

Abstract
The extracellular matrix (ECM) is a critical tissue component, providing structural support as well as important regulatory signaling cues to govern cellular growth, metabolism, and differentiation. The study of ECM proteins, however, is hampered by the low solubility of ECM components in common solubilizing reagents. ECM proteins are often not detected during proteomics analyses using unbiased approaches due to solubility issues and relatively low abundance compared to highly abundant cytoplasmic and mitochondrial proteins. Decellularization has become a common technique for ECM protein-enrichment and is frequently used in engineering studies. Solubilizing the ECM after decellularization for further proteomic examination has not been previously explored in depth. In this study, we describe testing of a series of protocols that enabled us to develop a novel optimized strategy for the enrichment and solubilization of ECM components. Following tissue decellularization, we use acid extraction and enzymatic deglycosylation to facilitate resolubilization. The end result is the generation of three fractions for each sample: soluble components, cellular components, and an insoluble ECM fraction. These fractions, developed in mass spectrometry-compatible buffers, are amenable to proteomics analysis. The developed protocol allows identification (by mass spectrometry) and quantification (by mass spectrometry or immunoblotting) of ECM components in tissue samples.
Biological significance
The study of extracellular matrix (ECM) proteins in pathological and non-pathological conditions is often hampered by the low solubility of ECM components in common solubilizing reagents. Additionally, ECM proteins are often not detected during global proteomic analyses due to their relatively low abundance compared to highly abundant cytoplasmic and mitochondrial proteins. In this manuscript we describe testing of a series of protocols that enabled us to develop a final novel optimized strategy for the enrichment and solubilization of ECM components. The end result is the generation of three fractions for each sample: soluble components, cellular components, and an insoluble ECM fraction. By analysis of each independent fraction, differences in protein levels can be detected that in normal conditions would be masked. These fractions are amenable to mass spectrometry analysis to identify and quantify ECM components in tissue samples. The manuscript places a strong emphasis on the immediate practical relevance of the method, particularly when using mass spectrometry approaches; additionally, the optimized method was validated and compared to other methodologies described in the literature.
Keywords: Extracellular matrix, Enrichment, Decellularization, Heart, Solubility, Matrix metalloproteinases
1. Introduction
The extracellular matrix (ECM) is a structural scaffold for cell adhesion and migration, as well as a signaling trigger for ECM receptors to regulate cell growth, metabolism, and differentiation [1,2]. Therefore, a complete understanding of cell biology requires an understanding of ECM biology. One particularly dynamic tissue remodeling process in which ECM becomes critically important is wound repair.
Studying the ECM, however, is not trivial. ECM components are organized into a complex fibrillar, three-dimensional matrix that cannot easily be solubilized or dissociated into component units. The interlocking mesh of collagens, elastic fibers, proteoglycans, and adhesive glycoproteins contains protein aggregates, cross-linked proteins, and glycosylated or other post-translationally modified proteins that make solubilizing the ECM particularly difficult [3,4]. The unusual physical properties of the ECM create challenges for characterization using standard tissue homogenization approaches because the insoluble components of ECM associate with fractions that are often discarded during sample preparation. Furthermore, the presence of highly abundant cytoplasmic and mitochondrial proteins can hamper detection of the relatively scarce ECM proteins. This is particularly a concern for tissues with high metabolical activity, such as the heart, in which there is an exceptionally high mitochondrial content.
Adverse remodeling of the left ventricle (LV) following myocardial infarction (MI) is a leading cause of congestive heart failure and cardiovascular morbidity and mortality. Post-MI, LV remodeling involves the removal of necrotic tissue and the formation of a myocardial scar. ECM turnover is a key component of LV remodeling. A proper balance is needed between synthesis and degradation of ECM in order to re-establish homeostasis for adequate scar formation. The replacement of the necrotic cells with an ECM scar increases myocardial stiffness and consequently leads to LV dysfunction [5,6]. Understanding the changes that occur in the ECM post-MI is, therefore, pivotal for identifying factors that tip the balance between favorable repair and adverse remodeling that can progress to heart failure. The functional properties of the complex ECM cannot be determined without studying the individual constituents and their individual functions.
In this manuscript, we describe the evaluation of several methodologies for the enrichment of cardiac ECM proteins to facilitate analysis by mass spectrometry (Fig. 1). In order to focus on the ECM components, we first analyzed proteins in the LV infarct area of wild type (WT) mice for comparison to the LV proteins in matrix metalloproteinase (MMP)-9 null mice at 7 days post-MI (Fig. 1, strategy 1). The 7 day time point was chosen because the infarct is approximately 30% scar by volume at this time, which provided a natural overabundance of ECM protein [7]. Proteins were analyzed by mass spectrometry (MS) and several ECM proteins were detected [8]. However, there was still a large number of cellular proteins (cytoplasmic and mitochondrial) in the preparation, which hampered relative quantification of the ECM components. To overcome this issue, we designed and tested a series of protocols for the enrichment and solubilization of ECM components. The final protocol uses a novel differential solubility-based, protein fractionation strategy that has been optimized for the analysis of cardiac ECM by eliminating the cellular proteins present in the LV and increasing ECM solubility.
Fig. 1.

Description of the evolution of strategies developed for the enrichment of cardiac ECM proteins to facilitate analysis by mass spectrometry.
2. Results
Using a step-wise protocol, we overcame several common issues in studying ECM proteins. Our two objectives were: 1) to reduce the presence of cellular proteins, thereby enriching for lower abundance ECM proteins; and 2) to efficiently solubilize and deglycosylate ECM proteins to facilitate their analysis by proteomic approaches.
