

Abstract
The major allergen domain (MA) is widely distributed in insects. The crystal structure of a single Bla g 1 MA revealed a novel protein fold in which the fundamental structure was a duplex of two subsequences (monomers), which had diverged over time. This suggested that the evolutionary origin of the MA structure may have been a homodimer of this smaller subsequence. Using publicly available genomic data, the distribution of the basic unit of this class of proteins was determined to better understand its evolutionary history. The duplication and divergence is examined at three distinct levels of resolution: 1) within the orders Diptera and Hymenoptera, 2) within one genus Drosophila, and 3) within one species Aedes aegypti. Within the family Culicidae, we have found two separate occurrences of monomers as independent genes. The organization of the gene family in A. aegypti shows a common evolutionary origin for its monomer and several closely related MAs. Molecular modeling of the A. aegypti monomer with the unique Bla g 1 fold confirms the distant evolutionary relationship and supports the feasibility of homodimer formation from a single monomer. RNAseq data for A. aegypti confirms that the monomer is expressed in the mosquito similar to other A. aegypti MAs after a blood meal. Together, these data support the contention that the detected monomer shares similar functional characteristics to related MAs in other insects. An extensive search for this domain outside of Insecta confirms that the MAs are restricted to insects.
Keywords: SDMA, molecular modeling, genome, gene family, tandem duplication, synteny, ortholog, gene expression, RNAseq, Bla g 1, ANG12, allergen
Introduction
The major allergen domain (MA) is the simplest member of a gene family that is widely distributed in insects (Fischer et al. 2008). The name major allergen comes from the characterization of the first member of the protein family, called Bla g 1, as a human allergen from the cockroach Blattella germanica (Helm et al. 1996; Pomés et al. 1998). Biochemical studies suggested that the function of the protein in cockroaches was related to nutrient uptake (Gore and Schal 2004; Suazo et al. 2009). Indeed, related proteins in the mosquitos Aedes aegypti and Anopheles gambiae were found upregulated after feeding (Nolan et al. 2011). In the Lepidopterans, multiple copies of two different types of the MA were found (Fischer et al. 2008). The first type was closely related to other insect MAs, while the other nitrile specifier protein (NSP) was more distantly related. The NSPs evolved to allow the larvae to detoxify nitrile compounds found in certain plants. To date, the MA was thought to only occur exclusively within the class Insecta.
Many other insect species contain MA proteins. In cases where an MA exists in a single copy, it is called a single domain major allergen (SDMA). When there are multiple MAs repeated on a single polypeptide, they are annotated as NDMA, where N indicates the number of MAs. One MA is defined by major protein domain databases as a 200-amino acid (aa) motif (Interpro: IPR010629, PFAM: PF06757, Ins_allergen_rp). However, there is some disagreement about the actual definition of the basic domain itself. Pomés et al. (1998) define the Bla g 1 of B. germanica as a tandem repeat of two distinct domains of 100 aa both by sequence homology and a dotplot analysis. Fischer et al. (2008) suggest that a minimal SDMA protein is a single domain of 200 aa and do not discuss an internal domain structure.
A recently solved crystal structure of Bla g 1 supports the Pomés definition (Mueller et al. 2013). Each of the 100 aa repeats form a nearly identical fold that resembles a planar pentagon with five alpha helices and a sixth helix displaced along the z axis that lies above the plane of the pentagon. The structure is the first of a new fold family. Here, we name the two 100-aa repeats sequentially α and β. The two pentagons stack on top of each other and interact via the rim of the pentagon creating a large cavity in the center of almost 3,000 Å3, which is lined exclusively with hydrophobic residues. When the structure of β was aligned with α, it appeared that many of the interactions along the rim would be maintained in a β/β homodimer. A similar thought experiment hinted that an α/α homodimer might be equally feasible. This analysis suggested that a primitive form of the protein may have existed as a homodimer, with a subsequent intragenic duplication that led to two subunits being expressed consecutively (the progenitor to the current SDMA) and subsequently diverging both in sequence and copy number. Based on this new structural data, we were interested in testing the hypothesis that SDMA genes may have evolved from one or more genes containing a 100-aa domain. To test this hypothesis, we searched the current genomic databases for SDMA relatives and utilized molecular modeling to verify that the sequence was compatible with the unique Bla g 1 fold.
Previous studies on the distribution of the MA gene family within insects have relied heavily on the generation of expressed sequence tags and genomic libraries of a limited number of orders within Insecta (Fischer et al. 2008). The ever-expanding nature of genomics now offers an opportunity to search a more diverse collection of completed genomes of a wide range of insect species for members of the MA gene family and thus present a more comprehensive analysis of this functionally important domain. Our primary interests were to survey available genomes, to examine the distribution of the MA in individual genomes, to distinguish between the competing hypotheses about the nature of the basic MA, and to examine whether the MA is truly exclusive to Insecta by searching more distantly related phyla.
Materials and Methods
Bioinformatics, Databases, and Search Algorithms
The genomes analyzed were Harpegnathos saltator, Camponotus floridanus (Bonasio et al. 2010), Heliconius melpomene (Heliconius Genome Consortium 2012), Apis mellifera (Honeybee Genome Sequencing Consortium 2006), Drosophila melanogaster (Adams et al. 2000), Culex quinquefasciatus (Arensburger et al. 2010), Daphnia pulex (Colbourne et al. 2011), Tetranychus urticae (Grbic et al. 2011), Anopheles gambiae (Holt et al. 2002), Pediculus humanus (Kirkness et al. 2010), Anopheles darlingi (Marinotti et al. 2013), Aedes aegypti (Nene et al. 2007), Acromyrmex echinatior (Nygaard et al. 2011), Tribolium castaneum (Richards et al. 2008), Pogomyrmex barbatus (Smith, Smith et al. 2011), Linepithema humile (Smith, Zimin et al. 2011), Atta cephalotes (Suen et al. 2011), Nasonia vitripennis (Werren et al. 2010), Solenopsis invicta (Wurm et al. 2011), Bombyx mori (Xia et al. 2004), Plutella xylostella (You et al. 2013), Danaus plexippus (Zhan et al. 2011), Acyrthosiphon pisum (International Aphid Genomics Consortium 2010), Ixodes scapularis, Megaselia scalaris (metazoan.ensembl.org), Rhodnius prolixus, An. stephensi, Glossina mortisans, M. scalaris, Lutzomyia longipalpis, and Phlebotomus papatasi (www.vectorbase.org, last accessed November 29, 2013).
All predicted protein sets were initially searched using the HMMER 3.0 package (http://hmmer.org/, last accessed November 29, 2013) function hmmsearch (Eddy 2011) with default settings using the Ins_allergen_rep.hmm for PF06757 (http://pfam.janelia.org/, last accessed November 29, 2013) as the query. Proteomes outside the Insecta were also searched with the HMMER 3.0 jackhmmer function as the PF06757 hmm is based solely on Insect representatives of the MA gene and thus has the potential to lose sensitivity over longer evolutionary distances. Signal peptide (SP) prediction for all SDMA gene family members was done using the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP, last accessed November 29, 2013) with default settings (Petersen et al. 2011). All protein sequence alignments were done with mafft 6.849 with default settings (mobyle.paster.fr) (Neron et al. 2009).
