

Abstract
Background
Previously, we have shown that the chloride channel ClC-2 modulates intestinal tight junction (TJ) barrier function. The aim of the present study was to investigate the role of ClC-2 in epithelial barrier function and recovery in the event of epithelial injury.
Methods
We investigated the role of ClC-2 in TJ barrier function in Dextran Sodium Sulfate (DSS)-induced colitis in ClC-2 knockout mice and ClC-2 knockdown intestinal Caco-2 cells. Barrier function was measured electrophysiologically and by transepithelial mannitol fluxes. Select tight junction and associated proteins were western blotted, cytokines were measured using qPCR and human colonic biopsies were examined with immunohistochemistry.
Results
ClC-2−/− mice had a higher disease activity index, higher histological scores, and increased paracellular permeability compared to wild type (WT) mice when treated with DSS. DSS-treated ClC-2−/− mice had increased claudin-2 expression, greater loss of occludin in the membrane, increased association of occludin with caveolin-1, and significantly increased TNFα and IL-1β mRNA. ClC-2 knockdown in human intestinal Caco-2 cells resulted in a greater loss of epithelial resistance in the event of epithelial injury. The restoration of colonic barrier function after DSS colitis was delayed in ClC-2−/− mice. In human colonic biopsies, the protein and mRNA expression of ClC-2 was found to be reduced in ulcerative colitis (UC) patients.
Conclusions
ClC-2 plays a critical role in experimental colitis in that its absence increases disease activity, reduces barrier function and recovery, and perturbs tight junctions. Furthermore, ClC-2 expression is markedly reduced in the colon of human patients with ulcerative colitis.
Keywords: Inflammatory Bowel Disease, Barrier Function, Tight Junction
Introduction
The ClC super family of voltage-gated Cl− channels consists of several isoforms that are expressed in a wide variety of tissues and organs. The nine ClC genes identified so far in mammals are categorized based on their localization and function in the plasma membrane (ClC-1, -2, -Ka/K1, and -Kb/K2), intracellular endosomal-lysosomal system (ClC-3, -4, and -5), and intracellular organelles and vesicles (ClC-6 and -7).1 The importance of ClC members in tissue homeostasis has been demonstrated by pathological conditions associated with either mutations or functional defects in ClC family members such as myotonia, epilepsy, Bartter syndrome type III-renal salt loss, Dent’s disease-proteinuria, and osteopetrosis.2 ClC-2 channels serve organ-and tissue-specific functional roles such as inhibition of GABA responses in neurons and ion homeostasis in the retina and the testis.3,4 The physiological role of ClC-2 in the intestine is not completely understood. Although ClC-2 has been suggested to mediate Cl− secretion in murine small intestine, recent studies have revealed that colonic electroneutral absorption of NaCl and KCl is altered in ClC-2−/− mice, supporting its role in colonic absorption rather than secretion.5 Localization of ClC-2, predominantly at the apical tight junctions (TJ) in the small intestine, is thought to be advantageous for its regulatory interactions with signaling molecules in the TJ complex.6,7 In recent studies, we have demonstrated that ClC-2 modulates TJ barrier function and is critical for repair of paracellular barrier function in the event of epithelial injury.8–11
TJs consist of an array of membrane spanning proteins (e.g. occludin and claudins) linked to cytoplasmic plaque proteins (e.g. zonula occludens-1, ZO-1) to the cytoskeleton. In the intestine where the epithelium regulates water, nutrient, and ion transport while providing a barrier against toxins and pathogenic organisms, the apical intercellular TJs are largely responsible for barrier function. Loss of intestinal barrier function is known to contribute to a number of critically important intestinal diseases such as the inflammatory bowel diseases (IBD), celiac disease, and ischemia/ reperfusion injury.12 Emerging evidence has also shown that intracellular vesicular membrane transport including caveolar transport is a key process in the formation of TJ domains.13,14 In previous in-vitro work we have shown that ClC-2 plays a vital role in shuttling the TJ protein occludin to the apical lateral membrane via its interaction with caveolin-1 and the small GTPase Rab5.9 As a basic model of the role of ClC-2 in injury and repair, we have also shown that ClC-2 knockout mice have impaired intestinal epithelial barrier recovery following a brief period of complete ischemia.11 In terms of intestinal permeability, understanding how the apical TJ barrier is regulated and repaired is critical for the development of targeted therapy of patients suffering from IBD.15 Considering the apparent pivotal role of ClC-2 in the regulation of the TJ, we hypothesized that ClC-2 regulation of barrier function is critical during the course of experimental colitis induced by dextran sodium sulfate (DSS). Our results show that ClC-2 plays a critical role in the progression and severity of DSS colitis, as well as mucosal recovery from DSS colitis because of its role in regulating the TJ barrier.