2.1. ECM enrichment by tissue decellularization
Intact LV samples were used to develop a method for the extraction of proteins present in the extracellular environment, including ECM proteins and proteins attached to the ECM. This method consisted of two steps: tissue decellularization to remove cellular proteins (Fig. 2a), and tissue homogenization with a protein extraction reagent. Tissue decellularization protocols have been in use since the early 1970s [9]. Ott and colleagues used multiple detergents to decellularize hearts by perfusion, including 1% polyethylene glycol (PEG), 1% sodium dodecyl sulfate (SDS), and 1% Triton X-100 [10]. SDS was the only one of those detergents that was able to fully decellularize the tissue. Ionic detergents, such as SDS, are regularly used for effective solubilization of membrane proteins in order to remove cellular components from tissues [11–13].
Fig. 2.
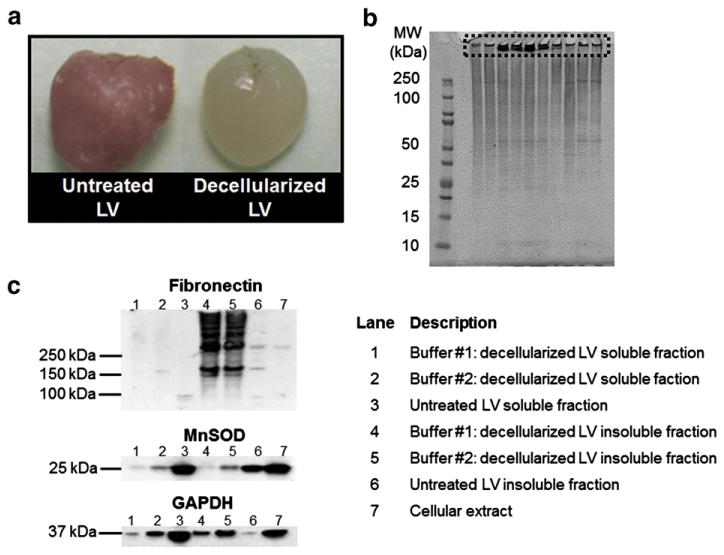
a. Image of decellularized and untreated mouse left ventricle (LV); b. proteins in decellularized LV samples from young (4 to 8 month old) wild type mice (n = 10 biological replicas) were visualized in a 1-D SDS-PAGE gel (strategy 4). Lane 1 is the molecular weight (MW) marker. Poor sample solubility caused insoluble proteins to stay trapped in the wells (boxed region), resulting in decreased electrophoretic mobility; c. immunoblots (5 μg total protein) of fibronectin, MnSOD, and GAPDH in soluble (lysis buffer) and insoluble (Protein Extraction Reagent 4) young LV protein extracts. Decellularization buffer # 1 (1% SDS): lanes 1 and 4. Decellularization buffer # 2 (1% SDS, 20 mM NH4OH): lanes 2 and 5. Untreated LV (lanes 3 and 6) and cellular extract (lane 7) were used as controls. Lane descriptions apply to Fig. 2c.
In our search for an optimized decellularization/ECM enrichment strategy, we explored a modification of the protocol described by Ott et al. [14]. Five decellularization buffers were tested (strategy 2). Buffer # 1 was 1% SDS; buffer # 2 was 1% SDS, 20 mM NH4OH; buffer # 3 was 1% Triton X-100, 20 mM NH4OH; buffer # 4 was 1% SDS, 1% Triton X-100; and buffer # 5 was 1% SDS, 1% Triton X-100, 20 mM NH4OH. In contrast to Ott and colleagues, we were not interested in keeping the intact heart as a scaffold. Therefore, the LV was cut into small pieces before decellularization. This substantially increased the tissue contact surface area resulting in faster decellularization (72 h versus 124 h). In addition, we included protease inhibitors (PI) in all of the decellularization buffers. This is an essential step to prevent proteolysis of ECM proteins by MMPs and other proteases. Only buffers that contained SDS as the only detergent completely decellularized the tissues (buffer # 1 and buffer # 2). Compared to other detergents, SDS has been shown to be more effective for removal of nuclear and cytoplasmic proteins [15]. SDS affords the dual advantage of efficiently extracting proteins and enhancing protein solubility. The denaturant properties of SDS are due to hydrophobic interactions between proteins and the long, flexible 12-carbon hydrocarbon tail that interacts with the hydrophobic regions of polypeptide chains, thereby interfering with intraprotein interactions. Moreover, the polar carboxylate head group of SDS associates with positively-charged side chains, thus disrupting ionic protein–protein interactions [16].
In order to extract protein from the decellularized tissue, two approaches using Protein Extraction Reagent 4 (Sigma Aldrich) were evaluated. This reagent contains thiourea (2 M) as a chaotrope in addition to urea (7 M) and the zwitterionic detergent, C7BzO, and has been shown to increase protein extraction and solubility compared to other detergents [17]. The methods tested included: 1) homogenization in a lysis buffer (0.25 M sucrose, 1 mM EDTA in 50 mM Tris with 1× PI) for isolation of soluble components, followed by centrifugation, and homogenization of the pellet (insoluble components) in Protein Extraction Reagent 4 with 1× PI (strategy 3); 2) direct homogenization in Protein Extraction Reagent 4 with 1× PI (strategy 4).
Although the use of SDS for decellularization efficiently removed cellular proteins, there were issues with the subsequent homogenization of the decellularized tissue. Visualization by 1-D SDS-PAGE indicated that although the protein profiles of replicate samples were consistent, there were aggregated proteins trapped in the wells of the gel (Fig. 2b). Immunoblot analysis of the soluble and insoluble components (tissue solubilization, strategy 3) was conducted to assess the degree of enrichment of ECM proteins, reduction of cellular proteins, and efficacy of the decellularization buffers. Fibronectin was chosen as the ECM protein target, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and manganese superoxide dismutase (MnSOD) as the cellular proteins. The decellularization buffer collected after the first day of incubation (cellular extract) was included as a negative control. Fibronectin was found to be enriched in the insoluble fraction of decellularized LV treated with buffers # 1 and # 2 compared to untreated LV (Fig. 2c, lanes 4 to 6). The cellular extract showed a low level of fibronectin present in that fraction (Fig. 2c lane 7), indicating that a very small amount of fibronectin was lost during the ECM enrichment process. The immunoblots for the cellular proteins showed significantly reduced levels of MnSOD and GAPDH both in the soluble and insoluble fractions of decellularized LV compared to the untreated (cellular) LV. Since MnSOD and GAPDH were not completely removed, the term ECM-enriched is used instead of ECM-purified to describe the resulting protein solution. Detection of strong immunoblot signals for MnSOD and GAPDH in the cellular extract (Fig. 2c lane 7) provided convincing evidence for the effective removal of cellular proteins by this treatment, supporting the idea that cellular proteins were removed during the ECM enrichment process. Since there were lower levels of cellular proteins remaining in the LV after decellularization with buffer # 1, compared to buffer # 2, we chose to use buffer # 1 for the tissue decellularization step in all further experiments. Additionally, fibronectin was mostly present only in the insoluble fraction; therefore, we elected to use only Protein Extraction Reagent 4 (strategy 4) for tissue homogenization.
