

Abstract
Environmental exposures during sensitive windows of development can reprogram normal physiological responses and alter disease susceptibility later in life in a process known as developmental reprogramming. For example, exposure to the xenoestrogen diethylstilbestrol (DES) during reproductive tract development can reprogram estrogen-responsive gene expression in the myometrium, resulting in hyper-responsiveness to hormone in the adult uterus and promotion of hormone-dependent uterine leiomyoma. We show here that the environmental estrogens genistein (GEN), a soy phytoestrogen, and the plasticizer bisphenol A (BPA), differ in their pattern of developmental reprogramming and promotion of tumorigenesis (leiomyomas) in the uterus. While both GEN and BPA induce genomic estrogen receptor (ER) signaling in the developing uterus, only GEN induced PI3K/AKT non-genomic ER signaling to the histone methyltransferase Enhancer of Zeste homolog 2 (EZH2). As a result, this “pre-genomic” signaling phosphorylates and represses EZH2, and reduces levels of H3K27 repressive mark in chromatin. Furthermore, only GEN caused estrogen-responsive genes in the adult myometrium to become hyper-responsive to hormone; estrogen-responsive genes were repressed in BPA exposed uteri. Importantly, this pattern of EZH2 engagement to decrease versus increase H3K27 methylation correlated with the effect of these xenoestrogens on tumorigenesis. Developmental reprogramming by GEN promoted development of uterine leiomyomas, increasing tumor incidence and multiplicity, while BPA did not. These data demonstrate that environmental estrogens have distinct non-genomic effects in the developing uterus that determines their ability to engage the epigenetic regulator EZH2, decrease levels of the repressive epigenetic histone H3K27 methyl mark in chromatin during developmental reprogramming, and promote uterine tumorigenesis.
Keywords: developmental reprogramming, xenoestrogens, uterine leiomyoma, genistein, bisphenol A, EZH2
INTRODUCTION
The term “developmental reprogramming” is used to describe the effects of early life exposures to adverse stimuli that can alter response to normal physiological signals and give rise to disease in adulthood. Numerous studies demonstrate that perinatal exposure to xenoestrogens, many found ubiquitously in the environment, can developmentally reprogram the female reproductive tract, causing alterations in morphology, hormonal milieu, and gene expression, and give rise to diseases such as obesity and cancer (1-3). Developmental reprogramming by in utero exposure to the xenoestrogen diethylstilbestrol (DES) is an early example of this phenomenon. Gestational exposure to DES results in malformations of the uterus, infertility, and vaginal cancers (e.g., clear cell vaginal adenocarcinoma) in daughters of women prescribed this drug during pregnancy. New evidence published in recent epidemiological reports has also shown a correlation between in utero DES exposure and an increased risk for breast cancer (4) as well as uterine leiomyomas (5), though the latter remains somewhat controversial (6).
Although DES is no longer in clinical use, other environmental xenoestrogens have the potential to affect the developing reproductive tract and induce developmental reprogramming. For example, bisphenol A (BPA), a chemical used in the production of plastics, food can linings, and dental sealants, can induce morphological abnormalities of the reproductive tract in rodents exposed neonatally to this xenoestrogen (7-9). BPA can induce precocious puberty, persistent vaginal cornification, absence of corpora lutea, cystic ovaries, cystic endometrial hyperplasia, and polyovular follicles in adult animals exposed perinatally to this compound (7, 10, 11). In the male reproductive tract, neonatal exposure to environmentally relevant micromolar BPA concentrations has been shown to promote neoplastic transformation, including formation of prostatic intraepithelial neoplasia (12). Genistein (GEN), a phytoestrogen in soybeans that is found in processed foods and soy-based infant formula, can also induce developmental reprogramming of the female reproductive tract in animals. Multiple studies report that neonatal exposure to environmentally relevant doses of GEN (e.g., 2.4-6.6 μM in plasma) (13) aberrantly reprograms reproductive function and morphology as evidenced by induction of ovarian and uterine morphological abnormalities, persistent estrus, accelerated vaginal opening, infertility, early reproductive senescence, multioocyte follicles, and uterine adenocarcinomas. In a recent epidemiological study of over 19,000 women, a correlation was found between early life soy formula consumption and increased risk of uterine leiomyomas (5), suggesting a link may also exist between environmental estrogen exposure and development of these tumors in women.
Uterine leiomyoma, commonly called fibroids, are benign gynecologic tumors of the uterine myometrium (14). Although these hormone-dependent tumors are the most frequent gynecologic tumor of women, little is known about how environmental exposures may contribute to the high incidence of this disease (2, 15). Previous studies from our group have demonstrated in genetically predisposed Eker rats that susceptibility to the development of uterine leiomyoma is modulated by developmental exposure to DES via developmental reprogramming of the reproductive tract and estrogen-responsive gene expression (16, 17). Exposure of neonatal Eker rats to DES during postnatal days 3 through 12 was sufficient to increase the penetrance of the tuberous sclerosis complex 2 (Tsc2) tumor suppressor gene defect, and increase spontaneous incidence of leiomyomas from 65% to 100% (16, 18). In these animals, DES also reprogrammed the morphology of the reproductive tract, giving rise to persistent vaginal cornification and ovaries that lacked corpora lutea. In addition, microarray analyses identified several estrogen-responsive genes developmentally reprogrammed by neonatal DES exposure, which became hyper-responsive to hormone in the uteri of adult animals prior to the onset of these tumors (19).
The mechanism(s) by which xenoestrogens might induce developmental reprogramming are not well understood. In the female reproductive tract, developmental reprogramming is mediated by estrogen receptor α (ERα), as demonstrated in ERα knock-out mice (ERKO), which are resistant to DES-induced developmental reprogramming (20). However, the role of genomic vs non-genomic ER signaling in developmental reprogramming has not been defined. The canonical pathway for genomic ER signaling (i.e transactivation of gene expression) can be induced by both natural endogenous estrogens (e.g. 17β-estradiol) as well as by xenoestrogens. The non-genomic pathway for rapid activation of membrane ER signaling is less well understood, though several studies demonstrate that rapid, non-genomic ER signaling via pathways such as PI3K and MAPK are important for blood vessel vasodilatation, neuron survival, bone loss prevention and reproductive function (21-24). Both endogenous and xenoestrogens can trigger non-genomic ER signaling through activation of PI3K/AKT, MAPK and PKA/PKC, albeit with tissue- and dose- specific effects on pathway activation (25, 26).
Both DNA methylation and histone modifications are thought to be epigenetic targets for developmental reprogramming by xenoestrogens. Histone modifications, including methylation, create binding sites for several regulators of gene expression that recognize these site-specific chromatin marks. Histone methylation, which is catalyzed by histone methyltransferases (HMTs), can be epigenetically inherited, repressing or activating gene expression. Importantly, non-genomic or more aptly “pre-genomic” ER signaling can change these epigenetic histone methyl marks. ER activation can modulate pre-genomic signaling through activation of the PI3K/AKT pathway, leading to AKT phosphorylation of the HMT, enhancer of zeste homologue 2 (EZH2). Phosphorylation of EZH2 by AKT results in decreased EZH2 activity and levels of tri-methylated lysine 27 on histone 3 (H3K27me3) (27). Reduction of H3K27me3 levels, a repressive mark for gene expression, results in increased expression of estrogen-responsive genes.
Here we demonstrate that the environmental estrogen GEN activates pre-genomic PI3K/AKT signaling to modulate EZH2 phosphorylation and decrease the repressive H3K27me3 methyl mark in the developing uterus, reprogramming estrogen-responsive genes to enhance responsiveness to hormone and increase leiomyoma incidence. In contrast, BPA, which could not increase uterine tumorigenesis, did not induce pre-genomic PI3K/AKT signaling in the neonatal uterus, increased rather than decreased H3K27me3 levels, and repressed, rather than enhanced, estrogen-responsive gene expression in the adult myometrium. Thus, activation of pre-genomic ER signaling to modulate EZH2 activity and reduce H3K27Me3 levels in the developing uterus distinguishes the genomic and non-genomic activity of xenoestrogens and their ability to induce developmental reprogramming of gene expression and tumorigenesis.
