

Abstract

ATP-binding cassette transporter G1 (ABCG1) mediates cholesterol and oxysterol efflux onto lipidated lipoproteins and plays an important role in macrophage reverse cholesterol transport. Here, we identified a highly conserved sequence present in the five ABCG transporter family members. The conserved sequence is located between the nucleotide binding domain and the transmembrane domain and contains five amino acid residues from Asn at position 316 to Phe at position 320 in ABCG1 (NPADF). We found that cells expressing mutant ABCG1, in which Asn316, Pro317, Asp319, and Phe320 in the conserved sequence were replaced with Ala simultaneously, showed impaired cholesterol efflux activity compared with wild type ABCG1-expressing cells. A more detailed mutagenesis study revealed that mutation of Asn316 or Phe 320 to Ala significantly reduced cellular cholesterol and 7-ketocholesterol efflux conferred by ABCG1, whereas replacement of Pro317 or Asp319 with Ala had no detectable effect. To confirm the important role of Asn316 and Phe320, we mutated Asn316 to Asp (N316D) and Gln (N316Q), and Phe320 to Ile (F320I) and Tyr (F320Y). The mutant F320Y showed the same phenotype as wild type ABCG1. However, the efflux of cholesterol and 7-ketocholesterol was reduced in cells expressing ABCG1 mutant N316D, N316Q, or F320I compared with wild type ABCG1. Further, mutations N316Q and F320I impaired ABCG1 trafficking while having no marked effect on the stability and oligomerization of ABCG1. The mutant N316Q and F320I could not be transported to the cell surface efficiently. Instead, the mutant proteins were mainly localized intracellularly. Thus, these findings indicate that the two highly conserved amino acid residues, Asn and Phe, play an important role in ABCG1-dependent export of cellular cholesterol, mainly through the regulation of ABCG1 trafficking.
ATP-binding cassette transporter G1 (ABCG1) belongs to the G branch of the ABC transporter superfamily that includes five half-transporters, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8. The putative structure of the G branch consists of one NH2-terminal nucleotide binding domain (NBD) and one COOH-terminal membrane-spanning domain (MSD) that contains six putative transmembrane α-helices (Figure.1A).1−4 Some of the G family members, such as ABCG1 and ABCG2, function as homodimers,5,6 whereas other family members, such as ABCG5 and ABCG8, function as heterodimers.7 ABCG1 is localized to the endoplasmic reticulum (ER) and Golgi and plasma membranes in macrophages and other cell types.5,8−11 The half-transporter mediates cholesterol efflux onto lipidated lipoproteins like high density lipoprotein (HDL) but not onto lipid poor apolipoprotein A-I (apoA-I).5,8,9,12,13 Mice lacking ABCG1 accumulate lipids in macrophages and in hepatocytes14 and show a significantly decreased level of plasma HDL after being fed a high cholesterol diet or treated with the liver X receptor (LXR) agonist T0901317.15
Figure 1.

Effect of the conservative sequence on ABCG1 function. Panel A: Predicted topology of ABCG1 and sequence alignment for ABCG family. Only part of sequence alignment that includes the conserved sequence (NPADF) is shown. The sequence alignment was performed by ClustalW2. Panel B: Expression of wild type and mutant ABCG1. Whole cell lysates were made from HEK293 cells transiently transfected with empty vectors (V) or vectors containing wild type ABCG1 (WT) or mutant ABCG1 (NPDF-A) cDNA and then subjected to SDS-PAGE and immunoblotting. Membrane was cut into halves along the 82 kDa based on the prestained protein standards (Life technology). The bottom part was probed with a polyclonal anti-ABCG1 antibody, H-65 (Santa Cruz), and the top part was detected with a polyclonal anticalnexin antibody. Antibody binding was detected using horseradish peroxidase-conjugated goat antimouse or with donkey antirabbit IgG (Sigma) followed by enhanced chemiluminescence detection (Pierce). The membranes were then exposed to Kodak BioMax MR films (Kodak). * indicates no specific band. Panel C: Cholesterol efflux. ABCG1-mediated cholesterol efflux onto lipidated apoA-I was carried out as described in Materials and Methods. HEK293 cells were transiently transfected with empty vector (V) or plasmid containing cDNA of wild type or mutant ABCG1, NPDF-A, in which Asn316, Pro317, Asp319, and Phe320 were replaced with Ala simultaneously. The cells were then labeled with [3H]-cholesterol. After washing, the cells were incubated with 5 μg/mL lipidated apoA-I. The radioactive content of the media and cells was measured separately. Sterol transfer was expressed as the percentage of the radioactivity released from the cells into the media relative to the total radioactivity in the cells plus media. * indicates p < 0.05; ** indicates p < 0.01. Values are mean ± SD of at least three independent experiments.
ABCG1 is highly expressed in macrophages and plays an important role in macrophage reverse cholesterol transport (RCT) in vivo. Overexpression of ABCG1 in macrophages significantly increases macrophage RCT in vivo, whereas knockdown or knockout of ABCG1 expression in macrophages markedly decreases macrophage RCT in vivo.12 However, the precise role of macrophage ABCG1 in the protection against the development of atherosclerosis remains uncertain. Transplantation of bone marrow lacking ABCG1 into low density lipoprotein receptor knockout (Ldlr–/–) mice shows conflicting effects. Out et al.16 observed a moderate increase in atherosclerosis, whereas two other independent groups reported that macrophages lacking ABCG1 decrease atherosclerosis in hyperlipidemic Ldlr–/– mice.17,18 The explanation for this discrepancy is not yet clarified. More recent findings have shown a complex role of ABCG1 during the progression of atherosclerosis, depending on the atherosclerotic stages examined. Lack of ABCG1 leads to a significant increase in early atherosclerotic lesion size but causes retarded lesion progression in the more advanced stages in Ldlr–/– mice.19 Interestingly, Schou et al. recently reported that a genetic variation in ABCG1 promoter (−376C > T) that reduces mRNA levels of ABCG1 by about 40% is associated with increased risk of myocardial infarction and ischemic heart disease.20
ABCG1 and ABCA1 have been shown to promote cellular cholesterol efflux synergistically.21,22 The efflux of cholesterol and phospholipids onto apoA-I mediated by ABCA1 converts apoA-I into nascent HDL, which can then act as an efficient acceptor for ABCG1-mediated cholesterol efflux. It has been demonstrated that ABCA1 and ABCG1, but not SR-BI, are responsible for macrophage RCT in vivo.12 Knockout of both ABCA1 and ABCG1 in mice leads to dramatic foam cell formation and acceleration of atherosclerosis.23−25 In addition, it has been shown that ABCG1-mediated cholesterol translocation plays an important role in pancreatic β-cell insulin secretion.26−28 ABCG1 is required for reconstituted HDL (rHDL)-promoted insulin secretion. Lack of ABCG1 expression dramatically reduces pancreatic β-cell insulin secretion both in vivo and in vitro. However, how ABCG1 mediates cholesterol efflux is unclear.
