

Abstract
Species divergence is typically thought to occur in the absence of gene flow, but many empirical studies are discovering that gene flow may be more pervasive during species formation. Although many examples of divergence with gene flow have been identified, few clades have been investigated in a comparative manner, and fewer have been studied using genome-wide sequence data. We contrast species divergence genetic histories across eight triplets of North American Sceloporus lizards using a maximum likelihood implementation of the isolation–migration (IM) model. Gene flow at the time of species divergence is modeled indirectly as variation in species divergence time across the genome or explicitly using a migration rate parameter. Likelihood ratio tests (LRTs) are used to test the null model of no gene flow at speciation against these two alternative gene flow models. We also use the Akaike information criterion to rank the models. Hundreds of loci are needed for the LRTs to have statistical power, and we use genome sequencing of reduced representation libraries to obtain DNA sequence alignments at many loci (between 340 and 3,478; mean = 1,678) for each triplet. We find that current species distributions are a poor predictor of whether a species pair diverged with gene flow. Interrogating the genome using the triplet method expedites the comparative study of species divergence history and the estimation of genetic parameters associated with speciation.
Keywords: 3s, gene flow, phylogeography, population genomics, speciation
Introduction
Estimating the population genetic parameters associated with species divergence is critical for understanding speciation. The coalescent times of alleles across species contain useful information about species divergence times, current and ancestral population sizes, and gene exchange (Kingman 1982a, 1982b; Beerli and Felsenstein 1999; Nielsen and Wakeley 2001). Speciation is typically thought to occur in the absence of gene flow, because genetic exchange constrains population differentiation and prevents the formation of reproductive isolation (Mayr et al. 1963; Coyne et al. 2004). However, strong disruptive selection can overwhelm genetic exchange, particularly when combined with factors that contribute to linkage disequilibrium, including reduced heterozygote fitness, tight linkage, assortative mating, or chromosomal rearrangements (Felsenstein 1981; Servedio 2008; Pinho and Hey 2010). The growing number of empirical examples supporting divergence with gene flow suggests that this mode of speciation might be more common than expected (Pinho and Hey 2010).
Identifying common trends in speciation requires a comparison of species divergence history across many replicate species pairs. Most studies aimed at investigating divergence with gene flow use the isolation–migration (IM) model (Nielsen and Wakeley 2001; Hey and Nielsen 2004) in which an ancestral population gives rise to two descendent populations, during which time there may be gene exchange between the two populations. The IM model provides a convenient statistical framework for comparing speciation models (i.e., divergence with or without gene flow), and the population data used in this approach have the added benefits of providing fine-scale phylogeographic information for mapping genetic diversity across space and for pinpointing areas of putative or actual genetic exchange. However, a focus on dense geographic sampling of populations has the drawback of diverting resources away from contrasting speciation histories across many replicate species pairs. The approach is computationally demanding (Hey 2010), and scaling-up to genomic data sets containing hundreds or thousands of loci does not seem feasible. The ease of acquiring comparative genomic data for non-model organisms is increasing steadily (Lemmon and Lemmon 2012; Peterson et al. 2012; Smith et al. 2013), and methods capable of analyzing these large, complex data sets are needed. The triplet method of Yang (2010) only requires one sample for each of three species, including a species pair and an outgroup for rooting the tree. By removing the need for phylogeographic sampling, the triplet method can help expedite the study of comparative species divergence across replicate species pairs.
The North American lizard genus Sceloporus is a large (95+ species) and diverse clade that is suitable for a comparative study of species divergence histories. Many species pairs are strictly allopatric or peripheral isolates (Sites et al. 1992), but others seem to have diverged along environmental gradients or are only narrowly sympatric along habitat gradients (Rosenblum et al. 2007; Leaché et al. 2010), which is suggestive of divergence with gene flow. Investigating species divergence histories in Sceloporus with the goal of identifying any common trends is relevant for understanding the general mode of speciation in the group. Increases in diversification rates in Sceloporus are correlated with chromosomal changes (Leaché and Sites 2010), and several episodes of rapid radiation have produced well-supported clades (species groups) containing as many as 18 species. Sister species are often distinguished by chromosomal rearrangements, and models of chromosomal evolution in Sceloporus include some degree of gene flow during species formation (Hall 2010). Out of the large number of speciation events available to study in Sceloporus, the few studies conducted that test speciation models all support divergence with gene flow (Leaché and Mulcahy 2007; Leaché 2011; Leaché et al. 2013).
In this study, we test models of divergence with gene flow in eight triplets of Sceloporus (fig. 1). The likelihood ratio test (LRT) used in the triplet method requires many loci to achieve statistical power, because the historical signature of gene flow is recorded as variable gene tree divergence times, and differences in divergence times might be subtle if speciation was recent or if gene flow only occurred for a short time interval following speciation. We sequence reduced representation libraries to acquire hundreds and thousands of homologous loci shared across closely related species. A comparison of species divergence histories across these eight triplets suggests that current geographic distributions alone are not reliable indicators of the model of species divergence.
Fig. 1.—

Time-calibrated species tree for the species groups of Sceloporus lizards used in the study (Leaché and Sites 2010) and the geographic distributions of the eight species triplets.
Materials and Methods
Sampling
We sampled 22 species of Sceloporus for comparative population divergence analysis (fig. 1 and table 1). From these, we compiled eight triplets, each containing two closely related species that may have diverged with gene flow and a third species (the outgroup) that is assumed not to have exchanged migrants with the other species or their common ancestor. Two species, Sceloporus clarkii and S. hunsakeri, were each used in two triplets. A time-calibrated species tree estimated using BEAST (Drummond and Rambaut 2007) with four nuclear protein-coding genes and one fossil calibration (Leaché and Sites 2010) was used to estimate the relationships among the species groups containing triplets used in this study (fig. 1). Nuclear loci support a species tree for Sceloporus that is at odds with the mitochondrial DNA (mtDNA) gene tree (Leaché 2010) as well as with those that concatenate mtDNA and nuclear loci (Wiens et al. 2010, 2013). Introgression of mtDNA across species boundaries is the likely cause for some instances of discordance (Leaché 2010), and we therefore avoided gene trees from mtDNA and concatenated nuclear + mtDNA phylogenies for triplet selection whenever possible. Detailed phylogeographic studies support the species pair selections in the following groups: magister group (Leaché and Mulcahy 2007), grammicus group (Marshall et al. 2006), undulatus group (Leaché 2009, 2011), spinosus group (Grummer JA, Calderon M, Smith E, Nieto Montes de Oca A, Leaché AD, in preparation), formosus group (Smith 2001) group. However, population substructure, species paraphyly, and species that are sister to clades containing multiple species could all pose significant challenges in Sceloporus triplet selection that could impact the accuracy of the method (see Discussion section).