Materials and Methods
Animals and treatments
Eker rats, from a closed colony at The University of Texas M. D. Anderson Cancer Center, were cared for in accordance with the guidelines of the M. D. Anderson Cancer Center Animal Care and Use Committee in an ALAC accredited facility. Females were given water and standard rat chow (Harlan Teklad 22/5 Rodent Diet) ad libitum and maintained on a 14:10 light-dark cycle, conditions historically associated with a 65% leiomyoma incidence in this animal model. Eker rats were treated on postnatal days 10-12 with BPA 50mg/kg (Acros Organics, Morris Plains, NJ), DES 1mg/kg (Sigma Chemical Co., St. Louis, MO) or GEN 50mg/kg (Sigma) in sesame oil, using a total of 50μl of this vehicle for each subcutaneous injection. A separate group of animals were given vehicle (VEH) only as controls. Upon weaning, all animals were genotyped for the presence of the Eker mutation (Tsc-2+/+ vs. Tsc-2Ek/+). For developmental reprogramming analysis, 61 animals were sacrificed at 3 months of age.
Acute response to xenoestrogen was evaluated by sacrificing PND 12 animals 1-12hrs after a single injection of GEN, BPA, or VEH. Upon determination that the peak of non-genomic signaling appears 6hrs following exposure in uterus, subsequent exposures to evaluate acute responses were 6hrs in duration. Low-dose BPA exposure studies were conducted in both PND 12 Eker (females only) and Sprague-Dawley rats (males and females). Neonatal Eker rats were given a single s.c. injection of BPA (50mg/kg, 50μg/kg, or 50ng/kg) or DES (1mg/kg) on PND 12 and sacrificed at 6hrs after exposure. In addition, Sprague-Dawley rats were given a single s.c. (10μg/kg) or oral (0.4-50μg/kg in sesame oil) dose of BPA and groups of 3-5 animals/time point/dose sacrificed 0.5 or 6hrs after exposure. Prostate studies were performed at PND 3 rather than 12, due to the earlier developmental reprogramming window of the rat prostate (PND 1-5) versus the rat uterus (PND 10-12).
Tissue collection, histologic studies and immunohistochemistry
Female rats were euthanized at 16 months of age, uteri were fixed before paraffin embedding and staining with hematoxylin-eosin or incubated with antibody directed against Calbindin D9k (1:2000; Swant, Bellinoza, Switzerland) as described previously (18). Tumors were also measured and sectioned for pathological examination, and a portion of each was snap-frozen in liquid nitrogen. Additionally, the uninvolved uterus was sectioned and analyzed for microscopic tumors, which together with quantitation of macroscopic lesions was used to calculate tumor incidence and multiplicity. Three-month-old animals were euthanized and their uteri were removed and scraped with a sterile scalpel in cold phosphate-buffered saline solution to remove endometrium from myometrium, which were snap-frozen in liquid nitrogen and stored at -80°C. To obtain adequate amounts of tissue for RNA extraction from animals euthanized 6hrs after treatment on PND 12, two to three uteri were pooled together.
Histological categorization of estrus
Reproductive staging was performed in accordance with the procedure described by Cook et al. (28). The 16-month-old rats were staged according to degree of reproductive senescence (pseudo pregnant, persistent estrus, or anestrus). 3-month-old rats were categorized as being in proestrus, estrus, metestrus, or diestrus stages of the estrus cycle.
Analytical method for BPA determinations
Quantification of BPA serum levels was performed as previously described (28). Briefly, serum was obtained from groups of 3-5 pooled animals treated with a single dose of BPA (0.4-50μg/kg), and 5ηg of deuterated-bisphenol A (d-BPA) was added as an internal standard. The final extract was concentrated and the solvent was re-constituted with 0.5 ml of methanol. This fraction is referred to as fraction 2 total BPA= free+bound BPA. BPA levels were quantified using a high-performance liquid chromatography (HPLC) coupled with API 2000 electrospray triple-quadrupole mass spectrometer (ESI-MS/MS). Data were acquired using multiple reaction monitoring (MRM) for the transitions of 227>212 for BPA and 241>223 for d-BPA. Quantification was based on external calibration curve prepared by injecting 10 mL of 0.02, 0.05, 0.1, 0.2, 0.5, 1, 5, 10, and 50ng/mL standards.
Real-time PCR (Q-PCR)
Frozen tumors, myometrium or uteri previously exposed to VEH, GEN (50mg/kg), or BPA (50mg/kg) on PND 10-12 were crushed under liquid nitrogen with mortar and pestle and RNA isolated and DNA removed by using the RiboPure™ Kit (Ambion Biosystems, Austin, TX) according to the manufacturer’s protocol. Following RNA extraction, cDNA was made by reverse-transcribing 1 μg of RNA through the Invitrogen Superscript™ First-Strand Synthesis III System for reverse transcriptase (RT)-PCR (Invitrogen, Carlsbad, CA). Q-PCR was performed using the 7900T Fast Real-Time detection system from Applied Biosystems (ABI, Foster City, CA). Fast Real-Time Taq-Man assays from ABI were used exclusively to analyze expression of Gdf10 (Rn00666937_m1), Calbindin D9k (Rn00560940_m1), Rasd2 (Rn00592054_m1), Sfrp2 (Rn01458836_m1), Krt19 (Rn01496867_m1), Nr2f2 (Rn00756178_m1), Gria2 (Rn00568514_m1), Igfbp5 (Rn00563116_m1), Spp1 (Rn00563571_m1), Car8 (Rn01473820_m1), Mmp3 (Rn00591740_m1), Tacst1 (Rn00684677_m1), Rps6k (Rn00667685_m1), Kcnk2 (Rn00572452_m1), Cspg2 (Rn01493763_m1), Aqp3 (Rn00581754_m1), Ramp1 (Rn00671666_m1) and Dio2 (Rn00581867_m1) genes. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or ribosomal 18s (18s) were used as endogenous controls. 18s was utilized for comparison to estrogen-responsive genes in the adults due to significant association of GAPDH expression with BPA exposure. The following set of conditions were used for each fast Q-PCR reaction: 95°C for 10min, followed by 40 cycles of 1sec at 95°C and 20sec at 60°C.
Q-PCR reactions were performed in triplicate and quantified by the -ΔΔCT method. A calibrator from each set of samples was chosen from which to subtract individual GEN, and BPA sample ΔCT values to obtain -ΔΔCT. The fold change for each sample was calculated in comparison to the calibrator by taking 2-ΔΔCT. Transcript levels were measured at 6hrs after the last injection for 12-day-old animals exposed to VEH, GEN or BPA. The calibrator for both 3-month-old and 12-day-old rats was VEH-treated myometrium or uteri. For the 3-month-old rats, an estrus-staged matched VEH myometrium was used.
Preparation of Tissue Lysates and Immunoblotting
Frozen tissue from PND 12 Eker or Sprague Dawley rats treated with a single injection of VEH, GEN or BPA and harvested at 1, 6 or 12hrs after the injection, as described above, was used for the preparation of protein lysates for immunoblotting. Uteri or prostates from 2-3 neonatal animals were pooled together in RSB buffer (10mM Tris HCl pH7-7.4, 10mM NaCl, 3mM MgCl2, 1mM PMSF, 1mM Na3VO4, 1mM NaF, and Complete Protease Inhibitor Cocktail (Roche Applied Science, Indianapolis, IN)) with 0.5 % NP-40. Tissue was dounce homogenized and incubated 10min on ice. Lysates were centrifuged 15min at 15,000 rpm at 4°C and supernatant collected. Protein concentration was determined using the Pierce BCA assay (Pierce Biotechnology, Rockford, IL). Tissue lysate was separated via 10% SDS-PAGE gels (BioRad Laboratories, Hercules, CA) and transferred to PVDF. Membranes were incubated with primary antibody and washed with TBS plus 0.5% Tween-20 followed by incubation with HRP-conjugated secondary antibody. Visualization of protein abundance was performed using Pierce ECL substrate or ECL plus (Thermo Fischer Scientific, Rockford, IL). Immunoblotting was performed with antibodies against phosphorylated Akt (S473) and (T308), Akt, phosphorylated S6 (S235/236), S6, and histone H3 obtained from Cell Signaling Technology (Montgomery, TX); H3K27me3 and EZH2 from Active Motif (Carlsbad, CA); and phosphorylated EZH2 (S21) from Bethyl Incorporated (Montgomery, TX).
Immunohistochemistry
Tissue embedded in paraffin were cut into 5μm sections and placed on slides for deparaffinization. Slides were heated at 60°C for 1hr and then in further de-paraffinized in Citrusolv for 5min 3 times. Slides were rinsed twice in 100% EtOH for 2min. Sections were progressively rehydrated in 90%, 80% and 70% EtOH for 2min each and rinsed with water for 1min. Slides were then boiled in antigen unmasking solution for 5min and allowed to cool for 30min, then rinsed twice with PBS for 5min. Endogenous peroxidase activity was blocked in 2% H2O2 for 30min and rinsed in PBS. Non-specific binding with blocked with Avidin D and Biotin for 15min and washed with PBS. Sections were incubated overnight at 4° in primary antibody, calbindin D9K (1:2000, Swant), in 5% goat serum plus 0.3% Triton-X, and then rinsed in 0.03% TBST for 10min twice and washed with PBS for 10min. Sections were incubated with horseradish peroxidase-conjugated secondary antibody for 1hr and visualized by staining with Tablet DAB (Sigma Chemical Company).
