

Abstract
Almost three decades ago Alec Jeffreys published his seminal Nature papers on the use of minisatellite probes for DNA fingerprinting of humans (Jeffreys and colleagues Nature 1985, 314:67–73 and Nature 1985, 316:76–79). The new technology was soon adopted for many other organisms including plants, and when Hilde Nybom, Kurt Weising and Alec Jeffreys first met at the very First International Conference on DNA Fingerprinting in Berne, Switzerland, in 1990, everybody was enthusiastic about the novel method that allowed us for the first time to discriminate between humans, animals, plants and fungi on the individual level using DNA markers. A newsletter coined “Fingerprint News” was launched, T-shirts were sold, and the proceedings of the Berne conference filled a first book on “DNA fingerprinting: approaches and applications”. Four more conferences were about to follow, one on each continent, and Alec Jeffreys of course was invited to all of them. Since these early days, methodologies have undergone a rapid evolution and diversification. A multitude of techniques have been developed, optimized, and eventually abandoned when novel and more efficient and/or more reliable methods appeared. Despite some overlap between the lifetimes of the different technologies, three phases can be defined that coincide with major technological advances. Whereas the first phase of DNA fingerprinting (“the past”) was dominated by restriction fragment analysis in conjunction with Southern blot hybridization, the advent of the PCR in the late 1980s gave way to the development of PCR-based single- or multi-locus profiling techniques in the second phase. Given that many routine applications of plant DNA fingerprinting still rely on PCR-based markers, we here refer to these methods as “DNA fingerprinting in the present”, and include numerous examples in the present review. The beginning of the third phase actually dates back to 2005, when several novel, highly parallel DNA sequencing strategies were developed that increased the throughput over current Sanger sequencing technology 1000-fold and more. High-speed DNA sequencing was soon also exploited for DNA fingerprinting in plants, either in terms of facilitated marker development, or directly in the sense of “genotyping-by-sequencing”. Whereas these novel approaches are applied at an ever increasing rate also in non-model species, they are still far from routine, and we therefore treat them here as “DNA fingerprinting in the future”.
Keywords: DNA fingerprinting, Genetic mapping, Genotyping-by-sequencing, Microsatellites, Plants, Population genetics, Single nucleotide polymorphisms, Systematics
Fingerprinting plants in the past
Telling plants apart in the olden days…
Many disciplines in botany are dependent on the ability to differentiate among plant genotypes, and/or to estimate the amount of diversity and relatedness in a set of genotypes. Traditionally, such tasks have been conducted mainly through data on morphological characteristics but these have certain limitations, including insufficient variation among the studied genotypes, subjectivity in the data collection and treatment, and plasticity due to environmentally induced variation. A more neutral and objective tool was offered by molecular markers based on isoenzymes; that is, enzymes that catalyze the same chemical reaction but differ in amino acid sequence and therefore also in the speed taken to travel through an electrophoretic gel. Isoenzymes were introduced into plant science in the early 1960s, and quickly increased in importance throughout the 1970s and 1980s. Co-dominant allozyme data (that is, allelic enzymes coded by genes at the same locus) soon became very popular for studies of, for example, population structure, gene flow, isolation-by-distance (IBD), mating systems and hybridization [1]. Protein extraction was, however, often a problem, especially for plants with high contents of polyphenols in their leaves. The analysis of plants growing in remote areas was also a problem, since proteins generally need to be isolated and purified within a short time from sampling. A third major problem was the often insufficient level of allozyme polymorphism among related genotypes.
Compared with proteins, the DNA molecule is very robust and easy to work with, and the potential for yielding polymorphic data is virtually inexhaustible. In the 1970s, the advent of the DNA-based restriction fragment length polymorphism (RFLP) technique enabled botanists to analyze samples collected from plants growing almost anywhere. Samples, usually leaves, were usually dried on silica gel before being transported to a laboratory, where they could be kept frozen until DNA isolation. The RFLP method was, however, rather time-consuming, with isolation of genomic DNA from the collected material, cutting the DNA samples with restriction enzymes, transferring the fragments with Southern blotting to a filter, hybridizing the filter-bound fragments with locus-specific probes, and finally utilizing for example autoradiography to detect the fragments. Still, the major constraint was the need for developing species-specific hybridization probes for these analyses. RFLP methodology was therefore applied mainly to economically important crop plants, with many active scientists and large grants. In these crops, RFLP markers constituted a highly appreciated tool for the development of genetic maps [2], and sometimes for cultivar identification and studies of genetic relatedness [3]. Nevertheless, a restricted availability of suitable loci often resulted in insufficient polymorphism also with the RFLP method.
In the 1980s, the RFLP methodology was also first applied to the chloroplast DNA (cpDNA) molecule. For this, DNA samples were digested with either single or combined restriction enzymes, and hybridized with radiolabelled cpDNA-specific probes from one of the universal libraries developed from, for example, Petunia. The obtained information was used to construct restriction site maps of the cpDNA molecule. Since the cpDNA molecule is highly conserved, there is very little intra-specific variation, and cpDNA-based RFLP studies have therefore mostly been conducted on an inter-specific level. By contrast, plant mitochondria have never been much used in molecular analyses. The major reason is that while the plant mitochondrial DNA (mtDNA) sequence is usually highly conserved, the size and structure of mtDNA molecules may vary widely even within individual plants [4]. Moreover, recent studies indicated that substitution rates of mtDNA genes can vary enormously even among closely related plant species [5].
Minisatellite and oligonucleotide DNA probes enable true plant fingerprinting
When Jeffreys and colleagues [6,7] published their groundbreaking papers on RFLP analyses with probes developed from tandemly repeated DNA sequences in human DNA, nobody expected that this new, so-called DNA fingerprinting technique, would revolutionize also the botanical science. However, since these new minisatellite probes showed a high potential for revealing individual-specific DNA fingerprints also in other mammals [8] as well as in birds [9], botanists soon decided to investigate the possibilities of applying this tool also in plants. In a paper appearing in 1988, Dallas [10] was able to distinguish among different rice cultivars, Oryza sativa, by hybridizing restriction-digested rice DNA with the human 33.6 minisatellite probe. The studied offspring from an individual rice plant proved to have identical fingerprints, which is the expected result since rice is self-pollinating and thus highly homozygous. In addition, Dallas was able to ascertain the Mendelian inheritance of DNA fragments from grandparents to the second-generation offspring (F2).
In the same year, two more papers reported on fingerprinting plant material with another minisatellite probe, this time derived from the genome of the bacteriophage M13. Whereas Ryskov and colleagues [11] obtained different DNA fingerprint patterns of two barley varieties, Hordeum vulgare, after hybridization with the M13 probe, Rogstad and colleagues [12] generated identical M13 fingerprints from separate branches of a cottonwood tree, Populus deltoides, as well as from a mother tree and its sucker plant, demonstrating somatic stability. These authors also showed that fingerprints obtained from the offspring from an inbred tomato, Solanum lycopersicon, were identical, while a high level of variation was encountered among sexually derived cottonwood trees, indicating that the degree of variation depends on the mode of reproduction. In the following years, the ability of minisatellite probes to distinguish between specimens derived by sexual recombination and specimens derived by vegetative propagation or apomixis (that is, seed set without prior fertilization) was demonstrated in numerous plants, including North American quaking aspen, Populus tremuloides[13], and various raspberry and blackberry species, Rubus spp. [14,15].
In cultivated plants, propagation is undertaken either through seeds (especially in annual and biennial crops) or vegetatively (in fruit crops, as well as in many woody ornamentals). In the latter, each cultivar is expected to consist of a single monomorphic genotype. DNA fingerprinting thus became a very efficient means of investigating identity as demonstrated in some Rubus cultivars [16]. In such crops, new and unique characters sometimes appear in, for example, a single branch of a tree through the occurrence of minor somatic mutations. Propagation of material collected from these deviating plant parts gives rise to new cultivars (known as, for example, sports in apple). Analysis of sports marketed under different names but all derived from the well-known 'Red Delicious’ apple, however, produced completely identical DNA fingerprints [17]. Obviously none of the minor DNA differences between these sports had been targeted by the M13 probe.
Still another set of RFLP hybridization probes was introduced in the early days of DNA fingerprinting, namely synthetic oligonucleotides such as (GACA)4 and (GATA)4[18]. These probes hybridized to short, tandem-repeated sequences (microsatellites; simple sequence repeats (SSRs)) in the genome, and produced polymorphic fragment patterns in, for example, cultivars of chickpea, Cicer arietinum[19], banana [20], tomato [21,22] and rice [23], in double-haploid lines of sugar beet (Beta vulgaris) [24], and in wild plants of the Chilean annual Microseris pygmaea[25]. As an experimental bonus, the oligonucleotide probes allowed hybridization directly within the dried gels, thus circumventing Southern blotting altogether. A typical banding pattern resulting from this ancient, so-called “oligonucleotide fingerprinting” methodology is shown in Figure 1.
Figure 1.
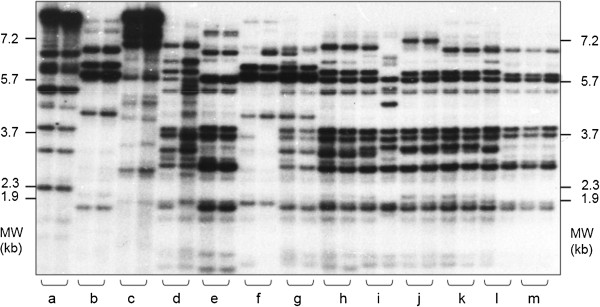
Hybridization-based restriction fragment length polymorphism fingerprints of tomato plants (Solanum lycopersicum). Five-microgram aliquots of genomic DNA from two individual plants each of three wild species (a to c) and 10 cultivars (d to m) were digested with the restriction enzyme HinfI, separated on a 1.4% agarose gel, and in-gel hybridized with the radioabelled oligonucleotide probe (GGAT)4 (Kaemmer and colleagues [22]). Banding patterns were visualized by autoradiography. Positions of size markers are indicated (kb = kilobase pairs). MW, molecular weight.
Technical issues of hybridization-based plant DNA fingerprinting
The successful application of minisatellite and oligonucleotide probes for DNA fingerprinting by Southern blot hybridization is dependent on the availability of relatively large quantities of very clean DNA in order for the restriction enzymes to produce clear fragment profiles. DNA isolation thus became a crucial step, and many different protocols were developed [26,27]. Other methodological advances such as the use of non-radioactive fragment detection - for example, digoxigenin-based labelling - were also described [28,29]. Since oligonucleotide probes sometimes yield a high background and can be sensitive to minute changes in temperature during hybridization, a PCR-based method for producing longer and more robust probes (typically 300 to 600 bp) but still with short repeated motifs such as, for example, (GACA)n was developed by Rogstad [30]. Hybridization of the same filter (stripped and re-hybridized consecutively) with nine of these so-called PCR-STR (synthetic tandem-repeat) probes produced polymorphic DNA fingerprints in turnip (Brassica rapa) plants, and allowed the verification of Mendelian fragment transmittal to the offspring [31].
Data evaluation remained relatively 'primitive’ in many of these early studies. Usually, the number of analyzed samples was well below 50, and numbers of polymorphic bands seldom reached more than 20 to 40 in each study. Moreover, experimentally induced differences in hybridization efficiency between the electrophoretic gels and filters often precluded the pooling of data from samples analyzed on different gels [32]. Therefore, manual comparisons of fragment profiles were usually performed for evaluating relationships among the studied samples. This information was then used to make deductions about, for example, the transfer of pollen among different cultivars in an apple orchard [33], the hybrid origin of a blackberry microspecies [34], and the mode of seed setting in experimentally produced blackberry hybrids [35]. For quantitative comparisons, the proportion of shared bands was usually calculated with Dice’s coefficient, also known as Nei and Li’s coefficient [36]. The results were compared with previous information on propagation and distribution of the investigated material. For example, Tzuri and colleagues [37] and Vainstein and colleagues [38] estimated variability among and within different groups of carnations, and obtained patterns that could be associated with mode of propagation (by seed or vegetatively) as well as the known origination (from Dianthus caryophyllus or from Dianthus hybrids). Analyses of genetic relatedness based on banding pattern similarity have been carried out also among, for example, Rubus cultivars [29,39] and among and within populations of wild plant species such as box elder, Acer negundo[40], and paw-paw, Asimina triloba[41].
Methods for assessing genetic diversity were soon improved, and reports on the use of DNA fingerprinting for estimating population genetics parameters, such as expected heterozygosity, Wright´s F-statistics and the number of migrants per population and generation, became increasingly common during the 1990s. Whenever tested, results obtained with these dominant multi-locus markers were usually consistent with those from previous studies using co-dominant allozyme markers. Using a resampling program, M13 fingerprinting-derived estimates for genetic identity within and between populations as well as population subdivision proved to be closely associated with the breeding system in three species of Plantago[42]. The selfing species P. major showed relatively little within-population variation compared to the mixed breeding P. coronopus and the outbreeding P. lanceolata. Interpopulation differentiation was, by contrast, more pronounced in the selfing species compared with the other two. Population genetics parameters from RFLP-based fingerprinting data were reported also in, for example, Gambel oak, Querus gambelii[32], common cattail, Typha latifolia[43], and two species of buckeye, Aesculus[44].
For more information on the methodology, applications and results obtained by hybridization-based fingerprinting with mini- and microsatellite complementary probes, see the reviews by Nybom [45,46], Weising and colleagues [26], Rogstad [47] and Weising and Kahl [48].
Present-day fingerprinting of plants
Method development and choice of markers
PCR-based multi-locus methods
Shortly after the invention of the ingenious PCR procedure by Saiki and colleagues [49], three PCR-based approaches to generate DNA fingerprints were published more or less at the same time. All of these methods used single oligonucleotide primers with arbitrary sequences to produce PCR fragments from genomic DNA, resulting in multi-locus banding patterns after electrophoretic separation and visualization by staining or radiography [50-52]. The so-called random amplified polymorphic DNA (RAPD) approach developed by Williams and colleagues [51] soon became the most popular variant of these methods. Major explanations for this immediate success include the small quantities needed of sample DNA, and the simple and fast procedures compared to the hybridization-based methods. Results from a typical RAPD experiment are illustrated in Figure 2.
Figure 2.
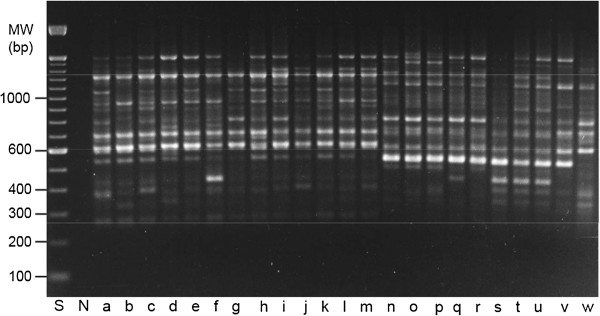
Random amplified polymorphic DNA (RAPD) fingerprints of Pelargonium. DNA aliquots from 13 cultivars of Pelargonium hortorum (lanes a to m), nine cultivars of P. peltatum (lanes n to v), and one individual of the wild subspecies P. peltatum ssp. dibrachya (lane w) were amplified with the arbitrary 10-mer primer OPG-5. RAPD products were separated on a 1.5% agarose gel and stained with ethidium bromide. Positions of size markers (lane S) are indicated in base bairs (bp). N, negative control (no template DNA in the PCR assay). MW, molecular weight.
A few years later, Zabeau and Vos [53] and Vos and colleagues [54] presented the amplified fragment length polymorphism (AFLP) technique, which represented an ingenious combination of RFLP and PCR methodology. AFLP analyses became soon very popular, mainly because of the large numbers of polymorphic bands obtained in a single experiment. The inter-simple sequence repeat (ISSR) method developed by Gupta and colleagues [55] and Zietkiewicz and colleagues [56] relied on microsatellite-complementary PCR primers that could be used in an anchored or unanchored version. RAPD, AFLP and ISSR are still much used nowadays, although RAPD especially has often been criticized for problems with reproducibility and competitive priming, as reviewed in Weising and colleagues [26]. These problems are less pronounced for AFLP and ISSR where more stringent PCR conditions can be applied. Nevertheless, all three methods usually arrived at quite similar estimates of genetic diversity and genetic distances when applied to the same plant material, as reviewed in Weising and colleagues [27].
Other less frequently used methods to generate multi-locus PCR fingerprints include the sequence-related amplified polymorphism (SRAP) technique that specifically amplifies polymorphic junction fragments between exons and the flanking intronic DNA [57,58], and the target region amplification polymorphism (TRAP) method [59]. Common features of SRAP and TRAP include the use of two primers of about 18 nucleotides length (one of which targets a protein-coding region), and non-stringent PCR conditions during the first five cycles. The so-called selective amplification of polymorphic microsatellite loci (SAMPL) is a variant of the AFLP technology that combines AFLP- and microsatellite-specific primers [60], whereas the direct amplification of minisatellite DNA (DAMD) utilizes primers that are specific for minisatellites rather than microsatellites [61]. Yet another approach, resistance gene-analog polymorphism (RGAP), makes use of PCR primers that bind to the conserved domains of plant resistance genes [62].
The Diversity Arrays Technology (DArT) is a high-throughput method based on the hybridization of fluorescent DNA probes to a set of target DNAs spotted onto a microarray [63,64]. The DNA is first digested with one or two restriction enzymes, followed by the ligation of specific adapters as in AFLP. Individual PCR products are spotted onto a grid to form an ordered microarray that represents hundreds of arbitrarily selected restriction fragments from all cultivars/species and various genomic regions of the gene pool of interest. Individual genomic DNA samples are pretreated in the same way as the pooled representatives (that is, restriction, ligation of adapter, and PCR with adapter-specific primers). Before being individually hybridized to the chip, each probe DNA is labelled with a fluorochrome to enable detection. Like AFLP and RAPD, DArT does not require previous sequence information. It allows simultaneous analysis of hundreds or even thousands of polymorphic loci, but the need to generate a microarray restricts the general use of the technique. By 2012, DArT technology has been developed for about 60 organisms, mostly crop and model plants [65], but also some wild plants such as the fern Asplenium viride[66].
Transposable elements and especially the retrotransposons bounded by long terminal repeats (LTRs) have proved to be useful for developing particularly sensitive multi-locus profiling techniques, either alone or in combination with other types of primers [67]. In the inter-retrotransposon amplified polymorphism (IRAP) approach developed by Kalendar and colleagues [68], primers are directed towards the LTRs of BARE-1, a retrotransposon of barley. The same authors also introduced retrotransposon-microsatellite amplified polymorphism (REMAP) which combines outward-facing LTR-specific primers with anchored microsatellite primers. Basically the same strategy, known as copia-SSR, was simultaneously developed by Provan and colleagues [69]. In the so-called sequence-specific amplification polymorphism (S-SAP) analysis, retrotransponson-specific primers are combined with AFLP primers [70]. S-SAP often produces highly variable fingerprints that are frequently more informative than AFLP. Related approaches have been developed for other plant transposons [71,72].
PCR-based single-locus methods
Because of their abundance, high polymorphism in the number of tandem repeats, co-dominant inheritance, excellent reproducibility and ease of use, PCR-amplified single-locus microsatellite markers have become the marker of choice for many applications, and presently remain more important than any of the other traditional DNA fingerprinting methods [73,74]. Typically, a pair of microsatellite-flanking primers is used to amplify the targeted locus by PCR, amplification products are separated by polyacrylamide or capillary electrophoresis, and banding patterns are monitored by radiography or fluorography. When locus-specific microsatellite analysis was first used in plants in 1992 [75], the need for developing species-specific microsatellite-flanking primers was still a serious drawback, requiring tedious cloning and enrichment strategies (see the reviews by Squirrell and colleagues [76] and Weising and colleagues [27]). Nowadays, this task has become relatively simple for (1) the increasing number of plant species with DNA sequence data in public databases and (2) the development of ultrafast “next generation sequencing” technologies that enable the identification of microsatellite loci and design of primers by random genomic sequencing (see "The future of DNA fingerprinting" below). A typical result from a microsatellite genotyping experiment is shown in Figure 3.
Figure 3.
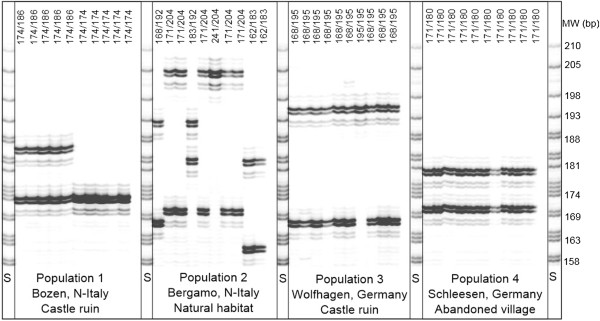
Locus-specific microsatellite analysis of four populations of the Lesser Periwinkle (Vinca minor) using a primer pair specific for locus Vimi43 (Möller, personal communication). For each population, ten samples were genotyped. Populations 1 and 2 were collected in northern Italy, whereas populations 3 and 4 were sampled in central Germany. Strong indications for clonality can be found in populations 1, 3 and 4. S, size standard: T-ladder derived from a chloroplast DNA fragment of Macaranga indistincta. Molecular weights (MW) of size markers are indicated in base pairs (bp).
Lately, expressed sequence tags (ESTs) have become a viable alternative to genomic DNA as a source for SSR loci, resulting in so-called EST-SSR markers that are either generated by cDNA cloning and sequencing [77] or, more commonly, by exploiting existing EST databases [78-80]. Database mining is often quite efficient, since EST-SSRs are surprisingly common and may be expected every 2 to 10 kb of EST sequence; for example, one per 6.3 kb in Hordeum vulgare[78]. These estimates of course depend on the search criteria and the search script used, most importantly on the minimum number of repeats used to define a microsatellite. Trinucleotide repeats commonly prevail in protein-coding regions of ESTs, whereas dinucleotide repeats are more frequent in 5′ and 3′ untranslated regions (UTRs). Expansions and deletions in coding regions can be tolerated for tri- and hexanucleotide repeats, because they do not perturb reading frames.
EST- and cDNA-derived SSRs have several important advantages over anonymous markers (see the review by Varshney and colleagues [81]). First, developing markers from already existing sequences is easy, fast and economical. Second, any type of microsatellite will be detected, whereas only SSRs with predefined motifs are captured by enrichment strategies. Third, EST-SSRs are physically linked to an expressed gene, which may encode a trait of interest. Finally, primer target sequences that reside in transcribed DNA regions are expected to be relatively conserved thus enhancing the chance of marker transferability across taxa. On the negative side, the association with coding regions sometimes limits the polymorphism of EST-derived SSR markers, resulting in fewer alleles and/or lower observed heterozygosity [78], but this is not necessarily the case. For example, Pashley and colleagues [79] compared the performance of 48 anonymous versus 48 EST-derived SSR markers from common sunflower, Helianthus annuus, and their transferability to two other Helianthus species. Their study showed that: (1) 73% of the EST-derived SSR markers were transferable among all species, compared with only 21% of the anonymous SSR markers; (2) EST-SSRs were on average only slightly less polymorphic that anonymous SSRs, both in the focal and the non-focal species; and (3) EST-SSRs located in coding regions were more readily transferable than those in untranslated regions - without differing significantly from the latter in terms of variability.
Locus-specific markers can also be developed from individual bands within multi-locus profiles, as exemplified by the sequence characterized amplified regions (SCARs). In the original description of the approach, specific primer pairs were designed for amplifying single bands of a RAPD profile [82]. SCARs have been used for cultivar identification in, for example, olive, Olea europaea[83], and sweet cherry, Prunus avium[84]. In the cleaved amplified polymorphic sequences (CAPS) approach [85], the resulting PCR product is treated with a restriction enzyme before scoring of fragments.
Single nucleotide polymorphisms
In the last decade, DNA fingerprinting methods based on single nucleotide polymorphisms (SNPs) have become increasingly important, especially in conjunction with microarray analyses that allow the simultaneous screening of very large numbers of SNP sites (see the review by Appleby and colleagues [86]). Among the many types of mutations occurring in genomes, single nucleotide exchanges stand out by their high absolute numbers as well as their biallelic nature, relatively low mutation rates, even distribution across the genome and relative ease of detection. In plants, one SNP is typically found per approximately 100 to 500 bp of DNA, but the average density depends on the studied species and the genomic region investigated. Numerous technologies have been developed for SNP discovery as well as for SNP genotyping [86]. Direct sequencing of multiple copies of the same genomic region is the most obvious method for SNP discovery, and has become very efficient after the development of high-throughput sequencing systems [87,88] (see "The future of DNA fingerprinting" below). Like SSRs, SNPs can also be mined from existing databases [89]. Practically all SNP genotyping assays are amenable to automation and therefore allow routine high-throughput analyses of large numbers of samples.
SNP markers are already well established in all major crop species [90,91], especially in those for which fully sequenced genomes are available. Recently, microarrays with typically 10,000 to 40,000 SNP markers (SNP-Chips) have been developed for many crops, and large-scale screenings of germplasm collections can now be undertaken at comparatively low costs [92]. Given that thousands of SNPs can be detected by novel sequencing approaches, SNP genotyping will receive increased attention, at least in economically important crop plants. However, poor transferability to related species may hamper their successful implementation for large-scale genotyping projects across genera.