Table 1.
Comparative Genomic Data for 22 Sceloporus Lizards
| Species | Vouchera | Total Reads (Million)b | de novo Contigs | N50c | de novo Coverage | Contigs Post-filterd | Average Coverage | Blast to WGS |
|---|---|---|---|---|---|---|---|---|
| adleri | UWBM 6608 | 58.0 | 368,090 | 474 | 13× | 99,530 | 39× | 54,687 |
| bicanthalis | UWBM 7307 | 47.7 | 247,932 | 495 | 16× | 67,201 | 48× | 29,136 |
| clarkii | MVZ 245876 | 47.0 | 59,562 | 376 | 55× | 57,269 | 57× | 33,998 |
| cowlesi | AMNH 154059 | 48.8 | 278,468 | 949 | 10× | 91,726 | 22× | 39,858 |
| edwardtaylori | UWBM 6588 | 44.6 | 272,080 | 495 | 12× | 71,730 | 36× | 36,772 |
| formosus | UWBM 6623 | 63.6 | 590,161 | 495 | 9× | 113,302 | 33× | 56,277 |
| gadoviae | UWBM 7309 | 54.3 | 288,885 | 383 | 14× | 90,567 | 32× | 45,720 |
| grammicus | UWBM 6585 | 46.4 | 258,309 | 539 | 13× | 93,738 | 30× | 49,602 |
| horridus | UWBM 6632 | 35.3 | 131,289 | 567 | 20× | 55,706 | 43× | 27,101 |
| hunsakeri | SDSNH 76079 | 41.8 | 158,212 | 533 | 17× | 53,719 | 44× | 26,557 |
| jalapae | UWBM 7318 | 65.1 | 741,561 | 467 | 8× | 102,957 | 33× | 41,309 |
| licki | SDSNH 76080 | 31.6 | 133,173 | 550 | 17× | 54,206 | 36× | 26,497 |
| magister | UWBM 7395 | 31.9 | 103,055 | 650 | 19× | 48,023 | 35× | 24,597 |
| occidentalis | UWBM 6281 | 409.2 | 955,511 | 2,967 | 29× | 834,098 | 27× | — |
| ochoterenae | UWBM 6641 | 58.2 | 292,345 | 533 | 15× | 105,403 | 34× | 45,955 |
| orcutti | UWBM 7654 | 36.6 | 154,480 | 514 | 15× | 51,898 | 39× | 26,519 |
| palaciosi | UWBM 7313 | 61.0 | 163,616 | 605 | 22× | 69,449 | 46× | 34,775 |
| scalaris | UWBM 6589 | 53.6 | 465,770 | 454 | 10× | 102,001 | 30× | 50,642 |
| spinosus | UWBM 6672 | 57.7 | 546,964 | 475 | 9× | 92,601 | 37× | 47,358 |
| taeniocnemis | MVZ 264322 | 45.0 | 74,107 | 388 | 41× | 69,169 | 45× | 41,755 |
| tristichus | AMNH 153948 | 53.1 | 311,638 | 937 | 10× | 93,465 | 24× | 37,253 |
| zosteromus | SDSNH 76081 | 21.6 | 88,389 | 628 | 16× | 43,057 | 28× | 20,664 |
Note.—RRLs were sequenced for all species, with the exception of one WGS library for Sceloporus occidentalis.
aFull specimen information is available on the arctos database: http://arctos.database.museum/SpecimenSearch.cfm, last accessed November 28, 2013.
bTotal reads = unfiltered reads.
cN50 = median contig size.
dContigs post-filter = contigs >8× average coverage and >250 bp.
Divergence with gene flow might be expected in species that have parapatric geographic distributions, and most of the species included here have this type of distribution (fig. 1). Exceptions include two species pairs with allopatric distributions, including S. hunsakeri and S. orcutti and S. jalapae and S. ochoterenae. We include one species pair, S. cowlesi and S. tristichus, that have different chromosomal rearrangements and are from opposite sides of a hybrid zone that may have formed as a result of either primary divergence or secondary contact (Leaché and Cole 2007; Leaché 2011).
Reduced Representation Libraries
To obtain homologous DNA sequences between species, we reduced the complexity of the genome using a reduced representation library (RRL) approach to library preparation (Van Tassell et al. 2008; Kerstens et al. 2009). First, whole genomic DNA was digested to completion in enzymatic reactions using StuI (AGGCCT). In silico computer experiments using empirical data from the Anolis carolinensis lizard genome directed our molecular lab protocols for selecting the appropriate restriction enzyme and identifying the specific size distribution of fragments to sequence. The in silico experiments suggested that a complete genome digest using the restriction enzyme StuI should produce approximately 31,000 fragments in the 1.5–2 kb size class (representing 2.7% of the genome) and provide >20× sequencing coverage. Second, a small subset of the whole-genome digest ranging in size from 1.5 to 2 kb was captured using agarose gel electrophoresis or using a Blue Pippin Prep (Sage Science). Third, this isolate of genomic DNA was purified and then sheared with a Bioruptor to produce genomic DNA fragments with a mean size of 300 bp. Finally, libraries were prepared using standard TruSeq multiplexing protocols supplied by Illumina. The quality of completed libraries (insert size and quantity) was verified using an Agilent 2100 Bioanalyzer. We conducted 100 bp, paired-end sequencing on 3.5 Illumina HiSeq2000 lanes at the QB3 facility at UC Berkeley.
Whole-Genome Shotgun
We conducted whole-genome shotgun (WGS) sequencing on the Western fence lizard, Sceloporus occidentalis, to provide a genome-wide scaffold to aid the downstream comparisons of the RRL data sets. As an alternative to investing in a low coverage whole-genome assembly, the RRL data could be assembled into a provisional reference genome using available techniques (Hird et al. 2011). Genomic DNA for S. occidentalis was sheared with a Bioruptor to produce genomic DNA fragments with a mean size of 300 bp. The WGS library was prepared using standard TruSeq protocols. Library quality was verified using an Agilent 2100 Bioanalyzer, and we conducted 100 bp, paired-end sequencing on one Illumina HiSeq2000 lane at the QB3 facility at UC Berkeley.