Materials and Methods
Experimental Animals and Induction of Colitis
Studies were approved by the North Carolina State University Institutional Animal Care and Use Committee. Breeding pairs of heterozygous mice (ClC-2+/−) were a kind gift of Dr. James E. Melvin (University of Rochester, Rochester, NY). ClC-2-null (ClC-2−/−) and WT (ClC-2+/+) mice (both of C57BL/6 background) ~8-weeks of age were used in this study. The generation of ClC-2 knockout (KO) mice and genotyping PCR has been previously described.10,16 Mice received 2.5% dextran sodium sulfate (DSS) (molecular mass, 36,000–50,000 daltons; MP Biomedicals, Santa Ana, CA) in autoclaved drinking water for 6 days.17 The body weights of mice were monitored daily, and the disease activity index and histological grading of colitis lesions were carried out, as described in the literature.17
Measurement of Transepithelial Electrical Resistance (TER) and Paracellular Flux
Colonic tissues were harvested immediately after euthanasia, cut longitudinally, and placed on 0.12-cm2-aperture Ussing chambers.18 Transepithelial electrical resistance (TER, Ω·cm2) was calculated from the spontaneous PD and short-circuit current and the mucosal paracellular permeability was assessed by [3H]mannitol fluxes.18
Gel Electrophoresis and Western Blotting
The lysates of colonic mucosa were prepared and processed for SDS-PAGE as described previously.9 Sucrose density gradients based on detergent insoluble fractions were isolated as described in the literature.19 Briefly, cell lysates were extracted in detergent buffer (50mM Tris, 25mM KCl, 5mM MgCl2.6H2O, 2mM EDTA, 40mM Sodium Fluoride, 4mM Sodium Orthovandate, pH7.4, 1% Triton X-100), and Protease Inhibitor Cocktail® (Roche Applied Science, Indianapolis, IN)). Ten fractions were collected by ultracentrifugation (250,000 g, 18 h at 4°C, SW28 rotor, Beckman Coulter, Brea, CA) of a 40%, 30%, 25%, 20%, and a 5% sucrose gradient (bottom to top). Equal amounts of each fraction were loaded in individual wells on the SDS-PAGE gel. After protein transfer to a polyvinylidene difluoride membrane, the membranes were probed using anti-occludin (Invitrogen, 71-1500), anti-flotillin-1 (Cell Signaling, catalog No. 3253), and anti-caveolin-1 (Cell Signaling, 3238S) antibodies.
Immunohistochemistry
Immunohistochemistry for ClC-2 on murine colonic tissues and human colonic biopsies was performed by standard methods. Intestinal tissues sections with no personal identification were obtained under an IRB exempt protocol at the Univeristy of North Carolina School of Medicine (Study #08-1151) from excess tissue after complete pathological evaluation. Heat activated antigen retrieval was performed in sodium citrate buffer (pH 7.4). Following inhibition of endogenous peroxidase and blocking in normal serum, the sections were incubated in primary antibody (Rabbit anti ClC-2, Almone labs, Jerusalem, Israel). Biotinylated secondary antibody, avidin-substrate, and peroxidase developing solutions were obtained from Vector Laboratories, Burlingame, CA.
Cell Culture and Transfections
C2bbe1 cells derived from human intestinal Caco-2 cells were obtained from ATCC (Manassas, VA) and grown in standard DMEM with 10% fetal bovine serum and 0.01 mg/ml human transferrin (GIBCO, Grand Island, NY). Cells were grown on cell culture treated surfaces of 12mm 0.4μm pore sized permeable supports (Corning, Corning, NY). Persistent knockdown of ClC-2 gene expression using short hairpin shRNA was achieved by transducing C2bbe1 cells with lentiviral particles encoding ClC-2 short hairpin (sh)RNA or control shRNA (catalog no. sc-42379-V and sc-108080; Santa Cruz Biotechnology). Selection of clones for stable expression of shRNA was performed using titrated concentrations of puromycin dihydrochloride (10 μg/ml for initial selection, and 6 μg/ml for maintenance). The confirmation of inhibition of ClC-2 expression was perfomed by western blotting and RT-PCR, as described previously.9 The transepithelial resistance (TER) measurements and permeability for FITC-labeled dextran (FD-4, Sigma-Aldrich, St Louis, MO) was assessed as described previously.9 In the experiments evaluating the effect of DSS on monolayer permeability, confluent monolayers were treated with 2% DSS on the apical surface. To evaluate the effect of caveolar disruption and DSS on TER, Methyl-β-cyclodextran (MβCD) was added (10mM, Sigma-Aldrich, St Louis, MO) one-hour prior to treatment with DSS. MβCD was removed from the media after two hours. To assess the effect of cytokines on TER, the cell monolayers were treated with IL-1β and TNFα (10ng/ml, Thermo Fisher Scientific, Waltham, MA) on both the apical and basal sides.