Organellar DNA-based methods
The most commonly used organelle for genetic studies in plants is the chloroplast. Since recombination is rare or absent in plastid genomes, all DNA polymorphisms for a certain individual can be combined to form a “haplotype”. Plastid DNA polymorphisms at the intra-specific level are relatively rare, and the numbers of detected band profiles (haplotypes) are therefore often considerably lower than those detected by nuclear markers. On the positive side, the high conservation of organellar DNA sequences has enabled the development of non-specific, so called universal, PCR primers that amplify cpDNA introns and intergenic spacers in a wide array of plant species [93,94]. Universal primers are also available for the amplification of SSR loci in the chloroplast genome [95]. Polymorphisms within the amplified fragments can be monitored by various approaches, including the detection of length variants by high-resolution electrophoresis, and the detection of sequence variants by sequencing, or by digesting the PCR products with restriction enzymes in an approach called PCR-RFLP.
Plastid DNA is especially useful in studies where a low mutation rate is desirable, such as in the analyses of phylogenetic and phylogeographic patterns. Often both plastid and nuclear markers are combined in the same study for complementary information. Since the mode of plastid inheritance is usually maternal in angiosperms and paternal in gymnosperms, these markers also have the potential for tracing uni-parental lineages over large distances in time and space.
Choice of method
The pros and cons of different molecular marker methods have been discussed in a number of comparative investigations (see [27,96]). The actual choice of method must of course take marker availability, costs, expertise, equipment and many other factors into consideration. Based on 292 papers published between mid-2006 and mid-2009 on discrimination among plant cultivars, locus-specific microsatellite analysis (SSR) was the most popular method (36%), followed by RAPD (27%), ISSR (13%), AFLP (11%), other nuclear DNA-based methods (10%, including for example CAPS, DAMD, IRAP, REMAP, SNPs, SCAR and SRAP) and organellar DNA-based methods (3%, mostly cpDNA) [97]. If the purpose of a study is to simultaneously discriminate both dissimilar and very similar entities, applying a whole battery of marker types may be the best solution.
While insufficient repeatability of DNA marker profiles can be regarded as a methodological artefact, insufficient germline stability of sequences corresponding to DNA markers can cause “biological artefacts” due to excessively high mutation rates. This problem is most likely to arise with the most sensitive types of markers, such as SSRs. The ability to merge data from different studies, even when developed in different laboratories, is a major asset of this method. The same is true for the other single-locus DNA markers, such as SNPs, SCARs and CAPS, but these are usually only biallelic. Nevertheless, the potential number of SNPs is virtually unlimited, and various SNP-based assay methods have already been developed (see above). In a comparative study on 58 maize inbred lines, SNPs outperformed SSRs both in terms of quality and quantity [98].
Exceptionally high mutation rates and reduced germline stability are often encountered when using retrotransposon-based markers [67]. Thus, several reports have indicated that S-SAP markers are especially useful for discriminating among clones derived by somatic mutations [99] or among genotypes derived by recombination among highly similar entities [100]. The S-SAP primers are usually designed according to species-specific sequence information but positive results have also been obtained by using universal retrotransposon-based sequences [101].
Besides their application for the identification of plant material and for the estimation of similarity and relatedness, DNA markers have been extensively used for the construction of genetic linkage-based genomic maps, with a major aim of identifying markers that are closely linked and therefore co-inherited with genes for specific traits (see "Genetic mapping" below). Dense linkage maps have been constructed for numerous plant species including all major crops using all kinds of markers. For ease of scoring when screening large numbers of progeny, singe-locus biallelic markers such as SNPs are usually preferred for this purpose.
Applications of present-day DNA fingerprinting in plants
Genotype identification
Since the humble beginnings in 1988, DNA fingerprinting has become an immensely important instrument for genotype identification in both wild plant species and their cultivated relatives. Plants differ widely in life history traits including reproductive parameters such as propagation method and, for those that propagate by seed, also in breeding system (selfing or cross-pollinated) and in the mode of pollen and seed dispersal. All these factors have profound influence on the amount and partitioning of genetic variability between and within various entities such as cultivars and populations. These differences affect the utilization of DNA markers for fingerprinting individual plants or genotypes.
In some cases, DNA-based estimates of similarity among a set of genotypes show a relatively close association with previous morphology-based estimates, but there are also considerable discrepancies in other cases. If the morphological characters are mostly quantitative in nature, correspondence with DNA marker estimates is generally quite high as compared to qualitative characters, which are more likely to reflect only a small number of mutation events. It has also been suggested that molecular data are better at differentiating cultivated genotypes as well as their wild relatives according to origin and pedigree, whereas conventional pomological characterization data are more closely associated with physiological properties [102].
Genotype identification in wild plants: the influence of life history traits
Proper identification of individual genotypes is an important basis for many wild-plant-based studies. As mentioned above, various life history traits affect the amount and partitioning of genetic variation. Inbreeding species are, for example, most useful for forensic applications, since they typically produce suitable-sized patches of genetically identical or almost identical plants [103]. By contrast, outcrossing species are characterized by a situation where every plant has a different genotype. While potentially very informative, it is usually extremely difficult to secure forensic evidence involving a particular, unique plant specimen. Clonal plants, whether due to extensive vegetative propagation or apomixis frequently produce large numbers of progeny with the same genotype (see also Figure 3). Such genotypes can cover large geographical areas and are thus not sufficiently accurate for tying botanical evidence to a certain location.
In other research areas, the variation in plant life history traits can, however, be regarded as a positive factor; a wide range of biological questions can be answered by choosing suitable material and methods. DNA marker analyses have thus been able to estimate genotype age in plant clones, which have often proved to be considerably larger - and therefore often also older - than expected from previous data. For example, Steinger and colleagues [104] studied Carex curvula, a sedge species found in the European Alps. RAPD analysis of 116 tillers from a small patch (2.0 × 0.4 m) identified a total of 15 multi-locus genotypes. More than half of the sampled tillers proved to belong to a single, large clone estimated to be around 2,000 years old. Invasive species sometimes produce particularly large clones, such as the Japanese knotweed, Fallopia japonica, and the alligator weed, Alternanthera philoxeroides, both of which displayed a single RAPD phenotype in spite of being sampled over very large areas [105,106]. In other cases, DNA marker analyses have revealed more heterogeneity than expected. Each of five investigated Chinese populations of the invasive water hyacinth Eichhornia crassipes were thus shown to consist of at least three different clones according to their RAPD profiles [107].
Information about clonal growth can be very helpful for determination of factors involved in shaping population structure. When a microsatellite-based study was carried out in the marine eelgrass Zostera marina, clonal size proved to be positively correlated with heterozygosity [108]. Outbreeding clones were larger and contained more flowering shoots, indicating that inbreeding depression had decreased vigor and fertility. An unexpectedly high degree of genetic homogeneity was recently described in the geophyte Gagea spathacea[109]. All but two of 138 examined specimens, representing 52 populations throughout the entire distributional area in northern, central and eastern Europe, had identical AFLP profiles. Probably this highly polyploid taxon has derived from a hybridogenic event, and has managed to attain its large area almost exclusively by bulbil production and spreading rather than by seed set and seedling establishment.
DNA fingerprinting has also helped to clarify the reproductive system in species that can produce seeds both sexually and asexually (that is, by apomixis). Many Taraxacum populations consist of triploid individuals that apparently reproduce through apomixis and therefore are clonal. Such clones can sometimes cover large areas as was demonstrated in an AFLP study [110]. A comparison of SSR and AFLP data showed that both marker types were able to discriminate among nine apomictic microspecies (defined on morphological characteristics) of Taraxacum, but that AFLP was more sensitive in detecting also small, mutation-derived differences within each microspecies [111]. By contrast, two dinucleotide repeat SSR loci detected considerably more variation than AFLP in apomictic lineages of Ranunculus carpaticola[112,113]. Evidence for an origin by mutation instead of by recombination was provided by the lack of allele segregation in the investigated SSR loci. Thus, in each lineage, the same number of alleles was always found within a locus, and these alleles also formed classes of related allele sizes within each lineage.
The availability of adequate tools to identify individual genotypes can be immensely useful in plant ecology. Thus, SSR-analysed Taraxacum clones were recently employed to investigate biodiversity and ecosystem functioning. In one study, five identified Taraxacum clones were used for setting up experimental plots where effects of levels of diversity in both favorable (fallow field) and unfavorable (mowed lawn) conditions could be quantified [114]. The genotypic diversity effects appear to be stronger in environments where intra-specific competition is more intense. In a parallel study, genotype × environment associations were studied in natural populations with the same set of Taraxacum clones [115]. Genotypes that produced poorly under favorable conditions instead showed the highest performance under stressful conditions.
Genotype identification in vegetatively propagated cultivars
All plants belonging to a particular cultivar of an asexually propagated crop are expected to share identical DNA fingerprints, except for rare mutations. By contrast, sexually derived cultivars are expected to exhibit non-uniform fingerprint patterns. However, there are crops for which the major breeding method involves selection amongst rather similar seedlings that originate from a very small number of widespread cultivars. This situation can be exemplified by peach, Prunus persica, which is self-fertile and self-pollinating to a large extent. In such crops, new cultivars sometimes have DNA fingerprints that are almost identical or at least very similar to those of the seed parent in spite of being derived through sexual recombination. By contrast, variation is sometimes encountered where one expected uniformity. Vegetatively propagated crops are usually still capable of producing sexually derived seeds, and these may germinate and develop into fertile but unnoticed plants in less well-tended fields and orchards. There is therefore an increased risk, especially for older cultivars, that a certain name is being used on several different entities, some of which have originated from seed setting.
Irrespective of propagation and breeding method, the value of accessions in plant genetic-material collections benefits tremendously from DNA marker-aided identification. This is, however, especially important in vegetatively propagated crops that must be grown in the field or maintained in greenhouses at high costs. Previous reviews [97,116] show that a higher number of mislabelled plant accessions are revealed using DNA markers (typically 25 to 30% mislabellings) as compared to traditional (pomological or ampelographic) characters (typically 5 to 10% mislabellings). Different categories of problems with synonyms and homonyms have been defined [116], and appear to be especially prevalent in locally grown and often older germplasm whereas well-known modern-day cultivars are correctly identified to a much higher extent.
For large-scale profiling of, for example, accessions in a genetic resources collection, SSR markers are usually preferred [117]. Although generally regarded as highly reproducible, problems are sometimes encountered with incorrect allele sizing, the occurrence of null alleles, allele drop-out (only one of two alleles is amplified at a heterozygous locus), false alleles (artefactual amplification products) and occasional amplification of isoloci (an isolocus is a similar but non-identical locus in the genome, common in allopolyploid species). While dinculeotide repeat SSRs are the most common type of microsatellite, less stuttering and improved allele sizing can be achieved with markers based on tri- or tetranucleotide repeats [117,118], although these are sometimes also less informative.
Zhang and colleagues [119] checked the accuracy and reliability of 15 SSR loci for clone identification in cacao, Theobroma cacao, and reported an average error rate of only 0.014 for allele drop out and 0.019 for false alleles. Some loci were more error-prone than others, suggesting that putative loci should be evaluated not only for their polymorphism but also for reliability prior to large-scale analyses. Vélez and Ibánez [120] checked 19 SSR loci in a study of more than 4,000 plants representing 19 grapevine cultivars. After removal of some minor technical artefacts, 99.8% of the samples matched to the expected genotype. Some loci, however, proved to be rather sensitive to the occurrence of chimeric mutations whereas others were not. Artefactual variation of SSR markers was also indicated in a study of olive [121]. Interestingly, SSR alleles that differed among olive samples from the same cultivar were only 2 bp (one repeat unit) apart, whereas samples from different cultivars usually exhibited larger size dissimilarity in the polymorphic alleles. If available, accurate pedigree information is very valuable for checking the reliability of marker profiles.
The possibility to merge SSR-derived data from different investigations is often reported as a major asset of this method. This option is, however, dependent on the use of identical SSR loci and suitable standardization procedures. Since absolute allele sizes of the SSR markers often differ when results from different laboratories are compared, a representative reference material with many different alleles should be used at all laboratories involved in the genotyping program, and the material for these standards should be harvested from predetermined plants in one collection only. By comparison with suitable standard alleles, sample alleles can then be defined according to relative number of core repeat units instead of relying only on the absolute fragment length in base pairs.
Increased attention has recently been paid to the use of SNP markers for genotype identification in vegetatively propagated cultivars. Advantages of SNPs are their potential abundance and the fact that they do not rely on fragment length variation like SSR, and therefore are easier to standardize across different laboratories and equipment. Numerous high- and low-density SNP arrays have recently been developed for different crops. For example, a set of 48 SNPs was developed in grapevine, Vitis vinifera, through resequencing of 11 genotypes [122]. High-throughput SNP genotyping can be conducted using bead arrays or microarrays (SNP chips) such as in, for example, Citrus[123]. Since the number of polymorphisms covered in these assays is usually several hundred to many thousand, the obtained data can also be used for detecting quantitative trait loci (QTL).
Genotype identification in seed-propagated cultivars
In seed-propagated crops, at least some genetic variation usually persists also within cultivars. This is especially pronounced in highly outcrossing species, thus making DNA-marker-aided cultivar identification considerably more difficult. The situation is further complicated by the fact that each seed production cycle can lead to the introduction of new genetic variation - for example, due to foreign pollen. A considerable influx of new alleles was thus demonstrated after 7 to 13 subsequent regenerations of open-pollinating rye, Secale cereale[124].
Even with all the precautions taken in connection with modern gene bank regenerations, changes in allele frequencies can result from just recombination and selection. This was clearly demonstrated in an AFLP analysis of 50 white cabbage, Brassica oleracea, accessions together with first-generation regeneration products from six of these accessions [125]. The genetic changes between original accessions and their respective regenerants were of the same magnitude as the differences among some of the more similar accessions. Moreover, while most alleles remained stable between generations, frequencies of some alleles instead changed considerably, suggesting that unintentional selection had taken place.
Obviously, a large number of markers are required for proper quantification of genetic changes between generations, and for efficient discrimination among outcrossing, seed-propagated cultivars. Such large numbers are, for example, provided by the DArT technology that proved very useful for distinguishing Festulolium cultivars (Festuca × Lolium experimentally produced hybrids) with 7,680 probes on a microarray [126]. In this study, each cultivar was represented by 20 individual plants. These plants were analyzed both as individual and bulked samples. In order to minimize the loss of low-frequency bands, bulks with only five plants in each were recommended.
Inbreeding crops are usually considered less problematic than outcrossers, since the cultivars are more homogeneous. However, some inbreeding crops still contain intra-varietal variability, especially in the case of primitive cultivars or landraces. Propagation cycles performed in a genebank with such material can cause prominent gene frequency changes due to gene flow and inadvertent selection. In these cases, pure-lining of the accessions may be necessary to avoid loss of diversity, as exemplified by the USDA Soybean Germplasm Collection [127]. In addition, selfing crops often contain a multitude of genetically very similar cultivars, thus necessitating the use of highly polymorphic markers for discrimination. While the commonly applied AFLP and SSR markers have produced sufficient results in many studies, the retrotransposon-based S-SAP method has been shown to resolve even very closely related plant accessions in, for example, wheat, Triticum aestivum. Nowadays, SNP markers receive increasing attention also in sexually propagated crops, mainly for the almost inexhaustible number of potential polymorphisms. Genome-specific SNPs have thus been developed from wheat gene intronic regions, and have proven highly useful for cultivar discrimination as well as enabling a quantification of genetic diversity at each of the genomes in this hexaploid crop [128].
Genotyping somatic mutations
Spontaneously occurring somatic mutations can give rise to so-called 'sports’. These deviate from the original cultivars in minor but economically important traits such as fruit color in fruit and berry crops, and flower or leaf color in ornamentals. Sports are difficult to distinguish with DNA fingerprinting since the markers usually cover only a minute part of the genome. In addition, chimeras are quite common - that is, mutations that occur in only one of the three meristematic cell layers in the apical meristem that differentiate into the various plant tissues. The existence of chimerism was very elegantly demonstrated in grapevine, Vitis vinifera, by Franks and colleagues [129]. Although grapevine is a diploid species, some SSR loci occasionally showed three alleles when different sports were analyzed. It turned out that plants regenerated from cell layers L1 and L2, respectively, had different SSR alleles as well as different phenotypic characteristics. SSR analysis was used to identify chimeric clones also in 'Cabernet Sauvignon’ [130], 'Grüner Veltliner’ [131] and 'Moscatel Galego Branco’ [132] while clones of 'Pinot’ were successfully distinguished with the S-SAP method [99]. In this study, three different retrotransposon-based primer pairs produced a total of 1,274 bands, one third of which were polymorphic and able to discriminate among all the 19 investigated clones.
S-SAP analysis has been successful for the genotyping of sports also in other crops, such as apple. Using 15 S-SAP primer combinations, five sports of 'Gala’ and one of 'Braeburn’ could be discriminated, both from each other and from the two original genotypes [133], whereas 24 SSR primer pairs generating a total of 64 alleles, and 35 AFLP primer combinations generating more than 1,000 bands, failed to do so. Based on two Ty1-copia LTR retrotransposons, a set of 19 bud sports of the apple cultivar 'Fuji’ were investigated with S-SAP [134]. All sports obtained unique DNA profiles. Other retrotransposon-based methods can also be quite useful. In 24 sports of clementine, Citrus reticulata, application of eight IRAP primers produced a total of five polymorphic bands whereas RAPD (26 primers), ISSR (16 primers), AFLP (8 primer combinations), S-SAP (9 primer combinations) and SSR (9 primer pairs) revealed, at the most, one (S-SAP) or two (RAPD) polymorphisms [135,136].
Some studies found surprisingly high levels of marker polymorphism within cultivars, such as in olive, where clones have been selected and subsequently multiplied by vegetative propagation for centuries. In one study, 27 putative clones of 'Verdeal-Transmontana’ could be differentiated with ISSR [137] while even higher levels of polymorphism were encountered with RAPD (50% polymorphic bands) and ISSR (54%) in the screening of 120 putative clones of 'Cobrançosa’ [138]. Possible explanations for these observations include a polyclonal origin, accumulation of somatic mutations over the long life-span of this woody species, and unnoticed establishment of sexual progeny in the orchards.
Genotyping in vitro-propagated material
Heritable somaclonal variation - that is, variation among regenerants due to somatic mutations - can be significantly enhanced by some micropropagation techniques. Although often regarded as an undesirable side-effect, these mutations can be valuable in crops that lack sexual reproduction (such as, for example, banana) or have very long generation cycles (such as, for example, palm trees). In general, axillary branches yield the most stable regenerants, followed by somatic embryogenesis and finally organogenesis. It is, however, impossible to predict whether markers will be able to find any variation in regenerated material, or what methods will prove to be most efficient.
Very few polymorphisms have generally been found in tissue culture regenerants. The extent of DNA marker polymorphism can, however, vary considerably between plant materials - even of the same species - as was shown by comparing the very uniform regenerants of the banana cultivar 'Prata Ana’ [139] with the highly variable regenerants of cultivar 'Valery’ [140]. When AFLP analysis was applied to regenerants of Helichrysum italicum, plantlets derived directly from leaves showed the same level of variability as plantlets that had passed through a callus stage [141]. Although only 6.2% of a total of 449 bands were polymorphic, almost all plantlets differed from the original genotype in at least one band. The same band polymorphism was encountered in several plantlets in some cases, suggesting a hot spot of DNA instability. In another study, plant material of date palm derived from asexual embryogenesis showed considerably more variability than plants derived from organogenesis when analyzed with AFLP markers [142].
Detailed sequence-based analysis of the molecular events responsible for SCAR marker polymorphism (for example, insertion or excision of transposons, microdeletion, recombination) between somaclones and sexual recombination-derived lines of maize, demonstrated that the same mechanisms apparently determine both in vitro and in vivo variability [143]. Therefore, it was concluded that cell culture only enhances the rate of heritable genomic changes which otherwise occur naturally in living organisms. Carrier and colleagues [144] studied somaclonal variation in the grapevine cultivar 'Pinot noir’ by high throughput sequencing and found that insertion polymorphism generated by transposable elements was responsible for most of the variation.
Forensic botany
In theory, DNA fingerprints obtained from plant fragments should be able to provide important evidence in crime investigations but success has been limited so far, probably due to problems with isolating DNA of sufficient quality from poorly preserved plant material. SSR markers are often chosen for forensic work since they work comparably well also with heavily degraded DNA. One famous early case, however, involved RAPD analysis of seed pods of the Palo Verde tree, Cercidium sp., recovered both from the crime site and from the pick-up truck of a suspect [145], while another case made use of SSR and RAPD analysis to compare fragments from clonally reproducing bryophytes (mosses) collected both on the crime site and on the suspect himself [146]. In subsequent experiments, a high likelihood of picking up fragments of bryophytes by walking outdoors wearing rubber boots was shown, as well as the ability to isolate DNA of sufficient quality after several months of storing bryophyte material under adverse conditions [147]. These facts together with the high level of clonality in many bryophyte species make them an ideal target for forensic analysis. In yet another criminal case, seedlings of the inbreeding herbaceous knotweed Polygonum aviculare obtained from germinating seeds found in the wheelhouse of a suspect’s car tire, and from a large number of soil samples taken at the crime site and various reference localities, were analyzed with AFLP [103].
Detection of adulterations of food, drink and medicinal products is another area for forensic botany. Licensing arrangements sometimes require that a specified clone, cultivar or landrace is utilized in the manufacturing of food and beverages. Thus, well-defined grapevine clones must be used to receive “appellation d'origine controllée” labelling in France. In one study, musts (that is, freshly pressed grape juice destined for wine-making) from two different grape cultivars could be identified using two SSR markers [148]. In another study, musts containing different proportions of two grape cultivars were analyzed with densitometry measurements of the SSR amplification products after separation and staining on polyacrylamide gels [149]. In Greece, Nemea wines are marketed with protected denomination of origin (PDO). Instead of using only the prescribed cultivar 'Agiorgitiko’ , the more productive 'Cabernet Sauvignon’ is sometimes added. DNA samples from fresh and fermented products, containing various mixtures of these two cultivars, were therefore subjected to a CAPS assay [150]. Presence of the adulterant could be detected down to 10% throughout the fermentation process.
Olive oil is also often marketed with PDO labelling. RAPD, ISSR and SSR analysis of Portuguese olive oils allowed the determination of geographic origin of the cultivars on which they had been based [151]. Similarly all 10 olive cultivars involved in samples of Italian oil samples could be identified with only one AFLP primer pair [152]. For rice, the adulteration of the expensive Basmati rice is an important issue, not only for European and US customs but also for consumers. Basmati cultivars have often been mixed with crossbred Basmati varieties and long-grain non-Basmati varieties. Several DNA-based markers have been proposed, and some were commercialized for adulteration tests, such as the multiplexed SSR markers developed by Archak and colleagues [153]. DNA analyses of various plant-based food products have similarly been used for authentication. The presence of the apple 'Annurca’ could thus be verified by SSR analysis in highly processed nectar and purée products [154]. Using relatively short SSR target sequences (below 160 bp), it was also possible to amplify genomic DNA from canned pear fruit and fruit juice while markers with longer target sequences failed [155].
Medicinal drugs constitute another important product area where adulterants cause major problems. Based on nine SNP sites, all populations except two could be distinguished in DNA isolated from the dried stems of the orchid Dendrobium officinale, which is a valuable source of 'Fengdou’ drugs used in traditional Chinese medicine [156]. The latter two populations could instead be distinguished using a more complex procedure known as suppression subtraction hybridization which involves PCR amplification, differential DNA fragment cloning and sequencing. Using these protocols, origination of the plant material could be determined for 50 drug samples obtained at a commercial market. For more information on DNA marker use in medicinal plants, see the reviews by Nybom and Weising [157] and Sarwat and colleagues [158].
A variety of DNA marker methods have been used to demonstrate infringement of Plant Breeder’s Rights, either in court or, in our experience much more common, leading to a settlement outside of court [159]. A related field concerns the identification of plants, the possession of which is considered illegal. Thus several studies have been published on the identification of Cannabis sativa specimens as part of drug enforcement [160]. In one approach, 15 SSR loci were combined into a single multiplex to enable fast and user-friendly discrimination between Cannabis genotypes [161]. One of the detected genotypes, however, proved to be very common in police seizure-derived evidence material, suggesting that many illicit growers had access to the same clone. This clonal propagation of course makes it difficult to determine the origination of a particular batch. A related DNA marker application concerns violation of trade restrictions. A special situation is encountered when products from protected trees are involved since woody tissue usually yields heavily degraded DNA. Nevertheless, a set of SNP markers derived from cpDNA intergenic spacers have proven useful for identification of tropical tree species using wood-derived DNA samples [162].
Genetic diversity, population structure and genetic relatedness
Discrimination among different genotypes is often only a starting point for the subsequent quantification of genetic variability among these genotypes and analysis of patterns of relatedness and gene flow. The extent of genetic variation in a species and its distribution among and within populations is determined by a large number of factors, such as the breeding system, historical events regarding, for example, habitat availability and immigration, population size, migration between populations and many biotic and abiotic ecological factors. Nybom and Bartish [163] compiled 106 RAPD-based studies and described the effects of several life history characters and sampling strategies on genetic diversity estimates. In another paper [96], 307 nuclear DNA marker studies (RAPD, AFLP and SSR) were compiled and investigated in a similar manner. One outcome of these surveys was that long-lived, outcrossing and late successional taxa retain most of their variation within populations, whereas annual, selfing and early successional taxa allocate more variation among populations. Within-population diversity is, in general, negatively correlated with the level of population differentiation.