De Novo Assembly
We used CLC Genomics Workbench v6 to quality filter and de novo assemble the RRL and WGS data sets. Raw data were imported into CLC using the Illumina import function, specifying paired-end reads with a minimum and maximum distance that matched the Bioanalyzer trace. Quality filtering followed the NCBI/Sanger or Illumina pipeline 1.8 and later function to trim low-quality reads and filter out failed reads. The remaining high-quality paired sequences were used for de novo assembly using scaffolding and autodetection of paired distances with default mapping options. CLC Genomics Workbench was used to visualize assembly quality and extract consensus sequences.
Bioinformatics
Following de novo assembly, the 21 RRL data sets were filtered, masked, and compared with the S. occidentalis WGS assembly. Individual 100 bp reads are phased, but the contigs that they form are not. Inability to phase large segments is a limitation of the short-read technology. Downstream population divergence analyses utilized unphased genotype data.
We retained consensus sequences with average coverage >8× and length >250 bp. As repetitive DNA is abundant in lizards (Janes et al. 2010; Alföldi et al. 2011), precautions were taken to exclude repetitive elements and potential chimeras from downstream analyses. Assembled contigs with excessive coverage discrepancies ≥3,000 were discarded. In addition, assemblies were scanned with RepeatMasker (http://www.repeatmasker.org/, last accessed November 28, 2013) against the Anolis genome to remove contigs identified as repeats or containing repetitive elements. Finally, we removed mtDNA using both RepeatMasker and Blast using the S. occidentalis mitochondrial genome as a reference library with default settings (Kumazawa and Nishida 1995).
We removed multiple copy loci by searching each RRL data set against itself using Blast+ (Camacho et al. 2009) and discarding sequences with multiple hits. Cross-species comparisons of loci utilized the S. occidentalis WGS as a reference genome. We used Blast+ to search S. occidentalis for hits to each single copy RRL locus. We generated homologous loci for triplets by merging three filtered and masked RRL data sets based on their mapping to S. occidentalis. Triplet loci containing ≥100 bp minimum overlap were subsequently aligned using MUSCLE v3.8.31 (Edgar 2004). Alignments were trimmed based on levels of missing data, allowing for internal gaps ≤20 bp. Alignments with ≤80% identical sites were also discarded. Finally, each locus was exported in PHYLIP format for downstream analyses.
Divergence with Gene Flow
We used the program 3s v2.1 (Yang 2010; Zhu and Yang 2012) to test models of divergence with gene flow for each triplet of Sceloporus. This program estimates gene-tree species-tree mismatch probabilities over time and compares three different population divergence models using LRTs (Zhu and Yang 2012). The three models include M0, speciation with no gene flow; M1, variable divergence times across the genome between sister species, which is interpreted as evidence for gene flow; and M2, the SIM3s model (Yang 2010; Zhu and Yang 2012), which includes an explicit migration parameter. All three models provide estimates of ancestral population sizes (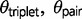 ) and divergence times (
) and divergence times (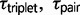 ). Additionally, model M1 estimates a q parameter, which allows the divergence time of the sister species to vary along a beta distribution (Yang 2010). The q parameter is inversely related to the variance in
). Additionally, model M1 estimates a q parameter, which allows the divergence time of the sister species to vary along a beta distribution (Yang 2010). The q parameter is inversely related to the variance in 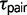 , and model M1 reduces to the null model of no migration (M0) when
, and model M1 reduces to the null model of no migration (M0) when 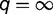 , which represents a constant
, which represents a constant 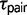 (Yang 2010). The M1 model is an approximation of divergence with gene flow, and because it is not a biological model the parameter estimates are unreliable (Yang 2010). The M2 model estimates the migration rate between sister species (
(Yang 2010). The M1 model is an approximation of divergence with gene flow, and because it is not a biological model the parameter estimates are unreliable (Yang 2010). The M2 model estimates the migration rate between sister species (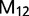 ), as well as
), as well as 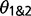 , the population size for species 1 and 2 (which is assumed to be equal for both species). The migration rate M12 is measured by the expected number of migrants from population 1 to population 2, with M21 defined similarly. The SIM3s model assumes M12 = M21 = M. The 3s program currently uses just one sequence per species at each locus, and it removes alignment gaps and ambiguous nucleotides from the alignment. Therefore, when using genotype data, this effectively reduces the information content of the data. The method also assumes that there is no recombination within a locus and free recombination between loci. Recombination can skew population genetic parameter estimates in the context of IM analysis (Strasburg and Rieseberg 2010), and ideally, we could accommodate recombination into the analytical framework (Becquet and Przeworski 2009). Under the SIM3s model, high recombination rates and large numbers of loci can lead to high false-positive rates for the LRTs (Zhu and Yang 2012). For each triplet, we ran ten replicates of 3s from random starting seeds to ensure convergence. Following recommendations in the 3s manual, we set the Gauss–Legendre quadrature to 32 points and the number of categories to discretize the beta distribution to 5. The Gauss–Legendre quadrature was increased up to 128 for some analyses to help convergence.
, the population size for species 1 and 2 (which is assumed to be equal for both species). The migration rate M12 is measured by the expected number of migrants from population 1 to population 2, with M21 defined similarly. The SIM3s model assumes M12 = M21 = M. The 3s program currently uses just one sequence per species at each locus, and it removes alignment gaps and ambiguous nucleotides from the alignment. Therefore, when using genotype data, this effectively reduces the information content of the data. The method also assumes that there is no recombination within a locus and free recombination between loci. Recombination can skew population genetic parameter estimates in the context of IM analysis (Strasburg and Rieseberg 2010), and ideally, we could accommodate recombination into the analytical framework (Becquet and Przeworski 2009). Under the SIM3s model, high recombination rates and large numbers of loci can lead to high false-positive rates for the LRTs (Zhu and Yang 2012). For each triplet, we ran ten replicates of 3s from random starting seeds to ensure convergence. Following recommendations in the 3s manual, we set the Gauss–Legendre quadrature to 32 points and the number of categories to discretize the beta distribution to 5. The Gauss–Legendre quadrature was increased up to 128 for some analyses to help convergence.
An LRT was used to compare the null model (M0) to alternative gene flow models M1 and M2. The test for the comparison between M0 and M1 uses the 5% critical value 2.71 (Yang 2010). The comparison between models M0 and M2 uses a χ2 distribution with two degrees of freedom, and the 5% critical value is 5.99 (Zhu and Yang 2012). Models M1 and M2 cannot be compared using an LRT, because they are not nested. Instead, we use the Akaike information criterion (AIC) to rank the M0, M1, and M2 models.