The LV decellularization protocol described above was able to effectively reduce the levels of cellular proteins and permitted detection of less abundant ECM proteins (monitored by fibronectin). However, complete protein solubilization was not achieved as evidenced by the residual material that was trapped in the wells of the gels. Additionally, there was inconsistency in the quantities of protein recovered from replicate samples, indicating a need to enhance the reproducibility of the solubilization step.
2.2. High molecular weight filtration, reduction and alkylation
Protein extracts processed by the SDS decellularization method using buffer # 1 followed by solubilization according to the method developed in strategy 4 were separated by 1-D SDS-PAGE. The proteins were digested in situ with trypsin and analyzed by HPLC–electrospray ionization–tandem mass spectrometry (HPLC–ESI–MS/MS). More than 150 proteins were identified, including titin. Titin is the largest known protein, with a molecular weight of 3906 kDa. It seemed likely that one of the proteins trapped in the 1-D gel wells was titin. Two methods were devised to eliminate this problem: proteins were reduced and alkylated prior to SDS-PAGE (strategy 5); and ultra-high molecular weight proteins were removed with a filtration device (strategy 6). Although reduction/alkylation appeared to improve sample solubility, the protein preparation that ensued was of lower quality with smeared and diffused protein bands (Fig. 3). When a 300 kDa cut-off membrane was used, 1-D SDS-PAGE analysis of the retentate (Fig. 3, lane 4) and flow-through (Fig. 3, lane 5) showed that although high molecular weight proteins were enriched in the retentate, the filtration device also removed proteins with molecular weights below 300 kDa (Fig. 3 boxed area). Filtration to remove ultra-high MW proteins did prevent proteins from remaining in the well (Fig. 3, lane 5), but the major problems were that the flow-through was quite diluted and protein recovery was low, indicating the need for a different approach. Accordingly, we focused on increasing protein solubility through enzymatic digestion.
Fig. 3.
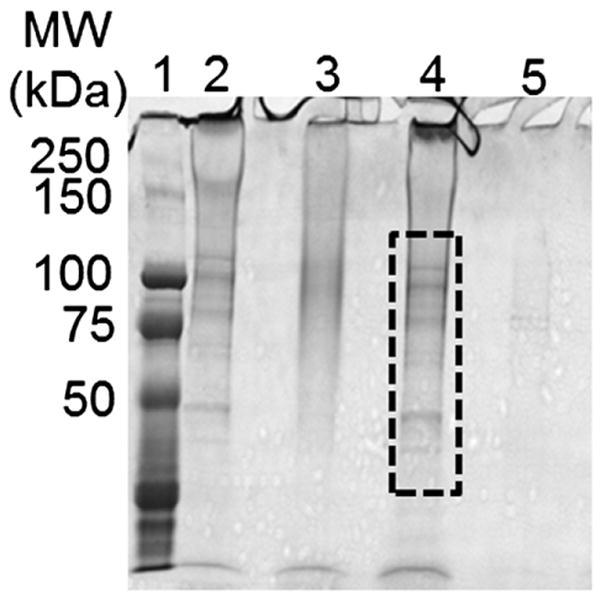
Two treatments were used to increase sample solubility and enhance protein resolution by electrophoresis: protein reduction/alkylation and filtration of high molecular weight proteins. The Coomassie Blue stained gel shows proteins in decellularized LV samples before and after treatments. Lane 1: molecular weight (MW) marker; lane 2: control untreated sample; lane 3: LV sample after reduction/ alkylation; lane 4: >300 kDa protein fraction of a decellularized LV sample after membrane filtration; lane 5: <300 kDa protein fraction of decellularized LV sample (same sample as in lane 4). Reduction and alkylation of the decellularized LV increased sample solubility but proteins were smeared, suggesting over-denaturation. Filtration to remove ultra-high MW proteins did prevent proteins from remaining in the well (<300 kDa, lane 5), but as observed in lane 4 the filtration device also removed proteins with molecular weights below 300 kDa (boxed area).
2.3. Acid extraction with pepsin digestion
Collagens are among the most abundant ECM components [18] and are highly insoluble in aqueous reagents. Digestion with pepsin under acidic conditions is one of the most commonly used methods to solubilize collagen [19,20]. As such, the insoluble pellets from the ECM-enriched protein homogenates were incubated in acetic acid containing increasing quantities of pepsin to release pepsin-resistant collagenous polypeptides [21] (strategy 7). We ran a control lane without pepsin (pellet in acid extraction buffer) in one of our initial analyses, but the no pepsin control sample was mostly stuck in the well. Pepsin digestion resulted in improved protein solubility, with minimal protein remaining at the top of the gel (Fig. 4). However, the sample-to-sample reproducibility was not acceptable.
Fig. 4.