The uniparentally inherited plastid genomes behave as a single, haploid character, and the effective population size for plastid markers is therefore only half of that of nuclear (diploid and biparentally inherited) markers. Consequently, population differentiation due to genetic drift occurs much faster for cpDNA markers than for nuclear markers. Because of their relatively high intra-specific variability, chloroplast and mitochondrial micro- and minisatellites are therefore very useful for studying genetic structure at a species-wide scale.
Population differentiation and gene flow
DNA markers have become a major tool for studying fundamental evolutionary influences of natural selection, mutation, gene flow and genetic drift on wild plant populations. While selection and colonization history is responsible mainly for large-scale structuring of genetic variation, gene flow and genetic drift operate also at a more narrow geographic scale. Among these factors, gene flow especially has received much attention since it is crucial in determining levels of species integrity and subdivision. As already mentioned, breeding system has a profound effect on gene flow and the partitioning of genetic variation between and within populations. The occurrence of IBD between populations has been demonstrated with DNA markers in many different kinds of outcrossing plant species such as, for example, the herb Saxifraga oppositifolia[164], the Brazilian peppertree Schinus terebinthifolius[165] and the Australian shrub Grevillea mucronulata[166]. IBD has been shown to occur, although much more seldom, also in selfing species such as wild emmer wheat, Triticum dicoccoides[167]. In accordance with these results, a correlation was found between collection distance and RAPD-based among-population diversity estimates for outcrossing taxa [163]. A corresponding association was, however, not found for selfing taxa.
In addition to the inherent dispersal capabilities of a species, gene flow is also affected by natural and anthropogenic habitat heterogeneity. Spatial autocorrelation analysis has thus become a valuable tool for studying spatial scale-dependent changes in DNA marker polymorphism within a population or group of closely occurring populations, and the impact of habitat characteristics on the resulting spatial genetic structure (SGS). Several computational methods have been used to calculate autocorrelation coefficients that measure the genetic similarity between individuals that fall within a defined distance class. A positive autocorrelation is frequently encountered over shorter distances, even if there is no overall linear correlation between geographic and genetic distances when calculated across the whole data set. Using RAPD data, Torres and colleagues [168] found significant autocorrelation in the first distance class (15 m) in populations of the endangered cliff specialist Antirrhinum microphyllum, suggesting a patchy distribution of genetic diversity. This is consistent with the territorial behavior of the main pollinator Rhodanthidium sticticum, short-distance seed dispersal, and a likewise patchy distribution of suitable habitats.
Many plant species comprise both central, so-called core populations as well as more or less peripheral populations. Such populations may experience considerable differences in the magnitude of operating evolutionary and ecological forces. For example, edge and core populations of the herb Pulmonaria officinalis exhibited strong differences in allelic and genotypic richness, expected heterozygosity and inbreeding coefficent when analyzed with SSR markers [169]. Similarly, an SSR analysis of eastern white cedar, Thuja occidentalis, showed that SGS could be detected over a six times larger distance (90 m) within peripheral populations compared to within core populations (15 m) [170].
Autocorrelation analysis has demonstrated IBD also in mainly selfing species but then usually at a very narrow scale, as was shown in the wild barley species Hordeum spontaneum[171].
Highly informative estimations of gene flow can be obtained by genotyping the same plant material using both nuclear and organellar markers. Since the former are biparentally inherited and the latter usually only maternally inherited, the resulting data provide an indication of the relative importance of pollen versus seed migration [172]. This ratio can vary by at least two orders of magnitude, and is typically much lower for insect- as compared to wind-pollinated plants [173]. In dioecious and therefore obligatory outcrossing plants, a mixture of autosomal and sex-linked SSR markers can provide direct evidence of the relative importance of seed versus pollen dispersal. Contrary to previous expectations, similar levels of pollen and seed dispersal were detected in the dioecius perennial plant Silene latifolia[174]. In selfing species, the lower incidence of inter-plant pollen transfer is expected to reduce the pollen to seed migration ratio, as verified by values well below unity at short distances within wild populations of Hordeum spontaneum[171].
While genes can move between populations by seed and/or pollen, colonization of new habitats is dependent on seed only. In coastal plants, seeds often have the potential to disperse over long distances by hydrochory. In a study of wild sea beet, Beta vulgaris subsp. Maritima, comprising more than a thousand plants from 33 populations along the French coast of the Anglo-Norman gulf, both mitochondrial and nuclear SSRs were applied [175]. Analysis of SGS and determination of zones of sharp genetic change demonstrated narrow IBD indicative of short-range dispersal, as well as genetic barriers fitting the orientation of marine currents and indicative of long-range seed dispersal.
Effects of an increased subdivision or fragmentation of natural plant habitats has received much attention lately; dispersal between populations is reduced as well as genetic diversity. Outcrossing species may especially suffer from enforced selfing or biparental inbreeding in fragmented habitats, and lose much of their potential for adaptation to changing environmental conditions. Using SSR markers, White and colleagues [176] compared fragmented versus continuous populations of the tropical tree Swietenia humilis in Honduras. Genetic variation was still high in all habitat fragments, but low-frequency alleles were more scarce, thus foreboding future genetic erosion. In another early study on tropical trees, Aldrich and Hamrick [177] reconstructed a population-level pedigree of Symphonia globulifera. Seedlings only occurred in primary and remnant forests, but not in pastures. Surprisingly, however, the majority of seedlings in fragmented forests proved to be derived from a few adult trees located in the open pasture land. Thus the genetic bottleneck experienced by the seedlings in remnant forest patches was caused by the reproductive dominance of a few spatially isolated trees in pasture land, in conjunction with unusually high levels of selfing in these trees.
Overall, tree species have been considered as comparatively resilient to fragmentation due to their often highly effective long-distance dispersal mechanisms. Recently, however, the wind-pollinated and wind-dispersed Andean tree Polylepis multijuga was analyzed with AFLP and shown to contain surprisingly little heterozygosity and to display SGS at short distances, suggesting that most seeds moved only a few meters [178]. This type of information is valuable when developing conservation plans for species protection and perhaps also for a possible reintroduction. Information about, for example, colonization and spreading behavior can be equally helpful when developing measures for stopping further growth of an invasive species. A combination of spatial genetic and geostatistical analyses of data from chloroplast and nuclear SSRs showed how the original two introductions of the invasive Brazilian peppertree Schinus terebinthifolius in western and eastern Florida, respectively, had spread and hybridized in little more than one century [165]. Since both long-distance jumps and short-distance diffusive spread could be demonstrated, highly concerted eradication efforts or the manufacturing of effective biocontrol agents are apparently called for.
Genetic relatedness
DNA fingerprinting data are often used to quantify levels of relatedness among genotypes or groups of genotypes, and numerous relatedness estimators have been described and compared. When wild plants are involved, the purpose is often to compare DNA marker-derived estimations of relatedness with current systematic treatment (see also "Applications of present-day DNA fingerprinting in plants" above). Other applications include parentage analysis, which is the most direct way to estimate gene flow. SSRs are the most commonly used markers for this purpose but data simulations have shown that multi-locus markers such as AFLP can also be used with high confidence, at least when the dominant alleles occur in frequencies of 0.1 to 0.4 [179]. Using SSR, a paternity analysis was conducted in a natural stand with two oak species, Quercus robur and Q. petraea[180]. The spatial distribution of male parents of the offspring from 13 maternal progeny arrays was determined, and the information used for calculation of pollen dispersal curves and analysis of gene flow. Similarly, gene flow was estimated from an SSR-based paternity analysis in the South American palm tree Euterpe edulis[181]. First, an exclusion analysis was performed by comparing adult and juvenile genotypes. After that, a paternity index was calculated among adults that could be the putative parents for a particular juvenile. Gene flow was shown to take place over longer distances than expected (up to 22 km), but it was not possible to distinguish between seed versus pollen transport. Since chloroplasts are paternally inherited in conifers, chloroplast simple sequence repeat (cpSSR) markers can, however, be very useful for direct estimates of paternity, as was demonstrated in white fir, Abies alba[182].
Access to correctly defined relationships can be very important in plant breeding for the calculation of heritability of specific traits. Various statistical formulae have therefore been developed for determining genetic relationships among individual plants. In a comparison of either purely marker-derived estimations of relationships or combined pedigree and marker-derived estimations, the latter proved to be more informative when analyzing Scots pine, Pinus sylvestris, offspring in a progeny test of open-pollinated genotypes in a seed orchard [183]. Surprisingly incongruent data were obtained when S-SAP markers, SNPs and pedigree data for a set of 35 wheat cultivars were compared [184]. The molecular methods produced similar estimates for the overall partitioning of genetic diversity between and within groups of cultivars, but the genetic similarities between pairs of cultivars were not correlated. SNP-based data were more closely associated with pedigree information than S-SAP-based estimates, probably because polymorphisms are strongly dependent on retrotransposon-related genomic rearrangements.
For cultivated crop plants, estimates of relatedness can provide valuable insights into the domestication process when material originating from different geographic areas is analyzed such as in, for example, Italian olive cultivars [121]. Relatedness among cultivated material on the one hand, and wild populations of the same or closely related species on the other hand, has also been addressed in, for example, apple using SSR, AFLP and cpDNA markers [185,186] (see also "Hybridization and introgression" below).
Estimating the true level of genetic relatedness among cultivars from nuclear DNA marker data is quite difficult, since the obtained information can usually only estimate identity by state (phenetic analysis) instead of the more desirable identity by descent (phylogenetic analysis). One interesting approach towards a true phylogenetic analysis has, however, been achieved within the HiDRAS project [187]. This project involves the analysis of specific chromosomal regions in genetically related apple cultivars using a large set of SSR markers that cover almost the whole genome. Thus, being able to accurately detect levels of genetic relatedness between different cultivars is very helpful for further analyses of, for example, QTL inheritance.
DNA marker-based procedures have frequently been applied to assess diversity and relatedness in collections of cultivated plant material - for example, gene banks. Interestingly, the anticipated loss of overall genetic diversity proved to be negligible when studied in 198 Nordic bread wheat landraces and cultivars that were developed during the last 100 years [100]. DNA markers are also highly useful for the purpose of setting up core collections within gene banks - that is, subsets of the entire plant material, chosen so as to preserve as much as possible of the initial diversity. Two main approaches have been used; the first with some kind of stratification using cluster analysis, and the second with methods for determination of genetic uniqueness. Numbers of retained SSR alleles can be maximized using a measure of uniqueness known as maximation strategy [188-190].
Genome constitution: hybridization, introgression and polyploidy
In contrast to animals, many plant groups are characterized by highly variable ploidy levels, often even within the same species. This addition of genomes has certain effects on DNA marker application and data treatment. Moreover, the formation of hybrids by fusion of gametes from two different entities (species, subspecies, and so forth) is also common in plants [191]. While homoploid hybridization takes place at the same ploidy level, most hybridization events instead involve the duplication of genomes, resulting in allopolyploid taxa.
Hybridization and introgression
In a series of classical studies, homoploid hybridization among American Iris species was investigated using a wide variety of DNA-based methods. First, Iris fulva and I. hexagona were shown to each have a species-specific rDNA profile [192]. Subsequently, DNA profiles indicated inter-specific hybridization as well as further introgression in both directions in populations where the two species co-occurred [193]. Diagnostic RAPD and cpDNA-CAPS markers were generated for these two species as well as for I. brevicaulis, and I. nelsonii was shown to have derived from hybridization between all three species [194].
Another important set of studies on homoploid hybridization has been undertaken in the sunflower genus, Helianthus. RAPD linkage maps were developed for the sympatric and hybridizing species H. petiolaris and H. annuus and subsequently used to analyze the genome of a recently formed hybrid species, H. anomalus, as well as of an artificially generated hybrid [191,195]. Later on, divergence between the two parental species was analyzed using 108 mapped SSR markers [196], and below average introgression was noted for SSR markers located close to QTLs for species differences when two parapatric species, H. annuus and H. debilis, were investigated [197,198]. Interestingly, gene flow was mainly in the direction from the hybrid back into these two parental species [199].
cpDNA-derived information has played a major role in elucidating many cases of homoploid hybridization and subsequent introgression. Studies of multiple taxa in several tree genera have thus shown that chloroplast haplotypes often are closer associated with geographic origin than with species affiliation - for example, in oak trees, Quercus[200], in Eucalyptus from Tasmania [201] and in the South East Asian pioneer tree genus Macaranga[202]. This introgression phenomenon has been coined “chloroplast capture” [203].
DNA markers are also commonly used for detecting both ancient and ongoing hybridization between crops and their wild relatives. Malus sieversii grows in Kazakhstan and has been suggested as progenitor of cultivated apple, M. domestica, based on morphological, historical and molecular evidence [204]. Nuclear SSR-based analyses have later been undertaken to investigate the genetic diversity and population structure in M. sieversii[205]. The origination of cultivated apple may, however, be more complicated. In another SSR-based study, three separate although partly overlapping gene pools were formed by (1) M. sieversii, (2) the European wild apple species M. silvestris, and (3) old and modern apple cultivars [186]. In the same plant material, analyses of chloroplast haplotypes produced rather unexpected results. Thus, M. sylvestris not only had the same common haplotypes as M. domestica, but there was also local sharing of uncommon haplotypes between the two species, suggesting recent inter-specific gene flow. A strong affinity between M. sylvestris and modern apple cultivars was likewise suggested in an SSR analysis of 839 genotypes collected from China to Spain, and representing four wild species as well as cultivated apple [206]. In this study, data were analyzed both with the computer program STRUCTURE, and with approximate Bayesian computation which offers a more historical perspective on gene flow.
Two variants of STRUCTURE, InStruct and NewHybrids, were used by Muranishi and colleagues [207] in a recent SSR-based study of Magnolia stellata and M. salicifolia together with putative F1 and F2 hybrids and backcrosses. The resulting clusters could be verified also with morphological trait analysis. Simultaneous application of cpDNA SSR markers showed that introgression was heavily asymmetric, with M. salicifolia being the seed parent of almost all hybrids and backcrosses.
In plant breeding, there can be good reasons for analyzing parental contributions in recently developed, experimental hybrids, especially if the breeding process has involved one or several generations of backcrossing. The amount of parental influence could thus be quantified using a microarray analysis with 7,680 probes simultaneously detecting SNPs, indels and methylation differences, in a set of intergeneric hybrids between the commercially important grass genera Festuca and Lolium[126]. The extent of similarity between the derived Festulolium cultivars and the parental genomes was clearly associated with the type of crossings performed - that is, F1, F2 or backcrosses.
Polyploidy
Polyploidy is very common in the plant kingdom. Although the same marker technologies can be used for genotyping diploid as well as polyploid samples, statistical analyses and interpretations are usually less straightforward when polyploid samples are involved. Many species are allopolyploids and have been derived from their diploid ancestors by hybridization. Moreover, molecular studies of allopolyploid taxa and their putative progenitor taxa have shown that multiple origination is the rule rather than the exception. While frequent gene flow between polyploid lineages and back-crossing to parental taxa can further confound this process, a more easily studied case is offered in apomictic species where the speciation event is more or less frozen in time. One such example is the North American allopolyploid cloak fern, Astrolepis integerrima, which was recently studied by cpDNA sequencing and AFLP analysis [208]. Six relatively localized cpDNA haplotypes were detected, some of which were further divided by AFLP. All in all, the results suggested that a total of 10 A. integerrima lineages have been formed through multiple independent hybridizations between A. cochisensis and A. obscura.
Identification of the putative progenitor species of polyploids can be attempted with various types of markers, including the internal transcribed spacer (ITS) region in the nuclear ribosomal RNA gene clusters which was sequenced and analyzed in, for example, polyploid rose cultivars and species [209]. Nair and colleagues [210] used IRAP primers to determine the genomic constitution in a set of mostly triploid banana cultivars. Primer sequences were derived from two different retrotransposons, one occurring in the A genome (Musa acuminata) and the other in the B genome (M. balbisiana). A more easily applied CAPS marker, obtained from PCR amplification of the ITS region followed by restriction with RsaI, has also been applied for this task [211]. More recently, application of 653 DArT markers similarly allowed the discrimination between A and B genomes, and the identification of these genomes within a set of banana cultivars [212].
Multi-locus based methods such as RAPD and AFLP are sometimes used for studying population genetics in species with different ploidy levels, but problems can arise due to a positive correlation between ploidy level and number of scored bands [213]. In addition, banding patterns may differ qualitatively between samples at different ploidy levels, and thus give rise to scoring errors. Single-locus markers such as SSRs and SNPs are also problematic due to the occurrence of multiple alleles and complex segregation ratios. SSR markers may be more or less genome (and species)-specific and therefore fit only one of the two homologous genomes of an allopolyploid hybrid, producing no amplification in the other (null alleles, or allele drop-out). Truly genome-specific SSR loci that consistently produce a maximum of only two alleles in each sample are rare, but can be quite useful as demonstrated in the hexaploid Mercurialis annua[214]. With these markers, population genetics parameters could be calculated as if the species instead were diploid.
For allopolyploid crops with intermediate levels of similarity among homologous genomes, such as tetraploid potato, SSR primers in general produce a variable number of bands per locus. For example, Fu and colleagues [215] found a total of 64 alleles when investigating 169 potato accessions with 36 SSR primer pairs. Even apparently diploid species such as apple may be “ancient polyploids” in which some primer pairs can produce a second set of alleles derived from an unrecognized duplicated genomic area [187]. Amplification of these supernumerary loci (isoloci) frequently varies with the experimental conditions, and can cause problems when data are being combined from several laboratories.
In autopolyploids and in allopolyploids with low genomic differentiation, SSR analyses usually produce multiple alleles of a single locus in each genotype, as demonstrated in the autopolyploid and apomictic Ranunculus kuepferi[216] and in allopolyploid species and cultivars in the genus Rosa[217-219]. To fully utilize the information content of the obtained DNA profiles, segregation patterns must be determined. This, however, requires the ability to score allele dosage, in contrast to just the presence or absence of an allele. The MAC-PR approach (microsatellite DNA allele counting - peak ratios) determines allele copy number based on quantitative differences between microsatellite allele peak ratios and therefore allows the precise determination of allelic configuration in each studied sample, as was shown in tetraploid roses [217,220]. Using this approach, inheritance patterns have been studied even in the absence of experimental crosses [221]. High-quality banding patterns are, however, needed for successful application of the MAC-PR, as well as repeatability of relative allelic amplification intensities among individuals and, thus, homology of microsatellite marker alleles within a species.
In crop plants with detailed pedigree information, the so-called microsatellite allele dose and configuration establishment (MADCE) procedure can be used to trace the transmittal of SSR alleles through documented generations of the investigated plant material, and determine the exact allele copy number in the target cultivars [222]. Originally, the MADCE procedure was applied in apple [222] but informative results have recently been obtained also for the Strawberry Crop Reference Set within the RosBreed research project (Bassil N, personal communication).
In many situations, allele dosage can, however, not be accurately scored with the methods chosen, and much plant material lacks or has only unsatisfactory pedigree information. Specialized programs have therefore been developed for analyzing polyploids with SSR markers, such as, for example, TETRA [223] and POLYSAT [224]. The fitTetra R package has been developed for enabling genotype calling in tetraploid species from biallelic marker data, and is especially useful for large-scale SNP analyses in material with high levels of polysomic allele segregation such as potato [225]. By contrast, the bead array MSV package [226] appears to be more useful for material with mainly disomic segregation.
A major drawback with any multi-locus approach is the loss of information about exact levels of heterozygosity and about genome inheritance. In addition, genetic distances between cultivars are exaggerated as compared to distances calculated on the basis of co-dominant data [220]. A method for calculating genetic distances that permits unbiased comparisons between different ploidy levels has, however, been described [227] and is available in the computer program package GENOTYPE/GENODIVE. Another approach is based on the formation of multi-locus allele phenotypes of each investigated individual, and calculation of phenotype-based estimates of genetic diversity and differentiation [228]. For more information on methods to describe the population genetics of polyploids, see Assoumane and colleagues [229].
Plant speciation, phylogeny and systematics
As the availability of DNA-based information increases, more attention is being paid to the genomic patterns of differentiation among plant species. According to the genic view of plant speciation, small “genomic islands” may be responsible for much of the differentiation between taxa through divergent selection or reproductive isolation barriers, while the remainder so-called “porous genome” is more permeable to gene flow [230,231]. In this context, the choice of molecular method becomes crucial for the ability to reflect genomic differentiation in a phylogenetically relevant perspective. To determine relative marker sensitivity in monitoring inter-specific differentiation, Scotti-Saintagne and colleagues [232] conducted a genome scanning experiment with 389 markers (allozymes, AFLPs, SCARs, SSRs and SNPs) on samples from pairs of populations of the sympatric oak species Quercus robur and Q. petraea. Distribution of markers according to their ability to detect inter-species diversity was clearly L-shaped; apparently only a few markers were located in genomic regions responsible for species differentiation. As expected, these markers were more likely to reside in coding regions than in non-coding regions. In another genome scan based on 88 mapped SSR loci, most loci again showed considerable migration between the analyzed taxa: sunflower species Helianthus annuus, H. debilis and their inter-specific hybrid [199]. The genomic regions that are responsible for genetic differentiation therefore appear to be small in these taxa, whether estimated as level of species differentiation or as migration rates.
When targeting differentiation at a larger taxonomic scale, DNA sequence information of the chloroplast genome and/or nuclear genic regions, is usually preferred over DNA fingerprinting. Proper elucidation of the complex puzzle of plant systematics is, however, often best achieved with a combination of different types of molecular information. In many plant groups, various types of multi-locus or single-locus markers have thus provided important pieces to the puzzle. So far, these marker loci have seldom been placed on a genomic map (but see, for example, [199,232]), and whether they reside in a conserved versus a “porous” part of the genome is usually unknown. Instead, choice of markers has mainly been based on the feasibility of obtaining a sufficiently large number of polymorphic bands.
Multi-locus DNA profiling methods such as AFLPs have become the most commonly used DNA fingerprinting tool in plant systematics, mainly in situations where DNA sequencing produces insufficient phylogenetic resolution [233]. In an early plant systematic study using AFLPs, 551 polymorphic bands were obtained with three primer combinations for 30 accessions from 19 taxa of Solanum section Petota and three taxa of Solanum section Lycopersicum[213]. Ploidy level was reflected in the profiles, with hexaploids exhibiting more bands than tetraploids and diploids. Mating system had, as expected, a large impact, with 40 to 60% intra-specific polymorphism detected in outcrossing taxa as compared to only 0 to 2% in selfing taxa. AFLP methodology was also employed to investigate phylogenetic relationships among 43 species of the paleotropic pioneer tree genus Macaranga[234]. About 30 of these species have a symbiotic relationship with specific ant partners. The resulting phenograms supported the monophyly of several sections and subsectional groups within the genus, and provided evidence for a polyphyletic origin of the ant-plant mutualism.
Besides the two species of cultivated rice, the genus Oryza also comprises of around 22 wild species that have received considerable attention due to their potential importance for rice breeding. Six diploid genomes (A, B, C, E, F and G) and four allotetraploids (BC, CD, HJ and HK) have been identified using, among other methods, total genomic DNA hybridization [235]. In another early study on rice, 77 samples representing 23 Oryza species were analyzed with AFLP [236]. Pairwise genetic distances showed a linear increase depending on the taxonomic level, with 0.02 to 0.21 within species, 0.2 to 0.35 between species sharing the same genome type, and >0.7 between species carrying different genomes. For the subsequent analysis of phylogenetic relationships among these genomes, more conserved markers were developed through the identification and sequencing of numerous rice genes [237]. Comparison of sequences for 142 such genes in six species, representing the six different diploid genomes, allowed the reconstruction of the rapid diversification in Oryza. In a follow-up study based on the sequences of 106 nuclear genes, divergence times and ancestral effective population sizes were also determined [238].
In the large and complex genus Rosa, several different DNA-based methods have been applied for phenetic and phylogenetic analyses, with mostly consistent results - for example SSR [239] and AFLP [240]. Two major clades were identified, with sections Carolinae, Cinnamomeae and parts of Pimpinellifoliae forming one clade and most of the other seven commonly recognized sections forming the other clade. As for the division into sections, Synstylae appears to be mainly monophyletic and rather closely affiliated with sections Indicae and Rosa[240]. Furthermore, section Pimpinellifoliae is apparently polyphyletic, and R. spinosissima should be separated from the other species in this section. In spite of its size (currently, about 50 species are acknowledged) and hybridogenous origination, the mainly European section Caninae (also known as dog roses) apparently constitutes a well-circumscribed monophyletic group. Another, very large AFLP study was recently conducted on >900 dog rose specimens sampled in a transect across Europe, with more than 200 non-dog rose samples analyzed for comparison [241]. Two lines of statistical analyses were applied: (1) an unstructured model with principal coordinate analysis and hierarchical clustering, and (2) a model with a superimposed taxonomic structure based on analysis of genetic diversity using a novel approach that combines assignment tests with canonical discriminant analysis. Support was found for five of the seven subsections, including the three major ones: Caninae, Rubigineae and Vestitae. Within the subsections, many species overlapped considerably, and geographic distances often appeared to be at least as important as the conventional taxonomy in explaining similarities between analyzed specimens. Complementary information on phylogeny in Rosa has also been obtained with DNA sequencing. Although sharing some ITS sequence types with species in other sections thereby confirming their hybridogenous origin, the Caninae species also have one unique ITS sequence type which is further evidence of their monophyly [209,242].