Most recently, we found that ABCG1 is palmitoylated at five cysteine residues. This covalent posttranslational modification is required for ABCG1-mediated cholesterol efflux.11 We also found that one highly conserved cysteine residue located at position 514 (Cys514) in the first putative transmembrane α helix plays an important role in ABCG1-mediated cholesterol efflux. Replacement of Cys514 with Ala or Ser essentially eliminates ABCG1-mediated cholesterol efflux.29 The transmembrane domains of the ABC transporters vary considerably between different ABC proteins. In contrast, the NBDs of the ABC transporters are highly conserved. Each NBD contains three highly conserved sequence elements (the Walker A motif, the Walker B motif, and a signature motif) that play critical roles in ATP binding and hydrolysis and in the provision of energy for the transporter to translocate its substrates across the cell membranes. In addition to the traditional conserved motifs, we found that a highly conserved sequence exists in the G branch of the ABC transporter superfamily (Figure 1A). The sequence is located between the NBD and MSD and contains five amino acid residues. Asn, Pro, Asp, and Phe are completely conserved among all of the five G subfamily members. The middle amino acid residue is an Ala in all subfamily members except for ABCG5, where it is a Phe (Figure 1A). To define its functional role, we replaced these amino acid residues with Ala simultaneously or individually. Human embryonic kidney 293 (HEK293) cells transiently or stably expressing wild type or mutant ABCG1 were labeled with 3H sterols. The efflux of sterol onto lipidated apoA-I and rHDL was examined. We found that mutations of Asn316 and Phe 320 within the conserved sequence impaired ABCG1 trafficking to the cell surface and reduced cellular sterol efflux conferred by ABCG1.
Materials and Methods
Materials
Fetal bovine serum (FBS) was obtained from Sigma. Complete EDTA-free protease inhibitors were purchased from Roche. [3H]-cholesterol (54.2 Ci/mmol) was purchased from PerkinElmer. [3H]-7-ketocholesterol (40–60 Ci/mmol) was obtained from American Radiolabeled Chemicals, Inc. Polyclonal anti-ABCG1 antibody, 4497, was made by Genscript using a peptide (TKRLKGLRKDSSSM, amino acids 374 to 387 of ABCG1) as an antigen as described in ref (11). Polyclonal anti-ABCG1 antibody, H-65, was purchased from Santa Cruz Biotechnology, Inc. Mouse monoclonal anticalnexin, GM-130, EEA1, Na+K+-ATPase, Rab11 antibodies were purchased from BD Bioscience. Mouse monoclonal anti-Myc antibody, 9E10, was purified from a hybridoma cell line CRL-1729 as described in ref (11). Polyclonal anti-HA antibody was purchased from Pierce. Lipofectamine 2000 and Directional pcDNA3.1 TOPO vectors were obtained from Life Technologies Inc. QuickChange Site-Directed Mutagenesis Kits were purchased from Agilent Technologies. All other reagents were obtained from Fisher Scientific unless otherwise indicated.
The lipidated apoA-I used in these experiments was purified from HEK 293S cells as described.29,30 rHDL was prepared as described in ref (31) with modifications. Briefly, human apoA-I cDNA that contained amino acid 25 to 267 and a His6 tag at the NH2-terminus was cloned into a pET-28a(+) vector and expressed in E. coli BL-21-DE3 cells. The His-tagged protein was purified using Ni-NTA agarose (Qiagene) according to the manufacturer’s protocol. The protein was concentrated and further purified using size-exclusion chromatography on a Tricorn Superose 12 10/300 fast-performance liquid chromatography column (GE Healthcare). Fractions containing recombinant apoA-I were concentrated using a 10 kDa-MW cutoff Centriplus filter. Protein purity was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie Brilliant Blue R-250 staining. Next, 5 mg of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids Inc., Pelham, AL) dissolved in chloroform was dried under nitrogen gas in a glass tube and suspended in 500 μL of phosphate-buffered saline (PBS). After that, 2 mg of recombinant apoA-I in 500 μL of PBS was added, and the sample was sonicated in a water-bath sonicator at 26 °C until clear.
Site-Directed Mutagenesis and Cell Culture
Human ABCG1 cDNA was cloned from human hepatocytes HepG2 as described29 and used as the template to generate mutations using QuickChange Site-Directed Mutagenesis Kits according to the manufacturer’s instructions. Oligonucleotides bearing mismatched bases at the residue to be mutated were synthesized by IDT, Inc. (Coralville, IA). The presence of the desired mutation and the integrity of each construct were verified by DNA sequencing (TAGC, Edmonton, Canada).
HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (glucose, 4.5 g/Liter) containing 10% (v/v) FBS under a 37 °C incubator with 5% CO2. The cell lines stably expressing wild type or mutant ABCG1 were generated as described.29,32 Several subclones were isolated, and the expression of ABCG1 was confirmed by Western Blot analysis using a rabbit anti-ABCG1 polyclonal antibody, H-65.
Immunoprecipitation and Immunoblot Analysis of ABCG1
The cells expressing wild type or mutant ABCG1 were collected and lysed in lysis buffer A (1% Triton X-100, 150 mM NaCl, 50 mM Tris–HCl, pH 7.4) containing 1X Complete EDTA-free protease inhibitors. Protein concentration was determined by BCA protein assay. The same quantities of total proteins were used to immunoprecipitate ABCG1 from the whole cell lysates. Anti-C-Myc antibody 9E10 and anti-ABCG1 antibody 4497-conjugated beads were used to immunoprecipitate C-Myc tagged and untagged ABCG1, respectively. The whole cell lysates and immunoprecipitated ABCG1 eluted from the beads by 1× SDS loading buffer were fractionated by 8% SDS-PAGE, which was followed by immunoblotting with specific antibodies indicated. Antibody binding was detected using IRDye-labeled goat antimouse or antirabbit IgG (Li-Cor Biosciences). The signals were detected by a Licor Odyssey Infrared Imaging System (Li-Cor Biosciences).
Sterol Efflux Assay
Sterol efflux assay was performed as described previously.9,13,29 Briefly, on day 1, HEK293 cells were seeded at 2.5 × 105 cells/well in a 12-well plate. After 24 h, the cells were transfected with plasmids expressing wild type or mutant ABCG1 using Lipofectamine 2000. After 48 h, the cells were directly labeled with [3H]-cholesterol or [3H]-7-ketocholesterol (2 μCi/ml) for 16 h and then washed three times in 1 mL of DMEM containing 0.02% bovine serum albumin (BSA). After, the cells were incubated with DMEM containing 0.02% BSA and 5 μg/mL lipidated apoA-I or rHDL. The media were collected. The cells were lysed in 0.5 mL of lysis buffer B (0.1 N NaOH, 0.01% SDS). The radioactive content of the media and cells was measured separately by scintillation counting. Sterol transfer was expressed as the percentage of the radioactivity released from the cells into the media relative to the total radioactivity in cells plus media.