Phylogenetic analyses were performed with MrBayes 3.1 (http://mrbayes.sourceforge.net, last accessed November 29, 2013) (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). For figure 2 (the SDMA gene family of A. aegypti) protein sequences, we used MEGA 5.10 to determine the appropriate substitution model (WAG) (Tamura et al. 2011). The analysis was repeated twice until the standard deviation of the prior probability was <0.01. The consensus tree was identical in each case. For supplementary figure S10, Supplementary Material online (the Formicidae SDMA tree), the protein sequences were aligned with mafft 6.849 and the appropriate substitution model was WAG with a discrete gamma distribution of five rate categories. The analysis was repeated twice until the standard deviation of the prior probability was <0.01.
Fig. 2.—

Phylogeny of the SDMA genes of Aedes aegypti. Names of genes are color-coded based on the clustering observed in the A. aegypti genome assembly at the supercontig level depicted in figure 5. Dots next to gene names denote exon–intron structure with numbering within the red dots corresponding to the grouping of the exon–intron structures as shown in supplementary figure S1, Supplementary Material online. Numbers at each of the nodes represent posterior probabilities from the two separate runs of the tree.
RNAseq Analysis
RNAseq data were downloaded from the short read archive (SRA) at NCBI for A. aegypti (SRP008153 and SRP003874 for the data in fig. 6). Fastqc 0.10.1 was used to QC these data sets (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, last accessed November 29, 2013). Replicates for both conditions for each strain were combined into one set (similar results were obtained for the independent replicates, data not shown). The single end reads were aligned to the A. aegypti reference sequence (AaegL1, August 2005) with TopHat 2.0.4 using default setting except the following: -g 1 (one unique mapping per read) -I 50000 (maximum intron size of 50 kb) (Kim et al. 2013). Cufflinks 2.0.2 was used to quantitate transcript levels from the TopHat output using a custom gtf annotation file for all SDMA genes derived from the complete A. aegypti gtf file (Trapnell et al. 2013). This was done to speed up analysis time and does not change the results. Both reference sequence and annotation files were downloaded from EnsemblMetazoa (http://metazoa.ensembl.org/info/data/ftp/index.html, last accessed November 29, 2013). The RNAseq data sets used in supplementary figure S11, Supplementary Material online, are also from SRA (SRP009679) and were analyzed with the same parameters.
Fig. 6.—

RNAseq analyses of expression of SDMA genes in Aedes aegypti females of three strains. Three strains of A. aegypti were sugar fed or blood fed. RPKM (fragments per kb per million reads) values for each SMDA homolog are shown on the y axis, and for those genes with multiple isoforms, only the most abundant isoform is shown. SDMA genes are organized by the evolutionary relatedness described in figure 2. AAEL010436 (β monomer) and AAEL010429 (AEG12) are highlighted. Control genes (AAEL002595, AAEL001216, AAEL000795, and AAEL002881) representing different RPKM levels are shown at right; none of the differences between the feeding conditions or strains are statistically significant in these controls. The data are collated from Bonizzoni et al. (2012).
Model Building and Optimization
Sequence alignments of various candidate sequences (AAEL010436-PA, ASTM006939-PA, ASTM009013-PA, PRNA_AY425622, and Tetur06g03260) with Bla g 1 were achieved using the sequence alignment tool, T-Coffee (Notredame et al. 2000). Using the X-ray crystal structure of Bla g 1 (pdb entry:4JRB) as the template, the starting structures of the selected candidate proteins were constructed with the help of Modeller 9.11 (Eswar et al. 2006). The starting structures were dynamically optimized by running Generalized Born (GB) simulations using the Sander module of Amber.12 (Case et al. 2012). GB simulations for optimizations and interaction energy calculations between the two domains were carried out using the FF12SB force filed of Amber.12.
Results
SDMA Homologs within Insecta
The increased availability of complete insect genomes led us to a reexamination of the distribution of SDMA within Insecta. We restricted our analysis to 26 non-Drosophila genomes which were publicly available and with proteomes and the 12 Drosophila genomes available at Flybase (www.flybase.org, last accessed November 29, 2013). These represent the more mature sequencing projects and a good baseline for a “complete” genome. These genomes also largely represent the Holometabolous insects. A summary of the genomes searched and a summary of the characteristics of the SDMA families discovered in each species is found in table 1. Our primary analysis includes only D. melanogaster; a summary of our analyses of other Drosophila genomes is in the supplementary text, Supplementary Material online. Figure 1A graphically summarizes the SDMA terminology introduced earlier. SDMA homologs were found in virtually all available insect genomes, an exception being the Hemiptera and Phthiraptera, although this absence may be due to the low sampling of genomes in these two orders or the incompleteness of the genomes available. The SDMA genes (86) in these genomes range in size from 156 to 514 aa. For 14 of the canonical SDMA genes, there is no signal peptide (SP) predicted. Among these canonical SDMA genes, there is a class of 11 SMDA genes that, either with or without a SP, contain an unusually long N-terminal amino acid extension of 49–288 aa prior to an intact SDMA; none of these extensions have any similarity to known motifs and all of these are in dipteran genomes. The 288 aa extension is exceptional; others range in size from 49 to 78 aa.
Table 1.
Primary Genomes Analyzed
| Groups | Order | Scientific Name | Common Name | SDMA Homologs, Type | Genes in Clusters |
|---|---|---|---|---|---|
| Outgroups | Crustacea | Daphnia pulex | Water flea | 0 | NAa |
| Chelicerata | Tetranychus urticae | Spider mite | 0 | NA | |
| Ixodes scapularis | Deer tick | 0 | NA | ||
| Insecta | |||||
| Hymenoptera | |||||
| Acromyrmex echinatior | Leafcutter ant | 2 SDMA, 1 2DMA | 3 | ||
| Apis mellifera | Honey bee | 4 SDMA, 1 2DMA, 1 3DMA | 2 | ||
| Atta cephalotes | Leafcutter ant | 2 SDMA, 1 2DMA | 2 | ||
| Camponotus floridanus | Carpenter ant | 3 SDMA, 1 2DMA | 2 | ||
| Harpegnathos saltator | Jumping ant | 3 SDMA, 1 2DMA | 4 | ||
| Linepithema humile | Argentine ant | 3 SDMA, 1 2DMA | 3 | ||
| Nasonia vitripennis | Parasitoid wasp | 1 7DMA | NA | ||
| Pogomyrmex barbatus | Red harvester ant | 3 SDMA, 1 2DMA | 3 | ||
| Solenopsis invicta | Fire ant | 4 SDMA, 1 2DMA | 3 | ||
| Coleoptera | |||||
| Tribolium castaneum | Flour beetle | 1 SDMA, 1 8DMA | 2 | ||
| Hemiptera | |||||
| Acyrthosiphon pisum | Pea aphid | 0 | NA | ||
| Rhodnius prolixus | Kissing bug | 0 | NA | ||
| Phthiraptera | |||||
| Pediculus humanus | Body louse | 0 | NA | ||
| Lepidoptera | |||||
| Bombyx mori | Silkworm | 3 SDMA | 2 | ||
| Danaus plexippus | Butterfly | 2 SDMA | None | ||
| Heliconius melpomene | Butterfly | 3 SDMA | 2 | ||
| Plutella xylostella | Moth | 0 | NA | ||
| Diptera | |||||
| Aedes aegypti | Mosquito | 20 SDMA, 1 βMA | 19, six clusters | ||
| Anopheles darlingi | Mosquito | 2 SDMA | None | ||
| Anopheles gambia | Mosquito | 5 SDMA | 2 | ||
| Anopheles stephensi | Mosquito | 2 SDMA, 1 αMA | None | ||
| Culex quinquefasciatus | Mosquito | 8 SDMA | 7, two clusters | ||
| Glossina mortisans | Tsetse fly | 2 SDMA, 1 2DMA | None | ||
| Drosophila melanogaster | Fruit fly | 7 SDMA | 3 | ||
| Megaselia scalaris | Scuttle fly | 0 | NA | ||
| Lutzomyia longipalpis | Sand fly | 4 SDMA | None | ||
| Phlebotomus papatasi | Sand fly | 0 | NA |
aEither no homologs or only one SDMA homolog in genome.