Plant systematic studies have occasionally been conducted also using SSR markers, especially when the focus has been on genetic differentiation among closely related taxa. For example, ten Puerto Rican populations of the cycad genus Zamia were analyzed with 31 SSR primer pairs [243]. These populations could be treated either as belonging to a single polymorphic species, Z. pumila, or as representing three more narrowly circumscribed taxa: Z. erosa, Z. portoricensis and Z. pumila sensu stricto. The SSR analysis showed that Z. erosa is strongly differentiated from the other two species, and thus may represent an independent introduction into Puerto Rico. The data are consistent with an allopatric speciation scenario with Z. portoricensis being the youngest taxon according to Bayesian coalescent analysis and effective population size, and still showing considerable admixture with Z. pumila.
Genetic relationships among 35 Arachis species from seven sections, including 11 accessions of cultivated peanut, A. hypogaea, were analyzed on the basis of allelic variation at 32 SSR loci [244]. A neighbor joining tree was generated on the basis of pairwise Dice distances between individual accessions, calculated from a binary presence/absence matrix of SSR alleles. Most con-specific accessions grouped together on the tree, as did species from the same section, with several exceptions that were attributed by the authors to either homoplasy in the dataset or extensive within-species variation.
From the relatively few studies available, it appears that SSR markers do have some potential not only for species delimitation, but also for the reconstruction of genetic relationships among closely related species groups that are only a few million years old. However, SSR markers are usually highly polymorphic and therefore multiallelic within a species. Accordingly, the within-population component of SSR variation is often much higher than the between-population or between-species component [245]. It is therefore a “must” that several accessions per species are included in any phylogenetic study that is based on SSRs, the more the better. Optimally, genetic distances between populations (or species) rather than genetic distances between individuals should be used to generate phenetic trees.
Phylogeography
Phylogeography aims to study the spatio-temporal history of a species on the basis of its intra-specific genetic variation [246]. In principle, phylogeographic studies can be based on information from either nuclear, mitochondrial or chloroplast DNA. In practice, organellar DNA is usually preferred since organelle-derived markers are more likely to retain information about biogeographical history than nuclear markers [247]. There are several reasons for this. First, the haploid genomes of plastids and mitochondria exhibit a smaller effective population size as compared with the diploid nuclear genome, resulting in stronger substructuring of fragmented populations under genetic drift. Second, organellar genomes are usually inherited uniparentally. In angiosperms, the plastid DNA is generally transmitted by seeds - that is, maternally. Given that plants can colonize a new habitat only by seeds, plastid-derived markers have the potential to provide information about past changes in species distribution that is unaffected by pollen flow. Third, intermolecular recombination is usually absent in plastid DNA, so that individual sequence polymorphisms can be combined into haplotypes that remain mostly unchanged when passed to the next generation.
Evolutionary relationships between cpDNA haplotypes are often depicted as networks [248], which can be superimposed on the geographic distribution of the sampled plants. One has to keep in mind, however, that a non-recombining DNA molecule behaves like a single gene. The phylogeographic pattern retrieved from a plastid haplotype network therefore only represents one out of several possible outcomes of the genealogical process [249]. This is why phylogeographic analyses based on other genes and genomes are becoming increasingly popular. In conifers, where plastid DNA (paternal) and mitochondrial DNA (maternal) show contrasting modes of transmission from parents to offspring, both genomes have often been analyzed side-by-side [250,251]. In addition, phylogeographic studies often employ nuclear ribosomal ITS sequences. In most plant species, the ribosomal genes are rapidly homogenized by concerted evolution and then behave like uniparentally inherited organellar DNA.
There is no clear division between phylogeography on the one hand and traditional population genetics on the other. Accordingly, the use of nuclear SSR markers to study genetic diversity, genetic subdivision and gene flow within and among extant species is sometimes also called “phylogeography” [252], and there have been numerous successful attempts to elucidate intra-specific phylogeographic patterns by multi-locus DNA profiling methods such as RAPD, ISSR and AFLP [253,254]. Multi-locus banding patterns are typically analyzed phenetically - that is, phenograms or networks are reconstructed on the basis of a pair-wise similarity matrix that is generated from a binary presence/absence matrix of band positions. The (groups of) genotypes depicted in the resulting phenogram or network are then compared with their geographic distribution [253].
The majority of plant phylogeographic studies still rely on plastid DNA polymorphisms that can be searched for by either PCR-RFLP, screening of length-variable plastid microsatellites (cpSSRs), or by comparative sequencing of PCR-amplified non-coding DNA [255]. Unique polymorphisms are then combined into distinct haplotypes, followed by the analysis of haplotype distribution and frequencies in different geographical regions, quantification of the genetic divergence between haplotypes, and the evaluation of genetic relationships between haplotypes - for example, in the form of a statistical parsimony network such as TCS [256]. The use of cpSSRs is, however, controversial, since their often high mutation rates can cause homoplasy [257,258].
Application areas of marker-based phylogeographic studies are diverse, and include, for example, the analysis of postglacial re-colonization patterns of the Central European landscape by trees and shrubs in the Quaternary [252,259], the identification of glacial refugia [260], the reconstruction of migration routes of halophytes along coastal and inland salty habitats [253], the investigation of the evolutionary history of tropical trees [255] and the historical biogeography of threatened species [251]. Increasingly important are comparative phylogeographies that involve numerous animals and plants from the same geographical region [261,262]. Such meta-analyses yield invaluable data on common evolutionary patterns across many biota from large geographical areas.
Genetic mapping
Linkage mapping and genetic maps
One prominent application of molecular markers is the generation of genetic maps which have been established for all major and many minor crops and other plants (for example, rice [263], barley [264], and maize [265], to name just a few). A genetic map is a graphic representation of a chromosome (or linkage group) onto which genetic elements (= loci, for example markers or genes) are aligned. The loci are arranged based on their co-segregation during meiosis, which depends on the frequency of recombination events. Genetic distances between loci are measured in centiMorgan (cM). One cM is defined as the distance that two loci have to each other, if in 100 meiotic events the loci are segregating only once (= 99% chance of co-segregation). As the extent of recombination varies in different genomes, this translates into varying physical distances. The recombination frequency also varies among different genomic regions - for example, recombination is suppressed near centromeres.
To estimate genetic distances among loci, the co-segregation of genetic elements is monitored in mapping populations or in association mapping approaches (see below). Mapping populations usually originate from a cross between two parental lines, which ideally can be distinguished by a large number of polymorphisms that are monitored in the progeny. Particularly convenient mapping populations consist of so-called Recombinant Inbred Lines (RILs), which are generated by selfing single-seed descent from different sibling F2 plants through six or more generations. The continuous selfing causes very high levels of homozygosity, and each RIL from a population of RILs hence conserves one particular recombinational event from the F1 cross. Design and construction of RILs have been reviewed by Pollard [266].
To identify loci that are very tightly linked with a specific trait, “fine-mapping” is performed by enriching the density of markers in proximity of the responsible genes or, in the best case, markers for the responsible genetic elements themselves. The most commonly applied technique for fine-mapping is Bulked Segregant Analysis (BSA), originally developed by Michelmore and colleagues [267]. In a BSA, all genotypes that show a specific phenotype (that is, a specific trait) are pooled and screened for polymorphisms that distinguish them from the remaining plants. All genetic elements that do not influence the bulk-trait are randomly distributed among all plants, whereas all genetic elements responsible for the trait are to be found preferentially if not only in the respective bulk. In consequence, any difference between the bulk and the remaining plants is likely to be linked with the trait of interest. The source of polymorphisms can be, for example, the metabolome, the proteome, the transcriptome or the genome. The latter two have profited enormously from the advent of high-throughput-sequencing technologies and are now the most widely used sources for genetic polymorphisms.
Different types of markers can be combined into integrated maps, which become more highly resolved (that is, saturated) with each newly added marker. Furthermore, data from different crosses can be integrated in the same map. For example, Wenzl and colleagues [264] published an integrated map for barley using DArT, SSR, RFLP and STS markers, altogether comprising 2,935 different loci. In the current era of genome sequencing, genetic maps are also a versatile tool for defining the order of assembled contigs from shotgun sequencing approaches, as has been done, for example, during the assembly of the recently published chickpea genome [268].
Association mapping
Association mapping (AM) aims at linking phenotypes to genotypes, independent of the kinship of the genotypes. The concept of AM has been implemented in humans and model organisms for many years (for example within the human HapMap project that started in 2002), and is now increasingly applied for plant genomes (see the reviews by Abdurakhmonov and Abdukarimov [269] and Soto-Cerda and Cloutier [270]). The major advantage of AM over linkage mapping (LM) or QTL mapping is that no mapping population is required. The establishment of good mapping populations is a time-consuming and costly task, especially for plants with long generation times and hence a limited number of meiotic recombinations. Furthermore, LM is usually restricted to a small subset of genotypes and to those loci that are polymorphic among these genotypes (= low allelic richness). In contrast, AM examines genotype-phenotype correlations in a large germplasm and hence monitors the historical meiotic recombination events that accumulated in natural populations and collections of landraces, breeding materials and varieties [270].
Association mapping is based on the occurrence of Linkage Disequilibrium (LD) between a particular trait and one or more alleles of a marker locus in a population. In contrast to LM, which refers to the combined inheritance of loci due to their close physical proximity on the same chromosome, LD refers to the non-random occurrence of allele combinations of loci in a population. Thus, the reason for LD can be linkage (and in most cases it is), but also other factors influence LD, such as selection, mutation, mating system, population structure, and so forth, which can result in significant LD even of alleles that are located on different chromosomes [270]. Because of this, AM is more complex than linkage mapping and might be biased by various factors.
More recently, AM-based analyses have been successfully carried out in many crops. In rice, for example, Zhao and colleagues [271] genotyped more than 44,000 SNPs across 413 accessions from 82 countries. Dozens of variants could be identified that influence numerous complex traits. In maize, a high-density analysis based on 56,110 SNPs was performed to analyze chilling tolerance in 375 inbred lines [272]. Nineteen highly significant association signals that explained between 5.7 and 52.5% of the phenotypic variance observed for early growth and chlorophyll fluorescence parameters were identified. An AM-based approach that was termed “landscape genomics” aims at simultaneously examining the effects of demographic history, migration and selection in a defined geographical site (see, for example, Sork and colleagues and references cited therein [273]).
Thanks to new, high-density genotyping methods such as the INfinumHD assay that assesses thousands of markers simultaneously, almost all major crops can now be subjected to AM [91]. High-density SNP arrays that comprise the information of several thousand loci were recently also developed for forest trees and horticultural plants, including white spruce, Picea glauca[274], peach [275], apple [276] and sweet and sour cherry [277], and are expected to greatly facilitate AM also in these plant species. A combination of LM and AM represents a particularly powerful tool for selection. Thus, Yu and colleagues [278] presented a so-called “nested association mapping” (NAS) approach in maize that involved crossing of 25 different variants and 5,000 offspring, whereas Kover and colleagues [279] performed a “Multiparent Advanced Generation Inter-Cross” in Arabidopsis. Nineteen variants were crossed in a random mating scheme, resulting in 527 F4 plants. The RILs originating from the maize NAS recently helped to identify important genes involved in maize kernel composition [280], resistance to northern leaf blight [281] and stalk strength [282]. In apple, Khan and colleagues [283] used an AM and LM combined approach to identify three important QTLs for fire blight resistance.
Marker-assisted breeding and genome-wide selection
One major aim of genetic linkage analysis in crop plants is marker-assisted breeding (see the review by Jiang [284]). A particularly promising current concept of marker-assisted breeding has been termed “genomic selection” (GS) or “genome-wide selection” (GWS) [285]. In contrast to the traditional marker assisted selection (MAS) concept, where only a subset of markers is considered, in GS all available markers are evaluated simultaneously for the calculation of a so-called breeding value. This is done by combining major and minor QTLs according to Meuwissen and colleagues [286]. In this way, QTLs with only minor positive and negative effects that are missed in traditional MAS are also taken into consideration for selection. The concept of GWS is widely used in livestock breeding and has been discussed as a future selection approach also for plants [287]. In their analysis that was based on a large data set of 25 nested association mapping populations, Guo and colleagues [285] found better predictions using the GWS approach as compared with MAS for flowering traits in maize (days to silking, days to anthesis and anthesis-silking interval). MAS was performed by composite interval mapping (see the review by Zou and Zeng [288]), and GS using a “ridge regression-best linear unbiased prediction” to calculate breeding values.
For genotyping moderate numbers of SNP loci in hundreds to thousands of samples, PCR-based approaches provide a more flexible alternative to microarray-based methods. Besides direct sequencing, three currently popular methods for SNP genotyping of PCR products are high-resolution melting (HRM) analysis, allele-specific PCR (ASP), and the TaqMan assay. In HRM analysis, the PCR product is continuously heated, and the separation of the two DNA strands is monitored in real time [289]. Polymorphic PCR products that differ slightly in length or sequence will have different melting temperatures. These differences can be measured with sensitive optics, which monitor the fluorescence-to-signal intensity of an intercalating dye. HRM analysis has, for example, been successfully applied to gene mapping in rice [290], to cultivar identification in sweet cherry [291], and to discriminate between closely related chloroplast DNA haplotypes in the wild species Arenaria ciliata and A. norvegica[292].
In ASP, the alternative alleles at a particular polymorphic site are amplified with allele-specific primers that are each labelled with a different fluorochrome. The presence of a particular allele is hence indicated by a diagnostic fluorochrome signal. If primers of different length are used, the ASP products can also be assessed by gel electrophoresis. The TaqMan™ assay dates back to the early 1990s [293]. It involves the fluorescence-based detection and quantification of a specific probe that is hybridized to the SNP site of interest. A light signal is only emitted when the probe is degraded by the exonuclease activity of the Taq DNA polymerase, which occurs only when the probe has specifically bound to its target site. The ASP and TaqMan™ assays can be assessed in regular quantitative PCR machines while HRM requires specific optics.
The status of traditional DNA fingerprinting: concluding remarks
Taken together, the last two decades have witnessed a prominent increase in the application of various DNA markers for plant DNA fingerprinting. In the beginning, multi-locus dominant markers, especially RFLP, RAPD, AFLP and ISSR, were most popular, but single-locus SSRs and eventually SNP markers rapidly caught up. The chip-based DArT technology is also still used. We believe that traditional multi-locus methods and their various spin-offs will still be employed a decade from now, but mostly for exploratory research that does not necessarily result in published papers. Locus-specific SSR markers will probably remain more popular, due to their co-dominant inheritance, ease of analysis and the fact that new data can easily be added to already existing files. Novel input to DNA fingerprinting was, however, provided by an unforeseen major breakthrough in DNA sequencing technology, which can be envisaged as the starting point for the future of DNA fingerprinting, and which is discussed in the following section.
The future of DNA fingerprinting
The advent of massively parallel high-throughput genomic sequencing about 10 years ago was a breath-taking step forward in the yet short history of molecular genetics and genomics (see, for example, the reviews by Mardis [294], Metzker [295], and Rothberg and Leamon [296]). The immense speed with which DNA sequences can be read by 454 pyrosequencing, SOLID, Illumina and other machines also had a considerable and two-fold impact on the development and use of molecular markers in general, and on DNA fingerprinting in particular. On the one hand, conventional markers such as microsatellites and SNPs can now be discovered with reduced cost and effort and at unprecedented rates. On the other hand, DNA marker technologies are currently successively complemented or even replaced by the sequencing process itself, as is expressed by the term “genotyping-by-sequencing” [87,297,298] (see also the Special Issue of Molecular Ecology 22(11), 2013).
Discovery of nuclear microsatellites by high-throughput DNA sequencing
Quite obviously, random high-throughput sequencing of genomic DNA offers itself as a useful strategy to identify all kinds of repetitive DNA in a genome, including microsatellites. It nevertheless took several years before this potential was realized. In one of the first reports on the use of next-generation DNA sequencing for microsatellite marker development, Abdelkrim and colleagues [299] used a 454 platform to produce 17,215 reads of unselected, fragmented genomic DNA of the blue duck (Hymenolaimos malacorhynchos), a waterfowl species endemic to New Zealand. Each read had an average size of 243 bp, adding up to a total of approximately 4.1 Mb. Using appropriate bioinformatic tools, microsatellites were detected in 231 reads. The number of suitable marker loci was, however, reduced to 24 by the necessity to design primers on either side of the microsatellite. Thirteen of the primer pairs displayed polymorphism, and 13 markers were thus generated in a single sequencing run.
Santana and colleagues [300] also used 454 technology to create microsatellite markers from a fungus (the pine pathogen Fusarium circinatum), an insect (the wasp Sirex noctilio) and a nematode (Deladenus siridicola). Two methods, ISSR-PCR and “fast isolation by AFLP of sequences containing repeats” (FIASCO), were used to enrich the template DNA for microsatellites prior to sequencing. Altogether, 1.2 to 1.7 Mb of DNA were sequenced, and 873 potentially amplifiable microsatellites were identified with sufficient flanking sequence available for primer design. A set of 28 SSR-flanking primer pairs were developed for Fusarium circinatum. Of these, 19 yielded single fragments in the expected size range, and 13 produced polymorphic amplicons from a set of fungal isolates. The authors also generated a traditional library from F. circinatum DNA enriched for microsatellites by the ISSR method. Sanger sequencing of 100 clones from this library yielded only eight potentially amplifiable microsatellites.
Again using a 454 platform, Allentoft and colleagues [301] searched for microsatellites in an ancient DNA source, a bone fragment of an extinct New Zealand moa species (a flightless bird). A total of 79,796 sequences were obtained with an average length of 112 bp. Of 195 di-, tri- and tetranucleotide repeats present in the data set, only one polymorphic microsatellite marker could eventually be generated. This low yield can be accounted for by the ancient source and therefore degraded state of the DNA and hence short read lengths.
The above three papers were published in the same issue of BioTechniques and indicated for the first time that high-throughput sequencing has a strong potential to isolate SSR markers from non-model organisms where no genomic information exists. The novel methods save a lot of time, and are also cost-efficient as compared to traditional enrichment cloning. For example, Csencsics and colleagues [302] spent approximately US$5,000 to develop microsatellite markers for the plant species Typha minima within 6 weeks. In their study, 307 di-, tri- and tetranucleotide repeats were found in a total of 76,692 sequence reads. One hundred loci were selected for primer design, 30 primer pairs were tested and yielded 17 polymorphic markers.
The relatively long read-lengths provided by 454 sequencing facilitate the identification of enough flanking sequence on either side of the SSR for the design of PCR primers. 454 technology was therefore initially preferred over other next-generation sequencing approaches for generating SSR markers. In plants, 454 sequencing was first applied to microsatellite isolation in Typha minima[302], Amaranthus tuberculatus[303] and Vigna radiata[304]. Many more studies followed, and genomic shotgun sequencing soon replaced traditional enrichment strategies as the method of choice for generating sets of microsatellite markers in any organism (see the review by Zalapa and colleagues [305]). More recently, SSR identification from paired-end genomic sequencing using the more economical Illumina platforms has been advocated by several working groups [306,307], especially since average lengths of Illumina reads have considerably increased over the years. For example, Castoe and colleagues [306] showed in one snake and two bird species that 454 and Illumina detect similar numbers of potentially amplifiable SSRs on a read-by-read basis, but the much lower costs per sequenced nucleotide are clearly favoring Illumina.
Given that sequencing methodology is still developing at a rapid pace, a further decrease of costs may be expected. The large amount of sequence data obtained in even a small-scale experiment allows a top-down selection of the most promising candidates (for example, only trinucleotide repeats, or only perfect repeats). Enrichment for microsatellite motifs prior to sequencing is sometimes advocated [307,308], but is probably unnecessary in most cases [306]. The use of (bar)coded adapters and primers for the amplification step enables the parallel sequencing of multiple templates at the same time [307,309], which further contributes to the efficiency of the method. Using barcoded adaptors (also known as multiplex identifier adaptors), Takayama and colleagues [310] were thus able to isolate a large number of microsatellites from only 10,000 to 20,000 genomic 454 reads each of six unrelated plant species at low cost.
Concerning the read-length of high-throughput sequencing platforms, a major breakthrough was recently reached by Pacific Bioscience, whose SMRT cells produce read-lengths of up to 15 kb. Sequence quality is, however, relatively low. It was therefore suggested to additionally produce reads with another sequencing platform (for example, Illumina) to correct the long PacBio reads in a hybrid sequencing approach [311]. Another alternative relies on the circularization of template molecules prior to single-molecule sequencing, and multipass sequencing of the same circular templates was reported to generate highly accurate consensus sequences [312]. Most recently, Grohme and colleagues [313] demonstrated for the first time the successful use of single-molecule circular consensus sequencing on a PacBio RS platform for discovering SSR markers in the genome of a goose, Anser albifrons. Whatever technology is used for sequencing, it is essential that appropriate bioinformatic tools and hardware are available to cope with the huge numbers of sequences that have to be screened.
An additional benefit of the use of next-generation sequencing for SSR marker development is the large amount of random nuclear and organellar sequence data generated as a “by-catch” of microsatellite identification. For example, 382 SSRs were sampled in the course of 454 sequencing of Amaranthus tuberculatus along with a contig representing an almost complete chloroplast genome, mitochondrial DNA fragments, transposable elements and numerous nuclear genes of interest [303]. Krapp and colleagues [314] assembled approximately 84% of the Dyckia marnier-lapostollei (Bromeliaceae) chloroplast genome from only 59,624 pyrosequencing reads. A total of 34 chloroplast microsatellites (cpSSRs) with a minimum number of 10 A´s or T´s were also found, and flanking primers were constructed that produced highly polymorphic amplification products in various Dyckia species.
High-throughput genotyping-by-sequencing
The radically increased throughput in sequencing capacity at 50 to 100,000 times lower costs per base as compared with traditional Sanger sequencing also had a large impact on DNA fingerprinting technology itself. High-throughput sequencing opened completely new and unprecedently fast avenues for genotyping (up to the highest possible resolution) the resequencing of the entire genomes of populations of plants (WGS, for example [315,316]). Whereas the list of fully sequenced plant genomes is constantly growing (for crop plants, see Bevan and Uauy [317]), most plant genomes are nevertheless too large, too complex, and too rich in repetitive DNA to be good candidates for WGS. This is why essentially all genotype-by-sequencing approaches aim at reducing the complexity of genomes to a smaller subset (“reduced representation sequencing”; for example, see [298,318-324]). By reducing the analyzed genome space to a manageable size, a sufficiently high sequence coverage can be attained that allows for the detection of polymorphisms (mostly SNPs) with necessary confidence, even in plants with very large genomes.
Several strategies were developed that aim to enrich particular portions of the genome prior to sequencing. One obvious approach is to sequence only the transcriptome that is available in ESTs or cDNA libraries (for details see "Transcriptome sequencing: RNAseq and related approaches" below). Other techniques rely on the prior amplification of certain genomic subsets using PCR with selected primers (for example, with SRAP primers [321]), or enrich certain parts of the genome by hybridization with a pre-cast set of oligonucleotides (for example, see [322]). In the currently most widely used technique, genomic DNA is first digested with a restriction enzyme, and only the regions on both flanking sites of the enzyme-recognition sites are sequenced (for details see "Restriction Enzyme Anchored Sequencing: RADseq and related approaches" below). This principle was first presented by Altshuler and colleagues [325] who described it as “reduced representation shotgun sequencing”, still using Sanger methodology. A different version of this approach, already involving high-throughput sequencing, was introduced by van Orsouw and colleagues [323] who referred to their technique as CRoPS (Complexity Reduction of Polymorphic Sequences). The most widely used term for the concept, including its many variants, was however suggested by Baird and colleagues [319] who coined their approach “Restriction Site Associated DNA Sequencing” (RADseq). As all of these techniques aim at sequencing restriction site-associated DNA, we will here use the more general term “Restriction Enzyme Anchored Sequencing” (REAS; see Figure 4).
Figure 4.

Schematic outline of a typical “Restriction Enzyme Anchored Sequencing” (REAS) approach. (1) DNA from different genotypes is digested with one or several restriction enzymes. (2) Biotinylated adapters that carry a barcode for distinguishing the different genotypes are ligated to the restriction site. (3) A second fragmentation step can be applied, either involving random shearing, or further digestion of the sample with a second restriction enzyme. Biotinylated fragments are then bound to a streptavidin matrix. A second adapter is ligated to the construct that is now ready for sequencing, usually after some PCR cycles. A size selection-step can be included after the first or the second restriction digestion to further reduce the complexity. A mix of both adapters or Y-adapters can also be ligated directly after the first or second digestion or after random shearing. (4) Samples are sequenced and (5) sorted according to barcode, and the tag-sequences are compared for single nucleotide polymorphism detection.
Restriction Enzyme Anchored Sequencing: RADseq and related approaches
Depending on its recognition sequence, any given restriction enzyme targets only a small proportion of the entire genome. Restriction digestion is therefore the first (and sometimes the only) step of reducing genomic complexity in REAS. As the restriction sites in genomes of closely related genotypes are usually shared, orthologous genetic regions are sequenced throughout the different genotypes. By choosing an appropriate (pair of) restriction enzyme(s), the extent of reduction of genome representation can be tailored experimentally. For example, a four-base cutter would (in a theoretical genome with a random base distribution) cut every 256 bp, and a six-base cutter every 4,096 bp. In order to focus the sequencing on the non-repetitive, transcriptionally active parts of the genome, most applications make use of methylation-sensitive enzymes that digest only non-methylated sites. Although different epigenetic regulation and accompanying methylation patterns might occur in the different genotypes of interest and hence there is the risk of sequencing and comparing different genetic regions in different genotypes, the advantages outweigh this risk.