Results
Genomic Data and Alignments
Multiplexed RRLs of up to 12 samples were successfully sequenced on single Illumina lanes with high average coverage (table 1). Sequenced libraries (RRLs) contained 21.6–65.1 million bp of sequence data before filtering (mean = 47.8 million bp) and resulted in assemblies of 59,562–741,561 de novo contigs (mean = 265,874) with high average coverage (9×–55×, mean = 17×; table 1). Quality filtering of assembly contigs for size and average coverage resulted in 43,057–113,302 (mean = 76,390) contigs. Raw read count is generally correlated with the number of assembled contigs. Quality filtering for coverage less than 8× was necessary to account for sequencing error associated with NGS data and resulted in an average loss of 62% of the de novo assembled contigs. The S. occidentalis WGS resulted in 409.2 million bp of data. CLC quality control filtering and de novo assembly followed by filtering for average coverage and sequence length resulted in 834,098 contigs with an N50 (median contig size) of 2,967 bp (table 1). The percentage of assembled contigs that were removed from all assemblies after repeat masking ranged from 2.1% to 3.0% (mean = 2.6%).
For each of the eight triplets, cleaned and filtered RRL library contigs were compared with the S. occidentalis WGS library to determine homology. The number of homologous fragments (after alignment and trimming) for a triplet varied between 340 and 3,478 loci (mean = 1,678) and ranged in length from 98 to 1,588 bp (mean = 506 bp; table 2). The number of postfiltered contigs that Blast to the WGS for the three species in a triplet was not a predictor for the number of overlapping loci (i.e., BSC average contigs = 37,925 for 340 loci vs. HOL average contigs = 26,524 for 3,478 loci). Expected time to common ancestry for a triplet was not a predictor of loci number. HOL has a more recent divergence than BSC, which may explain the increased number of overlapping loci, but this trend disappears when other triplets are included. It is difficult to predict the resulting data set size based on sequencing effort using the RRL approach. Sequence variation between sister species varied from 0.7% (CTO) to 4.4% (JOG) and increased to as high as 9.5% (JOG) when including the outgroup species (table 2).
Table 2.
Alignments for Eight Triplets of Sceloporus Lizards
| Triplet (Name, Species) | Species Pair Distribution | Loci | Length | % Variable Sites |
||
|---|---|---|---|---|---|---|
| Sister Pair | Triplet | |||||
| AFT | adleri | Parapatric | 458 | 338 (104–592) | 2.5 (0–3.6) | 4.1 (1–5.4) |
| formosus | ||||||
| taeniocnemisa | ||||||
| BSC | bicanthalis | Parapatric | 340 | 336 (102–635) | 3.5 (0–3.8) | 7.3 (1.9–7.6) |
| scalaris | ||||||
| clarkiia | ||||||
| CTO | cowlesi | Parapatric | 3,015 | 745 (236–1,588) | 0.7 (0–0.9) | 2.4 (0.8–2.5) |
| tristichus | ||||||
| occidentalisa | ||||||
| GPC | grammicus | Parapatric | 914 | 349 (98–644) | 1.7 (0–2.0) | 4.7 (2.0–5.4) |
| palaciosi | ||||||
| clarkiia | ||||||
| HOL | hunsakeri | Allopatric | 3,478 | 639 (172–1,391) | 2.0 (0.6–2.1) | 3.1 (1.2–3.2) |
| orcutti | ||||||
| lickia | ||||||
| HSE | horridus | Parapatric | 3,044 | 602 (152–1,296) | 1.9 (0–2.2) | 3.8 (2.0–3.9) |
| spinosus | ||||||
| edwardtayloria | ||||||
| JOG | jalapae | Allopatric | 533 | 454 (124–1,043) | 4.4 (1.6–4.7) | 9.5 (4.8–9.8) |
| ochoterenae | ||||||
| gadoviaea | ||||||
| MZH | magister | Parapatric | 1,644 | 587 (138–1,316) | 2.8 (0.7–2.9) | 3.9 (1.4–4.1) |
| zosteromus | ||||||
| hunsakeria | ||||||
aThe outgroup for each triplet.
Divergence with Gene Flow
3s results for each triplet are summarized in table 3. Based on the LRTs, a model of no gene flow during divergence is supported in three of the triplets, including AFT, CTO, and HOL. For each of these triplets, the 2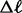 scores for the alternative gene flow models are 0.0. The five remaining triplets each support a model of gene flow during speciation with strong support exceeding the 5% critical value. The LRTs cannot distinguish between models M1 and M2, because they are not nested. The AIC results (table 4) provide ranks for the triplets that support the M1 and M2 models. The AIC results are consistent with the LRTs in their strong support for the migration models (AIC weights ≥ 0.05; table 4). Model M1 ranks higher than M2 for the triplets GPC, HSE, and JOG, but given that model M1 is an approximation of divergence with gene flow that does not explicitly estimate a migration rate parameter, we prefer to summarize parameter estimates from the M2 model.
scores for the alternative gene flow models are 0.0. The five remaining triplets each support a model of gene flow during speciation with strong support exceeding the 5% critical value. The LRTs cannot distinguish between models M1 and M2, because they are not nested. The AIC results (table 4) provide ranks for the triplets that support the M1 and M2 models. The AIC results are consistent with the LRTs in their strong support for the migration models (AIC weights ≥ 0.05; table 4). Model M1 ranks higher than M2 for the triplets GPC, HSE, and JOG, but given that model M1 is an approximation of divergence with gene flow that does not explicitly estimate a migration rate parameter, we prefer to summarize parameter estimates from the M2 model.
Table 3.
LRT Results of Species Divergence in Eight Triplets of Sceloporus
| Triplet | Loci | ℓ M0 | 2Δℓ M1a | 2Δℓ M2b |
|---|---|---|---|---|
| AFT | 458 | −25,960.3 | 0 | 0 |
| BSC | 340 | −37,126.1 | +34.1 | +39.2 |
| CTO | 3,015 | −290,344.7 | 0 | 0 |
| GPC | 914 | −71,165.1 | +49.9 | +43.0 |
| HOL | 3,478 | −335,756.5 | 0 | 0 |
| HSE | 3,044 | −368,037.6 | +90.9 | +67.1 |
| JOG | 533 | −100,221.5 | +74.0 | +57.5 |
| MZH | 1,644 | −201,949.2 | 0 | +7.0 |
Note.—Significant LRT results are in italic, and zero values indicate no difference in ℓ score.
a5% critical value = 2.71.
b5% critical value = 5.99.
Table 4.