Biotinylation Analysis of ABCG1
Cell surface proteins were biotinylated as described.33,34 Briefly, HEK293 cells transiently expressing wild type or mutant ABCG1 were biotinylated using EZ-Link Sulfo-NHS-LC-Biotin according to the manufacturer’s protocol (Pierce). The cells were lysed in 200 μL of lysis buffer A containing 1X protease inhibitors. A total of 40 μL of the cell lysate was saved and 150 μL of the lysate was added to 60 μL of a 50% slurry of NeutrAvidin agarose (Pierce). The mixture was rotated overnight at 4 °C. After centrifugation at 3000g for 5 min, the pellets were washed and the cell surface proteins were eluted from the beads by adding 2 × SDS loading buffer and incubated for 5 min at 85 °C. Proteins were then analyzed by 8% SDS-PAGE and immunoblotting.
Immunofluorescence of ABCG1
Confocal microscopy was carried out as described previously.29,35−37 Briefly, the cells growing on coverslips were fixed with 3% paraformaldehyde and permeabilized using cold methanol. After blocking, the cells were incubated overnight in 1XPBS containing 1% BSA and specific antibodies indicated. The cells were then incubated with Alexa Fluor 488 goat antirabbit IgG and Alexa Fluor 568 goat antimouse IgG (Life Technologies Inc.). After washing, coverslips were mounted on the slides with one drop of Antifade reagent containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Protein localization was determined using a Leica SP5 laser scanning confocal microscope (filters: 406 nm for DAPI, 488 nm for Fluor 488, and 543 nm for Fluor 568).
Subcellular Fractionation
The experiment was carried out as described in ref (38) with modifications. Briefly, two plates (100 mm) of 85% confluent cells stably expressing wild type or mutant ABCG1 were collected and resuspended in 2.0 mL of ice-cold, low ionic strength buffer C (10 mM Tris–HCl, pH 7.5, 0.5 mM MgCl2) containing 1X Complete EDTA-free protease inhibitors. The cells were then disrupted with 40 strokes in a Dounce homogenizer with a loose-fitting pestle. The homogenates were then made isotonic by the addition of 0.4 mL of 1.46 M sucrose in buffer C. The samples were centrifuged at 10 000g for 15 min at 4 °C. Next, 2 mL of the supernatant was loaded on top of 10 mL of linear sucrose gradient (10%–40%). Samples were centrifuged at 335 000g for 20 h at 4 °C. After centrifugation, 1 mL fractions were collected from the top to the bottom of the gradient, precipitated with TCA and subjected to SDS-PAGE (8–20%) and immunoblotting as described above.
Statistics
All statistical analyses were carried out by GraphPad Prism version 4.0 (GraphPad Software) and SPSS. Student’s t test was used to determine the significant differences between wild type ABCG1 and each mutant ABCG1. The significant differences between groups were also analyzed using one-way ANOVA. Significance was defined as p < 0.05. Results are presented as mean ± SD.
Results
Effect of the Conservative Sequence on ABCG1-Mediated Cellular Sterol Efflux
The G subfamily members are different from other ABC transporters. They contain an NH2-terminal cytoplasmic NBD. We found that, in addition to the traditional conserved motifs in the NBD, the five ABCG subfamily members contain one single highly conserved sequence existing between the NBD and the transmembrane domain. This sequence contains five amino acid residues (Asn, Pro, Ala, Asp, and Phe) in all G family members except for ABCG5, in which it is NPFDF (Figure 1A). This sequence is also completely conserved among different species of ABCG1. We replaced Asn 316, Pro 317, Asp 319, and Phe 320 in ABCG1 with Ala simultaneously (NPDF-A) to investigate their potential roles in ABCG1-mediated sterol efflux. As shown in Figure 1B, the expression levels of NPDF-A and wild type ABCG1 were comparable in transiently transfected HEK 293 cells. However, cells expressing mutant NPDF-A showed a significant reduction in cholesterol efflux compared with cells expressing wild type ABCG1 (Figure 1C).
Next, we substituted Asn316, Pro317, Asp319, and Phe320 with Ala individually to determine the roles of these amino acid residues in ABCG1-mediated cholesterol efflux. The wild type and mutant ABCG1 were transiently expressed in HEK293 cells and showed comparable protein levels (Figure 2A). Mutations P317A and D319A had no significant effect on ABCG1-mediated cholesterol efflux (Figure 2A, lanes 4 and 5). On the other hand, mutations N316A and P320A significantly reduced cellular cholesterol efflux compared with wild type ABCG1 (Figure 2A, lanes 3 and 6).
Figure 2.

Effects of individual residues within the conserved sequence on ABCG1-mediated cellular cholesterol efflux. Panels A and B: Cholesterol efflux to lipidated apoA-I. The experiments were carried out as described in Figure 1C. Samples tested are indicated in the figures. Values are mean ± SD of at least 3 independent experiments. The bottom figures in Panels A and B were representative figures showing protein levels. Expression of wild type and mutant ABCG1 in HEK293 cells transiently overexpressing wild type and mutant ABCG1 was determined as described in Figure 1B except that ABCG1 was detected with a polyclonal anti-ABCG1 antibody, 4497. Calnexin was detected with a mouse monoclonal anticalnexin antibody. Antibody binding was detected using IRDye-labeled goat antimouse or antirabbit IgG (Li-Cor). The signals were detected by a Licor Odyssey Infrared Imaging System. Panel C: 7-ketocholesterol efflux to lipidated apoA-I. The experiment was performed as described in the legend to Figure 1C except that the cells were labeled with [3H]7-ketocholesterol. Panel D: Cholesterol efflux to lipidated apoA-I or rHDL. The experiment was performed as described in the legend to Figure 1C. V: control cells. WT: wild type ABCG1-expressing cells. Panels E and F: Cholesterol and 7-ketocholesterol efflux to rHDL. The experiment was performed as described in the legend to Panels A and B except that the acceptor used was rHDL (5 μg/mL). Values are mean ± SD of one experiment that was performed in triplicate. * or # indicates p < 0.05; ** or ## indicates p < 0.01; *** or ### indicates p < 0.001. *Compared with the control cells that were transiently transfected with empty vector (V). #Compared with wild type ABCG1-expressing cells (WT). Similar results were obtained from at least one more independent experiment.
To further define how Asn316 and Phe320 affected ABCG1-mediated cellular cholesterol efflux, we mutated Asn316 to Asp (N316D) and Gln (N316Q) and mutated Phe320 to Ile (F320I) and Tyr (F320Y). We also made a double mutation N316AF320A, in which both Asn316 and Phe320 were mutated to Ala. A catalytic dead mutation K120M, in which the conserved lysine residue located at position 120 in the Walker A motif of ABCG1 was mutated to methionine, was used as a negative control because it virtually eliminates ABCG1-mediated cholesterol efflux.9,29 Western blot analysis of whole cell lysates isolated from transiently transfected HEK293 cells showed that wild type and mutant forms of ABCG1 were all expressed at similar levels (Figure 2B). Mutation F320Y showed the same phenotype as wild type ABCG1 (lane 8). However, when compared with cells expressing wild type ABCG1, cells expressing mutant N316AF320A, N316Q, N316D, F320I, or K120 M displayed reduced cholesterol efflux to lipidated apoA-I (Figure 2B, lanes 3 to 7).