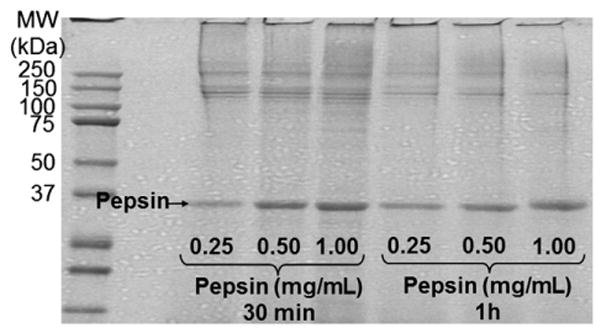
Coomassie Blue stained gel of decellularized LV extracts after acid extraction in conjunction with pepsin digestion. Pepsin treatment improved protein resolution on the gel. The same LV extract was treated with increasing quantities of pepsin for 30 min or 1 h. MW, molecular weight marker.
2.4. Texas 3-Step
We named the procedure that yielded the best results as the Texas 3-Step protocol, because it involved resolubilizing the sample in three buffers of increasing strength (strategy 8). LV samples are initially incubated in a neutral low salt, non-denaturing buffer (0.5 M NaCl, 10 mM Tris base pH 7.5 and 1× PI) for extraction of soluble proteins, including newly synthesized ECM proteins and degradation products (Step 1). The salts in the buffer interrupt ionic interactions between proteins, enabling the removal of non-covalently bound extracellular proteins [22]. The remaining tissue is then decellularized by treatment with SDS (Step 2). Finally, the insoluble protein fraction that remains is exposed to acid extraction, deglycosylation, and solubilization (Step 3).
Guanidine HCl (GnHCl) is a widely used protein denaturant that is very effective in solubilizing the majority of ECM proteins, including large aggregating proteoglycans, small proteoglycans, cell attachment matrix glycoproteins, and basement membrane components [3]. GnHCl simultaneously unfolds proteins and interrupts protein–protein interactions. GnHCl solubilizes ECM components by forming stable hydrogen-bonds to the protein backbone amides, which results in strong electrostatic repulsions, and destabilizes the ionic, disulfide-dependent protein conformations [23,24]. Following GnHCl treatment, the heavily cross-linked interstitial matrix, in particular collagens type I and III, and elastin remain insoluble [3].
ECM enrichment protocols that have stopped at the GnHCl extraction step are not suitable for proteomic analysis of insoluble ECM components. Mayr’s group reported the use of deglycosylation to remove glycosaminoglycan (GAG) side chains; this increased the recovery of proteins in the extracellular environment [3]. Deglycosylation appears to be critical for solubilizing the remaining ECM proteins, specifically proteoglycans, glycolipids, and GAGs. Chondroitinase ABC catalyzes the removal of polysaccharides containing 1 → 4-β-D-hexosaminyl and 1 → 3-β-D-glucuronosyl or 1 → 3-α-L-iduronosyl linkages to disaccharides containing 4-deoxy-β-D-gluc-4-enuronosyl groups [3]. This enzyme also acts on chondroitin-4-sulfate, chondroitin-6-sulfate, and dermatan sulfate glycosaminoglycan side chains. Keratanase cleaves internal 1 → 4-β-galactose linkages in unbranched, repeating poly-N-acetyl-lactosamine and keratan sulfate [25]. Heparinase II cleaves heparin and heparan sulfate [3,26].
Although the deglycosylation step significantly increased protein solubility of the decellularized LV, a small pellet of insoluble material was still visible. We dissolved this pellet in dimethyl sulfoxide (DMSO) to further solubilize the remaining ECM components. Following this step, all ECM proteins were in solution and no visible pellet was observed. DMSO is a well-known amphipathic solvent and hydrogen bond acceptor. By entering the aqueous region and interacting with hydrophobic proteins to form a large solvation shell, DMSO increases the number of sites available for hydrogen bonding and unfolds the protein [27], making the extended hydrogen-bond networks that stabilize aggregation of the ECM impossible [28]. By combining different reagents, in a step-by-step manner, we have been able to isolate and study the majority of the ECM proteins found in the LV. MS analysis of the insoluble protein fraction (Step 3) identified 157 proteins. This number of proteins was similar to the number of proteins identified previously with the strategy 4 protocol, when soluble proteins were excluded. However, only 18% of the proteins identified in the insoluble fraction of the Texas 3-Step protocol were mitochondrial, whereas when using Protein Extraction Reagent 4 to solubilize the LV 42% of the proteins identified by MS were mitochondrial. There were fewer ECM proteins identified in the insoluble fraction compared to the whole tissue (strategy 4 = 13% vs Texas 3-Step = 11%), since the Texas 3-Step analysis excluded the soluble ECM proteins present in the soluble fraction. Table 1 shows the ECM proteins identified by strategies 4 and 8.
Table 1.
ECM proteins identified in the LV infarcted tissue of WT mice, 5 days post-MI, by two methods: ECM-enrichment by SDS decellularization and solubilization with Protein Extraction Reagent 4 (strategy 4), and Texas 3-Step (Step 3, insoluble fraction only). Proteins identified by mass spectrometry are listed alphabetically.