AIC Comparison of Population Divergence Models
| Triplet | Model | -ℓ | Parameters | AIC | Rank | ΔAIC | Weight |
|---|---|---|---|---|---|---|---|
| BSC | M0 | 37,126.1 | 4 | 74,260 | 3 | 35.2 | 0 |
| M1 | 37,109.1 | 5 | 74,228 | 2 | 3.1 | 0.18 | |
| M2 | 37,106.5 | 6 | 74,225 | 1 | 0 | 0.82 | |
| GPC | M0 | 71,165.1 | 4 | 142,338 | 3 | 47.9 | 0 |
| M1 | 71,140.2 | 5 | 142,290 | 1 | 0 | 0.99 | |
| M2 | 71,143.6 | 6 | 142,299 | 2 | 8.9 | 0.01 | |
| HSE | M0 | 368,037.6 | 4 | 736,083 | 3 | 88.9 | 0 |
| M1 | 367,992.2 | 5 | 735,994 | 1 | 0 | 1.00 | |
| M2 | 368,004.1 | 6 | 736,020 | 2 | 25.8 | 0 | |
| JOG | M0 | 100,221.5 | 4 | 200,451 | 3 | 72 | 0 |
| M1 | 100,184.5 | 5 | 200,379 | 1 | 0 | 1.00 | |
| M2 | 100,192.8 | 6 | 200,397 | 2 | 18.5 | 0 | |
Maximum likelihood parameter estimates for the eight triplets are shown in figure 2. Speciation appears to be most recent in triplets AFT (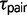 = 0.0003 ± 0.00089) and CTO (
= 0.0003 ± 0.00089) and CTO (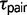 = 0.0003 ± 0.00004). These divergence times occurred in the Pleistocene around 300,000 years ago (±40,000 years) assuming a mutation rate in the order of 10−9 (Zhang and Hewitt 2003). However, without an accurate substitution rate for the RRL loci, it is not possible to obtain reliable parameter estimates on a demographic scale. Population size estimates
= 0.0003 ± 0.00004). These divergence times occurred in the Pleistocene around 300,000 years ago (±40,000 years) assuming a mutation rate in the order of 10−9 (Zhang and Hewitt 2003). However, without an accurate substitution rate for the RRL loci, it is not possible to obtain reliable parameter estimates on a demographic scale. Population size estimates  and
and 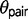 are generally unequal (fig. 2), and
are generally unequal (fig. 2), and  is typically larger. In one instance under the M0 model,
is typically larger. In one instance under the M0 model, 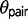 exceeds
exceeds  in the triplet AFT (
in the triplet AFT (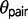 = 0.1313,
= 0.1313,  = 0.00101). Under the M2 model, the divergence time
= 0.00101). Under the M2 model, the divergence time 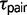 is exceptionally close to
is exceptionally close to 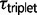 for triplets JOG, MZH, GPC, and BSC (fig. 2). Under the M0 model, the maximum likelihood estimates for
for triplets JOG, MZH, GPC, and BSC (fig. 2). Under the M0 model, the maximum likelihood estimates for 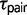 are more recent and indicate that speciation was not simultaneous in these triplets (fig. 2). This observed decrease in
are more recent and indicate that speciation was not simultaneous in these triplets (fig. 2). This observed decrease in 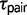 is accompanied by an increase in
is accompanied by an increase in 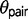 . Therefore, if gene flow exists between the species pair, then ignoring gene flow in M0 causes overestimation of
. Therefore, if gene flow exists between the species pair, then ignoring gene flow in M0 causes overestimation of 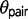 and underestimation of
and underestimation of 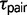 , because the model incorrectly attributes the excessive variation in divergence times among loci to a large ancestral population size
, because the model incorrectly attributes the excessive variation in divergence times among loci to a large ancestral population size 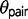 . The triplet parameters
. The triplet parameters  and
and 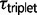 are stable across the M0 and M2 models (results not shown), although these estimates may be influenced by rate variation among loci.
are stable across the M0 and M2 models (results not shown), although these estimates may be influenced by rate variation among loci.
Fig. 2.—

Maximum likelihood estimates of population genetic parameters for eight triplets of Sceloporus. Divergence without gene flow (model M0) is supported in triplets HOL (B2), CTO (E), and AFT (G). The remaining triplets support divergence with gene flow and are shown with parameter estimates from model M2 (black branches and text) and M0 (gray branches and text). Estimates of θ and τ are scaled by 100.
Discussion
Testing Species Divergence
Empirical examples of divergence with gene flow span a wide array of organisms (Pinho and Hey 2010), including salamanders (Niemiller et al. 2008), lizards (Rosenblum et al. 2007), plants (Osborne et al. 2013), and butterflies (Stölting et al. 2013). Speciation with gene flow appears to be common among the great apes (Mailund et al. 2012; Prado-Martinez et al. 2013), including examples of admixture between modern humans and their recent Neandertal (Green et al. 2010) and Denisovan ancestors (Reich et al. 2011). The IM method (Nielsen and Wakeley 2001; Hey and Nielsen 2004) is the most commonly used approach for conducting statistical test of speciation models, because it offers a robust framework for model testing using the LRT (Hey and Nielsen 2007) or the AIC (Carstens et al. 2009). Explicit model testing is important for rigorous statistical phylogeography analysis (Knowles 2009; Carstens et al. 2013), and new methods that can handle large genomic data sets are becoming increasingly necessary to keep pace with the growing number of studies using next-generation sequencing data (Smith et al. 2013). The popular IM/IMa program has difficulty with large numbers of loci, and it is not quite able to scale-up to next-generation sequence data levels. By reducing the number of samples required for analysis, the triplet method (Yang 2010; Zhu and Yang 2012) provides a feasible approach for conducting comparative species divergence analysis using genomic data.
One of the limitations of the triplet method is that it cannot distinguish gene flow resulting from primary divergence versus secondary contact. The method quantifies variation in 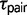 across loci, and it does not attempt to discern whether the variability in this parameter is reflective of gene flow during speciation or gene flow after divergence in allopatry (Yang 2010). This is important to consider when attempting to make inferences about the process of speciation supported by the LRT. New Bayesian phylogeography methods may be better suited for this purpose (Lemey et al. 2010), and complementing this approach with population genetic analyses can help distinguish allopatric divergence followed by secondary contact from primary intergradation (Pettengill and Moeller 2012).
across loci, and it does not attempt to discern whether the variability in this parameter is reflective of gene flow during speciation or gene flow after divergence in allopatry (Yang 2010). This is important to consider when attempting to make inferences about the process of speciation supported by the LRT. New Bayesian phylogeography methods may be better suited for this purpose (Lemey et al. 2010), and complementing this approach with population genetic analyses can help distinguish allopatric divergence followed by secondary contact from primary intergradation (Pettengill and Moeller 2012).