It has been reported that ABCG1 mediates oxysterol efflux.39,40 Thus, we investigated whether mutations of Asn316 and Phe320 affected the ability of ABCG1 to mediate cellular 7-ketocholesterol efflux, the most abundant nonenzymatic oxysterol in the atherosclerotic lesions.41 As shown in Figure 2C, when compared with cells transfected with empty vector, HEK 293 cells expressing wild type ABCG1 or F320Y showed similarly increased efflux of 7-ketocholesterol. However, mutations N316A, N316Q, F320A, and F320I significantly reduced cellular sterol efflux compared with wild type ABCG1.
Most recently, we made rHDL using purified recombinant apoA-I and examined the capacity of rHDL to mediate cholesterol efflux in HEK293 cells stably expressing ABCG1. We found that rHDL mediated ABCG1-dependent cholesterol efflux more efficiently than lipidated apoA-I (Figure 2D). Thus, we examined the effect of ABCG1 mutants on cellular cholesterol and 7-ketocholesterol efflux to rHDL. As shown in Figures 2E and 2F, when compared with cells expressing wild type ABCG1, cells expressing N316A, N316Q, N316D, and F320A displayed a significant reduction in sterol efflux to rHDL. Mutation F320I also reduced cellular sterol efflux to rHDL compared with wild type ABCG1, albeit to a lesser extend, whereas mutations P317A, D319A, and F320Y had no significant effect. These data were similar to what we observed in sterol efflux to lipidated apoA-I. Taken together, our findings reveal the important role of Asn at position 316 and Phe at position 320 of ABCG1 in cellular cholesterol and oxysterol efflux conferred by ABCG1.
Effects of Mutations N316Q and F320I on ABCG1 Stability
Next, we performed a pulse-chase experiment to determine if mutations on Asn316 and Phe320 affected ABCG1 stability. HEK 293 cells stably expressing wild type or mutant N316Q or F320I were metabolically labeled with [35S]-Met/Cys and then chased with excess Met/Cys for the indicated time periods. As shown in Figure 3, at the 1 h time point (lane 5), the 35S labeling of wild type (Figure 3A), N316Q (Figure 3B), and F320I (Figure 3C) was reduced to approximately 50% compared to the 0 time point. At 4h (lane 7), the 35S labeling of wild type and mutant ABCG1 was reduced to around 15%. Thus, mutations N316Q and F320I did not affect ABCG1 stability.
Figure 3.

Metabolic labeling of cells with 35S Cys/Met. The experiments were performed as described in Materials and Methods. Briefly, HEK293 cells stably expressing wild type ABCG1 or mutant N316Q or F320I were metabolically labeled with [35S]-Met/Cys (100 μCi/ml). ABCG1 was immunoprecipitated from same quantities of total proteins and then subjected to SDS-PAGE (8%). ABCG1 protein was detected by immunoblotting with a polyclonal anti-ABCG1 antibody, 4497. 35S signal was detected by X-ray film exposure. Similar results were obtained from two more independent experiments. Top figures in each panel were representative figures showing 35S signal levels and ABCG1 protein levels. Bottom figures in each panel were relative densitometry. The densitometry was determined using Image J Analysis Software. The relative densitometry was the ratio of the densitometry of 35S signal to that of ABCG1 protein signal at the same time point. Percentage of relative densitometry was the ratio of the relative densitometry of ABCG1 at different time points to that of ABCG1 at time 0. Values are mean ± SD of three independent experiments. The percentage of relative densitometry of ABCG1 at time 0 was defined as 100%.
Effect of Mutations of Asn316 and Phe320 on ABCG1 Oligomerization
ABCG1 is a half-transporter and functions as a homodimer.5 Thus, we studied whether mutations N316Q and F320I had any effect on ABCG1 oligomerization. Coexpression of half ABC transporters with two different tags, followed by immunoprecipitation is a common technique to determine ABCG transporter oligomerization.7,9,22 We employed the same technique to explore the homo-oligomerization of mutant ABCG1. ABCG1 was tagged with a C-Myc or HA tag at its COOH-terminus (ABCG1-Myc and ABCG1-HA). We first examined if C-Myc and HA tags affected trafficking of ABCG1 to the plasma membrane using confocal microscopy. As shown in Figure 4A, no ABCG1 signal was detected in mock-transfected HEK293 cells. The distribution patterns of ABCG1-Myc and ABCG1-HA in transiently transfected HEK293 cells were similar to that of untagged wild type ABCG1, colocalized with the plasma membrane marker, Na+/K+-ATPase (Figure 4A, right). Thus, C-Myc and HA tags did not affect ABCG1 trafficking to the cell surface. Next, ABCG1-Myc and ABCG1-HA were transiently cotransfected to HEK293 cells, and ABCG1-Myc was immunoprecipitated from whole cell lysates. We observed that ABCG1-HA was coimmunoprecipitated with ABCG1-Myc in the cotransfected cells (Figure.4B, lane 6), indicating that ABCG1 forms homo-oligomers, consistent with previous findings.9,22 Similar experiments were performed on cells cotransfected with C-Myc- and HA-tagged mutant N316Q or F320I. As shown in Figure 4B (lanes 7, 8), N316Q-HA and F320I-HA were efficiently coimmunoprecipitated with N316Q-Myc and F320I-Myc, respectively. Therefore, mutations N316D and F320I had no significant effect on ABCG1 homo-oligomerization.
Figure 4.

Effects of mutations of N316Q and F320I on ABCG1 oligomerization. Panel A: Immunofluorescence of ABCG1. The subcellular localization of tagged and untagged wild type ABCG1 in transiently transfected HEK 293 cells was determined by confocal microscopy as described in Materials and Methods. Briefly, 48 h after transfection, ABCG1 was detected using polyclonal anti-ABCG1 antibody (H-65). Location of ABCG1 is indicated in green. Nuclei were stained with DAPI and are shown in blue. The plasma membrane marker, Na+K+-ATPase, was detected using a monoclonal antibody and is shown in red. Transfectants tested expressed either untagged or tagged wild type ABCG1 as indicated in the figure. An x–y optical section of the cells illustrates the distribution of the wild type and mutant proteins between the plasma and intracellular membranes (magnification: 100×). Panel B: Immunoprecipitation of C-myc tagged ABCG1. HEK293 cells were transiently transfected with a combination of wild type or mutant ABCG1-Myc and ABCG1-HA. C-Myc tagged wild type or mutant ABCG1 was immunoprecipitated from same amount of whole cell lysates and then were fractionated by 8% SDS-PAGE, followed by immunoblotting with an anti-HA antibody to detect HA-tagged ABCG1. The same blot was also incubated with anti-ABCG1 antibody 4497 to detect total ABCG1 proteins. Similar results were obtained from at least two more independent experiments.