| ECM identified proteins | Molecular weight (kDa) | Strategy 4 | Texas 3-Step (insoluble fraction) |
|---|---|---|---|
| Collagen I alpha-1 | 138 | X | X |
| Collagen I alpha-2 | 130 | X | X |
| Collagen III alpha-1 | 139 | X | X |
| Collagen IV alpha-1 | 161 | X | |
| Collagen IV alpha-2 | 167 | X | X |
| Collagen V alpha-1 | 184 | X | X |
| Collagen VI alpha-1 | 108 | X | X |
| Collagen VI alpha-2 | 110 | X | X |
| Collagen VI alpha-3 | 352 | X | X |
| Emilin 1 | 108 | X | X |
| Fibrillin-1 | 312 | X | X |
| Fibronectin | 272 | X | X |
| Fibulin-2 | 132 | X | |
| Laminin alpha-2 | 343 | X | X |
| Laminin alpha-5 | 404 | X | |
| Laminin beta-1 | 197 | X | |
| Laminin beta-2 | 196 | X | |
| Laminin gamma-1 | 177 | X | X |
| Nidogen-1 | 137 | X | |
| Periostin | 93 | X | X |
| Perlecan | 398 | X | X |
| Tenascin C | 222 | X | |
| Total | 20 | 17 |
We used immunoblotting to quantify specific ECM proteins identified by MS in each fraction of the Texas 3-step protocol (Fig. 5) and compared control LV and LV infarct tissues. Distinctive fragments of collagen were observed in the soluble (Step 1) and insoluble (Step 3) fractions. Interestingly, cleaved mature collagen (70 kDa) was mainly observed in the protein fractions of Steps 1 and 2 (Fig. 5b). Fibronectin, an important ECM protein during LV remodeling post-MI, was predominantly present in the LVI samples and in Step 1 and 2 fractions. MnSOD, a cellular protein, was present only in the fractions from Steps 1 and 2, indicating that the Texas 3-Step protocol effectively reduced cellular components in the insoluble fraction (Step 3). Importantly, protein solubilization was greatly increased after Step 3, and insoluble proteins were not present in the wells (Fig. 5a boxed area). With this protocol, we detected increased levels of the 148 kDa soluble procollagen (Step 1) and reduced levels of the ~300 kDa soluble collagen molecule in LV infarct compared to control LV tissue. These changes would be masked by the high levels of collagen fragments in the insoluble fraction (Step 3) if a one-step traditional extraction protocols were used.
Fig. 5.
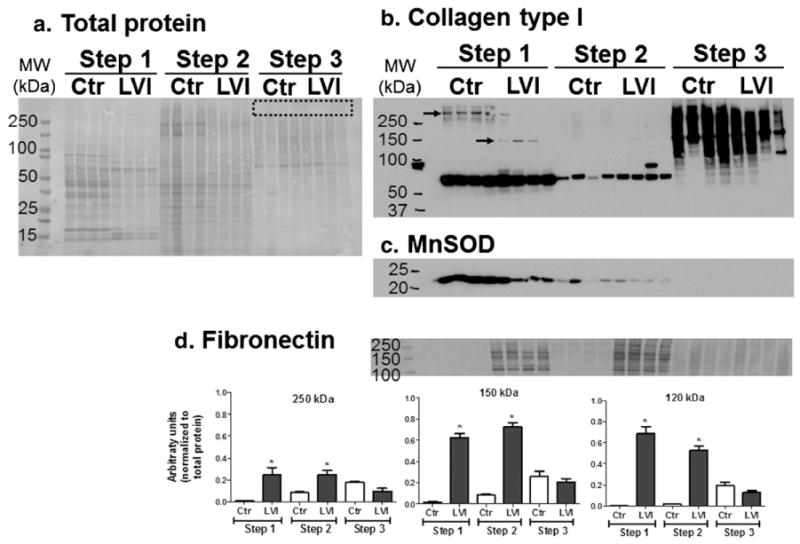
Protein fractions resulting from the Texas 3-Step protocol. a. Coomassie Blue stained gel (1 μg total protein per lane). No proteins were observed trapped in the wells, which shows an improved protein resolution compared to the previous tested strategies (boxed area). Immunoblots: b. collagen type I; c. fibronectin; and d) MnSOD; and d. fibronectin, densitometry analysis was normalized to total protein, *p < 0.05 versus respective control. By using a differential solubility-based, protein fractionation strategy, we unmasked protein differences (arrows) that otherwise would not be noticeable by a 1-fraction method. Step 1: soluble proteins; Step 2: cellular proteins; Step 3: insoluble proteins. MW: molecular weight; Ctr: LV control tissue from unoperated mice (n = 4); LVI: LV infarcted tissue 5 days post-MI (n = 4).
3. Discussion
ECM proteins generally have high molecular weights, are difficult to solubilize, are extensively post-translationally modified, and are usually present in low relative abundance, hampering comprehensive characterization and quantification [29]. Efficient sample preparation includes adequate protein solubilization, and avoiding the use of reagents that will cause downstream interference. We overcame issues with low abundance by enriching for ECM proteins with a thorough decellularization process. We subsequently focused on increasing protein solubility. After testing and combining several techniques and extraction buffers, our final optimized ECM-enrichment protocol, named the Texas 3-Step, separated the complex samples into three fractions: soluble, cellular, and insoluble proteins, the latter containing the majority of the ECM components. This method increased protein solubility considerably, allowed soluble and insoluble proteins to be evaluated separately, and resulted in the identification of ECM proteins in the insoluble fraction. One possible limitation of this method is the fact that some soluble ECM proteins may be present in the cellular fraction. In that case, these ECM proteins would likely be difficult to identify in mass spectrometry based protocols due to their relatively low abundance compared to the highly abundant cellular proteins. Our method is particularly useful for the study of insoluble ECM proteins but needs to be used cautiously when soluble ECM proteins are to be targeted. In that case, targeting immunoblotting may be a better approach to take.
4. Methods
All reagents were purchased from Sigma Aldrich (St Louis, MO) unless otherwise stated.
4.1. Animals and surgery
All animal procedures were conducted according to the “Guide for the Care and Use of Laboratory Animals” (NIH notice number: NOT-OD-12-020) and were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio. Male 4 to 8 month old C57BL6/J wild type (WT) mice were used in this study [30,31]. Animals were housed at constant temperature (22 ± 2 °C) on a 12 h light/dark cycle. They were fed standard laboratory mice chow ad libitum and had free access to tap water. MI was induced by permanent ligation of the left anterior descending coronary artery, as described previously [32]. At necropsy, mice were anesthetized with 2% isoflurane in a 100% oxygen mix. The coronary vasculature was flushed with cardioplegic solution (69 mM NaCl, 12 mM NaHCO3, 11 mM glucose, 30 mM 2,3-butanedione monoxime, 10 mM EGTA, 1 μM nifedipine, 50 mM KCl and 100 U heparin). The left ventricles were separated from the right ventricles, stained with 1% 2,3,5-triphenyltetrazolium chloride and photographed for measurement of infarct area. LV infarct (LVI) and remote tissues were separated and snap frozen in liquid nitrogen.