Immunoprecipitation
Tissue lysates were pre-cleared using protein A sepharose beads (GE healthcare, Piscataway, NJ) and rabbit IgG (Millipore, Billerica, MA). Pre-cleared lysates were incubated with antibody against phosphorylated EZH2 and protein A sepharose beads then washed with Cell Signaling Technology (CST) lysis buffer (20 mM Tris-HCl pH 7.5, 1 mM EGTA, 1 mM Na2EDTA, 150 mM NaCl, 1 mM beta-glycerophosphate, 2.5 mM sodium pyrophosphate, 1% Triton-X, 1 mM PMSF, 1 mM NaF and Roche Complete Protease Inhibitor Cocktail). Lysate from immunoprecipitation was separated and immunoblotted as describe above.
Acid Precipitation of Histones from Tissue Lysate
Cell pellets obtained from tissue lysates describe above were resuspended in a 1:1 ratio of 5mM MgCl2 and 0.8M HCl and sonicated for 20sec (30% power) followed by 1hr incubation on ice. Histone proteins were collected by centrifugation for 10min at 14,000rpm (4° C), supernantant transferred to a new tube and precipitated with trichloroacetic acid (50%) and ddH2O. Histone precipitants were collected after centrifiguation for 20min at 14,000rpm (4° C). Histone pellets were washed with cold acetone and allowed to dry before reconstituting in a solution of 1.5M Tris-HCl pH 8.8 and ddH2O. Histones were quantitated after separation on Tris-tricine gels (10-20%) and staining with coomassie. Total H3 was used to determine relative histone methylation levels. For determination of rapid changes in H3K27me3 levels in prostate and uterus, whole cell lysates (rather than acid precipitation of chromatin-associated histones), were used and levels of H3K27 methylation quantitated as described above.
Statistics
For analysis of Q-PCR data, a linear model analysis was applied to ΔCT values to determine the effects of gene and treatment, which were determined to be significant at p value of <0.05. In order to control for multiple comparisons the Benjamini and Hochberg test controlling for false discovery rate was used in determining significance level for each Q-PCR analysis. Analysis of tumor incidence was estimated using chi-square for the determination of significance between treatment groups. Tumor multiplicity was analyzed using the Poisson regression for comparing multiple tumors between VEH, GEN and BPA groups. A value of p < 0.05 was considered statistically significant.
Results
Xenoestrogen-specific activation of non-genomic signaling in the neonatal uterus
We previously identified 18 genes, containing known or putative estrogen response elements, which were differentially expressed in hormone-dependent uterine leiomyomas. Six of these genes became developmentally reprogrammed following neonatal exposure to DES in the adult uterine myometrium to promote tumorigenesis (19). To determine if the activity of DES could be generalized to other, more environmentally relevant, xenoestrogens, we first compared the ability of two environmental estrogens, GEN (50mg/kg) and BPA (50mg/kg), to induce genomic ER signaling (i.e. modulate transcription) in the developing uterus.
Neonatal rats were exposed to VEH, GEN and BPA (VEH n=9, GEN n=6, BPA n=6) and transcript levels were measured at 6hrs following the final of three exposures on PND 10-12, the period of uterine development most susceptible to developmental reprogramming (19). Q-PCR quantitation of transactivation/repression of these 18 hormone responsive genes revealed that the majority (12/18) were responsive to one or both environmental estrogens, being either induced or repressed by GEN and/or BPA compared to VEH controls (Table 1). GEN modulated the expression of Calbindin D9k, Dio2, Krt19, Gdf10, Car8, Gria2, Mmp3, Igfbp5, Spp1, Sfrp2, Rasd2, Nr2f2 and BPA modulated the expression of 8 of these same 12 genes (Calbindin D9k, Dio2, Gdf10, Car8, Gria2, Spp1, Sfrp2, Rasd2). While qualitatively similar in terms of their ability to induce or repress gene expression in the developing uterus, quantitative differences were observed between the three xenoestrogens evaluated. For example, CalbindinD9K expression increased 28-52-fold, with GEN>DES>BPA, Dio2 was induced 7-10-fold with BPA>DES>GEN, Gria2 was repressed 11-83-fold with DES>GEN>BPA and Gdf10 was repressed 5-fold with BPA=DES=GEN. Thus, although GEN and BPA exhibited xenoestrogen-specific patterns of gene expression in the developing rat uterus, both GEN and BPA engaged the ER to induce genomic ER signaling. Transactivation of gene expression in the neonatal uterus by GEN and BPA was confirmed at the protein level by examining induction of calbindin D9k protein expression in response to these xenoestrogens by immunohistochemistry (IHC) in neonatal uteri. Uteri from animals exposed to either GEN or BPA on PND 10-12 exhibited an increase in Calbindin D9k protein as shown in Supplementary Fig. S1, confirming that at the doses selected GEN and BPA both functioned as ER agonists, inducing genomic ER signaling and transactivating gene expression in the developing rat uterus.
Table 1.
Neonatal uterine gene expression in response to xenoestrogen exposure.
| Gene | Normal Estrogen Response | Neonatal DES Responsea | Neonatal GEN Response | Neonatal BPA Response |
|---|---|---|---|---|
|
| ||||
| Fold change±SEM | Fold change±SEM | Fold change±SEM | ||
| Calbindin D9k | Induced | 39.12±1.38 | 52.30±1.96 | 28.10±1.24 |
| Dio2 | Induced | 7.31±0.28 | 6.63±0.21 | 9.51±0.6 |
| Krt19 | Induced | 2.05±0.13 | NSD | |
| Gdf10 | Repressed | -4.78±0.06 | -4.78±0.08 | -5.00±0.06 |
| Car8 | Repressed | -7.14±0.05 | -20.1±0.05 | -12.5±0.04 |
| Gria2 | Repressed | -83.3±0.03 | -33.1±0.07 | -11.1±0.01 |
| Mmp3 | Repressed | -3.85±0.08 | -2.50±0.05 | NSD |
| Igfbp5 | Repressed | -3.53±0.09 | NSD | |
| Spp1 | Repressed | -2.97±0.13 | -3.58±0.07 | |
| Sfrp2 | Repressed | -9.13±0.08 | -4.35±0.13 | |
| Rasd2 | Repressed | -5.28±0.17 | -2.53±0.14 | |
| Nr2f2 | Repressed | -4.63±0.19 | NSD | |
Data previously reported in (Greathouse et al. 2008)
Fold change relative to vehicle, SEM; standard error of the mean.
Values statistically significant at p<0.05 using one-way ANOVA.
NSD: No significant difference
We next asked whether GEN and BPA were also equivalent in their ability to induce non-genomic ER signaling in the developing uterus. Activation of non-genomic PI3K signaling in response to GEN, BPA or VEH was determined at 1, 6 and 12hrs after xenoestrogen exposure. Uteri of rats exposed to GEN exhibited robust activation of PI3K/AKT signaling relative to VEH, as evidenced by phosphorylation of AKT at S473 and T308 and its downstream effector, S6 at S235/236 (Fig. 1). Activation of PI3K/AKT signaling by GEN in the developing uterus was transient, returning to baseline levels within 12 hours of exposure, consistent with the rapid kinetics of non-genomic signaling and previous observations of in vivo non-genomic signaling by DES (29). In contrast to GEN, BPA failed to activate PI3K/AKT signaling (Fig. 1). Thus, at doses where both GEN and BPA induced genomic ER signaling, GEN but not BPA activated non-genomic PI3K/AKT signaling in the developing rat uterus.
Figure 1. Xenoestrogen-specific modulation of non-genomic PI3K/AKT signaling.