IM analyses typically emphasize robust population sampling and assume that there are no unsampled populations exchanging genes with the sampled populations or their ancestors. Ancestral population subdivision can increase the frequency of incorrect gene trees (Slatkin and Pollack 2008) and lead to increased estimates of 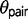 (Yang 2010). Some methods exploit this expectation of gene tree frequency differences to test for admixture between closely related populations (Durand et al. 2011). However, gene flow and ancestral population subdivision can produce similar coalescent times between two individuals from different populations, and distinguishing the two requires more than just one sample per species (Durand et al. 2011). The problems associated with population substructure could extend to triplets that include paraphyletic species or species pairs that include a focal species that is sister to a clade containing multiple species. The effect of population subdivision and species paraphyly on type I and type II error rates using the triplet method remains unstudied.
(Yang 2010). Some methods exploit this expectation of gene tree frequency differences to test for admixture between closely related populations (Durand et al. 2011). However, gene flow and ancestral population subdivision can produce similar coalescent times between two individuals from different populations, and distinguishing the two requires more than just one sample per species (Durand et al. 2011). The problems associated with population substructure could extend to triplets that include paraphyletic species or species pairs that include a focal species that is sister to a clade containing multiple species. The effect of population subdivision and species paraphyly on type I and type II error rates using the triplet method remains unstudied.
The use of two extra parameters into the M2 model, M12 and 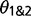 , have a major impact on the estimation of
, have a major impact on the estimation of 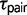 in some triplets. For example, we found that
in some triplets. For example, we found that 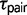 is nearly equal to
is nearly equal to 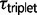 under the M2 model, but estimates for
under the M2 model, but estimates for 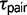 under the M0 model provide more recent estimates for speciation times. Estimates of
under the M0 model provide more recent estimates for speciation times. Estimates of 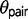 and of
and of 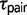 under models of gene flow implemented in 3s (M1 and M2) are unreliable due to the use of only three sequences at every locus, with only one sequence from each species. Zhu and Yang (2012) discussed the issue of nonidentifiability for
under models of gene flow implemented in 3s (M1 and M2) are unreliable due to the use of only three sequences at every locus, with only one sequence from each species. Zhu and Yang (2012) discussed the issue of nonidentifiability for 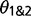 and M12, and even though
and M12, and even though 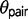 and
and 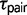 are identifiable, their estimates may be inaccurate due to a lack of information in the data. Extending the method to accommodate two or three sequences from the same species may increase the information content substantially, leading to more reliable parameter estimates. Despite the potential for poor parameter estimation, the method provides accurate LRT results (Yang 2010; Zhu and Yang 2012).
are identifiable, their estimates may be inaccurate due to a lack of information in the data. Extending the method to accommodate two or three sequences from the same species may increase the information content substantially, leading to more reliable parameter estimates. Despite the potential for poor parameter estimation, the method provides accurate LRT results (Yang 2010; Zhu and Yang 2012).
Comparative Species Divergence in Sceloporus
The new comparative genomic data sets collected for Sceloporus provide a robust statistical assessment of the model of species divergence history and the associated population genetic parameter estimates for the model. Three of the eight triplets of Sceloporus studied here support a history of speciation that does not include gene flow (table 3). Interestingly, one of these triplets (CTO) was found to support high rates of gene flow using multilocus DNA sequences (Leaché 2011). The sister pair in this triplet, S. cowlesi and S. tristichus, were sampled from opposite sides of a hybrid zone, and although the specific samples selected for this study have species-specific mtDNA, introgression has distributed S. cowlesi mtDNA haplotypes throughout the contact zone and into populations of S. tristichus (Leaché and Cole 2007). The recent divergence time for the species pair (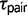 = 0.0003; fig. 2) suggests that the S. cowlesi sample used in this study may in fact be S. tristichus with introgressed mtDNA. Presumably, selecting different specimens from the hybrid zone that show some degree of admixture based on chromosomal polymorphisms or phenotypic traits would provide support for divergence with gene flow using the triplet method, even if the hybrid zone formed via secondary contact.
= 0.0003; fig. 2) suggests that the S. cowlesi sample used in this study may in fact be S. tristichus with introgressed mtDNA. Presumably, selecting different specimens from the hybrid zone that show some degree of admixture based on chromosomal polymorphisms or phenotypic traits would provide support for divergence with gene flow using the triplet method, even if the hybrid zone formed via secondary contact.
The two other triplets supporting speciation without gene flow include AFT and HOL, each contain a species pair with one widespread species and one species with a small and restricted distribution. In the formosus group, S. adleri is a high-elevation species that occurs in cool habitats above 2,183 m in the Sierra Madre del Sur (Smith and Savitzky 1974). The sister species S. formosus is more widely distributed at lower elevations, and we used a sample from an adjacent area on the same mountain range. The extrinsic environmental or intrinsic lineage-specific traits that contributed to the isolation of these species is unknown but occurred recently (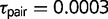 ; fig. 2). In the magister group, S. hunsakeri is restricted to the Cape Region of Baja California, Mexico, while the sister species S. orcutti is distributed throughout the Baja California Peninsula and into southern California. Divergence in the Baja California group is likely due to allopatric divergence resulting from the La Paz Embayment that isolated the Cape Region during the late Miocene/early Pliocene (Leaché and Mulcahy 2007), and this older divergence is supported by the estimate for
; fig. 2). In the magister group, S. hunsakeri is restricted to the Cape Region of Baja California, Mexico, while the sister species S. orcutti is distributed throughout the Baja California Peninsula and into southern California. Divergence in the Baja California group is likely due to allopatric divergence resulting from the La Paz Embayment that isolated the Cape Region during the late Miocene/early Pliocene (Leaché and Mulcahy 2007), and this older divergence is supported by the estimate for 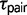 (0.0074; fig. 2).
(0.0074; fig. 2).
The five species pairs of Sceloporus that support divergence with gene flow have not been previously studied in the context of population divergence genetics. Many of these species are widespread generalists that occupy a wide diversity of environments and show extensive population substructure (Bryson et al. 2012). If the ancestral populations exhibited similar levels of substructure, then it is possible that the evidence for divergence with gene flow is an artifact of biases in 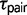 instead of gene flow. However, discovering triplets that support divergence with gene flow is not surprising given that chromosomal speciation is a dominant theme in Sceloporus diversification, and that models of chromosomal evolution involve stages of partial population subdivision that would facilitate continued gene flow (Sites et al. 1992; Hall 2010; Leaché and Sites 2010).
instead of gene flow. However, discovering triplets that support divergence with gene flow is not surprising given that chromosomal speciation is a dominant theme in Sceloporus diversification, and that models of chromosomal evolution involve stages of partial population subdivision that would facilitate continued gene flow (Sites et al. 1992; Hall 2010; Leaché and Sites 2010).