4.2. Tissue decellularization
The whole left ventricles of unoperated animals were incubated in distilled water with 1× protease inhibitor cocktail (PI) (cOmplete Mini tablets, Roche) at room temperature for 30 min. The water was decanted and replaced with decellularization buffer. Five decellularization buffers were tested: buffer # 1, 1% sodium dodecyl sulfate (SDS); buffer # 2, 1% SDS, 20 mM NH4OH; buffer # 3, 1% Triton X-100, 20 mM NH4OH; buffer # 4, 1% SDS, 1% Triton X-100; buffer # 5, 1% SDS, 1% Triton X-100, 20 mM NH4OH. All decellularization buffers were prepared in phosphate buffered saline (PBS) with 1× PI. Samples were left at room temperature in an orbital shaker until tissue was completely decellularized (three to four days). The decellularization buffer was collected daily and replaced with fresh decellularization buffer. When tissue looked translucent, samples were considered decellularized. Tissue was washed three times in distilled water with 1× PI for 5 min, and then left in fresh 1× PI/water overnight to remove all remnants of the decellularization buffer.
Two protocols were tested for homogenization of the decellularized samples. 1) Tissue samples were first homogenized (speed 6, 30 s, Power Gen 1000, Fisher Scientific) in soluble lysis buffer (0.25 M sucrose, 1 mM EDTA in 50 mM Tris with 1× PI), centrifuged at 14,000 RPM for 1 min, and the insoluble material homogenized (speed 6, 1 min 30 s, Power Gen 1000, Fisher Scientific) in Protein Extraction Reagent Type 4 (7.0 M urea, 2.0 M thiourea, 40 mM Trizma base, and 1.0% C7BzO, pH 10.4) and 1× PI. Both the soluble and the insoluble fractions were stored at −80 °C. 2) Tissue samples were directly homogenized (speed 6, 1 min 30 s, Power Gen 1000, Fisher Scientific) in Protein Extraction Reagent 4 (15 μL buffer per mg tissue) and stored at −80 °C.
Decellularization with buffer # 1 followed by homogenization with Protein Extraction Reagent 4 was used to prepare the samples discussed below.
4.3. High molecular weight filtration
Samples were applied to Nanosep centrifugal devices (Pall OD300C33) with a 300 kDa molecular weight cut-off. Each filter was sanitized with 70% ethanol and rinsed twice with deionized water before application of 175 μL of the sample. The total spin time for the samples was 4.5 min.
4.4. Reduction and alkylation
Samples were reduced by adding 1× PI, tributyl phosphine (TBP) to a final dilution of 1:40, and water to a final volume of 200 μL followed by incubation for 1 h in the dark at room temperature. Samples were alkylated with iodoacetamide (1:8 relative to the final volume) in water for 1 h in the dark at room temperature and then centrifuged at slow speed (2000 RPM) for 5 min. The supernatant was transferred to a new tube and proteins were precipitated with acetone added to a final concentration of 80%, and incubated at room temperature for 30 min. After centrifugation at 14,000 ×g for 10 min, the supernatant was discarded. The pellet was allowed to air dry for 5–10 min. The pellet was resuspended in Protein Extraction Reagent 4, vortexed, and incubated for 30 min at room temperature.
4.5. Acid extraction with pepsin digestion
Samples were centrifuged at 10,000 RPM, for 10 min at 4 °C. The supernatants were stored at −80 °C and the pellets used for the acid extraction step. Five hundred microliters of 0.5 M acetic acid, pH 2.5, was added to the pellet, mixed, and incubated overnight at 4 °C with agitation. On the next day, samples were centrifuged as described above, and the acid extraction repeated. After centrifugation at 10,000 RPM, for 10 min at 4 °C, 500 μL of acetic acid and pepsin (2500 U/mg) was added to the pellet. Three concentrations of pepsin were tested (0.25, 0.5 and 1 mg/mL), using incubation at 37 °C for either 30 min or 1 h. The reaction was stopped by addition of ~200 μL of 5 N NaOH to increase the pH to 8.0. Proteins were visualized by 1-D SDS-PAGE/Coomassie Blue staining.
4.6. Texas 3-Step protocol
After mincing samples, soluble proteins were extracted by incubation with Step 1 Buffer (0.5 M NaCl, 10 mM Tris base, pH 7.5, and 1× PI) overnight at 4 °C with agitation for 5 s at 800 RPM every 30 s (Triller Thermoshaker Incubator, PEQLAB Ltd.). After centrifugation at 14,000 RPM for 1 min, the supernatants (soluble proteins, Step 1) were stored at −80 °C and the pellets washed twice with Step 2 buffer (1% SDS in PBS and 1× PI). The samples were then incubated overnight in Step 2 buffer at room temperature, with agitation for 5 s at 800 RPM every 30 s. On the next day, the supernatants were removed and stored at −80 °C and fresh Step 2 buffer was added to the tissue. This process was repeated until the tissue was completely decellularized (three to five days). All supernatants from the decellularization process (containing mainly cellular proteins) were pooled in one tube (Step 2). The decellularized tissue samples were washed twice with deionized water before homogenization and sonication in Step 3 buffer (4 M GnHCl, 50 mM sodium acetate, pH 5.8 and 1× PI). The samples contain now mainly insoluble proteins; therefore, to increase solubility samples were maintained for 48 h in Step 3 buffer at room temperature, with agitation for 5 s at 1000 RPM every 30 s. After centrifugation at 4000 RPM for 25 min at 4 °C, the supernatants were removed and stored at −80 °C (Step 3a) and the pellets washed with 90% ethanol to remove any residual GnHCl. The pellets were resuspended in deglycosylation buffer [150 mM NaCl, 50 mM sodium acetate (pH 6.8) and 1× PI] and 0.05 U of each of the following deglycosylation enzymes: chondrotinase ABC from Proteus vulgaris, endo-β-galactosidase from Bacteroides fragilis, and heparinase II from Flavobacterium heparinum. The mixture was incubated overnight at 37 °C with agitation. If a pellet remained after deglycosylation, 20 μL of DMSO was added. These samples were pooled with Step 3a to combine all insoluble proteins into one fraction (Step 3). The insoluble fraction was precipitated with five volumes of cold acetone overnight at 4 °C and resolubilized in deglycolsylation buffer. This step is essential to remove the GnHCl and make samples amenable for mass spectrometry analysis. The total protein in each fraction from Steps 1, 2 and 3 was determined using the Quick Start Bradford Protein Assay (Bio-Rad). Proteins in each fraction were separated by 1-D SDS-PAGE and visualized by staining with Coomassie Blue.