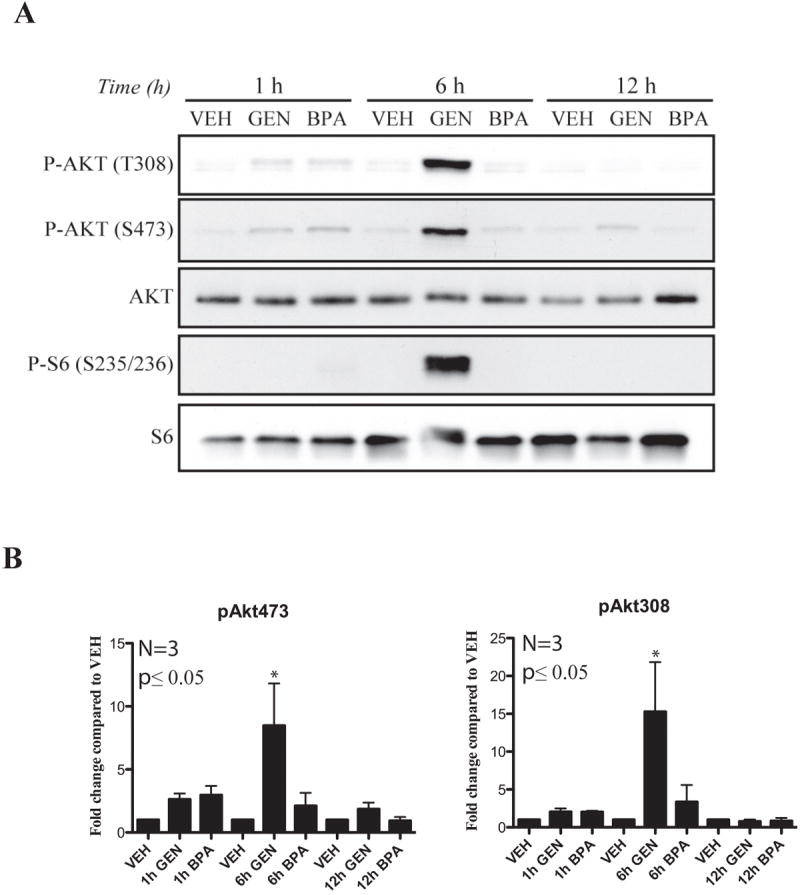
A. Western blot analysis of AKT phosphorylation (T308 and S473) and S6 phosphorylation in pooled (3) uteri of PND 10-12 Eker rats after a single exposure for 1-12hrs to VEH, GEN (50mg/kg) or BPA (50mg/kg) via s.c. injection.
B. Densitometric quantification of western blot analysis demonstrates significant differences in activation of AKT as represented by the ratio phospho-AKT T308 and S473 to total AKT in xenoestrogen-exposed Eker rats vs. VEH. Mean ± SEM is donated by error bars on each graph (n=3). The Student’s t-test was used to determine statistical significance, which was set at a value of *p≤0.05.
In some settings, BPA has been demonstrated to exhibit a U-shaped dose response curve, exhibiting stimulatory effects at low doses not evident at high(er) doses of this compound (30). To determine whether doses of BPA lower than those required for genomic ER activity could induce non-genomic signaling, we repeated these experiments at exposures 3 to 6-orders of magnitude lower. We observed that lower BPA doses (50μg/kg and 50ng/kg) were similarly ineffective at activating non-genomic PI3K/AKT signaling in the developing uterus relative to DES (1mg/kg, positive control) or VEH controls (Supplemental Fig. S2). To further investigate whether the inability of BPA to induce PI3K/AKT signaling in the developing uterus was caused by the use of subcutaneous rather than oral route of exposure, we next compared oral administration of BPA over a dose range of 0.4-50μg/kg to a subcutaneous (s.c.) injection of BPA (10μg/kg) previously established as able to induce developmental reprogramming and increase susceptibility to precancerous lesions and tumors in the prostate (12, 31). We found that oral BPA administered at 50μg/kg gave similar levels of circulating total BPA as 10μg/kg s.c. (5.19 and 4.82ng/ml (ppb) total serum BPA in males and 15.1 and 12.0ng/ml (ppb) total serum BPA in females, respectively). Importantly, over a dose range of 0.4, 2 and 50μg/kg, no activation of PI3K/AKT signaling was observed in the neonatal uterus, whereas both 2 and 50μg/kg doses of BPA activated PI3K/AKT signaling in the neonatal prostate (Supplemental Fig. S3A). This contrasted with what was observed for DES, which activated PI3K/AKT in both the uterus and prostate (Supplemental Fig. S3B).
Pre-genomic signaling to EZH2 in the developing uterus exhibits xenoestrogen-specificity
The HMT EZH2 is a target for AKT, with phosphorylation by this kinase at S21 reducing EZH2 activity and levels of the H3K27me3 repressive methyl mark in chromatin (27, 32). Differences in the ability of GEN and BPA to activate PI3K/AKT signaling suggested that these environmental estrogens might also differentially modulate the activity of this epigenetic regulator. Exposure of developing uteri to GEN increased phosphorylation of EZH2 at S21 with kinetics commensurate with activation of PI3K/AKT signaling (Fig. 2A). In contrast, doses of BPA ranging from 50mg/kg-50ng/kg failed to activate PI3K/AKT signaling and resulted in no increase in EZH2 phosphorylation relative to VEH controls (Fig. 2A and Supplemental Fig. S2). Importantly, as shown in Fig. 2B, H3K27me3 levels in chromatin were significantly reduced in GEN, but not in BPA exposed uteri, concordant with the ability of GEN to induce inhibitory phosphorylation of EZH2 at S21. In contrast, and consistent with induction of non-genomic PI3K/AKT signaling in the prostate, the same dose of BPA rapidly (0.5hr) reduced H3K27me3 levels (Supplemental Fig. S3C). These data indicate that rapid activation of non-genomic, or more appropriately “pre-genomic”, signaling of AKT to EZH2 in the developing uterus reduces H3K27me3 levels in chromatin, and exhibits both tissue- and xenoestrogen-specificity.
Figure 2. Xenoestrogen-specific modulation of EZH2 and H3K27me3.
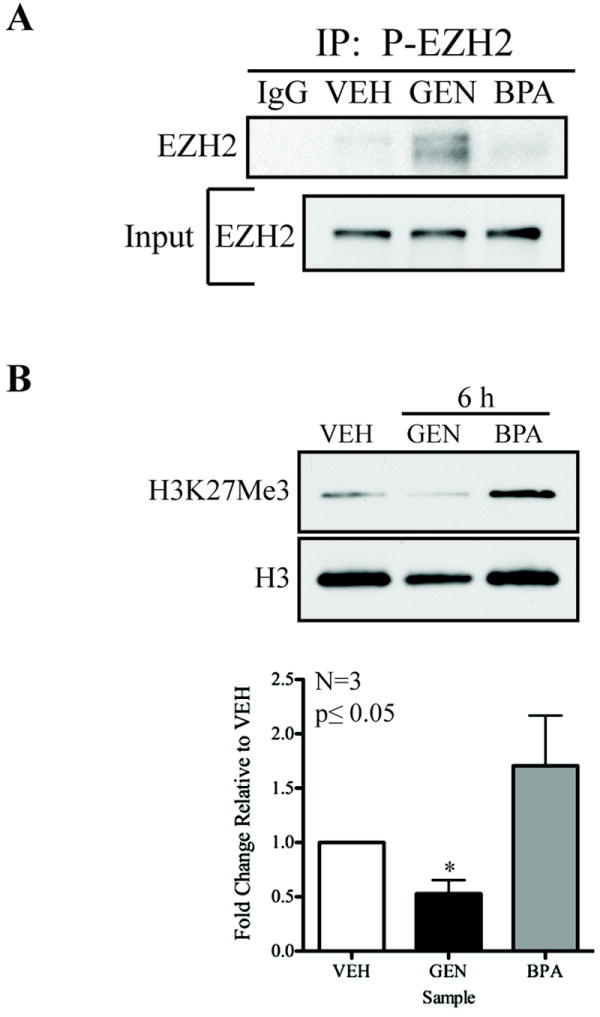
A. Neonatal PND 10-12 pooled (2-3) uteri from Eker rats were homogenized 6hrs after a single exposure to VEH, GEN (50mg/kg) or BPA (50mg/kg) and immunoprecipitated with anti-phospho-EZH2 atnibody. Western blot analysis of EZH2 from immunoprecipitants demonstrate a significant enrichment of phospho-EZH2 in uteri from GEN-exposed rats as compared to BPA and VEH.
B. Neonatal PND 10-12 pooled (2-3) uteri from Eker rats after a single exposure to VEH, GEN (50mg/kg) or BPA (50mg/kg) for 6hrs were used for acid precipitation of histones. Quantitation from western analysis of histone proteins after normalizing for total histone H3 levels shows a decrease in levels of H3K27me3 at 6hrs after exposure in GEN-exposed as compared to BPA- or VEH-exposed animals. Densitometeric analysis of western blot data illustrate significant differences in phosphorylation and methylation after xenoestrogen exposure in Eker rat uteri. The levels of phospho-EZH2 and H3K27me3 were normalized to total levels and were determined to be significantly different from VEH using the Student’s t-test, which was set at a value of *p≤0.05. Error bars represent the mean ± SEM in each graph (n=3).