Fig. 1.—
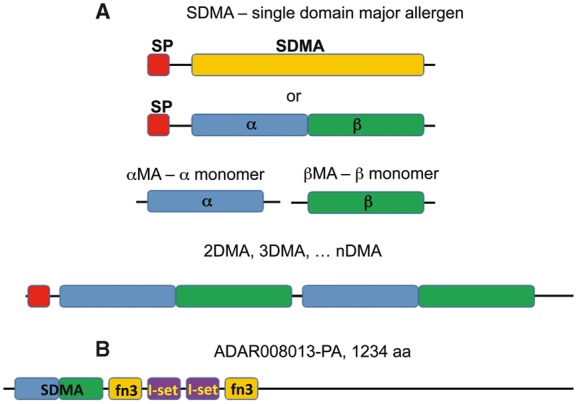
Terminology. (A) Two proposed alternative structures for the SDMA, either a single 200 aa domain (SDMA) or adjacent monomer domains (α and β). (B) Domain structure of ADAR008013 from Anopheles darlingi with a single SDMA followed by two pairs of fn3 (fibronectin type III, PF00041) and l-set (immunoglobulin I-set, PF07679) domains.
There is one exceptional peptide in this class with a conventional SP-SDMA N terminus but of 1,234 aa in length. In this protein, ADAR008013-PA from An. darlingi, the SDMA is followed by a Fibronectin type III domain, two Immunoglobulin I-set domains, and another Fibronectin type III domain and is the only SDMA-containing protein in any of the genomes queried here which shows an SDMA fused to any other known domains (fig. 1B).
Within the dipterans, we uncovered evidence for two smaller SDMA proteins, which we are proposing to call the α and β monomers (hereafter, αMA and βMA). One was found within An. stephensi (125 aa αMA; ASTM009013-PA) and A. aegypti (95 aa βMA; AAEL010436-PA). Each represents one half of the canonical SDMA (fig. 1A) and supports the hypothesis of Pomés that the canonical SDMA could represent a repeat of two smaller domains (Pomés et al. 1998). A more detailed analysis of these two genes is presented later.
Previous genomic analyses had shown the existence of multiple domain homologs of SDMAs (i.e., NSP and MA genes, 3DMA homologs) within the Pieridae subfamily of butterflies (Fischer et al. 2008). We find 11 new multiple domain proteins. These are widespread within the hymenopterans, wherein the species examined generally contain two to three canonical SDMA proteins and usually one additional multiple domain (2DMA) protein. A notable example within the Hymenoptera was N. vitripennis (parasitoid wasp), which contains a single 7DMA protein only, the most significantly expanded multiple domain protein aside from the previously described 8DMA protein of Tr. castaneum (Fischer et al. 2008). All of the 2DMA domain proteins found have single linker peptides of 15–25 aa in size between the continuous SDMAs. The 3DMA of Apis mellifera has unusually long linkers of 29 and 40 aa. The 7DMA of N. vitripennis and the previously identified 8DMA of Tr. castaneum both have regularly sized linkers (18–23 aa and 15–16 aa, respectively). An overview of all the SDMA genes used in this study with sequences is in supplementary table S1a–c, Supplementary Material online.
We also used the Bla g 1 protein sequence as a psi-Blast query of the NCBI nr protein data set (July 29, 2013), which found a further 83 complete and 18 partial SDMA homologs from an additional 29 species of insects not in our collection. One of these is from Pl. xylostella, one from Lu. longipalpis, and four from Ph. papatasi. Our primary genomic analysis failed to find these, highlighting the potential limitations of a predicted protein data set and why our summary in table 1 indicating that Pl. xylostella, Acy. pisum, R. prolixus, or M. scalaris contain SDMA genes is tentative. These are primarily canonical SDMA genes, but NDMA-type proteins, including some not identified previously, are also included. A list of these is in supplementary table S1d, Supplementary Material online.
Gene Families and Genome Organization of SDMA Genes I: Dipterans
Throughout most of the Insecta, there were small expansions of 2–8 copies of SDMA genes. One exceptional expansion of SDMAs was found in A. aegypti, which has 20 SDMA homologs. In five of the dipteran genomes examined (Drosophila spp. excluded), there is linkage between two or more of the SDMA genes. Culex quinquifasciatus has all 8 SDMA homologs on two separate supercontigs, whereas A. aegypti has 18 of its homologs organized on six supercontigs (table 1 and supplementary fig. S1, Supplementary Material online, for A. aegypti). This conclusion may underestimate the linkage, as whether or not the supercontigs themselves that contain these SDMA genes are part of a higher order linkage at the chromosomal level awaits a better assembly of the A. aegypti genome. For genomes that are of high enough quality to discern a genomic organization, linkage of SDMA genes is the rule, consistent with the hypothesis that many of these genes arose via gene duplications.
The SDMA gene structure varies across species. The most common structure in the dipterans is three exons and two introns. Specific exon–intron junctions of this class of SDMA genes are occasionally conserved between species and often conserved within a single species suggesting orthologous and paralogous relationships (see supplementary text, Supplementary Material online, for details).