One version of the many different REAS techniques is summarized in Figure 4, as a representative example. After digestion, an adapter is ligated to the restriction site, which can directly be used for sequencing on the desired high-throughput platform. The adapter contains a sample-specific barcode sequence which permits the simultaneous “multiplexed” high-throughput sequencing of numerous samples. The adapter can be biotinylated at the opposite end of the ligation site (as in Figure 4) or contain other modifications that inhibit concatemerization of the adapters. After barcoding, the DNA can be either further digested with another restriction enzyme, as in “Double Digest RADseq” [326] or can be randomly sheared to reach the optimal size for the desired sequencing platform. The latter approach was chosen in the original RADseq paper [319]. A second, platform-specific adapter is then ligated to the opposite end of the adapter-DNA fragment. A further reduction of complexity can be achieved by size-selection of the digested DNA. In this case, a frequently cutting restriction enzyme is used, as was proposed by Elshire and colleagues [320] who called their technique “genotyping-by-sequencing” and by Andolfatto and colleagues [318], who used the term “multiplexed shotgun genotyping”.
The primary data comprise the sequence information of the region flanking the restriction site to which the adapters were ligated. These restriction site-associated DNA sequences are referred to as “tags”, or “RAD tags”. Sequencing can be performed either by single end- or by paired-end sequencing. The latter is usually more cost efficient and facilitates mapping of the sequenced region to known related genomes [327,328]. By far the largest part of the restriction sites will usually represent orthologous loci throughout the different genomes. The tagged sequences can then directly be aligned and compared, allowing for the identification of SNPs or indels. The RAD sequencing approach is different from AFLPs or CAPS markers, which focus on differences in the recognition sites themselves. The genome space that is analyzed by an REAS is hence much bigger as compared to procedures that are based on restriction-site polymorphisms only. However, absence versus presence of restriction sites in the different organisms can also be exploited.
Provided that the genome under investigation is not too large, and an adequate sequence coverage is obtained, each particular RAD tag is resequenced many times. The data obtained from an REAS experiment can therefore be exploited for identifying huge numbers of SNP markers [329,330]. REAS data can also be directly used for generating linkage maps from a segregating population [331], or the polymorphic DNA sequences can be annotated to a reference genome, for genomic localization of the newly identified markers [92]. The RAD tags can also be annotated to genomes of plants with high levels of synteny as a cross-reference. This is a big advantage compared to many other DNA fingerprinting techniques, as, when a reference genome is available, the polymorphisms can often directly be localized, which facilitates, for example, fine mapping of QTL. The newly discovered SNPs and other polymorphisms can also be used as markers for fingerprinting or mapping in subsequent steps - for example, in microarrays or CAPS [330]. Several programs are available for the analyses of the data, such as “RADtools”, inaugurated by Baxter and colleagues [332], “Stacks” [333], “UNEAK” [334], “TASSEL” [335] and “Rainbow” [336]. To determine points of chromosomal recombination, a Hidden Markov Model was implemented by Andolfatto and colleagues [318].
Numerous applications of REAS have recently been described in plants (see the review by Poland and Rife [337]). For example, in artichoke, Cynara cardunculus, Scaglione and colleagues [330] discovered approximately 34,000 SNPs and nearly 800 indels from 9.7 million reads, corresponding to 1,000 Mb of sequence. Bus and colleagues [329] sequenced more than 113,000 RAD tags and identified more than 20,000 SNPs and indels in the allotetraploid rapeseed, Brassica napus. Yang and colleagues [338] used RADseq in a BSA to identify markers for anthracnose disease resistance in Lupinus angustifolius. Finally, Pfender and colleagues [331] used RADseq to genotype 193 F1 individuals from a cross between stem-rust resistant and susceptible parental lines of Lolium perenne and found several major QTLs for rust resistance.
Transcriptome sequencing: RNAseq and related approaches
Another widely used application of high-throughput sequencing for the discovery of polymorphisms is the sequencing of cDNA in an approach coined RNAseq (sequencing of transcribed regions). RNAseq is basically EST sequencing without prior cloning and on high-throughput sequencing machines. The technique and its applications in plants were recently reviewed by Martin and colleagues [88], and an “RNAseq tutorial” was presented by Wolf [339]. Several advantages make transcriptome sequencing very appealing. (1) After de novo assembly or annotation, the protein sequence as well as its variants are directly revealed. These polymorphisms represent the ideal markers for gene identification in a mapping approach, as the gene itself and not a co-segregating, anonymous genomic region is marked. (2) The method reduces the complexity of a genome to its transcriptionally active parts and thus allows the comparison of very large genomes. (3) RNAseq simultaneously allows both the determination of a genotype, and the assessment of quantitative gene expression, which permits the analysis of expression QTLs. This concept was described by Harper and colleagues [340] as “associative transcriptomics”. These authors used transcriptome sequencing in Brassica napus to correlate differences in gene sequences and gene expression on the one hand with trait variation on the other. The downside of the RNAseq approach is that, in order to be able to sequence a transcript, it must be expressed under the given circumstances - that is, in the investigated tissue - and at the time point of RNA isolation. Another problem is the uneven distribution of the transcript species, which either requires very deep sequencing, or normalization prior to sequencing. This problem especially concerns rare transcripts, such as those coding for transcription factors or other regulatory elements.
RNAseq has also been used in BSA. For example, Trick and colleagues [341] fine-mapped a cloned grain protein content gene, GPC-B1, in wheat, whereas Liu and colleagues [342] were able to directly locate, identify and clone the long-searched gene for glossy3 (gl3), which proved to be a putative myb transcription factor. A high-throughput sequencing-based, reduced complexity analysis strategy for plant transcriptomes was recently presented by Kahl and colleagues [343] who used an Illumina platform to sequence only the 3′-UTRs of the transcripts with Massive Analysis of cDNA Ends. As 3′-UTRs are the most polymorphic regions of a transcript, they provide a rich source of molecular markers. This strategy allows the sequencing of a larger number of genotypes in a multiplexed assay at sufficient coverage, when compared to RNAseq, but additionally reveals quantitative gene expression values for each transcript. To describe this concept, the term “TranSNiPtomics” was invented [343]. Unlike REAS, the technique does not depend on restriction enzymes, but targets a region of 100 to 500 bp upstream of the poly-A tail of each transcript molecule. The approach was recently successfully applied to fine-map an introgressed gene for yellow dwarf virus disease from H. bulbosum into barley [344].
Outlook
The large-scale identification of SNPs and SSRs via high-throughput sequencing approaches is now becoming routine, and highly multiplexed SNP-based genotyping-by-sequencing methods have already been used for numerous applications in many crop plants and model species. This is especially true for genetic mapping, as has been outlined in Genetic mapping. A steadily growing number of studies is also taking care of non-model wild species, where no reference genome is available [345,346]. In the field of population genetics, for example, the ability to sample the genome at much higher densities than ever before overcomes the limitations of traditional marker methodology, allowing for the first time the evaluation of genetic variation patterns across the whole genome [347,348]. The results and insights gained from such “population genomic” approaches will have an enormous impact on the fields of ecology and conservation, as was highlighted in a recent special issue of Molecular Ecology (see [349] and other papers in the same volume). Plant systematists will benefit from what may be called “next generation phylogenetics” [350], since plant phylogenies can now be set up on the basis of fully sequenced plastid genomes [351] and/or multiple sets of nuclear genes [352]. New and potentially useful nuclear markers can be identified by sequence comparisons over large taxonomic distances [353], and direct sequence analysis of barcoded individuals may resolve orthology problems in polyploid phylogenetics [352]. Finally, better-resolved phylogenies will help to elucidate species boundaries and species relationships in recent radiations (see the review by Harrison and Kidner [354]).
Where do we go from here? Most certainly, the full technological capabilities are still far from being attained, and “next-next-generation” single-molecule sequencing is currently replacing some of the pioneering methods such as 454 and SOLiD, which are already on the brink of extinction. Further improvements and innovations of sequencing methodology can be expected, and will soon allow for cost-effective genotyping-by-sequencing of any non-model plant species. Even whole-genome sequencing might become feasible for certain applications, such as the analyses of somaclonals or mutation screening. However, obtaining complete genomic sequence data for all individuals included in a study would be unnecessary in most situations, and only inflate the costs. The tremendously huge data sets generated by next-generation sequencing need to be handled and stored, and bioinformatics has already become a real challenge. An important question will therefore have to be asked at the onset of each study - that is, how to allocate the finite number of resources between (1) sequence coverage (that is, the number of times that a particular nucleotide is sequenced), (2) sampling density (that is, the number of individuals analyzed), and (3) the fraction of the genome that is subjected to sequencing [355]. We envisage that sequencing efforts will normally be confined to the necessary minimum also in the future, and that DNA fingerprints generated with an adequately tailored set of markers, obtained by low coverage sequencing of a well-selected representation of a genome, will remain the method of choice for most application areas in plant genotype identification, population genetics, relatedness studies and mapping. We therefore expect that the term “DNA fingerprinting”, once created by Alec Jeffreys for describing the unequivocal identification of human individuals by minisatellite hybridization, will survive in the long run, even in the era of brute force DNA sequencing.
Abbreviations
AFLP: Amplified fragment length polymorphism; AM: Association mapping; ASP: Allele-specific PCR; bp: Basepairs; BSA: Bulked Segregant Analysis; CAPS: Cleaved amplified polymorphic sequences; cDNA: copied DNA; cM: centiMorgan; cpDNA: chloroplast DNA; cpSSR: chloroplast simple sequence repeat; CRoPS: Complexity Reduction of Polymorphic Sequences; DAMD: Direct amplification of minisatellite DNA; DArT: Diversity Arrays Technology; EST: Expressed sequence tag; FIASCO: Fast isolation by AFLP of sequences containing repeats; GS: Genomic selection; GWS: Genome-wide selection; HRM: High-resolution melting; IBD: Isolation-by-distance; IRAP: Inter-retrotransposon amplified polymorphism; ISSR: Inter-simple sequence repeat; ITS: Internal transcribed spacer; LD: Linkage Disequilibrium; LM: Linkage mapping; LTR: Long terminal repeat; MAC-PR: Microsatellite DNA allele counting - peak ratios; MADCE: Microsatellite allele dose and configuration establishment; MAS: Marker-assisted selection; mtDNA: Mitochondrial DNA; NAS: Nested association mapping; PCR: Polymerase chain reaction; PDO: Protected denomination of origin; QTL: Quantitative trait loci; RAD: Restriction Site Associated DNA; RADseq: Restriction Site Associated DNA Sequencing; RAPD: Random amplified polymorphic DNA; rDNA: Ribosomal DNA; REAS: Restriction Enzyme Anchored Sequencing; REMAP: Retrotransposon-microsatellite amplified polymorphism; RFLP: Restriction fragment length polymorphism; RGAP: Resistance gene-analog polymorphism; RIL: Recombinant Inbred Line; RNAseq: Sequencing of transcribed regions; SAMPL: Selective amplification of polymorphic microsatellite loci; SCAR: Sequence characterized amplified region; SGS: Spatial genetic structure; SNP: Single nucleotide polymorphism; SRAP: Sequence-related amplified polymorphism; S-SAP: Sequence-specific amplification polymorphism; SSR: Simple sequence repeat; STR: Synthetic tandem repeat; TRAP: Target region amplification polymorphism; UTR: Untranslated region; WGS: Whole genome sequencing.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HN, KW and BR participated in the writing of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Hilde Nybom, Email: hilde.nybom@slu.se.
Kurt Weising, Email: weising@uni-kassel.de.
Björn Rotter, Email: rotter@genxpro.de.
Acknowledgements
We would like to thank Sina Möller for providing unpublished data on the genotyping of Vinca minor, and for help with preparing Figure 3. We also acknowledge the comments of two anonymous referees who helped to improve the paper.
References
- Hamrick JL, Godt MJW. In: Plant Population Genetics, Breeding and Genetic Resources. Brown AHD, Clegg MT, Kahler AL, Weir BS, editor. Sunderland: Sinauer Associates; 1989. Allozyme diversity in plant species; pp. 43–63. [Google Scholar]
- Tanksley SD, Young ND, Paterson AH, Bonierbale MW. RFLP mapping in plant breeding: new tools for an old science. Bio/Technology. 1989;7:257–264. doi: 10.1038/nbt0389-257. [DOI] [Google Scholar]
- Hubbard M, Kelly J, Rajapakse S, Abbott A, Ballard R. Restriction fragment length polymorphisms in rose and their use for cultivar identification. HortScience. 1992;27:172–173. [Google Scholar]
- Palmer JD, Herbon LA. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol. 1988;28:87–97. doi: 10.1007/BF02143500. [DOI] [PubMed] [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 2007;7:135. doi: 10.1186/1471-2148-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Wilson V, Thein SL. Hypervariable “minisatellite” regions in human DNA. Nature. 1985;314:67–73. doi: 10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Wilson V, Thein SL. Individual-specific “fingerprints” of human DNA. Nature. 1985;316:76–79. doi: 10.1038/316076a0. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Morton DB. DNA fingerprinting of dogs and cats. Anim Genet. 1987;18:1–15. doi: 10.1111/j.1365-2052.1987.tb00739.x. [DOI] [PubMed] [Google Scholar]
- Burke T, Bruford MW. DNA fingerprinting in birds. Nature. 1987;327:149–152. doi: 10.1038/327149a0. [DOI] [PubMed] [Google Scholar]
- Dallas JF. Detection of DNA “fingerprints” of cultivated rice by hybridization with a human minisatellite DNA probe. Proc Natl Acad Sci U S A. 1988;85:6831–6835. doi: 10.1073/pnas.85.18.6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskov AP, Jincharadze AG, Prosnyak MI, Ivanov PL, Limborska SA. M13 phage DNA as a universal marker for DNA fingerprinting of animals, plants and microorganisms. FEBS Letters. 1988;233:388–392. doi: 10.1016/0014-5793(88)80467-8. [DOI] [PubMed] [Google Scholar]
- Rogstad SH, Patton JC II, Schaal BA. M13 repeat probe detects DNA minisatellite-like sequences in gymnosperms and angiosperms. Proc Natl Acad Sci U S A. 1988;85:9176–9178. doi: 10.1073/pnas.85.23.9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogstad SH, Nybom H, Schaal BA. The tetrapod DNA fingerprinting M13 repeat probe reveals genetic diversity and clonal growth in quaking aspen (Populus tremuloides, Salicaceae) Pl Syst Evol. 1991;175:115–123. doi: 10.1007/BF00937841. [DOI] [Google Scholar]
- Nybom H, Schaal BA. DNA "fingerprints" reveal genotypic distributions in natural populations of blackberries and raspberries (Rubus, Rosaceae) Am J Bot. 1990;77:883–888. doi: 10.2307/2444504. [DOI] [Google Scholar]
- Antonius K, Nybom H. DNA fingerprinting reveals significant amounts of genetic variation in a wild raspberry Rubus idaeus population. Mol Ecol. 1994;3:177–180. doi: 10.1111/j.1365-294X.1994.tb00119.x. [DOI] [Google Scholar]
- Nybom H, Schaal BA, Rogstad SH. DNA "fingerprints" can distinguish cultivars of blackberries and raspberries. Acta Hortic. 1989;262:305–310. [Google Scholar]
- Nybom H. DNA fingerprints in sports of 'Red Delicious' apples. HortScience. 1990;25:1641–1642. [Google Scholar]
- Weising K, Weigand F, Driesel A, Kahl G, Zischler H, Epplen JT. Polymorphic simple GATA/GACA repeats in plant genomes. Nucleic Acids Res. 1989;17:10128–10128. doi: 10.1093/nar/17.23.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weising K, Kaemmer D, Weigand F, Epplen JT, Kahl G. Oligonucleotide fingerprinting reveals various probe-dependent levels of informativeness in chickpea (Cicer arietinum) Genome. 1992;35:436–442. doi: 10.1139/g92-065. [DOI] [Google Scholar]
- Kaemmer D, Afza R, Weising K, Kahl G, Nowak FJ. Oligonucleotide and amplification fingerprinting of wild species and cultivars of banana (Musa spp.) Bio/Technol. 1992;10:1030–1035. doi: 10.1038/nbt0992-1030. [DOI] [PubMed] [Google Scholar]
- Vosman B, Arens P, Ruskortekaas W, Smulders MJM. Identification of highly polymorphic DNA regions in tomato. Theor Appl Genet. 1992;85:239–244. doi: 10.1007/BF00222865. [DOI] [PubMed] [Google Scholar]
- Kaemmer D, Weising K, Kahl G, Beyermann B, Börner T, Epplen JT. Oligonucleotide fingerprinting of tomato DNA. Plant Breed. 1995;114:12–17. doi: 10.1111/j.1439-0523.1995.tb00751.x. [DOI] [Google Scholar]
- Ramakishana W, Lagu MD, Gupta VS, Ranjekar PK. DNA fingerprinting in rice using oligonucleotide probes specific for simple repetitive DNA sequences. Theor Appl Genet. 1994;88:402–406. doi: 10.1007/BF00223651. [DOI] [PubMed] [Google Scholar]
- Schmidt T, Boblenz K, Metzlaff M, Kaemmer D, Weising K, Kahl G. DNA fingerprinting in sugar beet (Beta vulgaris) – identification of double haploid breeding lines. Theor Appl Genet. 1993;85:653–657. doi: 10.1007/BF00225001. [DOI] [PubMed] [Google Scholar]
- Van Houten WHJ, Van Heusden AW, Rouppe Van Der Voort J, Raijmann L, Bachmann K. Hypervariable DNA fingerprint loci in Microseris pymaea (Asteraceae, Lactuceae) Bot Acta. 1991;104:252–255. [Google Scholar]
- Weising K, Nybom H, Wolff K, Meyer W. DNA Fingerprinting in Plants and Fungi. Boca Raton: CRC press; 1995. [Google Scholar]
- Weising K, Nybom H, Wolff K, Kahl G. DNA Fingerprinting in Plants: Principles, Methods and Applications. Boca Raton: Taylor & Francis/CRC press; 2005. [Google Scholar]
- Weising K, Beyermann B, Ramser J, Kahl G. Plant DNA fingerprinting with radioactive and digoxigenated oligonucleotide probes complementary to simple repetitive DNA sequences. Electrophoresis. 1991;12:159–169. doi: 10.1002/elps.1150120211. [DOI] [PubMed] [Google Scholar]
- Parent JG, Pagé D. Identification of raspberry cultivars by nonradoactive DNA fingerprinting. HortScience. 1992;27:1108–1110. [Google Scholar]
- Rogstad SH. Surveying plant genomes for variable number of tandem repeat loci. Molecular Evolution: Producing the Biochemical Data. 1993;224:278–294. doi: 10.1016/0076-6879(93)24021-l. [Methods in Enzymology, vol 224.] [DOI] [PubMed] [Google Scholar]
- Rogstad SH. Inheritance in turnip of variable number tandem-repeat genetic-markers revealed with synthetic repetitive DNA probes. Theor Appl Genet. 1994;89:824–830. doi: 10.1007/BF00224504. [DOI] [PubMed] [Google Scholar]
- Kumar A, Rogstad SH. A hierarchical analysis of minisatellite DNA diversity in Gambel oak (Quercus gambelii Nutt.; Fagaceae) Mol Ecol. 1998;7:859–869. doi: 10.1046/j.1365-294x.1998.00400.x. [DOI] [Google Scholar]
- Nybom H, Schaal BA. DNA "fingerprints" applied to paternity analysis in apples (Malus x domestica) Theor Appl Genet. 1990;79:763–768. doi: 10.1007/BF00224242. [DOI] [PubMed] [Google Scholar]
- Kraft T, Nybom H, Werlemark G. Rubus vestervicensis (Rosaceae) - its hybrid origin revealed by DNA fingerprinting. Nord J Bot. 1995;15:237–242. doi: 10.1111/j.1756-1051.1995.tb00148.x. [DOI] [Google Scholar]
- Nybom H. Evaluation of interspecific crossing experiments in facultatively apomictic blackberries (Rubus subgen. Rubus) using DNA fingerprinting. Hereditas. 1995;122:57–65. [Google Scholar]
- Nei M, Li W-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzuri G, Hillel J, Lavi U, Haberfeld A, Vainstein A. DNA fingerprint analysis of ornamental plants. Plant Sci. 1991;76:91–97. doi: 10.1016/0168-9452(91)90222-T. [DOI] [Google Scholar]
- Vainstein A, Hillel J, Lavi U, Tzuri G. Assessment of genetic relatedness in carnation by DNA fingerprint analysis. Euphytica. 1991;56:225–229. doi: 10.1007/BF00042368. [DOI] [Google Scholar]
- Nybom H, Hall HK. Minisatellite DNA "fingerprints" can distinguish Rubus cultivars and estimate their degree of relatedness. Euphytica. 1991;53:107–114. doi: 10.1007/BF00023790. [DOI] [Google Scholar]
- Nybom H, Rogstad SH. DNA "fingerprints" detect genetic variation in Acer negundo. Pl Syst Evol. 1990;173:49–56. doi: 10.1007/BF00937762. [DOI] [Google Scholar]
- Rogstad SH, Wolff K, Schaal BA. Geographical variation in Asimina triloba Dunal (Annonaceae) revealed by the M13 DNA fingerprinting probe. Am J Bot. 1991;78:1391–1396. doi: 10.2307/2445277. [DOI] [Google Scholar]
- Wolff K, Schaal BA, Rogstad SH. Population and species variation of minisatellite DNA in Plantago. Theor Appl Genet. 1994;87:733–740. doi: 10.1007/BF00222899. [DOI] [PubMed] [Google Scholar]
- Keane B, Pelikan S, Toth GP, Smith MK, Rogstad SH. Genetic diversity of Typha latifolia (Typhaceae) and the impact of pollutants examined with tandem-repetitive DNA probes. Am J Bot. 1999;86:1226–1238. doi: 10.2307/2656770. [DOI] [PubMed] [Google Scholar]
- Lim HW, Pelikan S, Rogstad SH. Genetic diversity among populations and size classes of buckeyes (Aesculus: Hippocastanaceae) examined with multilocus VNTR probes. Pl Syst Evol. 2002;230:125–141. doi: 10.1007/s006060200000. [DOI] [Google Scholar]
- Nybom H. In: DNA Fingerprinting: Approaches and Applications. Burke T, Dolph G, Jeffreys AJ, Wolff R, editor. Basel: Birkhäuser; 1991. Applications of DNA fingerprinting in plant breeding; pp. 294–311. [Google Scholar]
- Nybom H. In: DNA Fingerprinting: State of the Science. Pena SDJ, Chakraborty R, Epplen JT, Jeffreys AJ, editor. Basel: Birkhäuser; 1993. Applications of DNA fingerprinting in plant population studies; pp. 293–309. [Google Scholar]
- Rogstad SH. In: Genomes of Plants and Animals: 21st Stadler Genetics Symposium. Gustafson JP, Flavell RB, editor. New York: Plenum Press Publ. Corp; 1996. Assessing genetic diversity in plants with synthetic tandem repetitive DNA probes; pp. 1–14. [Google Scholar]
- Weising K, Kahl G. In: Markers: Protocols, Applications, and Overviews. Caetano-Anollés G, Gresshoff PM, editor. New York: Wiley-VCH; 1998. Hybridization-based microsatellite fingerprinting of plants and fungi; pp. 27–54. [Google Scholar]
- Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- Welsh J, McClelland M. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 1990;18:7213–7218. doi: 10.1093/nar/18.24.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990;18:6231–6235. doi: 10.1093/nar/18.21.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano-Anollés G, Bassam BJ, Gresshoff PM. High resolution DNA amplification fingerprinting using very short arbitrary oligonucleotide primers. Bio/Technology. 1991;9:553–557. doi: 10.1038/nbt0691-553. [DOI] [PubMed] [Google Scholar]
- Zabeau M, Vos P. Selective restriction fragment amplification: a general method for DNA fingerprinting. European Patent Office, publication. bulletin 93/13.
- Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995;23:4407–4414. doi: 10.1093/nar/23.21.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Chyi YS, Romero-Severson J, Owen JL. Amplification of DNA markers from evolutionary diverse genomes using single primers of simple-sequence repeats. Theor Appl Genet. 1994;89:998–1006. doi: 10.1007/BF00224530. [DOI] [PubMed] [Google Scholar]
- Zietkiewicz E, Rafalski A, Labuda D. Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics. 1994;20:176–183. doi: 10.1006/geno.1994.1151. [DOI] [PubMed] [Google Scholar]
- Li G, Quiros CF. Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet. 2001;103:455–461. doi: 10.1007/s001220100570. [DOI] [Google Scholar]
- Li G, McVetty PBE, Quiros CF. In: Plant Breeding from Laboratories to Fields. Andersen SB, editor. Rijeka, Croatia: InTech; 2013. SRAP molecular marker technology in plant science. DOI: 10.5772/54511. [Google Scholar]
- Hu J, Vick BA. Target region amplification polymorphism: a novel marker technique for plant genotyping. Plant Mol Biol Rep. 2003;21:289–294. doi: 10.1007/BF02772804. [DOI] [Google Scholar]
- Witsenboer H, Vogel J, Michelmore RW. Identification, genetic localization, and allelic diversity of selectively amplified microsatellite polymorphic loci in lettuce and wild relatives (Lactuca spp.) Genome. 1997;40:923–936. doi: 10.1139/g97-119. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Bebeli PJ, Somers DJ, Gustafson JP. Direct amplification of minisatellite-region DNA with VNTR core sequences in the genus Oryza. Theor Appl Genet. 1997;95:942–949. doi: 10.1007/s001220050645. [DOI] [Google Scholar]
- Chen XM, Line RF, Leung H. Genome scanning for resistance-gene analogs in rice, barley, and wheat by high-resolution electrophoresis. Theor Appl Genet. 1998;97:345–355. doi: 10.1007/s001220050905. [DOI] [Google Scholar]
- Jaccoud D, Peng K, Feinstein D, Kilian A. Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001;29:e25. doi: 10.1093/nar/29.4.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezar S, Myburg AA, Berger DK, Wingfield MJ, Wingfield BD. Development and assessment of microarray-based DNA fingerprinting in Eucalyptus grandis. Theor Appl Genet. 2004;109:1329–1336. doi: 10.1007/s00122-004-1759-9. [DOI] [PubMed] [Google Scholar]
- Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G. Diversity Arrays Technology: a generic genome profiling technology on open platforms. Meth Mol Biol. 2012;888:67–89. doi: 10.1007/978-1-61779-870-2_5. [DOI] [PubMed] [Google Scholar]
- James KE, Schneider H, Ansell SW, Evers M, Robba L, Uszynski G, Pedersen N, Newton AE, Russell SJ, Vogel J, Kilian A. Diversity Arrays Technology (DArT) for pan-genomic evolutionary studies of non-model organisms. PLoS One. 2008;3:e1682. doi: 10.1371/journal.pone.0001682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman AH. Molecular markers to assess genetic diversity. Euphytica. 2007;158:313–321. doi: 10.1007/s10681-006-9282-5. [DOI] [Google Scholar]
- Kalendar R, Grob T, Regina M, Suoniemi A, Schulmann A. IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Genet. 1999;98:704–711. doi: 10.1007/s001220051124. [DOI] [Google Scholar]
- Provan J, Thomas WTB, Forster BP, Powell W. Copia-SSR: a simple marker technique which can be used on total genomic DNA. Genome. 1999;42:363–366. [Google Scholar]
- Waugh R, McLean K, Flavell AJ, Pearce SR, Kumar A, Thomas BBT, Powell W. Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP) Mol Gen Genet. 1997;253:687–694. doi: 10.1007/s004380050372. [DOI] [PubMed] [Google Scholar]
- Casa A, Brouwer C, Nagel A, Wang L, Zhang Q, Kresovich S, Wessler SR. The MITE family Heartbreaker (Hbr): molecular markers in maize. Proc Natl Acad Sci U S A. 2000;97:10083–10089. doi: 10.1073/pnas.97.18.10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KC, Kim NH, Cho YS, Kang KH, Lee JK, Kim N-S. Genetic variations of AA genome Oryza species measured by MITE-AFLP. Theor Appl Genet. 2003;197:203–209. doi: 10.1007/s00122-003-1252-x. [DOI] [PubMed] [Google Scholar]
- Selkoe KA, Toonen RJ. Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett. 2006;9:615–629. doi: 10.1111/j.1461-0248.2006.00889.x. [DOI] [PubMed] [Google Scholar]
- Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit RJ. Current trends in microsatellite genotyping. Mol Ecol Resources. 2011;11:591–611. doi: 10.1111/j.1755-0998.2011.03014.x. [DOI] [PubMed] [Google Scholar]
- Akkaya MS, Bhagwat AA, Cregan PB. Length polymorphisms of simple sequence repeat DNA in soybean. Genetics. 1992;132:1131–1139. doi: 10.1093/genetics/132.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squirrell J, Hollingsworth PM, Woodhead M, Russell J, Lowe AJ, Gibby M, Powell W. How much effort is required to isolate nuclear microsatellites from plants? Mol Ecol. 2003;12:1339–1348. doi: 10.1046/j.1365-294X.2003.01825.x. [DOI] [PubMed] [Google Scholar]
- Tang S, Okashah RA, Cordonnier-Pratt MM, Pratt LH, Johnson VE, Taylor CA, Arnold ML, Knapp SJ. EST and EST-SSR marker resources for Iris. BMC Plant Biol. 2009;9:72. doi: 10.1186/1471-2229-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor Appl Genet. 2003;106:411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- Pashley CH, Ellis JR, McCauley DE, Burke JM. EST databases as a source for molecular markers: lessons from Helianthus. J Hered. 2006;97:381–388. doi: 10.1093/jhered/esl013. [DOI] [PubMed] [Google Scholar]
- Wöhrmann T, Weising K. In silico mining for simple sequence repeat (SSR) loci in a pineapple expressed sequence tag (EST) database and cross-species amplification of EST-SSR markers across Bromeliaceae. Theor Appl Genet. 2011;123:635–647. doi: 10.1007/s00122-011-1613-9. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Graner A, Sorrells ME. Genic microsatellite markers in plants: principles, methods, and applications. Trends Biotechnol. 2005;23:48–55. doi: 10.1016/j.tibtech.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Paran I, Michelmore RW. Development of reliable PCR-based markers linked to downy mildew resistance genes in lettuce. Theor Appl Genet. 1993;85:985–993. doi: 10.1007/BF00215038. [DOI] [PubMed] [Google Scholar]
- Bautista R, Crespillo R, Cánovas FM, Claros MG. Identification of olive-tree cultivars with SCAR markers. Euphytica. 2002;129:33–41. [Google Scholar]
- Turkec A, Sayar M, Heinze B. Identification of sweet cherry cultivars (Prunus avium L.) and analysis of their genetic relationships by chloroplast sequence-characterised amplified regions (cpSCAR) Genet Res Crop Evol. 2006;53:1635–1641. doi: 10.1007/s10722-005-2285-6. [DOI] [Google Scholar]
- Konieczny A, Ausubel FM. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 1993;4:403–410. doi: 10.1046/j.1365-313X.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- Appleby N, Edwards D, Batley J. New technologies for ultra-high throughput genotyping in plants. Methods Mol Biol. 2009;513:19–39. doi: 10.1007/978-1-59745-427-8_2. [DOI] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Rev Genet. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- Martin LB, Fei Z, Giovannoni JJ, Rose JK. Catalyzing plant science research with RNA-seq. Front Plant Sci. 2013;4:66. doi: 10.3389/fpls.2013.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picoult-Newberg L, Ideker TE, Pohl MG, Taylor SL, Donaldson MA, Nickerson DA, Boyce-Jacino M. Mining SNPs from EST databases. Genome Res. 1999;9:167–174. [PMC free article] [PubMed] [Google Scholar]
- Ganal MW, Altmann T, Röder MS. SNP identification in crop plants. Curr Opin Plant Biol. 2009;12:1–7. doi: 10.1016/j.pbi.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Ganal MW, Polley A, Graner EM, Plieske J, Wieseke R, Luerssen H, Durstewitz G. Large SNP arrays for genotyping in crop plants. J Biosci. 2012;37:821–828. doi: 10.1007/s12038-012-9225-3. [DOI] [PubMed] [Google Scholar]
- Nelson JC, Wang S, Wu Y, Li X, Antony G, White FF, Yu J. Single-nucleotide polymorphism discovery by high-throughput sequencing in sorghum. BMC Genomics. 2011;12:352. doi: 10.1186/1471-2164-12-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumolin-Lapègue S, Pemonge MH, Petit RJ. An enlarged set of consensus primers for the study of organelle DNA in plants. Mol Ecol. 1997;6:393–397. doi: 10.1046/j.1365-294X.1997.00193.x. [DOI] [PubMed] [Google Scholar]
- Shaw J, Lickey EB, Schilling EE, Small RL. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am J Bot. 2007;94:275–288. doi: 10.3732/ajb.94.3.275. [DOI] [PubMed] [Google Scholar]
- Weising K, Gardner RC. A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome. 1999;42:9–19. doi: 10.1139/g98-104. [DOI] [PubMed] [Google Scholar]
- Nybom H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol. 2004;13:1143–1155. doi: 10.1111/j.1365-294X.2004.02141.x. [DOI] [PubMed] [Google Scholar]
- Nybom H, Weising K. DNA-based identification of clonally propagated cultivars. Plant Breed Rev. 2010;34:221–295. [Google Scholar]
- Jones ES, Sullivan H, Bhattramaki D, Smith JSC. A comparison of simple sequence repeat and single nucleotide polymorphism marker technologies for the genotypic analysis of maize (Zea mays L.) Theor Appl Genet. 2007;115:361–371. doi: 10.1007/s00122-007-0570-9. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Blaich R, Forneck A. Retrotransposon-based mutations in clones of 'Pinot’ cultivars. Acta Hortic. 2009;827:83–86. [Google Scholar]
- Hysing S-C, Säll T, Nybom H, Liljeroth E, Merker A, Orford S, Koebner RMD. Temporal diversity changes among 198 Nordic bread wheat landraces and cultivars detected by retrotransposon-based S-SAP analysis. Plant Genet Resour Charact Utilizat. 2008;6:113–125. [Google Scholar]
- Wegscheider E, Benjak A, Forneck A. Clonal variation in Pinot noir revealed by S-SAP involving universal retrotransposon-based sequences. Am J Enol Vitic. 2009;60:104–109. [Google Scholar]
- Sorkheh K, Shiran B, Gradziel TM, Epperson BK, Martínez-Gomez P, Asadi E. Amplified fragment length polymorphism as a tool for molecular characterization of almond germplasm: genetic diversity among cultivated genotypes and related wild species of almond, and its relationship with agronomic traits. Euphytica. 2007;156:327–344. doi: 10.1007/s10681-007-9382-x. [DOI] [Google Scholar]
- Koopman WJM, Kuiper I, Klein-Geltink DJA, Sabatino GJH, Smulders MJM. Botanical DNA evidence in criminal cases: knotgrass (Polygonum aviculare L.) as a model species. Forensic Sci Internat Genet. 2012;6:366–374. doi: 10.1016/j.fsigen.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Steinger T, Korner C, Schmid B. Long-term persistence in a changing climate: DNA analysis suggests very old ages of clones of alpine Carex curvula. Oecologia. 1996;105:94–99. doi: 10.1007/BF00328796. [DOI] [PubMed] [Google Scholar]
- Hollingsworth ML, Bailey JP. Evidence for massive clonal growth in the invasive weed Fallopia japonica (Japanese knotweed) Bot J Linn Soc. 2000;133:463–472. [Google Scholar]
- Xu CY, Zhang WJ, Fu CZ, Lu BR. Genetic diversity of alligator weed in China by RAPD analysis. Biodivers Conserv. 2003;12:637–645. doi: 10.1023/A:1022453129662. [DOI] [Google Scholar]
- Ren MX, Zhang QG. Clonal diversity and structure of the invasive aquatic plant Eichhornia crassipes in China. Aquatic Bot. 2007;87:242–246. doi: 10.1016/j.aquabot.2007.06.002. [DOI] [Google Scholar]
- Hämmerli A, Reusch TBH. Inbreeding depression influences genet size distribution in a marine angiosperm. Mol Ecol. 2003;12:619–629. doi: 10.1046/j.1365-294X.2003.01766.x. [DOI] [PubMed] [Google Scholar]
- Pfeiffer T, Klahr A, Peterson A, Levichev IG, Schnittler M. No sex at all? Extremely low genetic diversity in Gagea spathacea (Liliaceae) across Europe. Flora. 2012;207:372–378. doi: 10.1016/j.flora.2012.03.002. [DOI] [Google Scholar]
- van der Hulst RGM, Mes THM, Den Nijs JCM, Bachmann K. Amplified fragment length polymorphism (AFLP) markers reveal that population structure of triploid dandelions (Taraxacum officinale) exhibits both clonality and recombination. Mol Ecol. 2000;9:1–8. doi: 10.1046/j.1365-294x.2000.00704.x. [DOI] [PubMed] [Google Scholar]
- Majeský L, Vasut RJ, Kitner M, Trávnicek B. The pattern of genetic variability in apomictic clones of Taraxacum officinale indicates the alternation of asexual and sexual histories of apomicts. PLoS One. 2012;7:e41868. doi: 10.1371/journal.pone.0041868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paun O, Greilhuber J, Temsch E, Hörandl E. Patterns, sources and ecological implications of clonal diversity in apomictic Ranunculus carpaticola (Ranunculus auricomus complex, Ranunculaceae) Mol Ecol. 2006;15:897–910. doi: 10.1111/j.1365-294X.2006.02800.x. [DOI] [PubMed] [Google Scholar]
- Paun O, Hörandl E. Evolution of hypervariable microsatellites in apomictic polyploid lineages of Ranunculus carpaticola: directional bias at dinucleotide loci. Genetics. 2006;174:387–398. doi: 10.1534/genetics.105.052761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond EBM, Vellend M. Genotypic diversity effects on the performance of Taraxacum officinale populations increase with time and environmental favorability. PLOS One. 2012;7:e30314. doi: 10.1371/journal.pone.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod KA, Scascitelli M, Vellend M. Detecting small-scale genotype-environment interactions in apomictic dandelion (Taraxacum officinale) populations. J Evol Biol. 2012;25:1667–1675. doi: 10.1111/j.1420-9101.2012.02549.x. [DOI] [PubMed] [Google Scholar]
- De Andres MT, Cabezas JA, Cervera MT, Borrego J, Martínez-Zapater JM, Jouve N. Molecular characterization of grapevine rootstocks maintained in germplasm collections. Am J Enol Viticult. 2007;58:75–86. [Google Scholar]
- Cipriani G, Marrazzo MT, Di Gaspero G, Pfeiffer A, Morgante M, Testolin R. A set of microsatellite markers with long core repeat optimized for grape (Vitis spp.) genotyping. BMC Plant Biol. 2008;8:127. doi: 10.1186/1471-2229-8-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song QJ, Quigley CV, Carter TE, Nelson RL, Boerma HR, Strachan J, Cregan PB. A selected set of trinucleotide simple sequence repeat markers for soybean cultivar identification. Plant Varieties Seeds. 1999;12:207–220. [Google Scholar]
- Zhang DP, Mischke S, Goenaga R, Hemeida AA, Saunders JA. Accuracy and reliability of high-throughput microsatellite genotyping for cacao clone identification. Crop Sci. 2006;46:2084–2092. doi: 10.2135/cropsci2006.01.0004. [DOI] [Google Scholar]
- Vélez MD, Ibánez J. Evaluation of the uniformity and stability of microsatellite markers in grapevine. Acta Hortic. 2009;827:163–168. [Google Scholar]
- Bracci T, Sebastiani L, Busconi M, Fogher C, Belaj A, Trujillo I. SSR markers reveal the uniqueness of olive cultivars from the Italian region of Liguria. Scientia Hortic. 2009;122:209–215. doi: 10.1016/j.scienta.2009.04.010. [DOI] [Google Scholar]
- Cabezas JA, Ibánez J, Lijavetzky D, Vélez D, Bravo G, Rodríguez V, Carreno I, Jermakow AM, Carreno J, Ruiz-García L, Thomas MR, Martinez-Zapater JM. A 48 SNP set for grapevine cultivar identification. BMC Plant Biol. 2011;11:153. doi: 10.1186/1471-2229-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Shamada T, Nonaka K, Kita M, Kuniga T, Endo T, Ikoma Y, Omura M. High-throughput genotyping in Citrus accessions using an SNP genotyping array. Tree Genet Genom. 2013;9:145–153. doi: 10.1007/s11295-012-0542-3. [DOI] [Google Scholar]
- Chebotar S, Röder MS, Korzun V, Saal B, Weber WE, Börner A. Molecular studies on genetic integrity of open-pollinating species rye (Secale cereale L.) after long-term genebank maintenance. Theor Appl Genet. 2003;107:1469–1476. doi: 10.1007/s00122-003-1366-1. [DOI] [PubMed] [Google Scholar]
- Van Hintum TJL, van de Wiel CCM, Visser DL, Van Treuren R, Vosman B. The distribution of genetic diversity in a Brassica oleracea gene bank collection related to the effects of regeneration as measured with AFLPs. Theor Appl Genet. 2007;114:777–786. doi: 10.1007/s00122-006-0456-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecký D, Bartos J, Christelová P, Cernoch V, Kilian A, Dolezel J. Genomic constitution of Festuca x Lolium hybrids revealed by the DArTFest array. Theor Appl Genet. 2011;122:355–363. doi: 10.1007/s00122-010-1451-1. [DOI] [PubMed] [Google Scholar]
- Nelson RL. Managing self-pollinated germplasm collections to maximize utilization. Plant Genet Resour Charact Utilizat. 2011;9:123–133. doi: 10.1017/S147926211000047X. [DOI] [Google Scholar]
- Chao S, Zhang W, Akhunov E, Sherman J, Ma Y, Luo MC, Dubcovsky J. Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.) cultivars. Mol Breeding. 2009;23:23–33. doi: 10.1007/s11032-008-9210-6. [DOI] [Google Scholar]
- Franks T, Botta R, Thomas MR. Chimerism in grapevines: implications for cultivar identity, ancestry and genetic improvement. Theor Appl Genet. 2002;104:192–199. doi: 10.1007/s001220100683. [DOI] [PubMed] [Google Scholar]
- Moncada X, Hinrichsen P, Pelsy F, Medinoglu D. Microsatellite markers as reproducible tools to identify chimerical polymorphisms in 'Cabernet Sauvignon’. Acta Hortic. 2009;827:259–264. [Google Scholar]
- Regner F, Hack R, Hanak K, Santiago JL. Variability in the cultivar 'Gruener Veltliner’. Acta Hortic. 2009;827:245–252. [Google Scholar]
- Castro I, Leal F, Guedes-Pinto H, Eiras Dias J, Veloso M, Pinto-Carnide O. Genomic variability in grapevine cultivars assessed by molecular markers. Acta Hortic. 2009;827:187–192. [Google Scholar]
- Venturi S, Dondini L, Donini P, Sansavini S. Retrotransposon characterisation and fingerprinting of apple clones by S-SAP markers. Theor Appl Genet. 2006;112:440–444. doi: 10.1007/s00122-005-0143-8. [DOI] [PubMed] [Google Scholar]
- Zhao G, Dai H, Chang L, Ma Y, Sun H, He P, Zhang Z. Isolation of two novel complete Ty1-copia retrotransposons from apple and demonstration of use of derived S-SAP markers for distinguishing bud sports of Malus domestica cv. Tree Fuji Genet Genom. 2010;6:149–159. doi: 10.1007/s11295-009-0236-7. [DOI] [Google Scholar]
- Bretó MP, Ruiz C, Pina JA, Asíns MJ. The diversification of Citrus clementina Hort. ex Tan., a vegetatively propagated crop species. Mol Phylogenet Evol. 2001;21:285–293. doi: 10.1006/mpev.2001.1008. [DOI] [PubMed] [Google Scholar]
- Liao Z-K, Zhang Q-M, Liu W-G, Ding W-P, Wang C-M. Identification of Citrus mutants by APLP technique. J Fruit Sci. 2006;23:486–488. [Google Scholar]
- Gomes S, Martins-Lopes PJ, Lima-Brito J, Meirinhos J, Lopes A, Martins A, Guedes-Pinto H. Evidence for clonal variation in 'Verdeal-Transmontana’ olive using RAPD, ISSR and SSR markers. J Hortic Sci Biotechnol. 2008;83:395–400. [Google Scholar]
- Martins-Lopes P, Gomes S, Lima-Brito J, Lopes J, Guedes-Pinto H. Assessment of clonal genetic variability in Olea europaea L. 'Cobrançosa’ by molecular markers. Scientia Hortic. 2009;123:82–89. doi: 10.1016/j.scienta.2009.08.001. [DOI] [Google Scholar]
- Guimaraes NCC, Torga PP, De Resende EC, Chalfun A, Paiva E, Paiva LV. Identification of somaclonal variants in 'Prata Ana’ banana using molecular and cytogenetic techniques [In Portuguese] Ciencia e Agrotecnologia. 2009;33:448–454. [Google Scholar]
- Sheidai M, Aminpoor H, Noormohammadi Z, Farahani F. RAPD analysis of somaclonal variation in banana (Musa acuminata L.) cultivar Valery. Acta Biol Szeged. 2008;52:307–311. [Google Scholar]
- Perrini R, Alba V, Ruta C, Morone-Fortunato I, Blanco A, Montemurro C. An evaluation of a new approach to the regeneration of Helichrysum italicum (Roth) G. Don, and the molecular characterization of the variation among sets of differently derived regenerants. Cell Molec Biol Lett. 2009;14:377–394. doi: 10.2478/s11658-009-0007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Kaabi HH, Zaid A, Shephard H, Ainsworth C. AFLP variation in tissue culture-derived date palm (Phoenix dactylifera L.) plants. Acta Hortic. 2007;736:135–159. [Google Scholar]
- Osipova ES, Lysenko EA, Troitsky AV, Dolgikh YI, Shamina ZB, Gostimskii SA. Analysis of SCAR marker nucleotide sequences in maize (Zea mays L.) somaclones. Plant Science. 2011;180:313–322. doi: 10.1016/j.plantsci.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Carrier G, Le Cunff L, Dereeper A, Legrand D, Sabot F, Bouchez O, Audeguin L, Boursiquot JM, This P. Transposable elements are a major cause of somatic polymorphism in Vitis vinifera L. PLoS One. 2012;7:e32973. doi: 10.1371/journal.pone.0032973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon CK. Botanical witness for the prosecution. Science. 1993;260:894–895. doi: 10.1126/science.8493521. [DOI] [PubMed] [Google Scholar]
- Korpelainen H, Virtanen V. DNA fingerprinting of mosses. J Forensic Sci. 2003;48:804–807. [PubMed] [Google Scholar]
- Virtanen V, Korpelainen H, Kostamo K. Forensic botany: usability of bryophyte material in forensic studies. Forensic Sci Int. 2007;172:161–163. doi: 10.1016/j.forsciint.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Plaza P, Gonzalez R, Moreno-Arribas MV, Polo MC, Bravo G, Martínez-Zapater JM, Martínez MC, Cifuentes A. Combining microsatellite markers and capillary gel electrophoresis with laser-induced fluorescence to identify the grape Vitis vinifera cultivar of musts. Eur Food Res Technol. 2006;223:625–631. doi: 10.1007/s00217-005-0244-2. [DOI] [Google Scholar]
- Faria MA, Nunes E, Oliveira MBPP. Relative quantification of Vitis vinifera L. cultivars in musts by microsatellite DNA analysis. Eur Food Res Technol. 2008;227:845–850. doi: 10.1007/s00217-007-0795-5. [DOI] [Google Scholar]
- Spaniolas S, Tsachaki M, Bennett MJ, Tucker GA. Toward the authentication of wines of Nemea denomination of origin through cleaved amplified polymorphic sequence (CAPS)-based assay. J Agric Food Chem. 2008;56:7667–7671. doi: 10.1021/jf801036f. [DOI] [PubMed] [Google Scholar]
- Martins-Lopes P, Gomes S, Santos E, Guedes-Pinto H. DNA markers for Portuguese olive oil fingerprinting. J Agric Food Chem. 2008;56:11786–11791. doi: 10.1021/jf801146z. [DOI] [PubMed] [Google Scholar]
- Montemurro C, Pasqualone A, Simeone R, Sabetta W, Blanco A. AFLP molecular markers to identify virgin olive oils from single Italian cultivars. Eur Food Res Technol. 2008;226:1439–1444. doi: 10.1007/s00217-007-0675-z. [DOI] [Google Scholar]
- Archak S, Lakshminarayanareddy V, Nagaraju J. High-throughput multiplex microsatellite marker assay for detection and quantification of adulteration in Basmati rice (Oryza sativa) Electrophoresis. 2007;28:2396–2405. doi: 10.1002/elps.200600646. [DOI] [PubMed] [Google Scholar]
- Melchlade D, Foroni I, Corrado G, Santangelo I, Rao R. Authentication of the 'Annurca’ apple in agro-food chain by amplification of microsatellite loci. Food Biotechnol. 2007;21:33–43. doi: 10.1080/08905430701191114. [DOI] [Google Scholar]
- Yamamoto T, Kimura T, Hayashi T, Ban Y. DNA profiling of fresh and processed fruits in pear. Breed Sci. 2006;56:165–171. doi: 10.1270/jsbbs.56.165. [DOI] [Google Scholar]
- Ding G, Zhang DZ, Feng ZY, Fan WJ, Ding XY, Li XX. SNP, ARMS and SSH authentication of medicinal Dendrobium officinale Kimura et Migo and application for identification of Fengdou drugs. Biol Pharmaceut Bull. 2008;31:553–557. doi: 10.1248/bpb.31.553. [DOI] [PubMed] [Google Scholar]
- Nybom H, Weising K. In: Medicinal Plant Biotechnology. Kayser O, Quax WJ, editor. Weinheim: Wiley-VCH Publishers; 2007. DNA profiling of plants; pp. 73–95. [Google Scholar]