Effects of Mutations on ABCG1 Trafficking
Several lines of evidence have shown that ABCG1 is localized to the plasma membrane.8,9,11,29,42 Thus, we carried out biotinylation experiments to investigate whether mutations on Asn316 and Phe320 influenced trafficking of ABCG1 to the plasma membrane. We were able to detect wild type ABCG1 but not calnexin in the cell surface proteins (Figure 5A, biotinylation, lane 2), suggesting that ABCG1 can be trafficked to the plasma membrane, consistent with previous reports.8,29 Mutants NPDF-A, N316AF320A, N316D, N316Q, and F320I all showed much less cell surface levels compared to the wild type ABCG1 (Figure 5A, lanes 3 to 7), even though the protein levels in whole cell lysates were similar (Figure 5A, whole cell lysate, lanes 2 to 8). However, mutation F320Y did not cause any reduction in the cell surface ABCG1 (lane 8). Thus, mutations of Asn316 and Phe320 but not F320Y affected ABCG1 trafficking to the cell surface.
Figure 5.

Effects of mutations of Asn316 and Phe320 on ABCG1 trafficking. Panel A: Biotinylation of cell surface proteins. HEK293 cells transiently expressing WT or mutant ABCG1 were biotinylated exactly as described in Materials and Methods. Biotinylated cell surface proteins (biotinylation) and total proteins from whole cell lysate were analyzed by 8% SDS-PAGE and immunoblotting. ABCG1 and calnexin were detected using a polyclonal anti-ABCG1 antibody, H-65, and a monoclonal anticalnexin, respectively. Similar results were obtained from at least two more independent experiments. Panel B: Immunofluorescence of wild type and mutant ABCG1. The subcellular localization of wild type and mutant ABCG1 in stably or transiently transfected HEK 293 cells was determined by confocal microscopy as described in the legend to Figure 4A. ABCG1 was detected with H-65 and indicated in green. Nuclei were stained with DAPI and shown in blue. The plasma membrane marker, Na+K+-ATPase was shown in red. Transfectants tested expressed either wild type or mutant ABCG1 as indicated in the figure (magnification: 100× ).
To further confirm the finding and determine the subcellular localization of these mutations, we performed confocal microscopy on HEK 293 cells stably expressing wild type, N316Q, or F320I. We also examined the subcellular localization of mutations N316A, P317A, D319A, and F320Y in transiently transfected HEK293 cells. ABCG1 was shown as green (Figure 5B, left panel). The plasma membrane marker, Na+/K+-ATPase, was shown as red (middle). No ABCG1 signal was detected in mock-transfected HEK293 cells. As shown in Figure 5B, a significant amount of wild type ABCG1, as well as mutations P317A, D319A, and F320Y that had no significant effect on ABCG1-mediated cellular cholesterol efflux, were detected on the cell periphery (left), colocalized with the plasma membrane marker Na+/K+-ATPase, and shown as yellow (right). The subcellular distributions of N316Q, F320I, and N316A were significantly different from that of the wild type protein. In the merged panel (right), N316Q, F320I, and N316A were not overlapped with Na+/K+-ATPase. Thus, taken together, mutations N316Q, N316A, N316D, F320A, and F320I impaired ABCG1 trafficking to the plasma membrane.
To further define the subcellular localization of mutant ABCG1, we next incubated the cells with an anticalnexin antibody (red, middle) and observed that there were significant quantities of F320I were colocalized with the ER marker (right), but the majority of N316Q were not overlapped with the ER calnexin (Figure 6A). Next, we employed a cell fractionation assay to further elucidate the subcellular distribution of mutant N316Q. HEK293 cells stably expressing the wild type or mutant transporter were disrupted and applied to a sucrose density gradient. The distribution patterns of subcellular organelles in control, wild type ABCG1, or mutant N316Q-expressing cells were the same. Thus, only the distribution of subcellular organelles obtained from N316Q-expressing cells was shown in Figure 6B. We observed that wild type ABCG1 was present in various fractions including the Golgi (GM130), the ER (calnexin), the plasma membrane (Na+K+-ATPase), the late endosome/lysosome (Rab11), and the early endosome (EEA1) (Figure 6B). The distribution pattern of N316Q was different from that of wild type ABCG1. The majority of N316Q resided in fractions 6 to 8, which were partially overlapped with the late endosome/lysosome, Rab11. Taken together, our data clearly show that mutations N316Q and F320I impair proper cellular trafficking of ABCG1. The mutant proteins were mainly localized intracellularly.
Figure 6.

Effect of mutations N316Q and F320I on ABCG1 trafficking. Panel A: Immunofluorescence of wild type and mutant ABCG1. The experiment was performed as described in the legend to Figure 4A. ABCG1 was detected with H-65 and indicated in green. Nuclei were stained with DAPI and shown in blue. The ER marker, calnexin, was shown in red (magnification: 100×). Panel B: Cell fractionation of wild type and mutant ABCG1. Cell lysates were fractionated as described in Materials and Methods. Briefly, HEK293 cells stably expressing wild type or mutant ABCG1 were disrupted by incubation with ice-cold, low ionic strength buffer C followed by homogenization with a Dounce homogenizer. The homogenates were made isotonic and centrifuged. The supernatant was loaded on top of linear sucrose gradient (10%–40%) and then subjected to centrifugation. After that, 1 mL fractions were collected from the top to the bottom of the gradient, precipitated with TCA, and subjected to SDS-PAGE (8–20%). After electro-transferring, the membranes were cut into halves between the 40 kDa and 30 kDa protein standards. The top membranes were blotted with a polyclonal antibody, 4497, and a monoclonal anti-GM130 antibody or with a polyclonal anticalnexin and a monoclonal anti-Na+K+-ATPase antibody. The bottom membranes were blotted with a monoclonal anti-EEA1 or a monoclonal anti-Rab11 antibody. Similar results were obtained from one more independent experiment.
It has been reported that substrates and modulators of P-glycoprotein can function as chaperones to facilitate processing of the misfolded P-glycoprotein mutants.43 On the other hand, the processing of misfolded cystic fibrosis transmembrane conductance regulator (CFTR) mutant, delta F508, can be corrected when the culture temperature is reduced to 26 °C.44 The direct substrates of ABCG1 are not well understood. Thus, we examined if trafficking of mutations N316Q and F320I was affected by reduced culture temperature. As shown in Figure 7, wild type ABCG1 was colocalized with Na+K+-ATPase. However, mutations N316Q and F320I were still mainly localized intracellularly when the cells were cultured at 26 °C. Thus, unlike CFTR delta F508, the processing of ABCG1 mutants N316Q and F320I could not be rescued by reduced culture temperature.
Figure 7.