To begin to understand the relationships between these genes in one species, we built a phylogenetic tree of all A. aegypti SDMA homologs including the candidate βMA using Bayesian inference (fig. 2). The figure is coded two ways, those genes with red balls represent the three conserved three exon groups described in supplementary figure S2, Supplementary Material online, blue balls the two variations on the two exon, and green balls the two variations of the four exon SDMA genes with N-terminal extensions. The different colored gene names represent the supercontig location of all genes described in supplementary figure S1, Supplementary Material online, with the two (AAEL001418 and AAEL005340) showing no linkage to each other or the other supercontigs underlined. It is clear that the phylogenetic relatedness of SDMA genes correlates very well with exon–intron structure and suggests six groups of paralogous genes. In contrast, different paralogs are occasionally found on different supercontigs, suggesting that none of these clusters of SDMA genes are entirely the result of local tandem duplications of a single ancestral SDMA gene. Such tandem duplications are likely in three cases with AAEL001611 and AAEL001621; AAEL013577-PA and AAEL013577-PB; and AAEL010431 and AAEL010429 each being likely cases of tandem duplication as shown by their proximity and shared sequence identity within the 5′ and 3′ untranslated DNA sequences within each of the above pairs (supplementary figs. S3–S5, Supplementary Material online). The proximity of several other gene pairs suggests tandem duplications. AAEL009165 and AAEL009166 (on supercontig 1.374) and the corresponding pair of AAEL013126 and AAEL013127 (on supercontig 1.793) are most clearly part of the larger 61 kb genomic duplication with AAEL01326/AAEL009165 (97.9% identity over 2.37 kb around the two genes) and AAEL01327/AAEL009166 (95.1% identity over 1.8 kb) most closely related. If AAEL009165/AAEL009166 and AAEL013126/AAEL013127 (each pair has >90% identity between the respective CDS regions but little in the 5′ and 3′ flanking region) are also the consequence of a tandem duplication, this likely occurred prior to the larger 61 kb genomic duplication as the lack of sequence conservation flanking these gene pairs is in stark contrast to the high level of genomic sequence conservation between the phylogenetically closer pairs.
The phylogenetic relationships and the distribution of the SDMA genes on two sets of supercontigs, c1.875 and c1.477 and c1.793 and c1.374, suggested the possibility that some SDMA genes could be part of larger duplication events. To test this, we compared the supercontigs involved using the wgVISTA analysis tool of the VISTA suite of programs for comparative genomic analysis (Frazer et al. 2004). Separately, the pair c1.374 (1.19 Mb) and c1.793 (591 kb) and the pair c1.477 (943 kb) and c1.875 (466 kb) were aligned. For the first pair, a duplication of 61 kb was identified between the two supercontigs, and for the second, one of 178 kb (supplementary fig. S6a and b, respectively, Supplementary Material online). Outside of these duplications, there is little similarity between these two pairs of supercontigs (supplementary figs. S7 and S8, Supplementary Material online). Within these duplications are our two clusters of SDMA genes, within a 16 kb region in the c1.374-c1.793 alignment and a 110 kb region within the c1.477-c1.875 alignment (fig. 3 and highlighted in supplementary fig. S6, Supplementary Material online). Within these two duplications, there are in addition to the conserved SDMA genes, extensive intergenic regions showing an extremely high level of sequence identity. The largest gaps within these regions are in the locations of two genes which are specific to one of the supercontigs, AAEL013118 (on c1.793) and AAEL010436, the β monomer (on c1.477).
Fig. 3.—

Duplications of SDMA genes in Aedes aegypti. The relevant regions of two separate alignments of four A. aegypti supercontigs are shown along with the locations of the SDMA genes on each supercontig. The peaks represent nucleotide identity, and only regions with between 50% and 100% identity are shown. The βMA AAEL010436 is highlighted with a black arrow.
Gene Families and Genome Organization of SDMA Genes II: Hymenopterans
The hymenopterans examined here have a distinctly different suite of SDMA genes compared with the dipterans that is very well conserved within this order. Except for N. vitripennis, the representatives of this order contain 2–4 SDMA genes and one 2DMA gene. In Hymenoptera, virtually all of the SDMA- and NDMA-containing genes are intronless. Only Apis mellifera contains a three domain gene similar to those previously described in Lepidoptera. Within the Formicidae, the prevalent organization is of three genes (two SDMA and one 2DMA) within 10 kb; C. floridanus, H. saltator, and Acr. echinatior show slight deviations from this common theme. This linked gene family potentially represents a set of orthologs within the ants, and this region is shown in supplementary figure S9, Supplementary Material online, with likely orthologs colored similarly. A phylogenetic tree of the Formicidae SDMA and 2DMA genes (supplementary fig. S10, Supplementary Material online) shows support for the orthology relationships shown in supplementary figure S9, Supplementary Material online, where likely orthologs are noted by colored dots corresponding to the shading seen in supplementary figure S10, Supplementary Material online. Most of these species also contain a loosely linked SDMA, separated by 200–500 kb from the conserved cluster but on the same supercontig. Supplementary figure S10, Supplementary Material online, also suggests that these (colored with red) are orthologs. A history of the gene duplications within the Formicidae is also suggested as the 2DMA genes are phylogenetically most similar to the immediately adjacent SDMA gene (clade A in supplementary fig. S10, Supplementary Material online). Separately, the other member of this cluster of SDMA genes branches with the loosely linked SDMA in each species, suggesting that this particular SDMA gene arose from a duplication in the common ancestor of these species (clade B in supplementary fig. S10, Supplementary Material online).
Modeling of Novel Minimal SDMAs
In order to support the genomic identification of SDMA monomer genes, we utilized molecular modeling of each of these α and β MA proteins. The crystal structure of Bla g 1 of B. germanica was recently solved (Mueller et al. 2013). It is the first structure of an SDMA protein to have been determined and with its unique fold represents the model for this gene family. The structure and models were examined for the two defining major characteristics: proper interactions between the two pentagonal structures and the large central cavity lined with hydrophobic residues.
To demonstrate the importance of these characteristics, we first investigated common evolutionary and biophysical properties of the SDMA motif. Figure 4 A and B introduce ribbon diagrams of the Bla g 1 structure (PDB code 4JRB) rotated 90° with respect to each other, where α is colored orange and β is colored green. Panels C and D simulate the in silico opening up of the structure to better reveal the interior characteristics of the α and β subunits in panels E–H, which are in the same orientation as panel D. In panel E, each residue of the structure of Bla g 1 is colored according to the residue conservation among canonical SDMA proteins using the program CONSURF (Celniker et al. 2013). Judging from the figure, there are not many strongly conserved sites throughout the SDMAs. We colored the structure according to other biophysical properties using the same SDMA sequence alignment and using the same color scheme developed by CONSURF. Panels F, G, and H highlight on the Bla g 1 structure the average at each residue for all SDMAs of the Chou–Fasman secondary structure propensity, the Kyte–Doolittle hydrophobicity score, and the Grantham residue polarity score, respectively (Grantham 1974; Chou and Fasman 1978; Kyte and Doolittle 1982). Panel F shows that there is very little conservation of secondary structure propensity, while Panel G shows there is a strong tendency to have very hydrophobic residues on the interior surface (cyan color). Panel H demonstrates that the inverse is also true: there is a very low probability of finding strongly polar residues on the interior (dark magenta color). Based on this comparison, the absence of polar residues on the interior surfaces appeared to be a good characteristic to decide whether new models were similar to the Bla g 1 fold.
Fig. 4.—

Introduction to the Bla g 1 structure and SMDA residue propensities. (A–D) Ribbon diagram of Bla g 1 (4JRB) where α is colored orange and β is colored green. The structure in B is rotated 90° with respect to A. Panels C and D simulate in silico opening up of the two halves. D–H are oriented with the interior of the protein facing the viewer. (E–H) Bla g 1 rendered as spheres color coded by the degree of sequence conservation (E, high conservation magenta), secondary structure propensity (F, high helical propensity magenta), hydrophobicity (G, more hydrophobic residues are cyan), and residue polarity (H, more polar residues are cyan). The scores are the average at each position for a clustlw alignment of SDMAs. The color scale is shown above panel E. The color scale was adapted from CONSURF (Celniker et al. 2013).