- Sarwat M, Nabi G, Das S, Srivastava PS. Molecular markers in medicinal plant biotechnology: past and present. Crit Rev Biotechnol. 2012;32:74–92. doi: 10.3109/07388551.2011.551872. [DOI] [PubMed] [Google Scholar]
- Kumar LD, Kathirvei M, Rao GV, Nagaraju J. DNA profiling of disputed chilli samples (Capsicum annum) using ISSR-PCR and FISSR-PCR marker assays. Forensic Sci Int. 2001;116:63–68. doi: 10.1016/S0379-0738(00)00350-9. [DOI] [PubMed] [Google Scholar]
- Gilmore S, Peakall R, Robertson J. Short tandem repeat (STR) DNA markers are hypervariable and informative in Cannabis sativa: implications for forensic investigations. Forensic Sci Internat. 2003;131:65–74. doi: 10.1016/S0379-0738(02)00397-3. [DOI] [PubMed] [Google Scholar]
- Köhnemann S, Nedele J, Schwotzer D, Morzfeld J, Pfeiffer H. The validation of a 15 STR multiplex PCR for Cannabis species. Int J Legal Med. 2012;126:601–606. doi: 10.1007/s00414-012-0706-6. [DOI] [PubMed] [Google Scholar]
- Höltken AM, Schröder H, Wischnewski N, Degen B, Magel E, Fladung M. Development of DNA-based methods to identify CITES-protected timber species: a case study in the Meliaceae family. Holzforschung. 2012;66:97–104. [Google Scholar]
- Nybom H, Bartish IV. Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Persp Plant Evol Ecol Syst. 2000;3:93–114. doi: 10.1078/1433-8319-00006. [DOI] [Google Scholar]
- Gabrielsen TM, Bachmann K, Jakobsen KS, Brochmann C. Glacial survival does not matter: RAPD phylogeography of Nordic Saxifraga oppositifolia. Mol Ecol. 1997;6:831–842. doi: 10.1111/j.1365-294X.1997.tb00137.x. [DOI] [Google Scholar]
- Williams DA, Muchugu E, Overholt WA, Cuda JP. Colonization patterns of the invasive Brazilian peppertree, Schinus terebinthifolius, in Florida. Heredity. 2007;98:284–293. doi: 10.1038/sj.hdy.6800936. [DOI] [PubMed] [Google Scholar]
- Forrest CN, Ottewell KM, Whelan RJ, Ayre DJ. Tests for inbreeding and outbreeding depression and estimation of population differentiation in the bird-pollinated shrub Grevillea mucronulata. Ann Bot. 2011;108:185–195. doi: 10.1093/aob/mcr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahima T, Sun GL, Beharav A, Krugman T, Beiles A, Nevo E. RAPD polymorphism in wild emmer wheat populations, Triticum dicoccoides, in Israel. Theor Appl Genet. 1999;98:434–447. doi: 10.1007/s001220051089. [DOI] [Google Scholar]
- Torres E, Iriondo JM, Escudero A, Pérez C. Analysis of within-population spatial genetic structure in Antirrhinum microphyllum (Scrophulariaceae) Am J Bot. 2003;90:1688–1695. doi: 10.3732/ajb.90.12.1688. [DOI] [PubMed] [Google Scholar]
- Meeus S, Honnay O, Jacquemyn H. Strong differences in genetic structure across disjunct, edge, and core populations of the distylous forest herb Pulmonaria officinalis (Boraginaceae) Am J Bot. 2012;99:1809–1818. doi: 10.3732/ajb.1200223. [DOI] [PubMed] [Google Scholar]
- Pandey M, Rajora OP. Higher fine-scale genetic structure in peripheral than in core populations of a long-lived and mixed-mating conifer – eastern white cedar (Thuja occidentalis L.) BMC Evol Biol. 2012;12:48. doi: 10.1186/1471-2148-12-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volis S, Zaretsky M, Shulgina I. Fine-scale spatial genetic structure in a predominantly selfing plant: role of seed and pollen dispersal. Heredity. 2010;105:384–393. doi: 10.1038/hdy.2009.168. [DOI] [PubMed] [Google Scholar]
- Ennos RA. Estimating the relative rates of pollen and seed migration among plant populations. Heredity. 1994;72:250–259. doi: 10.1038/hdy.1994.35. [DOI] [Google Scholar]
- Beebee T, Rowe G. An Introduction to Molecular Ecology. Oxford: Oxford University Press; 2004. [Google Scholar]
- Barluenga M, Austerlitz F, Elzinga JA, Texeira S, Goudet J, Bermasconi G. Fine-scale spatial genetic structure and gene dispersal in Silene latifolia. Heredity. 2011;106:13–24. doi: 10.1038/hdy.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievet V, Touzet P, Arnaud J-F, Cuguen J. Spatial analysis of nuclear and cytoplasmic DNA diversity in wild sea beet (Beta vulgaris ssp. maritima) populations: do marine currents shape the genetic structure? Mol Ecol. 2007;16:1847–1864. doi: 10.1111/j.1365-294X.2006.03208.x. [DOI] [PubMed] [Google Scholar]
- White GM, Boshier DH, Powell W. Genetic variation within a fragmented population of Swietenia humilis Zucc. Mol Ecol. 1999;8:1899–1909. doi: 10.1046/j.1365-294x.1999.00790.x. [DOI] [PubMed] [Google Scholar]
- Aldrich PR, Hamrick JL. Reproductive dominance of pasture trees in a fragmented tropical forest mosaic. Science. 1998;281:103–105. doi: 10.1126/science.281.5373.103. [DOI] [PubMed] [Google Scholar]
- Quinteros-Casaverde N, Flores-Negrón CF, Williams DA. Low genetic diversity and fragmentation in a wind-pollinated tree, Polylepis multijuga Plige (Rosaceae) in the high Andes. Conserv Genet. 2012;13:593–603. doi: 10.1007/s10592-011-0310-1. [DOI] [Google Scholar]
- Gerber S, Mariette S, Streiff R, Bodénès C, Kremer A. Comparison of microsatellites and amplified fragment length polymorphism markers for parentage analysis. Mol Ecol. 2000;9:1037–1048. doi: 10.1046/j.1365-294x.2000.00961.x. [DOI] [PubMed] [Google Scholar]
- Streiff R, Ducousso A, Lexer C, Steinkellner H, Gloessl J, Kremer A. Pollen dispersal inferred from paternity analysis in a mixed oak stand of Quercus robur L. and Q. petraea (Matt.) Liebl. Mol Ecol. 1999;8:831–841. doi: 10.1046/j.1365-294X.1999.00637.x. [DOI] [Google Scholar]
- Gaiotto FA, Grattapaglia D, Vencovsky R. Genetic structure, mating system, and long distance gene flow in heart of palm (Euterpe edulis Mart.) J Hered. 2003;94:399–406. doi: 10.1093/jhered/esg087. [DOI] [PubMed] [Google Scholar]
- Ziegenhagen B, Scholz F, Madaghiele A, Vendramin GG. Chloroplast microsatellites as markers for paternity analysis in Abies alba. Can J Forest Res. 1998;28:317–321. [Google Scholar]
- Korecký J, Klápste J, Lstiburek M, Kobliha J, Nelson CD, El-Kassaby YA. Comparison of genetic parameters from marker-based relationship, sibship, and combined models in Scots pine multi-site open-pollinated tests. Tree Genet Genom. 2013;9:1227–1235. doi: 10.1007/s11295-013-0630-z. doi:10.1007/s11295-013-0630-z. [DOI] [Google Scholar]
- Soleimani VD, Baum BR, Johnson DA. Analysis of genetic diversity in barley cultivars reveals incongruence between S-SAP, SNP and pedigree data. Genet Resour Crop Evol. 2007;54:83–97. doi: 10.1007/s10722-005-1886-4. [DOI] [Google Scholar]
- Coart E, Vekemans X, Smulders MJM, Wagner I, Van Huylenbroeck J, Van Bockstaele E, Roldán-Ruiz I. Genetic variation in the endangered wild apple (Malus sylvestris (L.) Mill.) in Belgium as revealed by amplified fragment length polymorphism and microsatellite markers. Mol Ecol. 2003;12:845–857. doi: 10.1046/j.1365-294X.2003.01778.x. [DOI] [PubMed] [Google Scholar]
- Coart E, Van Glabeke S, De Loose M, Larsen AS, Roldán-Ruiz I. Chloroplast diversity in the genus Malus: new insights into the relationship between the European wild apple (Malus sylvestris (L.) Mill.) and the domesticated apple (Malus domestica Borkh.) Mol Ecol. 2006;15:2171–2182. doi: 10.1111/j.1365-294X.2006.02924.x. [DOI] [PubMed] [Google Scholar]
- Patocchi A, Fernández-Fernández F, Evans K, Gobbin D, Rezzonico F, Boudichevskaia A, Dunemann F, Stankiewicz-Kosyl M, Mathis-Jeanneteau F, Durel CE, Gianfranceschi L, Vosta F, Toller C, Cova V, Mott D, Komjanc M, Barbaro E, Kodde L, Rikkerink E, Gessler C, van de Weg WE. Development and test of 21 multiplex PCRs composed of SSRs spanning most of the apple genome. Tree Genet Genom. 2009;5:211–223. doi: 10.1007/s11295-008-0176-7. [DOI] [Google Scholar]
- Escribano P, Viruel MA, Hormaza JL. Comparison of different methods to construct a core germplasm collection in woody perennial species with simple sequence repeat markers. A case study in cherimoya (Annona cherimola, Annonaceae), an underutilized subtropical fruit tree species. Ann Appl Biol. 2008;153:25–32. doi: 10.1111/j.1744-7348.2008.00232.x. [DOI] [Google Scholar]
- Escribano P, Viruel MA, Hormaza JL. Establishment of a core collection to optimise the conservation of cherimoya (Annona cherimola Mill.) genetic resources using SSR information. Acta Hortic. 2009;814:67–70. [Google Scholar]
- Le Cunff L, Fournier-Level A, Laucou V, Vezzulli S, Lacombe T, Adam-Blondon A-F, Boursiquot JM, This P. Construction of nested genetic core collections to optimize the exploitation of natural diversity in Vitis vinifera L. subsp. sativa. BMC Plant Biol. 2008;8:31. doi: 10.1186/1471-2229-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. The role of hybridisation in evolution: old wine in new skin. Am J Bot. 1995;82:944–953. doi: 10.2307/2445981. [DOI] [Google Scholar]
- Arnold ML, Bennett BD, Zimmer EA. Natural hybridisation between Iris fulva and Iris hexagona: pattern of ribosomal DNA variation. Evolution. 1990;44:1512–1521. doi: 10.2307/2409333. [DOI] [PubMed] [Google Scholar]
- Nason JD, Ellstrand NC, Arnold ML. Patterns of hybridization and introgression in populations of oaks, manzanitas and irises. Am J Bot. 1992;79:101–111. doi: 10.2307/2445203. [DOI] [Google Scholar]
- Arnold ML. Iris nelsonii (Iridaceae): origin and genetic composition of a homoploid hybrid species. Am J Bot. 1993;80:577–583. doi: 10.2307/2445375. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. Homology among RAPD fragments in interspecific comparisons. Mol Ecol. 1996;5:99–105. doi: 10.1111/j.1365-294X.1996.tb00295.x. [DOI] [Google Scholar]
- Yatabe Y, Kane NC, Scotti-Saintagne C, Rieseberg LH. Rampant gene exchange across a strong reproductive barrier between the annual sunflowers, H. annuus and H. petiolaris. Genetics. 2007;175:1883–1893. doi: 10.1534/genetics.106.064469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. Adaptive introgression of adaptive herbivore traits in the weedy sunflower Helianthus annuus. Am Nat. 2006;167:794–807. doi: 10.1086/504606. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. Adaptive introgression of abiotic tolerance traits in the sunflower Helianthus annuus. New Phytol. 2010;187:230–239. doi: 10.1111/j.1469-8137.2010.03234.x. [DOI] [PubMed] [Google Scholar]
- Scascitelli M, Whitney KD, Randell RA, King M, Buerkle CA, Rieseberg LH. Genome scan of hybridizing sunflowers from Texas (Helianthus annuus and H. debilis) reveals asymmetric patterns of introgression and small islands of genomic differentiation. Mol Ecol. 2010;19:521–541. doi: 10.1111/j.1365-294X.2009.04504.x. [DOI] [PubMed] [Google Scholar]
- Dumolin-Lapègue S, Demesure B, Fineschi S, Le Corre V, Petit RJ. Phylogeographic structure of white oaks throughout the European continent. Genetics. 1997;146:1475–1487. doi: 10.1093/genetics/146.4.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon GE, Vaillancourt RE, Jackson HD, Potts BM. Chloroplast sharing in the Tasmanian eucalypts. Evolution. 2001;55:703–711. doi: 10.1554/0014-3820(2001)055[0703:CSITTE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Bänfer G, Moog U, Fiala B, Mohamed M, Weising K, Blattner FR. A chloroplast genealogy of myrmecophytic Macaranga species (Euphorbiaceae) in Southeast Asia reveals hybridization, vicariance and long-distance dispersals. Mol Ecol. 2006;15:4409–4424. doi: 10.1111/j.1365-294X.2006.03064.x. [DOI] [PubMed] [Google Scholar]
- Tsitrone A, Kirkpatrick M, Levin DA. A model for chloroplast capture. Evolution. 2003;57:1776–1782. doi: 10.1111/j.0014-3820.2003.tb00585.x. [DOI] [PubMed] [Google Scholar]
- Harris SA, Robinson JP, Juniper BE. Genetic clues to the origin of the apple. Trends Genet. 2002;18:426–430. doi: 10.1016/S0168-9525(02)02689-6. [DOI] [PubMed] [Google Scholar]
- Richards CM, Volk GM, Reilley AA, Henk AD, Lockwood DR, Reeves PA, Forsline PL. Genetic diversity and population structure in Malus sieversii, a wild progenitor species of domesticated apple. Tree Genet Genomes. 2009;5:339–347. doi: 10.1007/s11295-008-0190-9. [DOI] [Google Scholar]
- Cornille A, Gladieaux P, Smulders MJM, Roldán-Ruiz I, Lauren F, Le Cam B, Nersesyan A, Clavel J, Olonova M, Feugey L, Gabrielyan I, Zhang XG, Tenaillon MI, Giraud T. New insight into the history of domesticated apple: secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genetics. 2012;8:e1002703. doi: 10.1371/journal.pgen.1002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranishi S, Tamaki I, Setsuko S, Tomaru N. Asymmetric introgression between Magnolia stellata and M. salicifolia at a site where the two species grow sympatrically. Tree Genet Genomes. 2013;9:1005–1015. doi: 10.1007/s11295-013-0612-1. doi:10.1007/s11295-013-0612-1. [DOI] [Google Scholar]
- Beck JB, Allison JR, Pryer KM, Windham MD. Identifying multiple origins of polyploid taxa: a multilocus study of the hybrid cloak fern (Astrolepis integerrima; Pteridaceae) Am J Bot. 2012;99:1857–1865. doi: 10.3732/ajb.1200199. [DOI] [PubMed] [Google Scholar]
- Ritz CM, Schmuths H, Wissemann V. Evolution by reticulation: European dogroses originated by multiple hybridization across the genus Rosa. J Hered. 2005;96:4–14. doi: 10.1093/jhered/esi011. [DOI] [PubMed] [Google Scholar]
- Nair AS, Teo CH, Schwarzacher T, Heslop-Harrison P. Genome classification of banana cultivars from South India using IRAP markers. Euphytica. 2005;144:285–290. doi: 10.1007/s10681-005-7321-2. [DOI] [Google Scholar]
- Ning A-P, Xu L-B, Lu Y, Huang B-Z, Ge X-J. Genome composition and genetic diversity of Musa germplasm from China revealed by PCR-RFLP and SSR markers. Scientia Hortic. 2007;114:281–288. doi: 10.1016/j.scienta.2007.07.002. [DOI] [Google Scholar]
- Sales EK, Butardo NG, Paniagua HG, Jansen H, Dolezei J. Assessment of ploidy and genome constitution of some Musa balbisiana cultivars using DArT markers. Philippine J Crop Sci. 2011;36:11–18. [Google Scholar]
- Kardolus JP, Van Eck HJ, Van den Berg RG. The potential of AFLPs in biosystematics: a first application in Solanum taxonomy (Solanaceae) Plant Syst Evol. 1998;210:87–103. doi: 10.1007/BF00984729. [DOI] [Google Scholar]
- Korbecka G, Rymer PD, Harris SA, Pannell JR. Solving the problem of ambiguous paralogy for marker loci: microsatellite markers with diploid inheritance in allohexaploid Mercurialis annua (Euphorbiaceae) J Hered. 2010;101:504–511. doi: 10.1093/jhered/esq026. [DOI] [PubMed] [Google Scholar]
- Fu Y-B, Peterson GW, Richards KW, Tarn TR, Percy JE. Genetic diversity of Canadian and exotic potato germplasm revealed by simple sequence repeat markers. Am J Potato Res. 2009;86:38–48. doi: 10.1007/s12230-008-9059-6. [DOI] [Google Scholar]
- Cosendai AC, Rodewald J, Hörandl E. Origin and distribution of autopolyploids via apomixis in the alpine species Ranunculus kuepferi (Ranunculaceae) Taxon. 2011;60:355–364. [Google Scholar]
- Esselink GD, Nybom H, Vosman B. Assignment of allelic configuration in polyploids using the MAC-PR (Microsatellite DNA Allele Counting - Peak Ratios) method. Theor Appl Genet. 2004;109:402–408. doi: 10.1007/s00122-004-1645-5. [DOI] [PubMed] [Google Scholar]
- Nybom H, Esselink DG, Werlemark G, Vosman B. Microsatellite DNA marker inheritance indicates preferential pairing between highly homologous genomes in polyploid and hemisexual dog-roses Rosa L. sect. Caninae. Heredity. 2004;92:139–150. doi: 10.1038/sj.hdy.6800332. [DOI] [PubMed] [Google Scholar]
- Nybom H, Esselink GD, Werlemark G, Leus L, Vosman B. Unique genomic configuration revealed by microsatellite DNA in polyploid dogroses. Rosa sect. Caninae. J Evol Biol. 2006;19:635–648. doi: 10.1111/j.1420-9101.2005.01010.x. [DOI] [PubMed] [Google Scholar]
- Babaei A, Tabaei-Aghdaei SR, Khosh-Khui M, Omidbaigi R, Naghavi MR, Esselink GD, Smulders MJM. Microsatellite analysis of Damask rose (Rosa damascena Mill.) accessions from various regions in Iran reveals multiple genotypes. BMC Plant Biol. 2007;7:12. doi: 10.1186/1471-2229-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán P, Segarra-Moragues JG, Palop-Esteban M, Moreno C, González-Candelas F. A Bayesian approach for discriminating among alternative inheritance hypotheses in plant polyploids: the allotetraploid origin of genus Borderea (Dioscoreaceae) Genetics. 2006;172:1939–1953. doi: 10.1534/genetics.105.042788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk T, Noordijk Y, Dubos T, Bink MCAM, Meulenbroek BJ, Visser RGF, van de Weg E. Microsatellite allele dose and configuration establishment (MADCE): an integrated approach for genetic studies in allopolyploids. BMC Plant Biol. 2012;12:25. doi: 10.1186/1471-2229-12-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W-J, Zhu B-R, Zeng Y-F, Zhang D-Y. TETRA: an improved program for population genetic analysis of allotetraploid microsatellite data. Mol Ecol Resources. 2008;8:1260–1262. doi: 10.1111/j.1755-0998.2008.02198.x. [DOI] [PubMed] [Google Scholar]
- Clark LV, Jasieniuk M. POLYSAT: an R package for polyploid microsatellite analysis. Mol Ecol Resources. 2011;11:562–566. doi: 10.1111/j.1755-0998.2011.02985.x. [DOI] [PubMed] [Google Scholar]
- Voorrips R, Gort G, Vosman B. Genotype calling in tetraploid species from bi-allelic marker data using mixture models. BMC Bioinformatics. 2011;12:172. doi: 10.1186/1471-2105-12-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidskehaug L, Kent M, Hayes BJ, Lien S. Genotype calling and mapping of multisite variants using an Atlantic salmon iSelect SNP array. Bioinformatics. 2011;27:303–310. doi: 10.1093/bioinformatics/btq673. [DOI] [PubMed] [Google Scholar]
- Bruvo R, Michiels NK, D'Souza TG, Schulenburg H. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol. 2004;13:2101–2106. doi: 10.1111/j.1365-294X.2004.02209.x. [DOI] [PubMed] [Google Scholar]
- Obbard DJ, Harris SA, Pannell JR. Simple allelic-phenotype diversity and differentiation statistics for allopolyploids. Heredity. 2006;97:296–303. doi: 10.1038/sj.hdy.6800862. [DOI] [PubMed] [Google Scholar]
- Assoumane A, Zoubeirou AM, Rodier-Goud M, Favreau B, Bezançon G, Verhaegen D. Highlighting the occurrence of tetraploidy in Acacia senegal (L.) Willd. and genetic variation patterns in its natural range revealed by DNA microsatellite markers. Tree Genet Genom. 2013;9:93–106. doi: 10.1007/s11295-012-0537-0. [DOI] [Google Scholar]
- Lexer C, Widmer C. The genic view of plant speciation: recent progress and emerging questions. Phil Trans R Soc. 2008;B363:3023–3036. doi: 10.1098/rstb.2008.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasburg JL, Sherman NA, Wright KM, Moyle LC, Willis JH, Rieseberg LH. What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Phil Trans Roy Soc Series B. 2012;367:364–373. doi: 10.1098/rstb.2011.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti-Saintagne C, Mariette S, Porth I, Goicoechea PG, Barreneche T, Bodénès C, Burg K, Kremer A. Genome scanning for interspecific differentiation between two closely related oak species [Quercus robur L. and Q. petraea (Matt.) Liebl.] Genetics. 2004;168:1615–1626. doi: 10.1534/genetics.104.026849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres L, Gielly L, Redoutet B, Taberlet P. Using AFLP to resolve phylogenetic relationships in a morphologically diversified plant species complex when nuclear and chloroplast sequences fail to reveal variability. Mol Phylogenet Evol. 2003;27:185–196. doi: 10.1016/S1055-7903(02)00445-1. [DOI] [PubMed] [Google Scholar]
- Bänfer G, Fiala B, Weising K. AFLP analysis of phylogenetic relationships among myrmecophytic species of Macaranga (Euphorbiaceae) and their allies. Pl Syst Evol. 2004;249:213–231. doi: 10.1007/s00606-004-0219-4. [DOI] [PubMed] [Google Scholar]
- Aggarwal RK, Brar DS, Khush GS. Two new genomes in the Oryza complex identified on the basis of molecular divergence analysis using total genomic DNA hybridization. Mol General Genet. 1997;254:1–12. doi: 10.1007/s004380050384. [DOI] [PubMed] [Google Scholar]
- Aggarwal RK, Brar DS, Nandi S, Huang N, Khush GS. Phylogenetic relationships among Oryza species revealed by AFLP markers. Theor Appl Genet. 1999;98:1320–1328. doi: 10.1007/s001220051198. [DOI] [Google Scholar]
- Zou XH, Zhang FM, Zhang JG, Zang LL, Tang L, Wang L, Sang T, Ge S. Analysis of 142 genes resolves the rapid diversification of the rice genus. Genome Biol. 2008;9:R49. doi: 10.1186/gb-2008-9-3-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou XH, Yang Z, Doyle JJ, Ge S. Multilocus estimation of divergence times and ancestral effective population sizes of Oryza species and implications for the rapid diversification of the genus. New Phytol. 2013;198:1155–1164. doi: 10.1111/nph.12230. [DOI] [PubMed] [Google Scholar]
- Scariot V, Akkak A, Botta R. Characterization and genetic relationships of wild species and old garden roses based on microsatellite analysis. J Am Soc Hort Sci. 2006;131:66–73. [Google Scholar]
- Koopman WJM, Vosman B, Sabatino GJH, Visser D, Van Huylenbroeck J, De Riek J, De Cock K, Wissemann V, Ritz CM, Maes B, Werlemark G, Nybom H, Debener T, Linde M, Smulders MJM. AFLP markers as a tool to reconstruct complex relationships in the genus Rosa (Rosaceae) Am J Bot. 2008;95:353–366. doi: 10.3732/ajb.95.3.353. [DOI] [PubMed] [Google Scholar]
- De Riek J, De Cock K, Smulders MJM, Nybom H. AFLP-based population structure analysis as a means to validate the complex taxonomy of dogroses (Rosa section Caninae) Mol Phylogenet Evol. 2013;67:547–559. doi: 10.1016/j.ympev.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Kovarik A, Werlemark G, Leitch AR, Souckova-Skalicka K, Lim KY, Khaitová L, Koukalova B, Nybom H. The asymmetric meiosis in pentaploid dogroses (Rosa sect. Caninae) is associated with a skewed distribution of rRNA gene families in the gametes. Heredity. 2008;101:359–367. doi: 10.1038/hdy.2008.63. [DOI] [PubMed] [Google Scholar]
- Meerow AW, Francisco-Ortega J, Calonje M, Griffith MP, Ayala-Silva T, Stevenson DW, Nakamura K. Zamia (Cycadales: Zamiaceae) on Puerto Rico: asymmetric genetic differentiation and the hypothesis of multiple introductions. Am J Bot. 2012;99:1828–1839. doi: 10.3732/ajb.1200494. [DOI] [PubMed] [Google Scholar]
- Koppolu R, Upadhyaya HD, Dwivedi SL, Hoisington DA, Varshney RK. Genetic relationships among seven sections of genus Arachis studied by using SSR markers. BMC Plant Biol. 2010;10:15. doi: 10.1186/1471-2229-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard M, Thorpe RS. Can microsatellites be used to infer phylogenies? Evidence from population affinities of the Western Canary Island Lizard (Gallotia galloti) Mol Phylogenet Evol. 2001;20:351–360. doi: 10.1006/mpev.2001.0981. [DOI] [PubMed] [Google Scholar]
- Avise JC. Phylogeography: The History and Formation of Species. Cambridge, MA: Harvard University Press; 2000. [Google Scholar]
- Schaal BA, Hayworth DA, Olsen KM, Rauscher JT, Smith WA. Phylogeographic studies in plants: problems and prospects. Mol Ecol. 1998;7:465–474. doi: 10.1046/j.1365-294x.1998.00318.x. [DOI] [Google Scholar]
- Posada D, Crandall KA. Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol. 2001;16:37–45. doi: 10.1016/S0169-5347(00)02026-7. [DOI] [PubMed] [Google Scholar]
- Lascoux M, Palmé AE, Cheddadi R, Latta RG. Impact of the Ice Ages on the genetic structure of trees and shrubs. Phil Trans R Soc Lond. 2004;B359:197–207. doi: 10.1098/rstb.2003.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burban C, Petit RJ. Phylogeography of maritime pine inferred with organelle markers having contrasted inheritance. Mol Ecol. 2003;12:1487–1495. doi: 10.1046/j.1365-294X.2003.01817.x. [DOI] [PubMed] [Google Scholar]