qPCR for Cytokine and ClC-2 Expression
Total RNA was isolated from colonic mucosal scrapings using an RNAeasy kit (Qiagen, Valencia, CA), quantified spectrophotometrically, and equal amounts of RNA were used for cDNA synthesis (SuperScript III First-Strand Synthesis, Invitrogen, Grand Island, NY). qPCR was performed using Platinum Taq DNA polymerase in SYBR Green SuperMix (Invitrogen, Grand Island, NY), the ABI StepOnePlus system (Applied Biosciences, Carlsbad, CA), and PCR primers (IL10: For-5′-AGTGGAGCAGGTGAAGAGT-3′, Rev-5′-TTCGGAGAGACGTACAAACG-3′; TGF-β1: For-5′-GCTACCATGCCAACTTCTGT-3′, Rev-5′-CGTAGTAGACGATGGGCAGT-3′; IL-1β: For-5′-CCCAACTGGTACATCAGCAC-3′, Rev-5′-TCTGCTCATTCACGAAAAGG-3′; TNF-α: For-5′-CCCACTCTGACCCCTTTACT-3′, Rev-5′-TTTGAGTCCTTGATGGTGGT-3′; IFN-γ: For-5′-CAAAAGGATGGTGACATGAA-3′, Rev-5′-TTGGCAATACTCATGAATGG-3′) obtained from Real Time Primers, Elkins Park, PA. The PCR conditions were followed as per recommended by manufacturer (Real Time Primers, Elkins Park, PA). Human colonic biopsy samples for qPCR analysis were obtained under the University of New Mexico Health Science Center Human Research Review Committee approved protocol (study #10-481), and were run in triplicate.
Statistical Analysis
Data are reported as means ± SE. Whenever needed, data were analyzed by using an ANOVA for repeated measures (SigmaStat, Systat Software, San Jose, CA). A Tukey’s test was used for post-hoc anlysis between treatments following ANOVA (P < 0.05).
Ethical considerations
Animal studies were approved by the Instituional Care and Use Committee of North Carolina State University. Studies on human tissues were approved by the Institutional Review Board of the Univeristy of North Carolina School of Medicine, and the University of New Mexico Health Science Center.
Results
Severity of Experimental Colitis in ClC-2−/− Mice
Considering our previous findings on the role of ClC-2 in the epithelial barrier,9–11 we compared the induction of experimental colitis in wild type (WT) and ClC-2−/− mice. We found that the severity of experimental colitis after 6 days of oral administration of 2.5% DSS was significantly higher in the ClC-2−/− mice as compared to the wild type mice. The loss of body weight (Figure 1A) and the disease activity index17 (Figure 1B) were significantly higher in ClC-2−/− mice compared to WT mice. Furthermore, the DSS-treated ClC-2−/− mice had extensive histopathological changes compared to WT DSS mice in terms of marked neutrophilic and mononuclear infiltration in the lamina propria, presence of inflammatory polyps, loss of crypts, and edema in the muscularis layer of the colon (Figure 2, histological score: 2 ± 0.28 and 3.33 ± 0.33 for WT and ClC-2−/− mice, respectively, on a scale of 0–4, p < 0.05).
Figure 1.
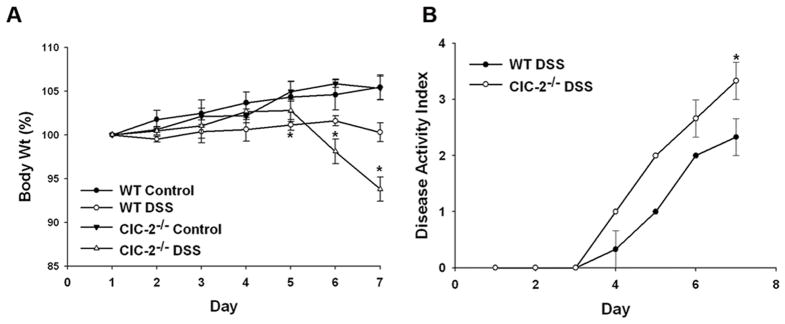
Severity of DSS colitis in ClC-2−/− mice. The loss of body weight (A) and disease activity index (B) during 6 days of DSS treatment were significantly higher in ClC-2−/− mice compared to wild type ClC-2+/+ mice (*p < 0.01). The data is representative of more than 3 independent experiemnts (n=3 for each group, in each experiment).
Figure 2.

Histological changes in DSS colitis. Histological examination revealed marked neutrophilic and mononuclear infiltration in the lamina propria, the presence of inflammatory polyps, disappearance of crypts, and edema in the muscularis in ClC-2−/− DSS colon (histological score: 2 ± 0.28 and 3.33 ± 0.33 for WT and ClC-2−/− mice, respectively (on a scale of 0–4, p < 0.01) (black bars = 50μM).
Paracellular Permeability in DSS colitis in ClC-2−/− mice
Because barrier function is a critical component of clinical as well as experimental colitis, we investigated intestinal permeability by mounting colonic tissues on Ussing chambers. In previous studies, we have demonstrated that the ClC-2−/− mice have increased baseline transepithelial resistance (TER) and reduced permeability to mannitol in the small intestine, due to structural changes in the TJs.10 Consistent with those observations, the average colonic TER in ClC-2−/− mice was found be significantly higher than the TER in WT mice (40 and 52 Ω·cm2 colonic TER in WT and ClC-2−/− mice, respectively). Similarily, the baseline mannitol permeability was found to be significantly lower in the colon of ClC-2−/− mice than WT mice (Jm-s mannitol, 0.78 and 0.36 μM·cm2·h−1 in WT and ClC-2−/− mice, respectively).