Next, we modeled the sequence of an NSP from Pieris rapae, which was suspected to have the same fold as Bla g 1 (25% sequence identity). For comparison, Bla g 1 is shown in figure 5. Panel A shows the ribbon diagram, and panel F is colored for residue polarity. The P. rapae model structure is rendered similarly in panels B and G. Note that in both structures there are few extraneous loops between the helices, and there are no significantly polar residues on the interior. The interaction energy between α and β is very good for both structures (table 2). Given the unique fold of Bla g 1 in which there is no canonical protein hydrophobic core, the two halves of the protein are primarily held together by the interactions between the rims of the pentagon. We also attempted to model ASTM00693-PA of An. stephensi, which was a potentially distantly related SDMA (E value = 0.05 in the hmmseach analysis described in Materials and Methods). In this case, there are many extra loops between the helices (compare fig. 5A–C), the interaction energy between α and β is very poor (table 2), and there are several polar residues that would need to be accommodated on the interior (fig. 5H). Hence, we conclude it is unlikely that ASTM00693-PA is an SDMA homolog.
Fig. 5.—

Structure of Bla g 1 and molecular models. Panels A–E are rendered as ribbon diagrams rotated 45 degrees compared to figure 4A. Panels F–J are rendered as spheres color coded by degree of polarity using the same scale as in figure 4H (polar residues are cyan and nonpolar magenta) and aligned to the same orientation as Bla g 1 in figure 3D–H. Bla g 1 is shown in panels A and F, the P. rapae NSP in B and G, ASTM00693 in C and H, AAEL0104236 in D and I, and tetur06g03260 in E and J.
Table 2.
Interactions Energies (kCal/mol) between the Two Domains Calculated Using the Optimized Structures with the FF12SB Force Field of Amber.12
| Total Energy of Domain α and β | Energy of Domain α | Energy of Domain β | Interaction Energy between Domains α and β | |
|---|---|---|---|---|
| Blg1 | −3207.8 | −732.0 | −1744.8 | −731.0 |
| AAEL010436 | −3260.8 | −1300.0 | −1310.5 | −650.3 |
| ASTM00693 | −3290.6 | −1700.0 | −1505.1 | −85.5 |
| ASTM009013 | −2674.7 | −1145.6 | −1339.3 | −189.8 |
| PRNA | −3417.3 | −1128.7 | −1193.1 | −1095.5 |
| Tetur06g0326 | −5290.7 | −2457.7 | −2550.6 | −282.4 |
Note.—The energies given in the table contain only the nonbonded (electrostatic and van der Walls) terms because the interaction energy between the domains results only from those two components under the current pairwise additive energy scheme.
Finally, we attempted to model the α and β “monomers” separately by threading both monomer sequences onto both the α and β subunit of Bla g 1 and minimizing the structure. Figure 5D and I shows the model of AAEL010436-PA of A. aegypti. The interaction energy between the βMAs is on a par with the Bla g 1 structure and the P. rapae model (table 2), and the interior of the structure is lined primarily with nonpolar residues (fig. 5I). In contrast, the modeling results of the possible α dimer from An. stephensi did not produce a viable Bla g 1-like protein (table 2). In summary, the modeling results do suggest that AAEL010436-PA (the β monomer) could self-associate into a dimer that looks like an SDMA structure.
Expression of SDMA Genes
Two SDMA genes, one in A. aegypti (AEG12/AEEL010429-RA) and one in Ano. gambiae (ANG12/AGAP006187-RA) are known to be expressed in the midgut of these mosquitos and induced after a blood feed (Shao et al. 2005). Nothing is known concerning the expression of any other SDMA genes in any of these species except D. melanogaster (see Discussion). We chose to more closely examine expression of the SDMA genes in A. aegypti, in light of the abundance of SDMA genes, the novel β monomer, and availability of existing RNAseq data sets. Primarily, we wished to determine whether there was any evidence for the expression of this monomer. In the SRA at NCBI, there are two RNAseq data sets (SRP008153 and SRP003874) derived from three strains of A. aegypti females. In each strain, RNA isolated after either a blood feed or, as a control, a sugar feed, was sequenced (Bonizzoni et al. 2012). As seen in figure 6, expression of many SDMA genes was induced to varying levels specifically in the blood-fed mosquitos; this induction of expression was seen in all three strains used. Specifically, the β monomer AAEL010436-RA does show regulated expression in all three strains. An independent RNAseq experiment from seven different life stages of the A. aegypti reference strain (Liverpool; designated LVR in fig. 6) also showed developmentally regulated expression of this gene family, including AAEL010436-RA, and confirms a high level of expression in a post-blood feed stage (supplementary fig. S11, Supplementary Material online). Many of the differences in expression of SDMA genes between the sugar- and blood-fed samples are statistically significant after a multiple test correction. A common set of 15 of these genes are significantly upregulated in the blood feed in all three strains, including AAEL010436-RA, which is the β monomer (supplementary table S1e, Supplementary Material online). Sixteen SDMA genes each are significantly upregulated in the Liverpool and Rex strains, while 18 are upregulated in the Chetumal strain. Only one (AAEL013585-RA) is uniquely upregulated in the Chetumal strain. This may simply be an artifact of generally higher expression of these SDMA genes in Chetumal compared with the other strains as this gene shows an extremely low level of expression compared with the other SDMA genes in all three strains. In their original analysis of this RNAseq data set, Bonizzoni et al. (2012) confirmed by reverse transcriptase-polymerase chain reaction the expression of four of these SDMA genes post-blood feed (AAEL01327-RB, AAEL009166-RA, AAEL013118-RA, and AAEL001621-RA).
SDMA Homologs in Other Lineages
To test whether SDMAs existed in more distantly related metazoans, we began a broader examination of available genomes, beginning with arthropods most closely related to insects, specifically crustaceans and cheliceratans, for which genomes are publicly available. As a query we again used the PFAM hmm model initially. One potential limitation of this query strategy is that the available hmm representing SDMA available from PFAM is based solely on previously identified MAs of insects, and thus when used to search more distantly related genomes, it may lose its sensitivity. We examined the proteomes of the three available genomes most closely related to Insects, the crustacean Daphnia pulex two cheliceratan species (I. scapularis and T. urticae). No potential hits were found in either Daphnia pulex or I. scapularis, but in T. urticae we found with our hmmsearch query two potential homologs to the β monomer region of the SDMA (tetur06g03260; 148 aa, and tetur06g03280; 138 aa) with identical amino acid sequences aside from a 10 aa insertion in tetur06g03260 immediately following the first methionine. For each of these, the match defined by the E value (7.5 × 10−4 and 7.8 × 10−4, respectively) was barely above the default threshold suggested by the HMM 3.0 program. Neither contained a potential SP as defined by the SignalP 4.0 server. Neither was found with iterative sequence search tools, either jackhammer (HMM 3.0) or psi-Blast using the A. aegypti β monomer AAEL010436-PA as query. These T. urticae proteins do, however, contain a KxDL domain (PF10241.4). This is a protein that is well conserved from yeast to humans (Hayes et al. 2011) and a search for this motif within the genomes listed in table 1 also found a single clear ortholog within most of the genomes examined. A recent study of the mouse KxDL ortholog suggests that it may be involved in the biogenesis of lysosome-related organelles (Yang et al. 2012). Multiple sequence alignments of these T. urticae proteins with either known KxDL orthologs downloaded from Ensembl or with SDMA proteins clearly show much more conservation to other eukaryotic KxDL proteins than to SDMA proteins (data not shown).