- Marchelli P, Baier C, Mengel C, Ziegenhage B, Gallo LA. Biogeographic history of the threatened species Araucaria araucana (Molina) K. Koch and implications for conservation: a case study with organellar DNA markers. Conserv Genet. 2010;11:951–963. doi: 10.1007/s10592-009-9938-5. [DOI] [Google Scholar]
- Cornille A, Giraud T, Bellard C, Tellier A, Le Cam B, Smulders MJM, Kleinschmit J, Roldán-Ruiz I, Gladieux P. Postglacial recolonization history of the European crabapple (Malus sylvestris Mill.), a wild contributor to the domesticated apple. Mol Ecol. 2013;22:2249–2263. doi: 10.1111/mec.12231. [DOI] [PubMed] [Google Scholar]
- Kadereit JW, Arafeh R, Somogyi G, Westberg E. Terrestrial growth and marine dispersal? Comparative phylogeography of five coastal plant species at a European scale. Taxon. 2005;54:861–876. doi: 10.2307/25065473. [DOI] [Google Scholar]
- Schönswetter P, Tribsch A, Niklfeld H. Phylogeography of the high Alpine cushion plant Androsace alpina (Primulaceae) in the European Alps. Plant Biol. 2003;5:623–630. doi: 10.1055/s-2003-44686. [DOI] [Google Scholar]
- Guicking D, Blattner FR, Fiala B, Slik F, Mohamed M, Weising K. Comparative chloroplast DNA phylogeography of two tropical pioneer tree species, Macaranga gigantea and M. pearsonii (Euphorbiaceae) Tree Genet Genomes. 2011;7:573–585. doi: 10.1007/s11295-010-0357-z. [DOI] [Google Scholar]
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Collevatti RG, Grattapaglia D, Hay JD. Evidences for multiple maternal lineages of Caryocar brasiliense populations in the Brazilian Cerrado based on the analysis of chloroplast DNA sequences and microsatellite haplotype variation. Mol Ecol. 2003;12:105–115. doi: 10.1046/j.1365-294x.2003.01701.x. [DOI] [PubMed] [Google Scholar]
- Vachon N, Freeland JR. Phylogeographic inferences from chloroplast DNA: quantifying the effects of mutations in repetitive and non-repetitive sequences. Mol Ecol Resources. 2011;11:279–285. doi: 10.1111/j.1755-0998.2010.02921.x. [DOI] [PubMed] [Google Scholar]
- Grivet D, Petit RJ. Chloroplast DNA phylogeography of the hornbeam in Europe: evidence for a bottleneck at the outset of postglacial colonization. Cons Genet. 2003;4:47–56. doi: 10.1023/A:1021804009832. [DOI] [Google Scholar]
- Valtuena FJ, Preston CD, Kadereit JW. Phylogeography of a Tertiary relict plant, Meconopsis cambrica (Papaveraceae), implies the existence of northern refugia for a temperate herb. Mol Ecol. 2012;21:1423–1437. doi: 10.1111/j.1365-294X.2012.05473.x. [DOI] [PubMed] [Google Scholar]
- Shafer ABA, Cullingham CI, Coté SC, Coltman DW. Of glaciers and refugia: a decade of study sheds new light on the phylogeography of northwestern North America. Mol Ecol. 2010;19:4589–4621. doi: 10.1111/j.1365-294X.2010.04828.x. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Morris AB, McLachlan JS, Manos PS, Soltis PS. Comparative phylogeography of unglaciated eastern North America. Mol Ecol. 2006;15:4261–4293. doi: 10.1111/j.1365-294X.2006.03061.x. [DOI] [PubMed] [Google Scholar]
- Harushima Y, Yano M, Shomura A, Sato M, Shimano T, Kuboki Y, Yamamoto T, Lin SY, Antonio BA, Parco A, Kajiya H, Huang N, Yamamoto K, Nagamura Y, Kurata N, Khush GS, Sasaki T. A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics. 1998;148:479–494. doi: 10.1093/genetics/148.1.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzl P, Li H, Carling J, Zhou M, Raman H, Paul E, Hearnden P, Maier C, Xia L, Caig V, Ovesná J, Cakir M, Poulsen D, Wang J, Raman R, Smith KP, Muehlbauer GJ, Chalmers KJ, Kleinhofs A, Huttner E, Kilian A. A high-density consensus map of barley linking DArT markers to SSR. RFLP and STS loci and agricultural traits. BMC Genomics. 2006;7:206. doi: 10.1186/1471-2164-7-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, Ware DH, Buckler ES. A first-generation haplotype map of maize. Science. 2009;326:1115–1117. doi: 10.1126/science.1177837. [DOI] [PubMed] [Google Scholar]
- Pollard DA. Design and construction of recombinant inbred lines. Methods Mol Biol. 2012;871:31–39. doi: 10.1007/978-1-61779-785-9_3. [DOI] [PubMed] [Google Scholar]
- Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci U S A. 1991;88:9828–9832. doi: 10.1073/pnas.88.21.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar'an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezel J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nature Biotechnol. 2013;31:240–246. doi: 10.1038/nbt.2491. [DOI] [PubMed] [Google Scholar]
- Abdurakhmonov IY, Abdukarimov A. Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int J Plant Genomics. 2008;2008:574927. doi: 10.1155/2008/574927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Cerda BJ, Cloutier S. Genetic Diversity in Plants. Mahmut Çalişkan: InTech; 2012. Association Mapping in Plant Genomes. DOI: 10.5772/33005. [Google Scholar]
- Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, McClung AM, Bustamante CD, McCouch SR. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun. 2011;2:467. doi: 10.1038/ncomms1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strigens A, Freitag NM, Gilbert X, Grieder C, Riedelsheimer C, Schrag TA, Messmer R, Melchinger AE. Association mapping for chilling tolerance in elite flint and dent maize inbred lines evaluated in growth chamber and field experiments. Plant Cell Environ. 2013;36:1871–1887. doi: 10.1111/pce.12096. [DOI] [PubMed] [Google Scholar]
- Sork VL, Aitken SN, Dyer RJ, Eckert AJ, Legendre P, Neale DB. Putting the landscape into the genomics of trees: approaches for understanding local adaptation and population responses to changing climate. Trees Genet Genomes. 2013;9:901–911. doi: 10.1007/s11295-013-0596-x. [DOI] [Google Scholar]
- Pavy N, Gagnon F, Rigault P, Blais S, Deschênes A, Boyle B, Pelgas B, Deslauriers M, Clément S, Lavigne P, Lamothe M, Cooke JE, Jaramillo-Correa JP, Beaulieu J, Isabel N, Mackay J, Bousquet J. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Ecol Resour. 2013;13:324–336. doi: 10.1111/1755-0998.12062. [DOI] [PubMed] [Google Scholar]
- Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, Gasic K, Micheletti D, Rosyara UR, Cattonaro F, Vendramin E, Main D, Aramini V, Blas AL, Mockler TC, Bryant DW, Wilhelm L, Troggio M, Sosinski B, Aranzana MJ, Arús P, Iezzoni A, Morgante M, Peace C. Development and evaluation of a 9 K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One. 2012;7:e35668. doi: 10.1371/journal.pone.0035668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagné D, Crowhurst RN, Troggio M, Davey MW, Gilmore B, Lawley C, Vanderzande S, Hellens RP, Kumar S, Cestaro A, Velasco R, Main D, Rees JD, Iezzoni A, Mockler T, Wilhelm L, Van de Weg E, Gardiner SE, Bassil N, Peace C. Genome-wide SNP detection, validation, and development of an 8 K SNP array for apple. PLoS One. 2012;7:e31745. doi: 10.1371/journal.pone.0031745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peace C, Bassil N, Main D, Ficklin S, Rosyara UR, Stegmeir T, Sebolt A, Gilmore B, Lawley C, Mockler TC, Bryant DW, Wilhelm L, Iezzoni A. Development and evaluation of a genome-wide 6 K SNP array for diploid sweet cherry and tetraploid sour cherry. PLoS One. 2012;7:e48305. doi: 10.1371/journal.pone.0048305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Holland JB, McMullen MD, Buckler ES. Genetic design and statistical power of nested association mapping in maize. Genetics. 2008;178:539–551. doi: 10.1534/genetics.107.074245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kover PX, Valdar W, Trakalo J, Scarcelli N, Ehrenreich IM, Purugganan MD, Durrant C, Mott R. A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 2009;5:e1000551. doi: 10.1371/journal.pgen.1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JP, McMullen MD, Holland JB, Tian F, Bradbury P, Ross-Ibarra J, Buckler ES, Flint-Garcia SA. Genetic architecture of maize kernel composition in the nested association mapping and inbred association panels. Plant Physiol. 2012;158:824–834. doi: 10.1104/pp.111.185033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland JA, Bradbury PJ, Buckler ES, Nelson RJ. Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc Natl Acad Sci U S A. 2011;108:6893–6898. doi: 10.1073/pnas.1010894108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer JA, Flint-Garcia SA, De Leon N, McMullen MD, Kaeppler SM, Buckler ES. The genetic architecture of maize stalk strength. PLoS One. 2013;8:e67066. doi: 10.1371/journal.pone.0067066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Zhao YF, Korban SS. Identification of genetic loci associated with fire blight resistance in Malus through combined use of QTL and association mapping. Physiol Plant. 2013;148:344–353. doi: 10.1111/ppl.12068. [DOI] [PubMed] [Google Scholar]
- Jiang G-L. In: Plant Breeding from Laboratories to Fields. Andersen SB, editor. InTech; 2013. Molecular markers and marker-assisted breeding in plants. DOI: 10.5772/52583. [Google Scholar]
- Guo Z, Tucker DM, Lu J, Kishore V, Gay G. Evaluation of genome-wide selection efficiency in maize nested association mapping populations. Theor Appl Genet. 2012;124:261–275. doi: 10.1007/s00122-011-1702-9. [DOI] [PubMed] [Google Scholar]
- Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Bosquet L, Crossa J, Von Zitzewitz J, Serret MD, Araus JL. High-throughput phenotyping and genomic selection: the frontiers of crop breeding converge. J Integr Plant Biol. 2012;54:312–320. doi: 10.1111/j.1744-7909.2012.01116.x. [DOI] [PubMed] [Google Scholar]
- Zou W, Zeng ZB. Statistical Methods for Mapping Multiple QTL. Int J Plant Genomics. 2008;2008:286561. doi: 10.1155/2008/286561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed GH, Kent JO, Wittwer CT. High-resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics. 2007;8:597–608. doi: 10.2217/14622416.8.6.597. [DOI] [PubMed] [Google Scholar]
- Li J, Wang X, Dong R, Yang Y, Zhou J, Yu C, Cheng Y, Yan C, Chen J. Evaluation of high-resolution melting for gene mapping in rice. Plant Mol Biol Rep. 2011;29:979–985. doi: 10.1007/s11105-011-0289-2. [DOI] [Google Scholar]
- Ganopoulos I, Tsaballa A, Xanthopoulou A, Madesis P, Tsaftaris A. Sweet cherry cultivar identification by high-resolution-melting (HRM) analysis using gene-based SNP markers. Plant Mol Biol Rep. 2013;31:763–768. doi: 10.1007/s11105-012-0538-z. [DOI] [Google Scholar]
- Dang X-D, Kelleher CT, Howard-Wiiliams E, Meade CV. Rapid identification of chloroplast haplotypes using High Resolution Melting analysis. Mol Ecol Resources. 2012;12:894–908. doi: 10.1111/j.1755-0998.2012.03164.x. [DOI] [PubMed] [Google Scholar]
- Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product by utilizing the 5′–3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci U S A. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies – the next generation. Nature Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Rothberg JM, Leamon JH. The development and impact of 454 sequencing. Nature Biotechnol. 2008;26:1117–1124. doi: 10.1038/nbt1485. [DOI] [PubMed] [Google Scholar]
- Byrne S, Czaban A, Studer B, Panitz F, Bendixen C, Asp T. Genome wide allele frequency fingerprints (GWAFFs) of populations via genotyping by sequencing. PLoS One. 2013;8:e57438. doi: 10.1371/journal.pone.0057438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Cezard T, Fuentes-Utrilla P, Eland C, Gharbi K, Blaxter ML. Special features of RAD sequencing data: implications for genotyping. Mol Ecol. 2013;11:3151–3164. doi: 10.1111/mec.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelkrim J, Robertson BC, Stanton J-AL, Gemmell NJ. Fast, cost-effective development of species-specific microsatellite markers by genomic sequencing. BioTechniques. 2009;46:185–192. doi: 10.2144/000113084. [DOI] [PubMed] [Google Scholar]
- Santana QC, Coetzee MPA, Steenkamp ET, Mlonyeni OX, Hammond GNA, Wingfield MJ, Wingfield BD. Microsatellite discovery by deep sequencing of enriched genomic libraries. BioTechniques. 2009;46:217–223. doi: 10.2144/000113085. [DOI] [PubMed] [Google Scholar]
- Allentoft ME, Schuster SC, Holdaway RN, Hale ML, McLay E, Oskam C, Gilbert TP, Spencer P, Willerslev E, Bunce M. Identification of microsatellites from an extinct moa species using high-throughput (454) sequence data. BioTechniques. 2009;46:195–200. doi: 10.2144/000113086. [DOI] [PubMed] [Google Scholar]
- Csencsics D, Brodbeck S, Holderegger R. Cost-effective, species-specific microsatellite development for the endangered dwarf bulrush (Typha minima) using next generation sequencing technology. J Hered. 2010;101:789–793. doi: 10.1093/jhered/esq069. [DOI] [PubMed] [Google Scholar]
- Lee R, Thimmapuram J, Thinglum KA, Gong G, Hernandez AG, Wright CL, Kim RW, Mikel MA, Tranel PJ. Sampling the waterhemp (Amaranthus tuberculatus) genome using pyrosequencing technology. Weed Sci. 2009;57:463–469. doi: 10.1614/WS-09-021.1. [DOI] [Google Scholar]
- Tangphatsornruang S, Somta P, Uthaipaisanwong P, Chanprasert J, Sangsakru D, Seehalak W, Sommanas W, Trangoonrung S, Srinives P. Characterization of microsatellites and gene contents from genome shotgun sequences of mungbean (Vigna radiata (L.) Wilczek) BMC Plant Biol. 2009;9:137. doi: 10.1186/1471-2229-9-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot. 2012;99:193–208. doi: 10.3732/ajb.1100394. [DOI] [PubMed] [Google Scholar]
- Castoe TA, Poole AW, De Koning APJ, Jones KL, Tomback DF, Oyler-McCance SJ, Fike JA, Lance SL, Streicher JW, Smith EN, Pollock DD. Rapid microsatellite identification from Illumina paired-end genomic sequencing in two birds and a snake. PLoS One. 2012;7:e30953. doi: 10.1371/journal.pone.0030953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings TN, Knaus BJ, Mullins TD, Haig SM, Cronn RC. Multiplexed microsatellite recovery using massively parallel sequencing. Mol Ecol Resources. 2011;11:1060–1067. doi: 10.1111/j.1755-0998.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- Lepais O, Bacles CFE. Comparison of random and SSR-enriched shotgun pyrosequencing for microsatellite discovery and single multiplex PCR optimization in Acacia harpophylla F. Muell. Ex Benth. Mol Ecol Resources. 2011;11:711–724. doi: 10.1111/j.1755-0998.2011.03002.x. [DOI] [PubMed] [Google Scholar]
- Binladen J, Gilbert MTP, Bollback JP, Panitz F, Bendixen C, Nielsen R, Willerslev E. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One. 2007;2:e97. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, López P, König C, Kohl G, Novak J, Stuessy TF. A simple and cost-effective approach for microsatellite isolation in non-model plant species using small-scale 454 pyrosequencing. Taxon. 2011;60:1442–1449. [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, Phillippy AM. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Chin C-S, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010;38:e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohme MA, Soler RF, Wink M, Frohme M. Microsatellite marker discovery using single molecule real-time circular consensus sequencing on the Pacific Biosciences RS. BioTechniques. 2013;55:253–256. doi: 10.2144/000114104. [DOI] [PubMed] [Google Scholar]
- Krapp F, Wöhrmann T, Pinangé D, Benko-Iseppon A, Huettel B, Weising K. A set of plastid microsatellite loci for the genus Dyckia (Bromeliaceae) derived from 454 pyrosequencing. Am J Bot Primer Notes & Protocols. 2012;99:e470–e473. doi: 10.3732/ajb.1200153. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, Wang X, Ott F, Müller J, Alonso-Blanco C, Borgwardt K, Schmid KJ, Weigel D. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan J, Fan D, Weng Q, Huang T, Dong G, Sang T, Han B. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MW, Uauy C. Genomics reveals new landscapes for crop improvement. Genome Biol. 2013;14:206. doi: 10.1186/gb-2013-14-6-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, Stern DL. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 2011;21:610–617. doi: 10.1101/gr.115402.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang J, Mou Y, Geng J, McVetty PB, Hu S, Li G. Integration of Solexa sequences on an ultradense genetic map in Brassica rapa L. BMC Genomics. 2011;12:249. doi: 10.1186/1471-2164-12-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitdewilligen JG, Wolters AM, D'hoop BB, Borm TJ, Visser RG, Van Eck HJ. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS One. 2013;8:e62355. doi: 10.1371/journal.pone.0062355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Orsouw NJ, Hogers RC, Janssen A, Yalcin F, Snoeijers S, Verstege E, Schneiders H, van der Poel H, Van Oeveren J, Verstegen H, Van Eijk MJ. Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes. PLoS One. 2007;2:e1172. doi: 10.1371/journal.pone.0001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature Methods. 2008;5:247–252. doi: 10.1038/nmeth.1185. [DOI] [PubMed] [Google Scholar]
- Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES. A SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double Digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter PD, Johnson E. RAD paired-end sequencing for local de novo assembly and SNP discovery in non-model organisms. Meth Mol Biol. 2012;888:135–151. doi: 10.1007/978-1-61779-870-2_9. [DOI] [PubMed] [Google Scholar]
- Willing E-M, Hoffmann M, Klein JD, Weigel D, Dreyer C. Paired-end RAD-seq for de-novo assembly and marker design without available reference. Bioinformatics. 2011;27:2187–2193. doi: 10.1093/bioinformatics/btr346. [DOI] [PubMed] [Google Scholar]
- Bus A, Hecht J, Huettel B, Reinhardt R, Stich B. High-throughput polymorphism detection and genotyping in Brassica napus using next-generation RAD sequencing. BMC Genomics. 2012;13:281. doi: 10.1186/1471-2164-13-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione D, Acquadro A, Portis E, Tirone M, Knapp SJ, Lanteri S. RAD tag sequencing as a source of SNP markers in Cynara cardunculus L. BMC Genomics. 2012;13:3. doi: 10.1186/1471-2164-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfender WF, Saha MC, Johnson EA, Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122:1467–1480. doi: 10.1007/s00122-011-1546-3. [DOI] [PubMed] [Google Scholar]
- Baxter SW, Davey JW, Johnston JS, Shelton AM, Heckel DG, Jiggins CD, Blaxter ML. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS ONE. 2011;6:e19315. doi: 10.1371/journal.pone.0019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Amores A, Hohenlohe P, Cresko W, Postlethwait J. Stacks: building and genotyping loci de novo from short-read sequences. Genes, Genomes, Genetics. 2011;1:171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, Casler MD, Buckler ES, Costich DE. Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013;9:e1003215. doi: 10.1371/journal.pgen.1003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Chong Z, Ruan J, Wu C-I. Rainbow: an integrated tool for efficient clustering and assembling RAD-seq reads. Bioinformatics. 2012;28:2732–2737. doi: 10.1093/bioinformatics/bts482. [DOI] [PubMed] [Google Scholar]
- Poland JA, Rife TW. Genotyping-by-sequencing for plant breeding and genetics. The Plant Genome. 2012;5:92–102. doi: 10.3835/plantgenome2012.05.0005. [DOI] [Google Scholar]
- Yang H, Tao Y, Zheng Z, Li C, Sweetingham M, Howieson J. Application of next-generation sequencing for rapid marker development in molecular plant breeding: a case study on anthracnose disease resistance in Lupinus angustifolius L. BMC Genomics. 2012;13:318. doi: 10.1186/1471-2164-13-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JBW. Principles of transcriptome analysis and gene expression quantification: an RNA-seq tutorial. Mol Ecol Resources. 2013;13:559–572. doi: 10.1111/1755-0998.12109. [DOI] [PubMed] [Google Scholar]
- Harper AL, Trick M, Higgins J, Fraser F, Clissold L, Wells R, Hattori C, Werner P, Bancroft I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nature Biotechnol. 2012;30:798–802. doi: 10.1038/nbt.2302. [DOI] [PubMed] [Google Scholar]
- Trick M, Adamski NM, Mugford SG, Jiang CC, Febrer M, Uauy C. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol. 2012;12:14. doi: 10.1186/1471-2229-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yeh C-T, Tang HM, Nettleton D, Schnable PS. Gene mapping via bulked segregant RNA-seq (BSR-seq) PloS One. 2012;7:e36406. doi: 10.1371/journal.pone.0036406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahl G, Molina C, Rotter B, Jüngling R, Frank A, Krezdorn N, Hoffmeier K, Winter P. Reduced representation sequencing of plant stress transcriptomes. J Plant Biochem Biotechnol. 2012;21(Suppl 1):119–127. [Google Scholar]
- Ruge-Wehling B, Wojacki J, Fischer K, Rotter B, Wehling P. Introgression of an effective resistance against BYDV from Hordeum bulbosum to cultivated barley [in German] Tagungsband der 63. Jahrestagung der Vereinigung der Pflanzenzüchter und Saatgutkaufleute Österreichs 19-21 November 2012. 2013. pp. 3–6. Raumberg-Gumpenstein, Austria. Resistenz gegen biotischen Stress in der Pflanzenzüchtung.
- Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2010;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeb JE, Carvalho G, Hauser L, Naish K, Roberts S, Seeb LW. Single-nucleotide polymorphism (SNP) discovery and applications of SNP genotyping in nonmodel organisms. Mol Ecol Resour. 2011;11(Suppl 1):1–8. doi: 10.1111/j.1755-0998.2010.02979.x. [DOI] [PubMed] [Google Scholar]
- Lasky JR, Des Marais DL, McKay JK, Richards JH, Juenger TE, Keitt TH. Characterizing genomic variation of Arabidopsis thaliana: the roles of geography and climate. Mol Ecol. 2012;21:5512–5529. doi: 10.1111/j.1365-294X.2012.05709.x. [DOI] [PubMed] [Google Scholar]
- Stölting KN, Nipper R, Lindtke D, Caseys C, Waeber S, Castiglione S, Lexer C. Genomic scan for single nucleotide polymorphisms reveals patterns of divergence and gene flow between ecologically divergent species. Mol Ecol. 2013;22:842–855. doi: 10.1111/mec.12011. [DOI] [PubMed] [Google Scholar]
- Narum SR, Buerkle CA, Davey JW, Miller MR, Hohenlohe PA. Genotyping-by-sequencing in ecological and conservation genomics. Mol Ecol. 2013;22:2841–2847. doi: 10.1111/mec.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JE, Faircloth BC. Next-generation phylogenetics takes root. Mol Ecol. 2013;22:19–21. doi: 10.1111/mec.12050. [DOI] [PubMed] [Google Scholar]
- Givnish TJ, Ames M, McNeal JR, McKain MR, Steele PR, DePamphilis CW, Graham SW, Pires JC, Stevenson DW, Zomlefer WB, Briggs BG, Duvall MR, Moore MJ, Heaney JM, Soltis DE, Soltis PS, Thiele K, Leebens-Mack JH. Assembling the tree of the monocotyledons: plastome sequence phylogeny and evolution of Poales. Ann Missouri Bot Gard. 2010;97:584–616. doi: 10.3417/2010023. [DOI] [Google Scholar]
- Griffin P, Robin C, Hoffmann A. A next-generation sequencing method for overcoming the multiple gene copy problem in polyploid phylogenetics, applied to Poa grasses. BMC Biol. 2011;9:19. doi: 10.1186/1741-7007-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JM, Wall PK, Edger PP, Landherr LL, Ma H, Pires JC, Leebens-Mack J, DePamphilis CW. Identification of shared single-copy nuclear genes in Arabidopsis, Populus, Vitis, and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol Biol. 2010;10:61. doi: 10.1186/1471-2148-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison N, Kidner CA. Next-generation sequencing and systematics: what can a billion base pairs of DNA sequence data do for you? Taxon. 2011;60:1552–1566. [Google Scholar]
- Buerkle CA, Gompert Z. Population genomics based on low coverage sequencing: how low should we go? Mol Ecol. 2013;22:3028–3035. doi: 10.1111/mec.12105. [DOI] [PubMed] [Google Scholar]