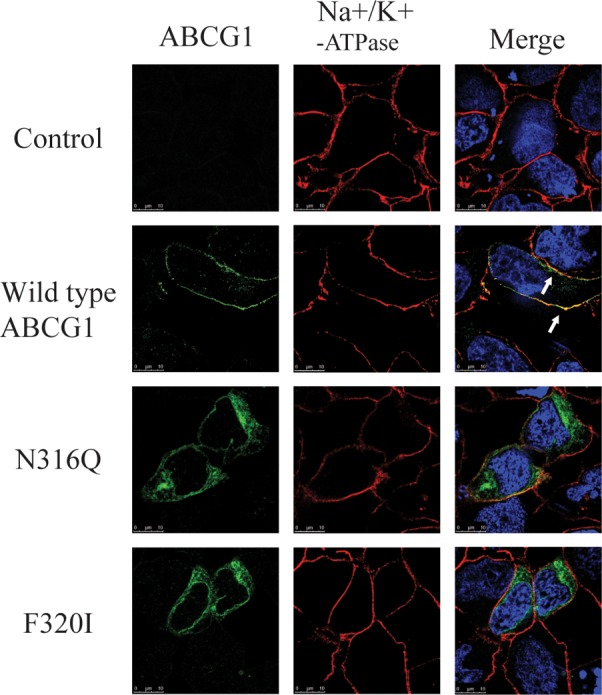
Immunofluorescence of ABCG1. The experiment was carried out as described in the legend to Figure 4A except that the cells were transiently transfected with wild type or mutant ABCG1 cDNA and cultured for 48 h after transfection under a 26 °C incubator with 5% CO2. ABCG1 was detected with H-65 and indicated in green. Nuclei were stained with DAPI and shown in blue. The plasma membrane marker, Na+K+-ATPase was shown in red (magnification: 100 × ).
Discussion
In the present study, we identified a highly conserved sequence present in all five ABCG family members and in different species of ABCG1. The sequence that is located between the NBD and MSD contains five amino acid residues, Asn, Pro, Ala/Phe, Asp, and Phe. Asn316 at the first position and Phe320 at the last position in the conserved sequence of ABCG1 play an important role in regulating ABCG1 trafficking. Replacement of Asn316 with Ala, Gln and Asp, or Phe320 with Ala and Ile affected trafficking of ABCG1 to the cell surface and impaired cellular cholesterol and 7-ketocholesterol efflux conferred by ABCG1. The mutant proteins were mainly localized intracellularly. However, mutation of Phe320 to Tyr had no significant effect on ABCG1 trafficking and sterol efflux activity.
Most recently, Tarling et al.45 reported that ABCG1 is located in endosomes but not in the plasma membrane. They proposed that ABCG1 functions as an intracellular cholesterol transporter to transfer sterols to the inner leaflet of intracellular vesicles, which then fuse with the plasma membrane and deliver cellular cholesterol to exogenous HDL. One unanswered question in this model is how ABCG1 is removed from vesicles before they fuse with the plasma membrane because no ABCG1 is detected on the cell surface in their studies. On the other hand, several independent groups have shown that ABCG1 is localized to the plasma membrane and to intracellular organelles such as the Golgi and endocytic recycling compartments in different cell types such as baby hamster kidney (BHK) cells, HEK293, HeLa, and THP-1 cells.8,9,11,29,42 Further, endogenous ABCG1 in macrophages is redistributed from intracellular compartments to the plasma membrane after treatment with an LXR agonist.42 Vaughan et al. reported that ABCG1 redistributes cholesterol to the outer leaflet of the plasma membrane, where cholesterol can be removed by HDL.8 Thus, it is possible that mutations on Asn316 and Phe320 disturb proper trafficking of ABCG1 and, consequently, impair ABCG1-mediated cellular sterol efflux.
How do these mutations affect ABCG1 trafficking? One simple explanation is that they may grossly disrupt ABCG1 structure and cause misprocessed proteins. Mutations that significantly perturb protein structure are unlikely to escape the surveillance machinery that traps misfolded proteins in the ER and targets them for degradation.46,47 For example, misfolded mutants of the human P-glycoprotein and CFTR are retained in the ER and rapidly degraded, resulting in lower protein levels.43,48 However, mutations N316A and F320I did not affect ABCG1 stability (Figure 3). Thus, mutations of Asn316 and Phe320 may not result in a major perturbation of the structure of ABCG1. It has been reported that heterodimerization of ABCG5 and ABCG8 is required for trafficking of ABCG5 and ABCG8 from the ER to the Golgi.7,49,50 Here, we found that mutations N316Q and F320I had no effect on ABCG1 homo-oligomerization (Figure 4B). Studies in ABCA1 and ABCA3 transporters have suggested that a conserved xLxxKN signal sequence located in the NH2-terminal cytoplasmic domain is required for the export of the two transporters from the Golgi.51 Ala mutation of the conserved signal sequence causes ER retention. The author proposed that xLxxKN signal sequence functions as a Golgi exit signal, targeting ABCA transporters to a post-Golgi sorting station for further selective delivery. The NPADF sequence is completely conserved in all species of ABCG1. Thus, it is possible that this sequence may function as a selective trafficking signal to target ABCG1 to a proper sorting station. The NPADF sequence is also completely conserved in other ABCG family members, except for ABCG5 that has NPFDF. The cellular localization of ABCG4 is unclear. However, ABCG2 is mainly localized on the cell surface.52 ABCG5 and ABCG8 form a heterodimer in the ER and are transported to the apical membranes.50 The potential role of the NPADF sequence in trafficking of these ABCG transporters remains the subject of ongoing investigation. We also checked the presence of NPADF in several ER proteins and found that they do not contain the NPADF sequence. Interestingly, they all contain at least one NxxxF motif. For instance, ER resident protein 44 contains N83QVVF and N174YRVF, protein disulfide-isomerase has N189SDVF and N224LLDF, eukaryotic translation initiation factor 2-alpha kinase 3 contains N39LEGF and N204IANF, calreticulin-3 has N42DSRF, endoplasmin has N96KEIF, and calnexin contains N175LDQF and N404PDFF. Unlike ABCG1, which does not contain an ER retention signal, these ER proteins have an ER retention signal, HDEL/KDEL/HVEL. It will be of interest to investigate the potential role of the NxxxF sequence in trafficking of these proteins.
ER cholesterol levels play a critical regulatory role in the process of sterol regulatory element-binding protein-2 (SREBP-2). The transport of SREBP-2 from the ER to the Golgi for further processing is inhibited when ER cholesterol levels are more than 5% of total ER lipids.53 ABCG1 has been shown to facilitate cholesterol transfer out from the ER.45 Overexpression of ABCG1 enhances process of SREBP-2 and increases levels of nuclear form SREBP-2.45 Thus, regulation of ER-to-Golgi trafficking of ABCG1 may play an important role in maintaining cellular cholesterol homeostasis. To the best of our knowledge, we are the first to demonstrate the important role of the conserved sequence NPADF in ABCG1 trafficking.