Developmental reprogramming of gene expression in the adult myometrium by environmental estrogens
Developmental reprogramming of the female rodent reproductive tract can result in morphological and histological alterations including persistent vaginal cornification, endometrial hyperplasia, and polycystic ovaries lacking corpora lutea (18, 33, 34). Examination of the uterus, ovary, and vagina of adult female rats exposed neonatally to BPA (n=17), GEN (n=14), or VEH (n=34) revealed that the effect of these xenoestrogens on reproductive tract morphology differed substantially from that previously observed with DES (Supplementary Fig. S4 and (18)). Relative to DES, adult females exposed neonatally to BPA or GEN had a more normal reproductive tract morphology as illustrated by an absence of persistent vaginal cornification, the presence of corpora lutea in the ovary, luminal epithelium cell height in the endometrium, and apparently normal estrus cyclicity (Supplementary Fig. 4c-l). Thus, compared to the potent pharmacological xenoestrogen DES, neonatal exposure to the environmental estrogens GEN and BPA did not induce dramatic morphological alterations of the reproductive tract in female rats. Additionally, we examined the effect of neonatal exposure to BPA, GEN or VEH on animal weight at 11 and 16 months of age and found no significant difference for either GEN or BPA as compared to VEH animal weights (data not shown).
To determine if environmental estrogens had induced developmental reprogramming at the molecular level, we examined expression of estrogen-responsive genes in adult uteri of animals exposed neonatally to GEN and BPA. For this analysis, we defined a developmentally reprogrammed gene as one that displays altered hormone responsiveness in adult rat myometrium as a result of developmental xenoestrogen exposure. To control for differences in estrogen levels associated with different stages of the estrus cycle, adult VEH-, GEN- or BPA-exposed female rats were grouped into the high-estrogen (proliferative) phase, corresponding to animals in proestrus or estrus, or low estrogen (secretory) phase, corresponding to animals in metestrus and diestrus. Q-PCR was used to quantitate gene expression, with average fold change in expression compared to VEH controls and normalized to expression of ribosomal 18s.
As shown in Table 2, of the 18 estrogen-responsive genes examined, three were developmentally reprogrammed by both GEN and BPA: CalbindinD9k, Gdf10 and Gria2. Similar to what was seen previously with DES, GEN reprogrammed these genes to become hyper-responsive to estrogen (i.e. expression was significantly increased during the proliferative phase of the estrus cycle relative to VEH controls). In the 3 month-old myometrium, CalbindinD9k, Gdf10 and Gria2 expression became elevated by 4-, 3-, and 4-fold, respectively, relative to stage-matched vehicle controls. In contrast, BPA had the opposite effect on these genes, reprogramming them to become repressed by estrogen, with expression significantly decreased during the proliferative phase of the estrus cycle. In 3 month-old rats exposed neonatally to BPA, CalbindinD9k, Gdf10 and Gria2 expression each decreased by 3-fold relative to stage-matched VEH controls.
Table 2.
Adult myometrial gene expression in animals exposed neonatally to GEN or BPA exposure.
| Gene | Normal Estrogen Response | Adult Response (neonatal GEN) | Adult Response (neonatal BPA) |
|---|---|---|---|
|
| |||
| Fold change±SEM | Fold change±SEM | ||
| Calbindin D9k | Induced | 3.8±0.15# | -3.30±0.21* |
| Gdf10 | Repressed | 2.9±0.19# | -2.8±0.21* |
| Gria2 | Repressed | 4.4±0.16# | -2.8±0.33* |
| Di02 | Induced | -2.1±0.09# | NSD |
| Krt19 | Induced | 2.3±0.13# | NSD |
| Igfbp5 | Repressed | -13.9±0.12# | NSD |
| Spp1 | Repressed | -2.2±0.13# | NSD |
| Sfrp2 | Repressed | NSD | -2.0±0.19* |
| Rasd2 | Repressed | NSD | 6.3±0.28* |
Fold change relative to vehicle, SEM; standard error of the mean.
Values statistically significant at
p<0.01 and
p<0.05 using one-way ANOVA.
NSD: No significant difference
In addition to the three genes developmentally reprogrammed by all xenoestrogens, GEN and BPA each reprogrammed a distinct set of genes, albeit with a trend similar to that observed with CalbindinD9k, Gdf10 and Gria2. As shown in Table 2, neonatal GEN exposure reprogrammed expression of Dio2, Krt19, Igfbp5 and Spp1 [only one of which, Dio2, was also reprogrammed by DES (19)], causing the majority (but not all) of these genes to become hyper-responsive to hormone. BPA reprogrammed Sfrp2 and Rasd2, neither of which was reprogrammed by DES or GEN. Though this is not an exhaustive list of all possible genes differentially expressed or developmentally reprogrammed by exposure to GEN and BPA, Table 2 serves as representation of the patterns of developmental reprogramming in estrogen-responsive genes following neonatal exposure.
Developmental Reprogramming by GEN but not BPA promotes uterine tumorigenesis
Eker rats carrying a Tsc2 tumor suppressor gene defect (Tsc2Ek/+) develop uterine leiomyomas by 16 months of age with a historical tumor incidence of 65% (35). The differential ability of GEN and BPA to induce pre-genomic signaling to EZH2 to modulate H3K27me3 levels and increase estrogen-responsive gene expression led us to ask if this differential engagement of EZH2 by these environmental estrogens translated to differences in their impact on uterine tumorigenesis in this animal model. Female Tsc2Ek/+ rats were exposed on PND 10-12 to GEN, BPA or VEH and tumor incidence and multiplicity determined in adult females at 16 months. Neonatal GEN exposure significantly increased tumor incidence at 16 months to 93% (GEN [n=14] vs. VEH controls [n=17], p <0.05) (Fig. 3A). Tumor multiplicity was also increased by neonatal GEN exposure vs. VEH controls (1.6/rat vs. 0.6/rat, respectively) (Fig. 3B). In contrast to GEN, neonatal BPA did not significantly increase tumor incidence or multiplicity relative to VEH controls, differing significantly from the effects of both DES (16) and GEN on tumorigenesis in this animal model (GEN vs BPA [n=30], p <0.01). Thus, induction of “pre-genomic” signaling, inhibition of EZH2 activity, decreased H3K27me3 methylation, increased estrogen-responsive gene expression, and impact on tumorigenesis were concordant, with GEN but not BPA engaging this pathway to developmentally reprogram the developing uterus and increase uterine tumorigenesis.
Figure 3. Tumor incidence and multiplicity of Eker rats exposed neonatally to GEN, BPA or VEH.
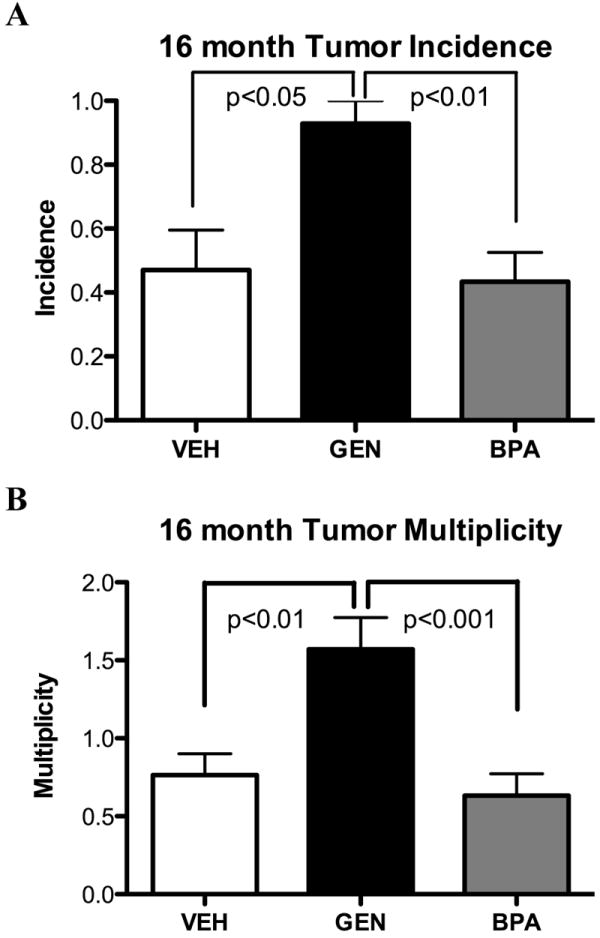
Incidence and multiplicity of uterine leiomyomas in Eker rats after xenoestrogen exposure.