Molecular modeling was done with terur06g03260 to further test whether it was potentially an SDMA. Figure 3E and J shows the modeling of KxDL motif, and table 2 shows the interaction energy between the two pentagons. The interaction energy is poor and despite our best possible alignment, the interior cavity does not show a consistently hydrophobic surface like the Bla g 1 model. Hence, we conclude that the KxDL motif is probably not an SDMA fold.
To further query noninsect genomes, we surveyed 13 additional genomes representing a diverse range of basal metazoans cataloged in supplementary table S1f, Supplementary Material online. No additional candidate SDMA homologs were found by any of the above methods. We created custom hmm models for both the α and β MAs based on the available canonical SDMA proteins as a novel search query; neither model revealed any additional SDMA homologs in any of the above-mentioned genomes. As an aside, the same hmm models failed to identify additional monomers in the insect genomes in table 1. The failure to identify SDMA genes by any of the approaches outside of Insecta reinforces the conclusions of previous research that this domain is specific to Insecta.
Discussion
We performed a comprehensive search of insect genomes for homologs to SDMA genes. We confirm that this domain is limited to the Insecta. More importantly, our results support the hypothesis of Pomés et al. (1998, 2007) that what has been defined as the SDMA is either a tandem duplication of, or gene fusion between, two domains of approximately 100 aa. Examples of independent monomers have been found in separate dipteran genomes and the molecular modeling of one shows strong similarity to the structure defined for the SDMA gene Bla g 1 of B. germanica. This suggests possible evolutionary models for the origin of the SDMA gene. We show that the genomic organization of the SDMA gene families within two orders of Insecta (Diptera and Hymenoptera) is very different, and evidence for the history of duplication and divergence of SDMA genes both within a specific species and within an order can be found.
Novel Major Allegen Monomers in Dipterans
We found within the genomes of two mosquitos possible examples of a single α and β monomer gene that together comprise the canonical SDMA gene structure. Specifically, an α monomer of 125 aa in An. stephensi and a β monomer of 95 aa in A. aegypti. This is based on sequence homology searching methods and is further supported by the molecular modeling in the case of the β monomer of A. aegypti. Searches of genomes outside Insecta found no evidence for the allergen motif aside from the crustacean T. urticae, where two genes with a low level of sequence homology to the SDMA model were found. However, based on their greater sequence similarity to the KxDL proteins and the inconclusive modeling result, we interpret these T. urticae proteins as representing KxDL orthologs and are not similar to MAs.
Origin and Evolution of the SDMA Gene
Our results suggest one of two hypotheses for the origin and evolution of this SDMA gene family. Either a tandem duplication of a single β-like progenitor (preferred over the α based on the modeling results) into an ancestral SDMA followed by divergence into two related α and β domains or an independent origin of an α and β progenitor followed by a gene fusion to form the basic α-β SDMA unit that is currently seen as the canonical SDMA gene in insects are possible (fig. 7). The rarity of the monomers in extant species may be due to a selective advantage for the two subdomain SDMA gene, as this structure offers advantages in terms of functionality. We speculate that two monomers that are fused as a functional dimer have a better chance of interacting when expressed on the same polypeptide chain as opposed to separately. If expressed alone, a monomer likely leaves a significant hydrophobic surface exposed, which could lead to aggregation or instability until the monomer finds a partner or appropriate ligands. To be clear, from the current data we cannot definitively distinguish between the two models in figure 7, and the existence of an expressed βMA only demonstrates the feasibility of our hypothesis. It does not imply that the exant βMA is the progenitor of the SDMA genes. However, we believe that given the history of duplication events, the duplication and divergence model is the most parsimonious.
Fig. 7.—
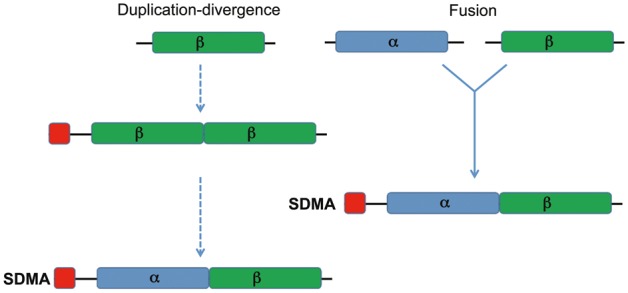
Models for the origin of the canonical SDMA gene. Two plausible scenarios are explored. The first is a tandem intragenic duplication of either a β-like monomer ancestor (most well supported by modeling and thus the most likely ancestral gene) followed by sequence divergence between the two while retaining their three-dimensional structural similarity. The second is an independent origin of an α and β progenitor comprising the basic unit of the SDMA gene, which arose by a fusion of these two genes.
SDMA Gene Families and Genomic Organization
Our analysis of the available genomic data shows that the SDMA gene likely occurs within most genera of Holometabolous insects and there is often divergence of SDMA genes within some species into extensive gene families. Tandem duplications of the basic α-β SDMA unit into multiple MA-containing genes occur frequently throughout the insects beyond the Lepidoptera, primarily in the Hymenoptera. The completeness of most of the genomes analyzed clearly shows that close physical linkage of SDMA genes within a species is nearly universal.
The two insect orders containing the most sequenced genomes, the dipterans and the hymenopterans, show two very different histories of SDMA gene family diversification. The dipterans show extensive variation between genera in the composition of the SDMA gene family, although within a specific genus (Drosophila) the composition of the SDMA gene family is fairly static, and clear orthologous relationships can be seen (supplementary text and supplementary figs. S14–S16, Supplementary Material online). Whether this is representative of other Dipteran genera requires more in-depth sequencing of other genera. Several conclusions can be drawn about relationships between SDMA genes in other species. There is conservation of several types of gene structures in terms of conservation of exon number and splice junctions. Within A. aegypti, which contains the most diverse SDMA gene family, a conserved three exon SDMA gene structure predominates with several subgroups of splice junctions. This helps reconstruct the duplication history within A. aegypti but also illustrates between-species conservation of some gene structures. A specific subgroup of 1st–2nd exon splice junction is also seen in Cu. quinquefasciatus, while a specific subgroup of 2nd–3rd exon splice junction is conserved between A. aegypti, Cu. quinquefasciatus, and An. gambiae. This, along with the phylogenetic tree of the SDMA genes in the genomes we have examined (supplementary fig. S13, Supplementary Material online), shows clear examples of orthologous genes in several dipteran species.