4.7. Immunoblotting
An aliquot of each sample (1 or 5 μg protein) was loaded onto a 4–12% Bis–Tris gel and separated by electrophoresis. Proteins were transferred to a nitrocellulose membrane which was hybridized overnight at 4 °C with primary antibodies against the following: collagen type I (#CL50141AP-1, Cedarlane), fibronectin (#F6140, Sigma), GAPDH (#ab9485, Abcam), and MnSOD (#SOD-111, Assay Designs).
4.8. Mass spectrometry
Proteins (12.75 μg) were separated in a 4–12% Bis–Tris gel. The gel region containing visually detectable proteins was excised into three slices. Each slice was separately destained and dehydrated and the proteins digested in situ with trypsin (Promega). The digests were analyzed by capillary HPLC–electrospray ionization tandem mass spectrometry (HPLC–ESI–MS/MS) on a Thermo Fisher LTQ Orbitrap Velos mass spectrometer fitted with a New Objective Digital PicoView 550 NanoESI source. Online HPLC separation of the digests was accomplished with an Eksigent/AB Sciex NanoLC-Ultra 2-D HPLC system: column, PicoFrit™ (New Objective; 75 μm i.d.) packed to 15 cm with C18 adsorbent (Vydac; 218MS 5 μm, 300 Å). Precursor ions were acquired in the Orbitrap in profile mode at 60,000 resolution (m/z 400); data-dependent collision-induced dissociation (CID) spectra of the six most intense ions in the precursor scan above a set threshold were acquired sequentially in the linear trap at the same time as the precursor ion scan. Mascot (version 2.3.02; Matrix Science) was used to search the CID spectra against a combination of the mouse subset of the NCBInr database [3/ 12/2011 version; Mus. (145,083 sequences)] and a database of common contaminants (179 sequences). Methionine oxidation was considered as a variable modification; trypsin was specified as the proteolytic enzyme, with one missed cleavage allowed. The mass tolerances used for the searches were: precursor, 20 ppm; product ion, 0.8 Da. [The majority of identified peptides were detected with a mass error of ≤5 ppm]. The Mascot data files were combined in Scaffold (Proteome Software; version 3) for a subset search of the CID spectra using X! Tandem, cross correlation of the X! Tandem, Mascot results, determination of protein, and peptide identity probabilities. The thresholds for acceptance of peptide and protein assignments in Scaffold were 95% and 99.9%, respectively, and minimum of two unique peptides.
Acknowledgments
We acknowledge support from NIH/NHLBI HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center, POST14350034 for KYD, NIH 1K99AT006704-01 for GVH, and R01 HL075360, HL051971, and the Veteran’s Administration (Merit) to MLL. We acknowledge support from the University of Texas Health Science Center at San Antonio for the Institutional Mass Spectrometry Laboratory.
Footnotes
Author contributions
L.E.C.B. and M.L.L. designed the research. K.Y.D., Y.M., and G.V.H. performed the animal experiments. L.E.C.B., T.A.R., Y.A.C., and Q.D. performed the ECM-enrichment experiments. K.H. and S.T.W. performed the MS experiments and analysis. L.E.C.B., T.A.R., K.Y.D., and M.L.L. wrote the manuscript and all authors edited the manuscript.
Conflict of interests
The authors declare no competing financial interests.
References
- 1.Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200:423–8. doi: 10.1002/path.1437. http://dx.doi.org/10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- 2.Scott-Burden T. Extracellular matrix: the cellular environment. NiPS. 1994;9:110–5. [Google Scholar]
- 3.Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048–62. doi: 10.1074/mcp.M110.001693. http://dx.doi.org/10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–78. doi: 10.1242/dmm.004077. http://dx.doi.org/10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994;26:279–92. doi: 10.1006/jmcc.1994.1036. http://dx.doi.org/10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- 6.Brilla CG. The cardiac structure-function relationship and the renin–angiotensin–aldosterone system in hypertension and heart failure. Curr Opin Cardiol. 1994;9(Suppl 1):S2–S10. doi: 10.1097/00001573-199407000-00002. discussion S10–11. [DOI] [PubMed] [Google Scholar]
- 7.Lindsey ML, Yoshioka J, MacGillivray C, Muangman S, Gannon J, Verghese A, et al. Effect of a cleavage-resistant collagen mutation on left ventricular remodeling. Circ Res. 2003;93:238–45. doi: 10.1161/01.RES.0000085580.45279.60. [DOI] [PubMed] [Google Scholar]
- 8.Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, et al. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 2010;10:2214–23. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown PR, Miech RP. Comparison of cell extraction procedures for use with high-pressure liquid chromatography. Anal Chem. 1972;44:1072–3. doi: 10.1021/ac60314a033. [DOI] [PubMed] [Google Scholar]
- 10.Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, et al. Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart. Nat Med. 2008;14:213–21. doi: 10.1038/nm1684. http://dx.doi.org/10.1038/nm1684. [DOI] [PubMed] [Google Scholar]
- 11.Korossis SA, Wilcox HE, Watterson KG, Kearney JN, Ingham E, Fisher J. In-vitro assessment of the functional performance of the decellularized intact porcine aortic root. J Heart Valve Dis. 2005;14:408–21. discussion 422. [PubMed] [Google Scholar]