Compared with similar approaches for estimating population parameters, the triplet method requires not only a minimal number of samples but also a large number of loci for statistical power. Acquiring large numbers of loci for non-model organisms is no longer a challenge when utilizing emergent genomic techniques. An obvious trade-off associated with scanning triplets for evidence of divergence with gene flow is the loss of phylogeographic information within a species. However, developing the large numbers of nuclear loci necessary for the triplet test has the benefit of creating a wealth of new comparative genomics information for subsequent phylogeographic investigations.
Acknowledgments
The authors thank T. Gill and D. Reid for technical assistance with data collection and Z. Yang for his assistance with the 3s program. The authors also thank A. Gottscho and J. Lemos-Espinal for collecting specimens from Baja California, Mexico, and A. Nieto Montes de Oca, M. McElroy, L. Gray, and J. Grummer for help acquiring specimens from mainland Mexico. The manuscript benefitted from constructive comments offered by C. Ané, T. Mailund, Z. Yang, the SDSU Herpetology Discussion Group, and one anonymous reviewer. This work was supported by grants from the National Science Foundation (DEB-1144630), the University of Washington Royalty Research Fund (A61649) awarded to A.D.L., and National Science Foundation (BIO-1202754) awarded to C.W.L.
Literature Cited
- Alföldi J, et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477:587–591. doi: 10.1038/nature10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becquet C, Przeworski M. Learning about modes of speciation by computational approaches. Evolution. 2009;63:2547–2562. doi: 10.1111/j.1558-5646.2009.00662.x. [DOI] [PubMed] [Google Scholar]
- Beerli P, Felsenstein J. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics. 1999;152:763–773. doi: 10.1093/genetics/152.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson RW, Jr, García-Vázquez UO, Riddle BR. Relative roles of neogene vicariance and quaternary climate change on the historical diversification of bunchgrass lizards (Sceloporus scalaris group) in Mexico. Mol Phylogenet Evol. 2012;62:447–457. doi: 10.1016/j.ympev.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Camacho C, et al. Blast+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens BC, Stoute HN, Reid NM. An information-theoretical approach to phylogeography. Mol Ecol. 2009;18:4270–4282. doi: 10.1111/j.1365-294X.2009.04327.x. [DOI] [PubMed] [Google Scholar]
- Carstens BC, et al. Model selection as a tool for phylogeographic inference: an example from the willow Salix melanopsis. Mol Ecol. 2013;22:4014–4028. doi: 10.1111/mec.12347. [DOI] [PubMed] [Google Scholar]
- Coyne JA, et al. Speciation. Sunderland (MA): Sinauer Associates; 2004. [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239–2252. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Skepticism towards Santa Rosalia, or why are there so few kinds of animals? Evolution. 1981;35:124–138. doi: 10.1111/j.1558-5646.1981.tb04864.x. [DOI] [PubMed] [Google Scholar]
- Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall WP. Chromosome variation, genomics, speciation and evolution in Sceloporus lizards. Cytogenet Genome Res. 2010;127:143–165. doi: 10.1159/000304050. [DOI] [PubMed] [Google Scholar]
- Hey J. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol Biol Evol. 2010;27:921–933. doi: 10.1093/molbev/msp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci U S A. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird SM, Brumfield RT, Carstens BC. PRGmatic: an efficient pipeline for collating genome-enriched second-generation sequencing data using a ‘provisional-reference genome’. Mol Ecol Res. 2011;11:743–748. doi: 10.1111/j.1755-0998.2011.03005.x. [DOI] [PubMed] [Google Scholar]
- Janes DE, Organ CL, Fujita MK, Shedlock AM, Edwards SV. Genome evolution in Reptilia, the sister group of mammals. Annu Rev Genomics Hum Genet. 2010;11:239–264. doi: 10.1146/annurev-genom-082509-141646. [DOI] [PubMed] [Google Scholar]
- Kerstens H, et al. Large scale single nucleotide polymorphism discovery in unsequenced genomes using second generation high throughput sequencing technology: applied to turkey. BMC Genomics. 2009;10:479. doi: 10.1186/1471-2164-10-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman JF. The coalescent. Stoch Process Appl. 1982a;13:235–248. [Google Scholar]
- Kingman JF. On the genealogy of large populations. J Appl Probab Stat. 1982b:27–43. [Google Scholar]
- Knowles LL. Statistical phylogeography. Annu Rev Ecol Evol Syst. 2009;40:593–612. [Google Scholar]
- Kumazawa Y, Nishida M. Variations in mitochondrial tRNA gene organization of reptiles as phylogenetic markers. Mol Biol Evol. 1995;12:759–772. doi: 10.1093/oxfordjournals.molbev.a040254. [DOI] [PubMed] [Google Scholar]
- Leaché AD. Species tree discordance traces to phylogeographic clade boundaries in North American fence lizards (Sceloporus) Syst Biol. 2009;58:547–559. doi: 10.1093/sysbio/syp057. [DOI] [PubMed] [Google Scholar]
- Leaché AD. Species trees for spiny lizards (genus Sceloporus): identifying points of concordance and conflict between nuclear and mitochondrial data. Mol Phylogenet Evol. 2010;54:162–171. doi: 10.1016/j.ympev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Leaché AD. Multi-locus estimates of population structure and migration in a fence lizard hybrid zone. PLoS One. 2011;6:e25827. doi: 10.1371/journal.pone.0025827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaché AD, Cole CJ. Hybridization between multiple fence lizard lineages in an ecotone: locally discordant variation in mitochondrial DNA, chromosomes, and morphology. Mol Ecol. 2007;16:1035–1054. doi: 10.1111/j.1365-294X.2006.03194.x. [DOI] [PubMed] [Google Scholar]
- Leaché AD, Helmer D-S, Moritz C. Phenotypic evolution in high-elevation populations of western fence lizards (Sceloporus occidentalis) in the Sierra Nevada mountains. Biol J Linn Soc. 2010;100:630–641. [Google Scholar]