Induction of DSS colitis led to reduction in the colonic TER in WT as well as ClC-2−/− mice. The relative reduction in TER in ClC-2−/− mice after induction of colitis was significantly higher than WT mice (Figure 3A, p < 0.01). Similarly, the relative increase in the paracellular permeability of mannitol after induction of DSS colitis was more than 2-fold higher in ClC-2−/− mice compared to WT mice (Figure 3B, P < 0.01). Thus, there was relatively increased loss of colonic paracellular barrier function with DSS colitis in ClC-2−/− mice compared to WT mice.
Figure 3.
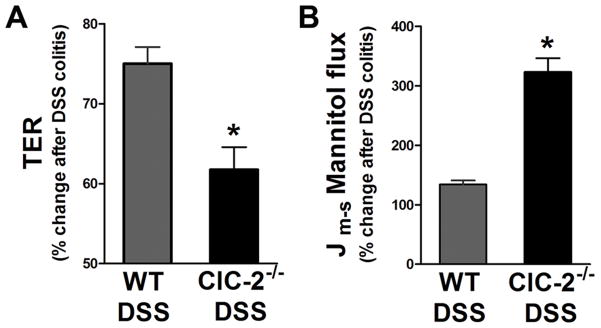
Epithelial permeability in DSS colitis. A: In Ussing chambers experiments, the percent reduction in the TER from control colon was significantly higher in ClC-2−/− mice than WT mice (*p < 0.05). B: After DSS treatment, the mucosal-to-serosal paracellular flux of mannitol showed 3-fold increases in the colon of ClC-2−/− mice compared to that of WT mice (*p < 0.01).
Changes in TJ during DSS Colitis in ClC-2−/− Mice
To examine the basis of changes in the barrier function in ClC-2−/− mice, we first studied total protein expression of select TJ proteins. The total protein expression of occludin (Figure 4A) was not different in the colon of DSS WT or ClC-2−/− mice while the total expression of claudin-2, a pore forming TJ protein, was elevated in both WT and ClC-2−/− mice. The increase in claudin-2 expression after DSS administration was significantly higher in ClC-2−/− mouse colon compared to WT mice colon (Figure 4B, p < 0.05). In confocal immunofluorescence, occludin staining on the surface epithelium was diffuse and subapically distributed in the colon of ClC-2−/− DSS mice as compared to the colon of WT DSS mice (Figure 4C). The confocal immunofluorescence staining of claudin-2 was found to be increased in colonic crypts after DSS administration. The DSS-induced increase in the intensity of claudin-2 staining was higher in ClC-2−/− DSS mice compared to WT DSS mice (Figure 4C). The total protein expression of claudin-1, claudin-4, and ZO-1 was not different in the colon of DSS WT or ClC-2−/− mice (data not shown).
Figure 4.

TJ analyses in DSS colitis. A: Western analysis showed increased total expression of claudin-2 protein in the DSS colon of ClC-2−/− mice. B: Densitometry analysis demonstrates a significant increase in total expression of claudin-2 in DSS colon of ClC-2−/− mice (*, different from all other groups, p < 0.05). C: In confocal immunofluorescence, the occludin (green) staining on surface epithelium was diffuse and subapically distributed in the colon of ClC-2−/− DSS mice compared to the colon of WT DSS mice. The staining of claudin-2 (red) was found to be increased in colonic crypts after DSS administration. The DSS-induced increase in the intensity of claudin-2 staining was higher in ClC-2−/− DSS mice compared to WT DSS mice. White bar = 50μM.
Since TJ proteins, particularly occludin, are known to be trafficked between the cytosol and cell membrane, we next examined the membrane localization of occludin by preparing subcellular sucrose density gradient fractions. In this method, low-density, detergent-insoluble fractions represent lipid raft and tight junction membranes.19,20 During initial experiments, sucrose density gradient based fractions were probed for flotillin-1 as a marker of lipid rafts (Figure 5A).19 The majority of flotilin-1 protein was present in low density fractions in control WT and ClC-2−/− mouse colon (fractions 2–5). However, in DSS colitis, flotillin-1 protein content was shifted to high density detergent soluble fractions (fractions 6–10) indicating the dispersion of proteins from lipid rafts in the membrane fractions to the cytosol. In further analyses using sucrose density gradients, there was a greater expression of occludin that was found to have shifted from the low density, detergent insoluble fractions to high density detergent soluble fractions of DSS-treated ClC-2−/− mice, compared to DSS-treated WT mice (Figure 5B & C). Therefore, although total expression of occludin in the colon was not different between DSS-treated ClC-2−/− mice and DSS-treated WT mice, occludin content in the membrane was reduced in DSS-treated ClC-2−/− mice.
Figure 5.
