar]
- 30.Sheedy FJ, O’Neill LAJ. Adding fuel to fire: microRNAs as a new class of mediators of inflammation. Ann Rheum Dis. 2008;67:iii50–iii55. doi: 10.1136/ard.2008.100289. [DOI] [PubMed] [Google Scholar]
- 31.Sloane JA, Blitz D, Margolin Z, Vartanian T. A clear and present danger: endogenous ligands of Toll-like receptors. Neuromolecular Medicine. 2010;12:149–163. doi: 10.1007/s12017-009-8094-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonkoly E, Stahle M, Pivarcsi A. MicroRNAs and immunity: Novel players in the regulation of normal immune function and inflammation. Seminars in Cancer Biology. 2008;18:131–140. doi: 10.1016/j.semcancer.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Stevens SL, Lueng PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. Multiple Preconditioning Paradigms Converge on Interferon Regulatory Factor-Dependent Signaling to Promote Tolerance to Ischemic Brain Injury. The Journal of Neuroscience. 2011;31:8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stevens SL, Stenzel-Poore MP. Toll-like receptors and tolerance to ischaemic injury in the brain. Biochem Soc Trans. 2006;34:1352–1355. doi: 10.1042/BST0341352. [DOI] [PubMed] [Google Scholar]
- 35.Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang W, He Q, Deng M, Liu S, Li G, Li Y, Zhou G, Xie P, Xie X, Duan Z, Hu J. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform {alpha} (tap63{alpha}) J Biol Chem. 2011;286:15918–28. doi: 10.1074/jbc.M110.178798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Empel VPM, Bertrand ATA, Hofstra L, Grijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovascular Research. 2005;67:21–29. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Ha T, Liu L, Zou J, Zhang X, Kalbfleisch J, Gao X, Williams D, Li C. Increased expression of microRNA-164a decreases myocardial ischemia/reperfusion injury. Cardiovascular Research. 2013;97:432–442. doi: 10.1093/cvr/cvs356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, Medvedovic M, Hu Z, Fan GC. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardial injury. Circulation. 2010;122:1308–1318. doi: 10.1161/CIRCULATIONAHA.110.964684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, Xiang J, Li JC, Fang CQ, Wang JZ. HMBG1 mediates ischemia-reperfusio injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab. 2011;31:593–605. doi: 10.1038/jcbfm.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu M, Levine SJ. Toll-like receptor 3, RIG-I-like receptors and the NLRP3 inflammasome: Key modulators of innate immune responses to double-stranded RNA viruses. Cytokine & Growth Factor Reviews. 2011;22:63–72. doi: 10.1016/j.cytogfr.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang G, Ghosh S. Toll-like receptor-mediated NF-kB activation: a phylogenetically conserved paradigm in innate immunity. The Journal of Clinical Investigation. 2001;107:13–19. doi: 10.1172/JCI11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, Xiong W, Li G, Lu J, Fodstad O, Riker AI, Tan M. MicroRNA-125b Confers the Resistance of Breast Cancer Cells to Paclitaxel through Suppression of Pro-apoptotic Bcl-2 Antagonist Kiler 1 (BAK1) Expression. Journal of Biological Chemistry. 2010;285:21496–21507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]