A. Tumor incidence in 16 month old Eker rats (gross and microscopic tumors) after GEN (50mg/kg)(n=14), BPA (50mg/kg)(n=30) or VEH (n=17) exposure for three consecutive days, PND 10-12.
B. Tumor multiplicity in 16 month old Eker rats (gross and microscopic tumors). Statistical significance for incidences was determined by chi square for incidence and by Poisson regression for multiplicity analysis.
Discussion
ER-mediated pre-genomic signaling can activate the PI3K/AKT signaling pathway to phosphorylate the HMT EZH2, modulating its activity and gene expression regulated by H3K27me3 methylation (27). We found that environmental estrogens engage this pathway during developmental reprogramming of gene expression and tumorigenesis in the uterus, and that xenoestrogens have distinct pre-genomic signaling profiles and effects on this epigenetic regulator that correlate with their ability to induce developmental reprogramming. Rapid activation of AKT, which repressed the activity of EZH2 and levels of H3K27me3 in chromatin, distinguished GEN from BPA, despite the fact that both xenoestrogens were able to induce genomic ER signaling and transactivate the expression of estrogen-responsive genes in the developing uterus. Importantly, these data demonstrate that the pathway by which DES interferes with epigenetic programming in the developing uterus, i.e. via activation of PI3K/AKT, phosphorylation of histone methyltransferase EZH2 and reduction in H3K27 methylation (29), is engaged by GEN but not BPA, and that disruption of this pathway during development correlates with the effect of these endocrine disruptors on tumorigenesis in myometrium.
Importantly while GEN and BPA both elicited a genomic ER response [modulation of expression of estrogen-responsive genes in the neonatal uterus (Table 1)], this genomic response did not correlate with the ability to induce developmental reprogramming in the adult myometrium. Several genes that exhibited a genomic response to GEN and/or BPA (i.e. were induced or repressed) in the neonate failed to become reprogrammed in the adult myometrium of exposed animals and/or exhibited differences in estrogen responsiveness, for example exhibiting elevated expression in GEN animals and decreased expression in BPA animals. Similarly, Sfrp2 exhibited a genomic response to GEN in the neonatal uterus but was not reprogrammed in adulthood, and while BPA induced Dio2 expression in the neonatal uterus, it did not reprogram this gene in the adult myometrium. Thus, induction of a genomic response by xenoestrogens in the developing uterus was insufficient to induce developmental reprogramming in adulthood, and did not predict xenoestrogen-specificity for reprogramming of gene expression.
Differences in the ability of xenoestrogens to induce developmental reprogramming are likely driven by several intrinsic differences between these xenoestrogens, for example binding to specific ER subtypes. DES, GEN and BPA have distinct binding efficiencies for ERα and ERβ, with DES and GEN binding with a higher affinity to ERα than BPA (DES>GEN>BPA), while BPA binds with a much higher affinity to ERβ than ERα (36). Additionally, some actions of BPA may be mediated via a non-classical G-protein-coupled membrane receptor that activates non-genomic signaling through PKA, phosphorylating targets such as CREB in vitro (37). Importantly, in the female reproductive tract, ERα is required for DES-induced developmental reprogramming, as αERKO mice are resistant to DES-induced developmental reprogramming (18) and overexpression of ERα exacerbates the effects of DES on developmental reprogramming. We also have previously demonstrated that αERKO mice are resistant to the DES-induced activation of pre-genomic PI3K/AKT signaling to EZH2 (29). Xenoestrogen-specific affinity for ERα vs ERβ has also been demonstrated in prostate tissue from male αERKO and βERKO mice exposed neonatally to DES (38). In that study, DES exposure induced prostate abnormalities in the βERKO mice, which express ERα. Conversely, αERKO mice that lack the ERα receptor were resistant to the effect of DES exposure. Our findings that DES and GEN but not BPA, developmentally reprogrammed uterine tumorigenesis is consistent with ERα being the predominate ER subtype in development of the female reproductive tract (39).
Histone methylation and cytosine methylation are heritable chromatin modifications that regulate gene expression. These epigenetic marks have been shown to each influence each other, although the exact mechanism(s) by which histone methyl marks direct patterns of DNA methylation and visa versa are just beginning to be understood (40-42). H3K27me3 methylation of chromatin by EZH2 is required for several key functions during development, including X inactivation, bivalent chromatin maintenance and silencing of HOX genes, which have been shown to be critical to proper murine uterine differentiation (43, 44). EZH2 can also directly silence estrogen-responsive genes, such as progesterone receptor, by inducing H3K27me3 methyl marks that promote DNA methylation via recruitment of the histone deacetylase HDAC1, followed by recruitment of DNA methyltransferases (45). It is not known at present if reduction in H3K27me3 methyl marks by xenoestrogens in the developing uterus alters gene expression in the adult uterus directly as a result of decreases in histone methylation, or by directing changes in DNA methylation of estrogen-responsive genes.
While we have focused on developmental reprogramming of tumorigenesis by environmental estrogens in the myometrium, xenoestrogens have been shown to lead to persistent changes in gene expression that are associated with neoplastic transformation in other tissues. In the endometrium, xenoestrogen exposure during critical windows of uterine development (i.e PND 1-5), permanently alters the expression of the estrogen-responsive genes lactoferrin and c-fos in the adult endometrium. Altered expression is associated with site-specific DNA hypomethylation, rather than promoter hypermethylation, which persists in uterine adenocarcinomas that develop in these animals (46). While the mechanism by which xenoestrogens may induce changes in DNA demethylation is not well understood, it was shown that parathyroid hormone can engage pre-genomic signaling via PKC, leading to phosphorylation of methyl binding protein 4 (MBD4) (47). MBD4 recruits DNA base excision repair machinery, resulting in demethylation of the vitamin D receptor-dependent promoter of the cytochrome p450 27B1 (CYP27B1) gene. Thus, chromatin remodeling via pre-genomic signaling pathways can result in changes in both histone methyl marks and DNA methylation and may function as a general pathway by which activation of nuclear hormone receptors engage the cell’s epigenetic machinery.
Overall, the patterns of developmental reprogramming that emerged from this analysis revealed that reprogramming by environmental estrogens exhibits xenoestrogen-specificity. GEN developmentally reprogrammed, Gdf10, Calbindin D9k, Gria2, Dio2, Krt19, Igfbp5, and Spp1; the first five of which were also reprogrammed by DES (19). In contrast, BPA reprogrammed Gdf10, Calbindin D9k, Gria2, Rasd2, and Sfrp2. Unlike DES and GEN, BPA reprogramming resulted in decreased gene expression (with the exception of Rasd). Therefore, in general, the effect of BPA on gene expression was the opposite of DES and GEN, repressing rather than enhancing estrogen-responsiveness. These findings are reminiscent of those reported by Dolinoy et al. (48), where developmental exposure to GEN and BPA had opposite effects on DNA methylation and subsequent gene expression of the agouti gene. Furthermore, we have now shown that the pattern of xenoestrogen-induced developmental reprogramming in genes targeted by GEN, BPA and DES correlated with the ability of these xenoestrogens to increase tumorigenesis. Both neonatal DES and GEN caused increased expression of estrogen-responsive genes in adult animals relative to VEH-exposed rats, promoting tumor formation in DES and GEN exposed animals (19). In contrast, neonatal BPA caused a general repression of estrogen-responsive genes, and did not increase tumor incidence or multiplicity compared to VEH. Together with our previous report (19) these data indicate that xenoestrogens such as DES, BPA or GEN have both shared and distinct effects on developmental reprogramming of target genes that reflect their effect on uterine tumorigenesis. It is important to note that BPA tumorigenicity studies were performed at a dose of 50mg/kg, the minimum required to elicit an equivalent genomic response to GEN and DES. Although our mechanistic studies clearly demonstrate that even at doses 6-orders of magnitude lower (i.e. 50ng/kg), BPA did not induce pre-genomic PI3K/AKT signaling to EZH2 in the developing uterus, it is still a formal possibility that effects on tumorigenicity and reprogramming of gene expression not evident at 50mg/kg could occur at lower-dose BPA exposures via other mechanisms.