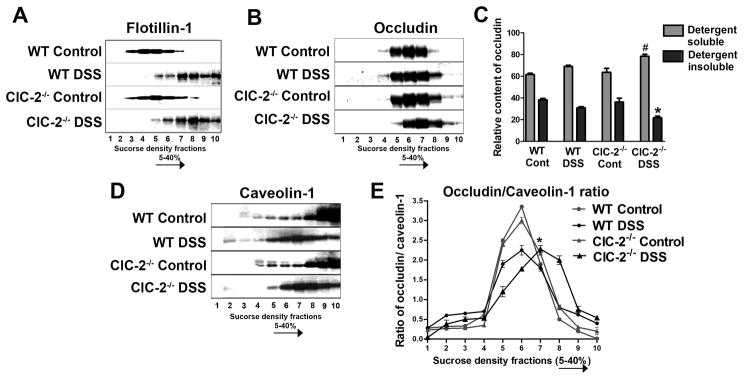
A. Sucrose density gradient based fractions were prepared, as detailed in methods, and were probed for flotillin-1 as a marker for lipid rafts. The majority of flotilin-1 protein was present in low density fractions in control WT and ClC-2−/− mouse colon (fractions 2–5). In DSS colitis, flotillin-1 protein content was shifted to high density detergent soluble fractions (fractions 6–10). B. In sucrose density gradient based fractionation studies, expression of occludin protein showed a significant shift from detergent insoluble fractions to detergent soluble fractions in DSS-treated ClC-2−/− colon when compared to DSS-treated WT colon. C: The relative content of occludin in low density detergent insoluble (fractions 2–5) and high density detergent soluble (fractions 6–10) fractions was calculated by densitometry. #, *, p<0.05 vs. respective WT DSS. D: In similar sucrose density gradient studies, the presence of caveolin-1 within high density sucrose fractions in control WT and ClC-2−/− colon was found to be shifted to medium density sucrose fractions after induction of DSS colitis. E: The densitometric values of occludin and caveolin-1, obtained from sucrose density fractions as in B and D, were used to calculate the ratio of occludin/caveolin-1 contents in respective sucrose fractions. In control WT and ClC-2−/− colon, the occludin/caveolin-1 ratio showed a peak in the middle sucrose density fractions. Alternatively, the peak occludin/caveolin-1 ratio in DSS treated ClC-2−/− was displaced toward high density, detergent soluble fractions, indicating higher content of occludin in association with caveolin-1 in these fractions (*, different from respective fraction of WT DSS group, p< 0.01, representation of n=6).
In our previous cell culture studies we have demonstrated that ClC-2 modulates barrier function via intracellular caveolar trafficking of occludin. To examine if ClC-2 modulates TJ barrier in DSS colitis via the same mechanism, expression of caveolin-1 protein was examined in the sucrose density gradient fractions. The presence of caveolin-1 within high density sucrose fractions in control WT and ClC-2−/− mice colon was found to be shifted toward medium density sucrose fractions after induction of DSS colitis (Figure 5D). We further calculated the ratio of occludin/caveolin-1 in the sucrose density fractions to determine the association between occludin and caveolin-1 during DSS colitis. As noted in Figure 5E, in control WT and ClC-2−/− mice, the occludin/caveolin-1 ratio had peak values in the middle of the sucrose density gradient. However, in DSS-treated colonic tissues, the peak occludin/caveolin-1 ratio was reduced with a more widespread dispersion of occludin and caveolin-1 toward detergent soluble fractions. This was particularly notable in DSS-treated ClC-2−/− mouse colon, where the peak of occludin/caveolin-1 ratio was displaced toward high density sucrose fractions (fractions 7 to 10) indicating a higher content of occludin associated with caveolin-1 in the detergent soluble fractions within the cytoplasm.
TJ Barrier in ClC-2 Knockdown Cells
We further examined the role of ClC-2 in caveolae-dependent modulation of TJ barrier injury and recovery in a cell culture model. Considering the increased baseline TER and reduced paracellular permeability in the small intestine 10 and colon of ClC-2−/− mice (as shown above), first, we examined if knockdown of ClC-2 expression in Caco-2 cells results in similar changes to the TJ barrier. Indeed, in Caco-2 cells, inhibition of ClC-2 expression using shRNA increased baseline TER significantly over the Caco-2 cells transfected with a scrambled shRNA (control shRNA) (Figure 6A). Consistent with the increased TER, ClC-2 shRNA expressing cells had reduced paracellular permeability to dextran (Figure 6B). When exposed to DSS, ClC-2 shRNA cells had significantly greater loss of TER compared to control shRNA cells (Figure 6C). To explore the role of caveolae in ClC-2-mediated barrier function, we studied the effect of caveolar disruption on DSS-induced loss of barrierfunction in Caco-2 cells. Pretreatment of cells with 10mM methyl-beta-cyclodextrin (MβCD, which selectively disrupts caveolae) before addition of DSS led to significantly greater and acute loss of TER in ClC-2 shRNA cells (Figure 6C). Additionally, after replacement of MβCD, there was evidence of recovery of TER in control but not ClC-2 shRNA cells. Thus, DSS-induced loss of the TJ barrier in Caco-2 cells was found to be exacerbated in the absence of ClC-2 and ClC-2 appeared to mediate the TJ barrier in response to DSS in a caveolae dependent manner.