As to the physical origin of the AAEL010436 β monomer in A. aegypti, it is clearly most closely related to the Group 2 three-exon two-intron genes. It is located between AAEL010429 and AAEL010431 on supercontig 1.477 (fig. 3), these two being a clear case of a tandem gene duplication based on the extent of sequence identity within and around these two genes (supplementary fig. S3a, Supplementary Material online). AAEL010436 has two introns itself, and the 2nd–3rd exon junction of AAEL010436 is identical to that of the other SDMA genes in this group (supplementary fig. S2c, Supplementary Material online), although its 1st–2nd exon junction is novel. Examination of the DNA sequence alignment immediately 3′ of the stop codon for each of these three genes shows much more sequence identity between AAEL010431 and AAEL010436, suggesting that the latter is derived from this gene. While sequence identity extends for 172 bp downstream of the open reading frame between each of the three genes, identity extends a further 223 bp between AAEL010431 and AAEL010436 and overall there is 100% identity over 395 bp between these two genes (supplementary fig. S3b, Supplementary Material online). This identity and its location immediately downstream of AAEL010431 suggest that it arose as the result of a tandem duplication of AAEL010431. All of the SDMA genes discussed above are within a single clade highlighted in red in supplementary figure S13, Supplementary Material online, and thus this single clade accounts for almost all of the diversification of this gene family in both A. aegypti and Cu. quinquefasciatus.
In contrast, within the hymenopterans, the Formicidae show a very conserved, clearly orthologous, set of 3–5 SDMA/2DMA genes which suggests a duplication and divergence history outlined in supplementary figure S12, Supplementary Material online. Using the L. humile SDMA gene family as an example, the conserved chromosomal organization and phylogenetic relationships of the Formicidae SDMA genes is consistent with an initial duplication (1) of an ancestral SDMA gene (this could be either the LH23975 or LH23978 gene in the example) followed by a duplication of each of these; a duplication (2) of LH23978 to a distant but physically linked locus (LH0984), and a duplication (3) of the LH23975 to an adjacent location (LH23975′) followed by a tandem intragenetic duplication (4) into a 2DMA domain organization seen in LH23974. Here, we suggest that duplication (2) precedes (3) but this is simply one illustration, and the order and timing cannot be determined with the genomes available. While this is the most common arrangement, H. saltator shows evidence of further rearrangements, while C. floridanus and Atta cephalotes have lost a copy of one SDMA gene.
Function and Expression of SDMA Genes
Two previously characterized members of the SDMA gene family have known functions. Within the Pieirinea subfamily of Lepidoptera (butterflies), the NSP gene (a 3DMA gene) is involved in defense response. Pieirinea larvae feed on plants and NSP is involved in detoxification of the glucosinolates produced by the Brassicales on which they feed (Fischer et al. 2008). Bla g 1 of the German cockroach, a canonical SDMA gene, is a potent allergen in humans (Gruchalla et al. 2005). The protein in cockroaches is localized to the gut and is involved in lipid binding and transport through the gastrointestinal tract, eventually being excreted (Gore and Schal 2005; Mueller et al. 2013). modENCODE data for D. melanogaster suggest that two of its SDMA genes are expressed predominantly in the gut (FBpp0072030 in the midgut and FBpp00787785 in the hindgut), with others being expressed during different developmental stages (www.flybase.org). SDMA gene expression was found only in the insect gut, suggesting the original role for these genes in the process of digestion, as AEG12 of A. aegypti and ANG12 of An. gambiae were both shown to be induced after a blood feed (Shao et al. 2005). The finding of a unique SDMA fused to a fibronectin domain in An. darlingi raises interesting questions about the biochemical function of the protein, especially in light of the Bla g 1 function. Immunogold staining of AEG12 localized this SDMA to the microvilli in the gut in A. aegypti (Shao et al. 2005). This would be more consistent with AEG12 being tethered to a membrane-associated protein, which is common for fibronectin domains. We infer that the SDMA–fibronectin fusion is possibly a lipid receptor tethered to a membrane.
Our analysis of the available RNAseq data in A. aegypti suggests a role in feeding for some of the SDMA homologs as indicated by the high level of expression of many SDMA genes 24 h post-blood feed compared with the sugar feed control (Bonizzoni et al. 2012). This is seen in three different strains of A. aegypti with a core set of 15 SDMA genes showing statistically significant upregulation during the blood feed in each strain; this extends the conclusions of several previous studies suggesting a role in digestion for these genes in insects and confirms the finding of Shao et al. (2005) that AEG12/AAEL010429-RA is highly expressed after a blood feed. Importantly, the β monomer protein is expressed and under conditions similar to the related SDMA proteins.
3DMA genes, such as the MA and NSP genes previously identified in the Lepidoptora, are rare outside this order. Of the complete genomes, only Apis mellifera appears to contain one. The psi-Blast query of the NCBI nr data set (supplementary data, Supplementary Material online) shows two others outside the Lepidoptora (one each in Bombus impatiens and B. germanica) and two within the Pieridae family (one 3DMA each in Anthocharis cardamines and Pontia daplidice). None of the other lepidopterans examined here, including the two butterflies (Dan. plexippus and He. melpomene), contain a potential MA or NSP homolog, further evidence suggesting a very specific adaptation of these 3DMA genes to the Pieirinea butterfly lineage.
It is possible that the specific diversifications of the SDMA gene families observed in other orders may have evolved to provide some selective advantage. Hymenoptera, at least the Formicidae representatives, have a fairly static pattern of a set of 2–4 SDMA genes and one copy of a 2DMA gene physically linked. Does this simply reflect the close evolutionary relationship of these species or was this an ancestral adaptation necessary to the lifestyle of all ants? The Hemiptera and Phthiraptera genomes appear to have no SDMA genes. Whether this has functional significance within these orders or is an artifact of a small sample size and/or sequencing depth in the available genomes is unclear. These orders are outside the Holometabolous orders; this could represent a specific loss of SDMA genes within this lineage. Dipterans show a wide variation in their SDMA gene family number, although aside from the novel α and β monomers we found, all the available dipteran genomes contain only SDMA genes, with the exception of G. mortisans, which contains one 2DMA gene. Assuming that the variation in the diversification within this order implies adaptations to specific environments inhabited by these species, there should be functional implications. Assuming a function for these genes in the gut as with NSP and Bla g 1 in other species also, these differences in SDMA gene complements among the orders could be due to the different feeding strategies and/or environments in which these various insect species exist. Alternatively, the relative difference in the beginnings of the radiations of the dipteran and hymenopteran lineages could also contribute to the differences in the diversification of SDMA gene families between orders. The Formicidae radiation has been dated back to 140–186 Ma, while the dipteran lineage is dated to be much older, at least 260 Ma, giving the dipterans more evolutionary time to diverge from one another (Moreau et al. 2006; Wiegmann et al. 2011). One possible explanation for the extensive diversification of SDMA genes in A. aegypti is suggested by its genome size. The dipteran genomes examined here range from 113 Mb (An. darlingi) to 490 Mb (M. scalaris), while the A. aegypti genome size is 1.3 Gb. The initial analysis of the A. aegypti genome suggested that a significant expansion of transposons relative to related genera accounted for this (Nene et al. 2007). For the SDMA gene family, we see two clear examples of genomic duplications involving SDMA genes. While a whole-genome duplication may not have occurred, this suggests that the possibility of smaller duplication events could account for some of the expansion of this genome.