Glossary
- ABC
ATP-binding cassette transporter
- NBD
nucleotide-binding domain
- MSD
membrane-spanning domain
- RCT
reverse cholesterol transport
- ER
endoplasmic reticulum
- apoA-I
apolipoprotein A-I
- HDL
high density lipoprotein
- Ldlr
low density lipoprotein receptor
- HEK
human embryonic kidney
- rHDL
reconstituted HDL
- LXR
liver X receptor
- FBS
fetal bovine serum
- DMEM
Dulbecco’s modified Eagle’s medium
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- BSA
bovine serum albumin
- PBS
phosphate-buffered saline
- DAPI
4′,6-diamidino-2-phenylindole
- CFTR
cystic fibrosis transmembrane conductance regulator
- BHK
baby hamster kidney
- SREBP-2
sterol regulatory element-binding protein-2
Author Contributions
# These authors contributed equally.
This research was supported by grants from the Canadian Institutes of Health Research to D.W.Z. (MOP 93794). D.W.Z. is a Scholar of the Alberta Heritage Foundation for Medical Research and supported in part by a Canadian Institutes of Health Research New Investigator Award. Zhang laboratory is supported by Canadian Foundation for Innovation.
The authors declare no competing financial interest.
References
- Higgins C. F. (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757. [DOI] [PubMed] [Google Scholar]
- Dean M. C. (2002) The human ATP-binding cassette (ABC) transporter superfamily, National Center for Biotechnology Information, Bethesda, MD. [Google Scholar]
- Sharom F. J. (2008) ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 9, 105–127. [DOI] [PubMed] [Google Scholar]
- Li G.; Gu H. M.; Zhang D. W. (2013) ATP-binding cassette transporters and cholesterol translocation. IUBMB Life 65, 505–512. [DOI] [PubMed] [Google Scholar]
- Engel T.; Bode G.; Lueken A.; Knop M.; Kannenberg F.; Nofer J. R.; Assmann G.; Seedorf U. (2006) Expression and functional characterization of ABCG1 splice variant ABCG1(666). FEBS Lett. 580, 4551–4559. [DOI] [PubMed] [Google Scholar]
- Leimanis M. L.; Georges E. (2007) ABCG2 membrane transporter in mature human erythrocytes is exclusively homodimer. Biochem. Biophys. Res. Commun. 354, 345–350. [DOI] [PubMed] [Google Scholar]
- Graf G. A.; Yu L.; Li W. P.; Gerard R.; Tuma P. L.; Cohen J. C.; Hobbs H. H. (2003) ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J. Biol. Chem. 278, 48275–48282. [DOI] [PubMed] [Google Scholar]
- Vaughan A. M.; Oram J. F. (2005) ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 280, 30150–30157. [DOI] [PubMed] [Google Scholar]
- Kobayashi A.; Takanezawa Y.; Hirata T.; Shimizu Y.; Misasa K.; Kioka N.; Arai H.; Ueda K.; Matsuo M. (2006) Efflux of sphingomyelin, cholesterol, and phosphatidylcholine by ABCG1. J. Lipid Res. 47, 1791–1802. [DOI] [PubMed] [Google Scholar]
- Xie Q.; Engel T.; Schnoor M.; Niehaus J.; Hofnagel O.; Buers I.; Cullen P.; Seedorf U.; Assmann G.; Lorkowski S. (2006) Cell surface localization of ABCG1 does not require LXR activation. Arterioscler., Thromb., Vasc. Biol. 26, e143–144author reply e145.. [DOI] [PubMed] [Google Scholar]
- Gu H. M.; Li G.; Gao X.; Berthiaume L. G.; Zhang D. W. (2013) Characterization of palmitoylation of ATP binding cassette transporter G1: effect on protein trafficking and function. Biochim. Biophys. Acta 1831, 1067–1078. [DOI] [PubMed] [Google Scholar]
- Wang X.; Collins H. L.; Ranalletta M.; Fuki I. V.; Billheimer J. T.; Rothblat G. H.; Tall A. R.; Rader D. J. (2007) Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 117, 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Lan D.; Chen W.; Matsuura F.; Tall A. R. (2004) ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 101, 9774–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M. A.; Barrera G. C.; Nakamura K.; Baldan A.; Tarr P.; Fishbein M. C.; Frank J.; Francone O. L.; Edwards P. A. (2005) ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 1, 121–131. [DOI] [PubMed] [Google Scholar]
- Wiersma H.; Nijstad N.; de Boer J. F.; Out R.; Hogewerf W.; Van Berkel T. J.; Kuipers F.; Tietge U. J. (2009) Lack of Abcg1 results in decreased plasma HDL cholesterol levels and increased biliary cholesterol secretion in mice fed a high cholesterol diet. Atherosclerosis 206, 141–147. [DOI] [PubMed] [Google Scholar]
- Out R.; Hoekstra M.; Hildebrand R. B.; Kruit J. K.; Meurs I.; Li Z.; Kuipers F.; Van Berkel T. J.; Van Eck M. (2006) Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler., Thromb., Vasc. Biol. 26, 2295–2300. [DOI] [PubMed] [Google Scholar]
- Baldan A.; Pei L.; Lee R.; Tarr P.; Tangirala R. K.; Weinstein M. M.; Frank J.; Li A. C.; Tontonoz P.; Edwards P. A. (2006) Impaired development of atherosclerosis in hyperlipidemic Ldlr–/– and ApoE–/– mice transplanted with Abcg1–/– bone marrow. Arterioscler., Thromb., Vasc. Biol. 26, 2301–2307. [DOI] [PubMed] [Google Scholar]
- Ranalletta M.; Wang N.; Han S.; Yvan-Charvet L.; Welch C.; Tall A. R. (2006) Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1–/– bone marrow. Arterioscler., Thromb., Vasc. Biol. 26, 2308–2315. [DOI] [PubMed] [Google Scholar]
- Meurs I.; Lammers B.; Zhao Y.; Out R.; Hildebrand R. B.; Hoekstra M.; Van Berkel T. J.; Van Eck M. (2012) The effect of ABCG1 deficiency on atherosclerotic lesion development in LDL receptor knockout mice depends on the stage of atherogenesis. Atherosclerosis 221, 41–47. [DOI] [PubMed] [Google Scholar]
- Schou J.; Frikke-Schmidt R.; Kardassis D.; Thymiakou E.; Nordestgaard B. G.; Jensen G.; Grande P.; Tybjaerg-Hansen A. (2012) Genetic variation in ABCG1 and risk of myocardial infarction and ischemic heart disease. Arterioscler., Thromb., Vasc. Biol. 32, 506–515. [DOI] [PubMed] [Google Scholar]
- Vaughan A. M.; Oram J. F. (2006) ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J. Lipid Res. 47, 2433–2443. [DOI] [PubMed] [Google Scholar]
- Gelissen I. C.; Harris M.; Rye K. A.; Quinn C.; Brown A. J.; Kockx M.; Cartland S.; Packianathan M.; Kritharides L.; Jessup W. (2006) ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler., Thromb., Vasc. Biol. 26, 534–540. [DOI] [PubMed] [Google Scholar]
- Out R.; Jessup W.; Le Goff W.; Hoekstra M.; Gelissen I. C.; Zhao Y.; Kritharides L.; Chimini G.; Kuiper J.; Chapman M. J.; Huby T.; Van Berkel T. J.; Van Eck M. (2008) Coexistence of foam cells and hypocholesterolemia in mice lacking the ABC transporters A1 and G1. Circ. Res. 102, 113–120. [DOI] [PubMed] [Google Scholar]