Figure 6.
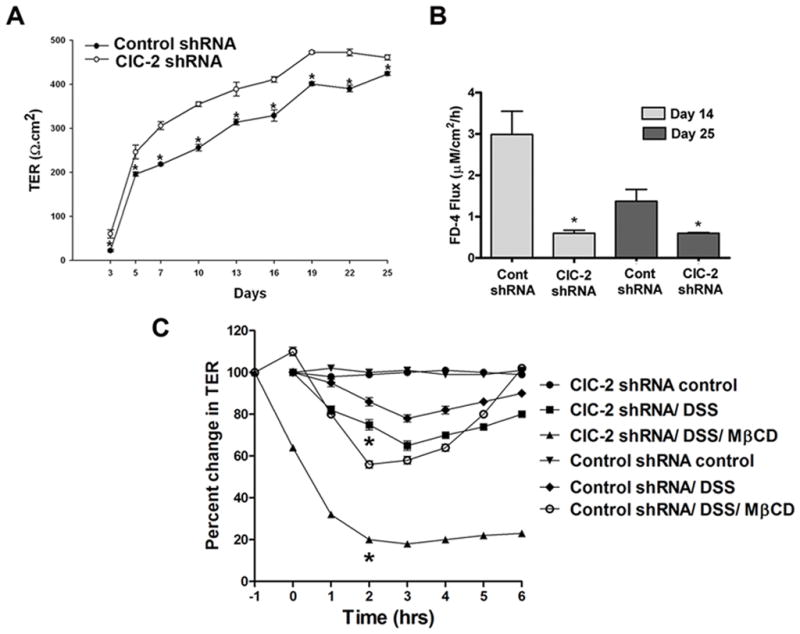
Barrier function in ClC-2 knockdown Caco-2 cells. A: The baseline TER in Caco-2 ClC-2 shRNA cells was significantly higher than control shRNA cells (representative of n = 6, multiple independent sets of observations, *p < 0.01). B: Consistent with increased TER measurements, ClC-2 shRNA cells had reduced paracellular permeability to dextran (4kD) as measured on day 14 and day 25 after plating (*, different from control shRNA at respective time points, p < 0.01, n=6). C: In spite of having reduced baseline permeability, ClC-2 shRNA Caco-2 cells, when treated with 2% DSS, showed significantly higher loss of the TER compared to control shRNA cells (*p < 0.01 vs. control shRNA DSS). Disruption of caveolae with methyl-beta-cyclodextrin (MβCD) aggravated the DSS-induced loss of TER. In ClC-2 shRNA cells, DSS treatment in the presence of MβCD led to profound and acute loss of TER, as compared to control shRNA cells. MβCD was added to the media 1 hour before treatment with DSS and MβCD media was replaced after two hours of treatment. After replacement of MβCD, the TER was found to have recovered in control but not ClC-2 shRNA cells. (*, p<0.01 vs. control shRNA/DSS/MβCD, n=6).
Inflammatory Cytokines and the Role of ClC-2 in DSS-Induced Loss of the TJ Barrier
Since anti- and pro-inflammatory cytokines play pivotal roles in the course of inflammatory changes, we quantified select cytokines in DSS colitis tissues. The qPCR studies on colonic mucosa showed the mRNA expression of IFNγ and IL-10 to be increased and that of TGF-β decreased in DSS colon of WT as well as ClC-2−/− mice (Figure 7A). The mRNA expression of TGF-β was found to be significantly lower in DSS ClC-2−/− mice compared to DSS WT mice. Furthermore, there were 8-fold and 2-fold increases for IL-1β and TNF-α mRNA, respectively, in DSS colon of ClC-2−/− mice verses DSS colon of WT mice. Thus, consistent with the severity of inflammatory changes in the DSS colon of ClC-2−/− mice, the mRNA expression of pro-inflammatory cytokines TNF-α and IL-1β was significantly elevated over DSS colon of WT mice. In further experiments, the effect of treatment with TNF-α or IL-1β n the TER of control and ClC-2 shRNA cells was studied. There was significantly greater loss of TER in ClC-2 shRNA cells compared to control shRNA cells, after the treatment with either TNFα or IL-1β (Figure 7B). To be certain that these results were not altered by baseline cytokine expression by Caco-2 cells, we performed qPCR for IL-10, TGF-β, IL-1β, and TNG-α and found that there was no significant difference between control and ClC-2 shRNA Caco-2 cells (data not shown). In further experiments, we also assessed treated cells for changes in claudin-2 expression, as we had seen in ClC-2−/− DSS mice. However, in cytokine-exposed cells there was no change in expression of claudin-2 (data not shown), suggesting a possible role of inflammation in up regulating claudin-2 expression during colitis.