In summary, the genomic evidence establishes that the monomer and the dimer are evolutionarily related. The molecular modeling results suggest that a monomer can form a homodimer with a fold similar to the Bla g 1 structure. The RNAseq data further established that the monomer is unlikely to be a pseudogene or genome assembly artifact because it is expressed and regulated similarly to conventional SDMA genes. Taken together, the evidence suggests that the MA evolved from a homodimeric ancestor.
Supplementary Material
Supplementary text, figures S1–S16, and table S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Jim Mason and Lee Pedersen for a critical reading of the manuscript. This research was supported by Research Project Number Z01-ES102885-01 to R.E.L. and Z01-ES043010-28 to L.P. in the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health.
Literature Cited
- Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Arensburger P, et al. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science. 2010;330:86–88. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329:1068–1071. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, et al. Strain variation in the transcriptome of the dengue fever vector, Aedes aegypti. G3. 2012;2:103–114. doi: 10.1534/g3.111.001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, et al. San Francisco (CA): University of California; 2012. AMBER 12. [Google Scholar]
- Celniker G, et al. ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr J Chem. 2013;53:199–206. [Google Scholar]
- Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- Colbourne JK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Accelerated profile HMM searches. PLoS Comp Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter. 2006;5 doi: 10.1002/0471250953.bi0506s15. Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer HM, Wheat CW, Heckel DG, Vogel H. Evolutionary origins of a novel host plant detoxification gene in butterflies. Mol Biol Evol. 2008;25:809–820. doi: 10.1093/molbev/msn014. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore JC, Schal C. Gene expression and tissue distribution of the major human allergen Bla g 1 in the German cockroach, Blattella germanica L. 2004 doi: 10.1603/0022-2585-41.5.953. (Dictyoptera: Blattellidae). J Med Entomol. 41:953–960. [DOI] [PubMed] [Google Scholar]
- Gore JC, Schal C. Expression, production and excretion of Bla g 1, a major human allergen, in relation to food intake in the German cockroach, Blattella germanica. Med Vet Entomol. 2005;19:127–134. doi: 10.1111/j.0269-283X.2005.00550.x. [DOI] [PubMed] [Google Scholar]
- Grantham R. Amino-acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Grbic M, et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 2011;479:487–492. doi: 10.1038/nature10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruchalla RS, et al. Inner City Asthma Study: relationships among sensitivity, allergen exposure, and asthma morbidity. J Allergy Clin Immunol. 2005;115:478–485. doi: 10.1016/j.jaci.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Hayes MJ, Bryon K, Satkurunathan J, Levine TP. Yeast Homologues of three BLOC-1 subunits highlight KxDL proteins as conserved interactors of BLOC-1. Traffic. 2011;12:260–268. doi: 10.1111/j.1600-0854.2010.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heliconius Genome Consortium. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm R, et al. Isolation and characterization of a clone encoding a major allergen (Bla g Bd90K) involved in IgE-mediated cockroach hypersensitivity. J Allergy Clin Immunol. 1996;98:172–180. doi: 10.1016/s0091-6749(96)70240-3. [DOI] [PubMed] [Google Scholar]
- Holt RA, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010;8:e1000313. doi: 10.1371/journal.pbio.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkness EF, et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc Natl Acad Sci U A. 2010;107:12168–12173. doi: 10.1073/pnas.1003379107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Marinotti O, et al. The genome of Anopheles darlingi, the main neotropical malaria vector. Nucleic Acids Res. 2013;41:7387–7400. doi: 10.1093/nar/gkt484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau CS, Bell CD, Vila R, Archibald SB, Pierce NE. Phylogeny of the ants: diversification in the age of angiosperms. Science. 2006;312:101–104. doi: 10.1126/science.1124891. [DOI] [PubMed] [Google Scholar]
- Mueller GA, et al. Novel structure of cockroach allergen Bla g 1 has implications for allergenicity and exposure assessment. J Allergy Clin Immunol. 2013;132:1420–1426. doi: 10.1016/j.jaci.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nene V, et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316:1718–1723. doi: 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neron B, et al. Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan T, et al. Analysis of two novel midgut-specific promoters driving transgene expression in Anopheles stephensi mosquitoes. PLoS One. 2011;6:e16471. doi: 10.1371/journal.pone.0016471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Nygaard S, et al. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 2011;21:1339–1348. doi: 10.1101/gr.121392.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Pomés A, Wunschmann S, Hindley J, Vailes LD, Chapman MD. Cockroach allergens: function, structure and allergenicity. Protein Pept Lett. 2007;14:960–969. doi: 10.2174/092986607782541178. [DOI] [PubMed] [Google Scholar]
- Pomés A, et al. Novel allergen structures with tandem amino acid repeats derived from German and American cockroach. J Biol Chem. 1998;273:30801–30807. doi: 10.1074/jbc.273.46.30801. [DOI] [PubMed] [Google Scholar]
- Richards S, et al. The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452:949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Shao L, Devenport M, Fujioka H, Ghosh A, Jacobs-Lorena M. Identification and characterization of a novel peritrophic matrix protein, Ae-Aper50, and the microvillar membrane protein, AEG12, from the mosquito, Aedes aegypti. Insect Biochem Mol Biol. 2005;35:947–959. doi: 10.1016/j.ibmb.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Smith CD, Zimin A, et al. Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile) Proc Natl Acad Sci U S A. 2011;108:5673–5678. doi: 10.1073/pnas.1008617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, Smith CD, et al. Draft genome of the red harvester ant Pogonomyrmex barbatus. Proc Natl Acad Sci U S A. 2011;108:5667–5672. doi: 10.1073/pnas.1007901108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suazo A, Gore C, Schal C. RNA interference-mediated knock-down of Bla g 1 in the German cockroach, Blattella germanica L., implicates this allergen-encoding gene in digestion and nutrient absorption. Insect Mol Biol. 2009;18:727–736. doi: 10.1111/j.1365-2583.2009.00912.x. [DOI] [PubMed] [Google Scholar]
- Suen G, et al. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 2011;7:e1002007. doi: 10.1371/journal.pgen.1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327:343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmann BM, et al. Episodic radiations in the fly tree of life. Proc Natl Acad Sci U S A. 2011;108:5690–5695. doi: 10.1073/pnas.1012675108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurm Y, et al. The genome of the fire ant Solenopsis invicta. Proc Natl Acad Sci U S A. 2011;108:5679–5684. doi: 10.1073/pnas.1009690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori) Science. 2004;306:1937–1940. doi: 10.1126/science.1102210. [DOI] [PubMed] [Google Scholar]
- Yang Q, et al. The BLOS1-interacting protein KXD1 is involved in the biogenesis of lysosome-related organelles. Traffic. 2012;13:1160–1169. doi: 10.1111/j.1600-0854.2012.01375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nature Genet. 2013;45:220–225. doi: 10.1038/ng.2524. [DOI] [PubMed] [Google Scholar]
- Zhan S, Merlin C, Boore JL, Reppert SM. The monarch butterfly genome yields insights into long-distance migration. Cell. 2011;147:1171–1185. doi: 10.1016/j.cell.2011.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.