Distinctions between the effects of these environmental estrogens on the developing reproductive tract also were observed. Morphological reprogramming of the reproductive tract following neonatal GEN and BPA exposure was not observed, unlike what was seen in this model with DES (18). However, other studies have reported reproductive abnormalities as a result of perinatal exposure to GEN and BPA (2, 9, 49, 50). While other mechanisms have been demonstrated for the developmental reprogramming effects of GEN and BPA, including disruption of the HPG axis (51, 52), our previous work (18) from ovariectomized (or sham) Eker rats exposed neonatally to DES (or VEH) we observed abolishment of calbindin D9k and significantly reduced progesterone receptor expression compared to sham controls, indicating that the genes had become hyper-responsive to endogenous hormones as opposed to constitutively active. Thus, in our model of uterine tumorigenesis, we believe that ovarian hormones are required for the developmental reprogramming effects of GEN and BPA to be observed in the adult animals – though we did not specifically test the effects of GEN and BPA on the HPG axis. Several possibilities exist for these discordant observations including differences in dose, route of administration, timing of administration, and model species. Regardless, data obtained in this rat model system clearly demonstrate that reprogramming of tumor susceptibility and estrogen-responsive gene expression at the molecular level can occur at doses of xenoestrogens that do not cause overt morphological changes in the female reproductive tract. Similar to our observations, Adachi et al. demonstrated that molecular alterations in the testis can occur in the absence of morphological alterations (53). For example, while neonatal DES exposure induced both morphological and molecular alterations in the testis, GEN reprogrammed genes in the adult testes without inducing morphological reprogramming. Therefore, the effects of developmental reprogramming may manifest in the absence of histological or morphological alterations, pointing to the need to develop molecular biomarkers to detect reprogramming even in morphologically “normal” appearing tissues.
In summary, this study demonstrates that neonatal exposure to GEN, in contrast to BPA, activates pre-genomic signaling to AKT, phosphorylating EZH2 and reducing H3K27me3 levels in the developing uterus. The disparate ability of GEN and BPA to activate pre-genomic signaling to EZH2 was reflected in the adult myometrium by different patterns of developmental reprogramming of estrogen-responsive genes and differences in the ability of these xenoestrogens to increase tumorigenesis in this tissue. In addition to highlighting what are likely important intrinsic differences between xenoestrogens, this study also identifies a pathway by which xenoestrogens can engage the cell’s epigenetic machinery during the process of developmental reprogramming, an important first step towards understanding how early life xenoestrogen exposures can increase susceptibility to hormone-dependent tumors in adulthood.
Supplementary Material
Photomicrographs of uteri stained with either hematoxylin and eosin or calbindin D9K from VEH-, DES-, GEN-, or BPA-exposed females (1, 50 or 50mg/kg, respectively) sacrificed on PND 12, 6hrs after the final of three exposures (all photomicrographs were taken at 20X). The scale bar is 100 μm.
Lysates were collected from pooled (2-3) Eker rat uteri on PND 12 after a single exposure to 50mg/kg, 50μg/kg or 50ng/kg of BPA, DES (1mg/kg) or VEH for 6hrs. A) Western blot analysis revealed no significant activation of pS6 by BPA as compared to DES or VEH. B) Densitometry of western blots revealed no significant activation of the PI3K/AKT pathway. Data are represented as the average ratio of phopsho-AKT/AKT of three independent experiments.
(A) Activation of rapid non-genomic PI3K/AKT signaling in Sprague Dawley rats 0.5hr after a single exposure to doses of BPA shown. Western analysis and quantification of ratio of phospho-S6 to total S6 from a representative experiment (N=2) using tissues pooled from groups of 2-3 animals. (B) Quantification of induction of non-genomic PI3K/AKT signaling 6hr after exposure to 1mg/kg DES. (C) Western analysis to detect changes in H3K27me3 levels in the developing prostate in response to BPA demonstrating a reduction in H3K27me3 0.5hrs after exposure.
Effects of neonatal exposure to DES, GEN, or BPA (1, 50 or 50mg/kg, respectively for three consecutive days, PND 10-12) on adult uterine morphology. Shown are photomicrographs of hematoxylin and eosin–stained histological sections of uteri, ovaries, and vaginas from adult (3-5 months old) rats exposed neonatally to VEH (a, b and c), DES (d, e and f), GEN (g, h and i), or BPA (j, k and l) (20X). The length of scale bar is 200 μm.
Acknowledgments
GRANT AND FELLOWSHIP SUPPORT: Support for this work was funded by grants from the National Institute of Environmental Health Sciences (P30ES006096, RC2ES018789, P30ES007784, R01ES008263), National Institute of Child Health and Development (HD046282) and the National Cancer Institute (P30CA016672). K. Leigh Greathouse was supported by the Schissler Foundation Fellowship in Human Genetics and Cancer from the Graduate School of Biomedical Sciences, University of Texas.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose except for Tiffany Bredfeldt, Shuk-mei Ho, and Cheryl Walker who have received NIH grants for this research in an amount greater than $10,000
References
- 1.Kyung-Chul C, Eui-Bae J, Leung PCK. Impact of Environmental Endocrine Disruption on the Reproductive System for Human Health. Immunology, Endocrine & Metabolic Agents - Medicinal Chemistry. 2006;6:3. [Google Scholar]
- 2.Newbold RR, Banks EP, Bullock B, Jefferson WN. Uterine adenocarcinoma in mice treated neonatally with genistein. Cancer Res. 2001;61:4325–8. [PubMed] [Google Scholar]
- 3.Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, et al. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1509–14. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 4.Camp EA, Coker AL, Robboy SJ, Noller KL, Goodman KJ, Titus-Ernstoff LT, et al. Breast cancer screening in women exposed in utero to diethylstilbestrol. J Womens Health (Larchmt) 2009;18:547–52. doi: 10.1089/jwh.2007.0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Aloisio AA, Baird DD, DeRoo LA, Sandler DP. Association of intrauterine and early-life exposures with diagnosis of uterine leiomyomata by 35 years of age in the Sister Study. Environ Health Perspect. 2010;118:375–81. doi: 10.1289/ehp.0901423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wise LA, Palmer JR, Rowlings K, Kaufman RH, Herbst AL, Noller KL, et al. Risk of benign gynecologic tumors in relation to prenatal diethylstilbestrol exposure. Obstet Gynecol. 2005;105:167–73. doi: 10.1097/01.AOG.0000147839.74848.7c. [DOI] [PubMed] [Google Scholar]
- 7.Newbold RR, Jefferson WN, Padilla-Banks E. Long-term adverse effects of neonatal exposure to bisphenol A on the murine female reproductive tract. Reprod Toxicol. 2007;24:253–8. doi: 10.1016/j.reprotox.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature. 1999;401:763–4. doi: 10.1038/44517. [DOI] [PubMed] [Google Scholar]
- 9.Schonfelder G, Friedrich K, Paul M, Chahoud I. Developmental Effects of Prenatal Exposure to Bisphenol A on the Uterus of Rat Offspring. Neoplasia. 2004;6:584. doi: 10.1593/neo.04217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato H, Ota T, Furuhashi T, Ohta Y, Iguchi T. Changes in reproductive organs of female rats treated with bisphenol A during the neonatal period. Reprod Toxicol. 2003;17:283. doi: 10.1016/s0890-6238(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 11.Nikaido Y, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. Effects of prepubertal exposure to xenoestrogen on development of estrogen target organs in female CD-1 mice. In Vivo. 2005;19:487–94. [PubMed] [Google Scholar]
- 12.Prins GS, Tang WY, Belmonte J, Ho SM. Developmental exposure to bisphenol A increases prostate cancer susceptibility in adult rats: epigenetic mode of action is implicated. Fertil Steril. 2008;89:e41. doi: 10.1016/j.fertnstert.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Isoflavone content of infant formulas and the metabolic fate of these phytoestrogens in early life. Am J Clin Nutr. 1998;68:1453S–61S. doi: 10.1093/ajcn/68.6.1453S. [DOI] [PubMed] [Google Scholar]
- 14.Walker CL, Stewart EA. Uterine fibroids: the elephant in the room. Science. 2005;308:1589–92. doi: 10.1126/science.1112063. [DOI] [PubMed] [Google Scholar]
- 15.Jefferson WN, Padilla-Banks E, Newbold RR. Disruption of the female reproductive system by the phytoestrogen genistein. Reprod Toxicol. 2007;23:308. doi: 10.1016/j.reprotox.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Cook JD, Davis BJ, Cai SL, Barrett JC, Conti CJ, Walker CL. Interaction between genetic susceptibility and early-life environmental exposure determines tumor-suppressor-gene penetrance. Proc Natl Acad Sci U S A. 2005;102:8644–9. doi: 10.1073/pnas.0503218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker CL, Hunter D, Everitt JI. Uterine leiomyoma in the Eker rat: A unique model for important diseases of women. Genes, Chromosomes and Cancer. 2003;38:349–56. doi: 10.1002/gcc.10281. [DOI] [PubMed] [Google Scholar]