- Out R.; Hoekstra M.; Habets K.; Meurs I.; de Waard V.; Hildebrand R. B.; Wang Y.; Chimini G.; Kuiper J.; Van Berkel T. J.; Van Eck M. (2008) Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arterioscler., Thromb., Vasc. Biol. 28, 258–264. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L.; Ranalletta M.; Wang N.; Han S.; Terasaka N.; Li R.; Welch C.; Tall A. R. (2007) Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Invest. 117, 3900–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryirs M. A.; Barter P. J.; Appavoo M.; Tuch B. E.; Tabet F.; Heather A. K.; Rye K. A. (2010) Effects of high-density lipoproteins on pancreatic beta-cell insulin secretion. Arterioscler., Thromb., Vasc. Biol. 30, 1642–1648. [DOI] [PubMed] [Google Scholar]
- Sturek J. M.; Castle J. D.; Trace A. P.; Page L. C.; Castle A. M.; Evans-Molina C.; Parks J. S.; Mirmira R. G.; Hedrick C. C. (2010) An intracellular role for ABCG1-mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic beta cells. J. Clin. Invest. 120, 2575–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruit J. K.; Wijesekara N.; Westwell-Roper C.; Vanmierlo T.; de Haan W.; Bhattacharjee A.; Tang R.; Wellington C. L.; LutJohann D.; Johnson J. D.; Brunham L. R.; Verchere C. B.; Hayden M. R. (2012) Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired beta-cell function. Diabetes 61, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Gu H.; Li G.; Rye K. A.; Zhang D. W. (2012) Identification of an amino acid residue in ATP-binding cassette transport G1 critical for mediating cholesterol efflux. Biochim. Biophys. Acta 1821, 552–559. [DOI] [PubMed] [Google Scholar]
- Zhang D. W.; Garuti R.; Tang W. J.; Cohen J. C.; Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. U. S. A. 105, 13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C. I.; Beckstead J. A.; Thompson A.; Hafiane A.; Wang R. H.; Ryan R. O.; Kiss R. S. (2012) Tweaking the cholesterol efflux capacity of reconstituted HDL. Biochem. Cell Biol. 90, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. W.; Nunoya K.; Vasa M.; Gu H. M.; Cole S. P.; Deeley R. G. (2006) Mutational analysis of polar amino acid residues within predicted transmembrane helices 10 and 16 of multidrug resistance protein 1 (ABCC1): effect on substrate specificity. Drug Metab. Dispos. 34, 539–546. [DOI] [PubMed] [Google Scholar]
- Zhang D. W.; Graf G. A.; Gerard R. D.; Cohen J. C.; Hobbs H. H. (2006) Functional asymmetry of nucleotide-binding domains in ABCG5 and ABCG8. J. Biol. Chem. 281, 4507–4516. [DOI] [PubMed] [Google Scholar]
- Gu H. M.; Adijiang A.; Mah M.; Zhang D. W. (2013) Characterization of the role of EGF-A of low-density lipoprotein receptor in PCSK9 binding. J. Lipid Res. 54, 3345–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. W.; Cole S. P.; Deeley R. G. (2001) Identification of a nonconserved amino acid residue in multidrug resistance protein 1 important for determining substrate specificity: evidence for functional interaction between transmembrane helices 14 and 17. J. Biol. Chem. 276, 34966–34974. [DOI] [PubMed] [Google Scholar]
- Zhang D. W.; Cole S. P.; Deeley R. G. (2001) Identification of an amino acid residue in multidrug resistance protein 1 critical for conferring resistance to anthracyclines. J. Biol. Chem. 276, 13231–13239. [DOI] [PubMed] [Google Scholar]
- Zhang D. W.; Lagace T. A.; Garuti R.; Zhao Z.; McDonald M.; Horton J. D.; Cohen J. C.; Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612. [DOI] [PubMed] [Google Scholar]
- Li Y.; Ge M.; Ciani L.; Kuriakose G.; Westover E. J.; Dura M.; Covey D. F.; Freed J. H.; Maxfield F. R.; Lytton J.; Tabas I. (2004) Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 279, 37030–37039. [DOI] [PubMed] [Google Scholar]
- Wang N.; Yvan-Charvet L.; Lutjohann D.; Mulder M.; Vanmierlo T.; Kim T. W.; Tall A. R. (2008) ATP-binding cassette transporters G1 and G4 mediate cholesterol and desmosterol efflux to HDL and regulate sterol accumulation in the brain. FASEB J. 22, 1073–1082. [DOI] [PubMed] [Google Scholar]
- Terasaka N.; Wang N.; Yvan-Charvet L.; Tall A. R. (2007) High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc. Natl. Acad. Sci. U. S. A. 104, 15093–15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. J.; Jessup W. (1999) Oxysterols and atherosclerosis. Atherosclerosis 142, 1–28. [DOI] [PubMed] [Google Scholar]
- Wang N.; Ranalletta M.; Matsuura F.; Peng F.; Tall A. R. (2006) LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler., Thromb., Vasc. Biol. 26, 1310–1316. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1997) Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. J. Biol. Chem. 272, 709–712. [DOI] [PubMed] [Google Scholar]
- Denning G. M.; Anderson M. P.; Amara J. F.; Marshall J.; Smith A. E.; Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764. [DOI] [PubMed] [Google Scholar]
- Tarling E. J.; Edwards P. A. (2011) ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl. Acad. Sci. U. S. A. 108, 19719–19724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton R. Y. (2002) ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell. Biol. 14, 476–482. [DOI] [PubMed] [Google Scholar]
- Meusser B.; Hirsch C.; Jarosch E.; Sommer T. (2005) ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772. [DOI] [PubMed] [Google Scholar]
- Cheng S. H.; Gregory R. J.; Marshall J.; Paul S.; Souza D. W.; White G. A.; O’Riordan C. R.; Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T.; Kono T.; Niibori A.; Harada K.; Kusuhara H.; Takada T.; Shuto T.; Suico M. A.; Sugiyama Y.; Kai H. (2006) Calreticulin facilitates the cell surface expression of ABCG5/G8. Biochem. Biophys. Res. Commun. 347, 67–75. [DOI] [PubMed] [Google Scholar]
- Graf G. A.; Li W. P.; Gerard R. D.; Gelissen I.; White A.; Cohen J. C.; Hobbs H. H. (2002) Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J. Clin. Invest. 110, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers M. F.; Hawkins A.; Shuman H.; Zhao M.; Newitt J. L.; Maguire J. A.; Ding W.; Mulugeta S. (2011) A novel conserved targeting motif found in ABCA transporters mediates trafficking to early post-Golgi compartments. J. Lipid Res. 52, 1471–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Lee E. W.; Cai X.; Ni Z.; Zhou L.; Mao Q. (2008) Membrane topology of the human breast cancer resistance protein (BCRP/ABCG2) determined by epitope insertion and immunofluorescence. Biochemistry 47, 13778–13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A.; Goldstein J. L.; McDonald J. G.; Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 8, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]