- Leaché AD, Mulcahy DG. Phylogeny, divergence times and species limits of spiny lizards (Sceloporus magister species group) in western North American deserts and Baja California. Mol Ecol. 2007;16:5216–5233. doi: 10.1111/j.1365-294X.2007.03556.x. [DOI] [PubMed] [Google Scholar]
- Leaché AD, Palacios JA, Minin VN, Bryson RW., Jr Phylogeography of the Trans-Volcanic bunchgrass lizard (Sceloporus bicanthalis) across the highlands of southeastern Mexico. Biol J Linn Soc. Forthcoming. 2013;110:852–865. [Google Scholar]
- Leaché A, Sites J., Jr Chromosome evolution and diversification in North American spiny lizards (genus Sceloporus) Cytogenet Genome Res. 2010;127:166–181. doi: 10.1159/000293285. [DOI] [PubMed] [Google Scholar]
- Lemey P, Rambaut A, Welch JJ, Suchard MA. Phylogeography takes a relaxed random walk in continuous space and time. Mol Biol Evol. 2010;27:1877–1885. doi: 10.1093/molbev/msq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon AR, Lemmon EM. High-throughput identification of informative nuclear loci for shallow-scale phylogenetics and phylogeography. Syst Biol. 2012;61:745–761. doi: 10.1093/sysbio/sys051. [DOI] [PubMed] [Google Scholar]
- Mailund T, et al. A new isolation with migration model along complete genomes infers very different divergence processes among closely related great ape species. PLoS Genet. 2012;8:e1003125. doi: 10.1371/journal.pgen.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, Arévalo E, Benavides E, Sites JL, Sites JW. Delimiting species: comparing methods for Mendelian characters using lizards of the Sceloporus grammicus (squamata: Phrynosomatidae) complex. Evolution. 2006;60:1050–1065. [PubMed] [Google Scholar]
- Mayr E, et al. Animal species and evolution. Cambridge (MA): Harvard University Press; 1963. [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemiller ML, Fitzpatrick BM, Miller BT. Recent divergence with gene flow in Tennessee cave salamanders (plethodontidae: Gyrinophilus) inferred from gene genealogies. Mol Ecol. 2008;17:2258–2275. doi: 10.1111/j.1365-294X.2008.03750.x. [DOI] [PubMed] [Google Scholar]
- Osborne OG, Batstone TE, Hiscock SJ, Filatov DA. Rapid speciation with gene flow following the formation of Mt. Etna. Genome Biol Evol. 2013;5:1704–1715. doi: 10.1093/gbe/evt127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettengill JB, Moeller DA. Phylogeography of speciation: allopatric divergence and secondary contact between outcrossing and selfing Clarkia. Mol Ecol. 2012;21:4578–4592. doi: 10.1111/j.1365-294X.2012.05715.x. [DOI] [PubMed] [Google Scholar]
- Pinho C, Hey J. Divergence with gene flow: models and data. Annu Rev Ecol Evol Syst. 2010;41:215–230. [Google Scholar]
- Prado-Martinez J, et al. Great ape genetic diversity and population history. Nature. 2013;499:471–475. doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 2011;89:516–528. doi: 10.1016/j.ajhg.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum EB, Hickerson MJ, Moritz C. A multilocus perspective on colonization accompanied by selection and gene flow. Evolution. 2007;61:2971–2985. doi: 10.1111/j.1558-5646.2007.00251.x. [DOI] [PubMed] [Google Scholar]
- Servedio M. The role of linkage disequilibrium in the evolution of premating isolation. Heredity. 2008;102:51–56. doi: 10.1038/hdy.2008.98. [DOI] [PubMed] [Google Scholar]
- Sites J, Jr, Archie J, Cole C, Flores Villela O. A review of phylogenetic hypotheses for lizards of the genus Sceloporus (Phrynosomatidae): implications for ecological and evolutionary studies. Bull Am Mus Nat Hist. 1992;213:1–110. [Google Scholar]
- Slatkin M, Pollack JL. Subdivision in an ancestral species creates asymmetry in gene trees. Mol Biol Evol. 2008;25:2241–2246. doi: 10.1093/molbev/msn172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BT, Harvey MG, Faircloth BC, Glenn TC, Brumfield RT. Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Syst Biol. Forthcoming 2013 doi: 10.1093/sysbio/syt061. Advance Access published September 10, 2013, doi: 10.1093/sysbio/syt061. [DOI] [PubMed] [Google Scholar]
- Smith EN. Species boundaries and evolutionary patterns of speciation among the malachite lizards (Formosus group) of the genus Sceloporus (Squamata: Phrynosomatidae). [PhD thesis] [Arlington (TX)]: University of Texas at Arlington; 2001. [Google Scholar]
- Smith HM, Savitzky AH. Another cryptic associate of the lizard Sceloporus formosus in Guerrero, Mexico. J Herpetol. 1974;8:297–303. [Google Scholar]
- Stölting KN, et al. Genomic scan for single nucleotide polymorphisms reveals patterns of divergence and gene flow between ecologically divergent species. Mol Ecol. 2013;22:842–855. doi: 10.1111/mec.12011. [DOI] [PubMed] [Google Scholar]
- Strasburg JL, Rieseberg LH. How robust are “isolation with migration” analyses to violations of the IM model? A simulation study. Mol Biol Evol. 2010;27:297–310. doi: 10.1093/molbev/msp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tassell CP, et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008;5:247–252. doi: 10.1038/nmeth.1185. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Kozak KH, Silva N. Diversity and niche evolution along aridity gradients in North American lizards (Phrynosomatidae) Evolution. 2013;67:1715–1728. doi: 10.1111/evo.12053. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Kuczynski CA, Arif S, Reeder TW. Phylogenetic relationships of phrynosomatid lizards based on nuclear and mitochondrial data, and a revised phylogeny for Sceloporus. Mol Phylogenet Evol. 2010;54:150–161. doi: 10.1016/j.ympev.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Yang Z. A likelihood ratio test of speciation with gene flow using genomic sequence data. Genome Biol Evol. 2010;2:200. doi: 10.1093/gbe/evq011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D-X, Hewitt GM. Nuclear DNA analyses in genetic studies of populations: practice, problems and prospects. Mol Ecol. 2003;12:563–584. doi: 10.1046/j.1365-294x.2003.01773.x. [DOI] [PubMed] [Google Scholar]
- Zhu T, Yang Z. Maximum likelihood implementation of an isolation-with-migration model with three species for testing speciation with gene flow. Mol Biol Evol. 2012;29:3131–3142. doi: 10.1093/molbev/mss118. [DOI] [PubMed] [Google Scholar]