- 12.Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ, Christman KL. Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering. Biomaterials. 2009;30:5409–16. doi: 10.1016/j.biomaterials.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang N, Chen R, Young N, Wishart D, Winter P, Weiner JH, et al. Comparison of SDS- and methanol-assisted protein solubilization and digestion methods for Escherichia coli membrane proteome analysis by 2-D LC–MS/MS. Proteomics. 2007;7:484–93. doi: 10.1002/pmic.200600518. http://dx.doi.org/10.1002/pmic.200600518. [DOI] [PubMed] [Google Scholar]
- 14.Ott HC, Clippinger B, Conrad C, Schuetz C, Pomerantseva I, Ikonomou L, et al. Regeneration and orthotopic transplantation of a bioartificial lung. Nat Med. 2010;16:927–33. doi: 10.1038/nm.2193. [DOI] [PubMed] [Google Scholar]
- 15.Vavken P, Joshi S, Murray MM. TRITON-X is most effective among three decellularization agents for ACL tissue engineering. J Orthop Res. 2009;27:1612–8. doi: 10.1002/jor.20932. http://dx.doi.org/10.1002/jor.20932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Speers AE, Wu CC. Proteomics of integral membrane proteins—theory and application. Chem Rev. 2007;107:3687–714. doi: 10.1021/cr068286z. http://dx.doi.org/10.1021/cr068286z. [DOI] [PubMed] [Google Scholar]
- 17.Chevallet M, Santoni V, Poinas A, Rouquie D, Fuchs A, Kieffer S, et al. New zwitterionic detergents improve the analysis of membrane proteins by two-dimensional electrophoresis. Electrophoresis. 1998;19:1901–9. doi: 10.1002/elps.1150191108. http://dx.doi.org/10.1002/elps.1150191108. [DOI] [PubMed] [Google Scholar]
- 18.Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. http://dx.doi.org/10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 19.Miyahara T, Murai A, Tanaka T, Shiozawa S, Kameyama M. Age-related differences in human skin collagen: solubility in solvent, susceptibility to pepsin digestion, and the spectrum of the solubilized polymeric collagen molecules. J Gerontol. 1982;37:651–5. doi: 10.1093/geronj/37.6.651. [DOI] [PubMed] [Google Scholar]
- 20.Markiewicz M, Asano Y, Znoyko S, Gong Y, Watson DK, Trojanowska M. Distinct effects of gonadectomy in male and female mice on collagen fibrillogenesis in the skin. J Dermatol Sci. 2007;47:217–26. doi: 10.1016/j.jdermsci.2007.05.008. http://dx.doi.org/10.1016/j.jdermsci.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson R, Diseberg AF, Gordon L, Zivkovic S, Tatarczuch L, Mackie EJ, et al. Comprehensive profiling of cartilage extracellular matrix formation and maturation using sequential extraction and label-free quantitative proteomics. Mol Cell Proteomics. 2010;9:1296–313. doi: 10.1074/mcp.M000014-MCP201. http://dx.doi.org/10.1074/mcp.M000014-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mason RM, Mayes RW. Extraction of cartilage protein-polysaccharides with inorganic salt solutions. Biochem J. 1973;131:535–40. doi: 10.1042/bj1310535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien EP, Dima RI, Brooks B, Thirumalai D. Interactions between hydrophobic and ionic solutes in aqueous guanidinium chloride and urea solutions: lessons for protein denaturation mechanism. J Am Chem Soc. 2007;129:7346–53. doi: 10.1021/ja069232+. http://dx.doi.org/10.1021/ja069232+ [DOI] [PubMed] [Google Scholar]
- 24.Sajdera SW, Hascall VC. Proteinpolysaccharide complex from bovine nasal cartilage. A comparison of low and high shear extraction procedures. J Biol Chem. 1969;244:77–87. [PubMed] [Google Scholar]
- 25.Holland JW, Meehan KL, Redmond SL, Dawkins HJ. Purification of the keratan sulfate proteoglycan expressed in prostatic secretory cells and its identification as lumican. Prostate. 2004;59:252–9. doi: 10.1002/pros.20002. http://dx.doi.org/10.1002/pros.20002. [DOI] [PubMed] [Google Scholar]
- 26.Ferreira TM, Medeiros MG, Dietrich CP, Nader HB. Structure of heparan sulfate from the fresh water mollusc Anomantidae sp. : sequencing of its disaccharide units. Int J Biochem. 1993;25:1219–25. doi: 10.1016/0020-711x(93)90070-u. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Allen HC. Interactions of dimethylsulfoxide with a dipalmitoylphosphatidylcholine monolayer studied by vibrational sum frequency generation. J Phys Chem A. 2009;113:12655–62. doi: 10.1021/jp905066w. http://dx.doi.org/10.1021/jp905066w. [DOI] [PubMed] [Google Scholar]
- 28.Santos NC, Figueira-Coelho J, Martins-Silva J, Saldanha C. Multidisciplinary utilization of dimethyl sulfoxide: pharmacological, cellular, and molecular aspects. Biochem Pharmacol. 2003;65:1035–41. doi: 10.1016/s0006-2952(03)00002-9. http://dx.doi.org/10.1016/S0006-2952(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 29.Pflieger D, Chabane S, Gaillard O, Bernard BA, Ducoroy P, Rossier J, et al. Comparative proteomic analysis of extracellular matrix proteins secreted by two types of skin fibroblasts. Proteomics. 2006;6:5868–79. doi: 10.1002/pmic.200402040. [DOI] [PubMed] [Google Scholar]
- 30.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–22. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin MD, Carter KJ, Jean-Philippe SR, Chang M, Mobashery S, Thiolloy S, et al. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008;68:6251–9. doi: 10.1158/0008-5472.CAN-08-0537. http://dx.doi.org/10.1158/0008-5472.CAN-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290:H232–9. doi: 10.1152/ajpheart.00457.2005. http://dx.doi.org/10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]