Figure 7.
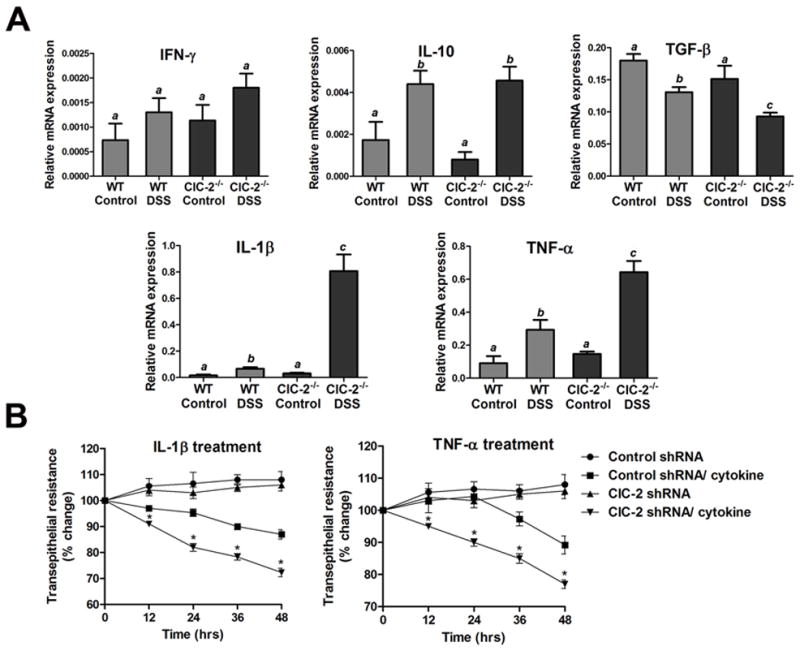
Inflammatory cytokines in DSS-induced colitis and loss of TJ barrier function. A: The qPCR analysis of DSS colitis tissue showed decreased mRNA expression of TGF-β and markedly increased expression of mRNA of TNF-α and IL-1β in ClC-2−/− DSS colon verses WT DSS colon (a,b, and c indicates significant difference compared to WT control, p < 0.01, n=4). The mRNA expression of respective cytokines was normalized to mRNA expression of GAPDH. B: Treatment of Caco-2 cells with IL-1β and TNF-α (both 10ng/ml) reduced the TER over a period of 48 hours. The reduction in the TER with either IL-1β or TNF-α was significantly higher in ClC-2 shRNA cells compared to control scrambled shRNA cells (*p < 0.01 vs. control shRNA, n=6).
Intestinal TJ Barrier Recovery after DSS Colitis
Our previous studies 8,9,11 clearly indicated that ClC-2 plays a critical role in intestinal paracellular barrier recovery. Thus, next we investigated TJ barrier recovery after DSS colitis in the ClC-2−/− mice. Following 6-days of DSS treatment, the DSS was withdrawn from the drinking water, and the mice were offered plain drinking water for the next 48-hours. The harvested colonic tissues were then mounted in Ussing chambers. We found significant barrier recovery in WT mice in terms of TER and mannitol permeability being comparable to control values (Figure 8A). However, in ClC-2−/− mice, there was no evidence of barrier recovery. There was persistent loss of TER and significantly elevated mannitol permeability in ClC-2−/− mice after 48-hours of DSS withdrawl (Figure 8B). On histological examination, WT mice showed amelioration of inflammation after two-days of DSS withdrawl (Figure 8C). The surface epithelium regained a normal columnar appearance with minimal presence of inflammatory cells and edema in the lamina propria. In contrast, ClC-2−/− mice had abnormally proliferating crypts, crypt abscesses, and persistence of inflammatory cells and edema in the submucosa (Figure 8C; histological score of 0.75 ± 0.14 and 2.52 ± 0.24 in WT and ClC-2−/− mice, respectively, on a scale of 0–4, p < 0.01). This data clearly shows that resolution of intestinal inflammation and intestinal TJ barrier recovery is diminished in the absence of ClC-2.
Figure 8.