- 18.Cook JD, Davis BJ, Goewey JA, Berry TD, Walker CL. Identification of a sensitive period for developmental programming that increases risk for uterine leiomyoma in Eker rats. Reprod Sci. 2007;14:121–36. doi: 10.1177/1933719106298401. [DOI] [PubMed] [Google Scholar]
- 19.Greathouse KL, Cook JD, Lin K, Davis BJ, Berry TD, Bredfeldt TG, et al. Identification of uterine leiomyoma genes developmentally reprogrammed by neonatal exposure to diethylstilbestrol. Reprod Sci. 2008;15:765–78. doi: 10.1177/1933719108322440. [DOI] [PubMed] [Google Scholar]
- 20.Couse JF, Korach KS. Estrogen receptor-alpha mediates the detrimental effects of neonatal diethylstilbestrol (DES) exposure in the murine reproductive tract. Toxicology. 2004;205:55–63. doi: 10.1016/j.tox.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 21.Glidewell-Kenney C, Hurley LA, Pfaff L, Weiss J, Levine JE, Jameson JL. Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc Natl Acad Sci U S A. 2007;104:8173–7. doi: 10.1073/pnas.0611514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen JR, Plotkin LI, Aguirre JI, Han L, Jilka RL, Kousteni S, et al. Transient versus sustained phosphorylation and nuclear accumulation of ERKs underlie anti-versus pro-apoptotic effects of estrogens. J Biol Chem. 2005;280:4632–8. doi: 10.1074/jbc.M411530200. [DOI] [PubMed] [Google Scholar]
- 23.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–6. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 24.Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–6. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson CS, Bulayeva NN, Wozniak AL, Alyea RA. Xenoestrogens are potent activators of nongenomic estrogenic responses. Steroids. 2007;72:124–34. doi: 10.1016/j.steroids.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Zhang S, Safe S. Activation of kinase pathways in MCF-7 cells by 17[beta]-estradiol and structurally diverse estrogenic compounds. The Journal of Steroid Biochemistry and Molecular Biology. 2006;98:122–32. doi: 10.1016/j.jsbmb.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-Induced Regulation of EZH2 and Histone Methylation via Estrogen Receptor Signaling to PI3K/AKT. Mol Endocrinol. doi: 10.1210/me.2009-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prins GS, Ye SH, Birch L, Ho SM, Kannan K. Serum bisphenol A pharmacokinetics and prostate neoplastic responses following oral and subcutaneous exposures in neonatal Sprague-Dawley rats. Reprod Toxicol. 2011;31:1–9. doi: 10.1016/j.reprotox.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol. 2010;24:993–1006. doi: 10.1210/me.2009-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect. 2008;116:1642–7. doi: 10.1289/ehp.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho SM, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006;66:5624–32. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–10. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 33.Burroughs CD, Mills KT, Bern HA. Long-term genital tract changes in female mice treated neonatally with coumestrol. Reprod Toxicol. 1990;4:127–35. doi: 10.1016/0890-6238(90)90007-i. [DOI] [PubMed] [Google Scholar]
- 34.Katsuda S, Yoshida M, Watanabe G, Taya K, Maekawa A. Irreversible effects of neonatal exposure to p-tert-octylphenol on the reproductive tract in female rats. Toxicol Appl Pharmacol. 2000;165:217–26. doi: 10.1006/taap.2000.8940. [DOI] [PubMed] [Google Scholar]
- 35.Walker CL, Cesen-Cummings K, Houle C, Baird D, Barrett JC, Davis B. Protective effect of pregnancy for development of uterine leiomyoma. Carcinogenesis. 2001;22:2049–52. doi: 10.1093/carcin/22.12.2049. [DOI] [PubMed] [Google Scholar]
- 36.Routledge EJ, White R, Parker MG, Sumpter JP. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) alpha and ERbeta. J Biol Chem. 2000;275:35986–93. doi: 10.1074/jbc.M006777200. [DOI] [PubMed] [Google Scholar]
- 37.Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect. 2009;117:1053–8. doi: 10.1289/ehp.0800367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prins GS, Birch L, Couse JF, Choi I, Katzenellenbogen B, Korach KS. Estrogen imprinting of the developing prostate gland is mediated through stromal estrogen receptor alpha: studies with alphaERKO and betaERKO mice. Cancer Res. 2001;61:6089–97. [PubMed] [Google Scholar]
- 39.Mowa CN, Iwanaga T. Developmental changes of the oestrogen receptor-alpha and -beta mRNAs in the female reproductive organ of the rat--an analysis by in situ hybridization. J Endocrinol. 2000;167:363–9. doi: 10.1677/joe.0.1670363. [DOI] [PubMed] [Google Scholar]
- 40.Sarraf SA, Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. 2004;15:595–605. doi: 10.1016/j.molcel.2004.06.043. [DOI] [PubMed] [Google Scholar]
- 41.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–40. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 42.Esteve PO. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–5. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 44.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 45.Leu YW, Yan PS, Fan M, Jin VX, Liu JC, Curran EM, et al. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res. 2004;64:8184–92. doi: 10.1158/0008-5472.CAN-04-2045. [DOI] [PubMed] [Google Scholar]
- 46.Li S, Washburn KA, Moore R, Uno T, Teng C, Newbold RR, et al. Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res. 1997;57:4356–9. [PubMed] [Google Scholar]
- 47.Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, et al. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009;461:1007–12. doi: 10.1038/nature08456. [DOI] [PubMed] [Google Scholar]
- 48.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–61. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal Exposure to Low Doses of Bisphenol A Affects Body Weight, Patterns of Estrous Cyclicity, and Plasma LH Levels. Environ Health Perspect. 2001;109:675. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newbold WNJ. Elizabeth Padilla-Banks Prenatal Exposure to Bisphenol A at Environmentally Relevant Doses Adversely Affects the Murine Female Reproductive Tract Later in Life. Environ Health Perspect. 2009:117. doi: 10.1289/ehp.0800045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faber KA, Hughes CL., Jr Dose-response characteristics of neonatal exposure to genistein on pituitary responsiveness to gonadotropin releasing hormone and volume of the sexually dimorphic nucleus of the preoptic area (SDN-POA) in postpubertal castrated female rats. Reprod Toxicol. 1993;7:35–9. doi: 10.1016/0890-6238(93)90007-t. [DOI] [PubMed] [Google Scholar]
- 52.Fernandez M, Bianchi M, Lux-Lantos V, Libertun C. Neonatal exposure to bisphenol a alters reproductive parameters and gonadotropin releasing hormone signaling in female rats. Environ Health Perspect. 2009;117:757–62. doi: 10.1289/ehp.0800267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adachi T, Ono Y, Koh K-B, Takashima K, Tainaka H, Matsuno Y, et al. Long-term alteration of gene expression without morphological change in testis after neonatal exposure to genistein in mice: toxicogenomic analysis using cDNA microarray. Food & Chemical Toxicology. 2004;42:445. doi: 10.1016/j.fct.2003.10.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Photomicrographs of uteri stained with either hematoxylin and eosin or calbindin D9K from VEH-, DES-, GEN-, or BPA-exposed females (1, 50 or 50mg/kg, respectively) sacrificed on PND 12, 6hrs after the final of three exposures (all photomicrographs were taken at 20X). The scale bar is 100 μm.
Lysates were collected from pooled (2-3) Eker rat uteri on PND 12 after a single exposure to 50mg/kg, 50μg/kg or 50ng/kg of BPA, DES (1mg/kg) or VEH for 6hrs. A) Western blot analysis revealed no significant activation of pS6 by BPA as compared to DES or VEH. B) Densitometry of western blots revealed no significant activation of the PI3K/AKT pathway. Data are represented as the average ratio of phopsho-AKT/AKT of three independent experiments.
(A) Activation of rapid non-genomic PI3K/AKT signaling in Sprague Dawley rats 0.5hr after a single exposure to doses of BPA shown. Western analysis and quantification of ratio of phospho-S6 to total S6 from a representative experiment (N=2) using tissues pooled from groups of 2-3 animals. (B) Quantification of induction of non-genomic PI3K/AKT signaling 6hr after exposure to 1mg/kg DES. (C) Western analysis to detect changes in H3K27me3 levels in the developing prostate in response to BPA demonstrating a reduction in H3K27me3 0.5hrs after exposure.
Effects of neonatal exposure to DES, GEN, or BPA (1, 50 or 50mg/kg, respectively for three consecutive days, PND 10-12) on adult uterine morphology. Shown are photomicrographs of hematoxylin and eosin–stained histological sections of uteri, ovaries, and vaginas from adult (3-5 months old) rats exposed neonatally to VEH (a, b and c), DES (d, e and f), GEN (g, h and i), or BPA (j, k and l) (20X). The length of scale bar is 200 μm.