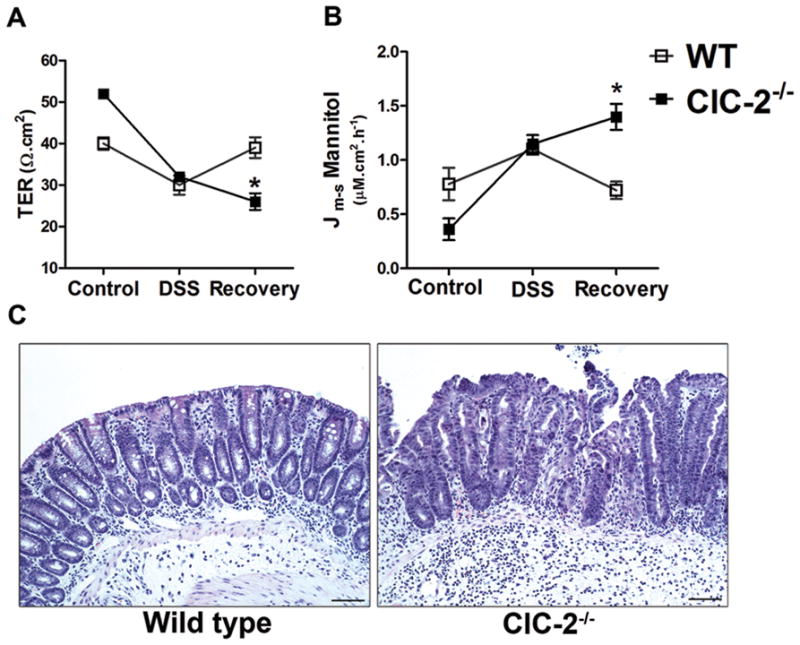
The role of ClC-2 in post-DSS barrier recovery. After 6 days of DSS treatment, the DSS was withdrawn for 48 hours after which the colonic tissues were harvested and mounted on Ussing chambers. A: WT mice showed significant recovery of TER, whereas ClC-2−/− mice showed continued loss of TER (*p < 0.01 vs. WT). B: Consistent with the lack of recovery of TER, mannitol permeability in ClC-2−/− colon remained significantly increased for 48 hours following removal of DSS (*p < 0.01 vs. WT). C: In histological examination, WT mice revealed re epithelization with minimal presence of inflammatory cells and oedema in the lamina propria of the colon. In ClC-2−/− mice, abnormally proliferating crypts, crypt abscesses, and persistence of inflammatory cells and oedema in the submucosal was seen. Bar = 25 μM.
ClC-2 Immunolocalization in Murine DSS colitis
Immunohistochemical examination of murine colon revealed the presence of ClC-2 principally at the apical lateral membrane and in the supranuclear cytoplasmic region of surface epithelium (Figure 9A & B). ClC-2 staining was completely absent in the crypts. In DSS colitis tissues, the ClC-2 staining revealed altered localization; the ClC-2 staining on the lateral membranes was minimal and prominent cytoplasmic aggregations were noted (Figure 9C & D). In western analyses, the total expression of ClC-2 was, however, found not to be significantly altered after DSS colitis (data not shown).
Figure 9.
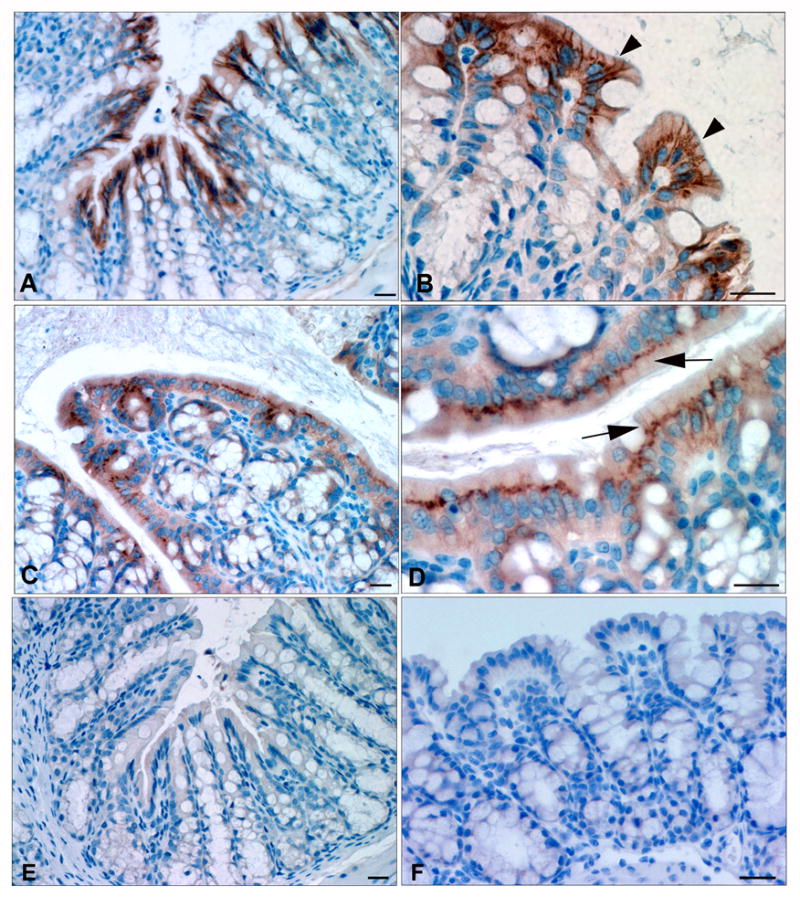
Immunohistochemical examination of murine colon. ClC-2 staining (brown color) was present at the apical lateral membrane and supranuclear cytoplasmic location in the surface epithelium (A & B, arrowheads). No staining was detected in the crypts. In DSS colitis tissues, ClC-2 staining on the lateral membranes was weak and prominent cytoplasmic aggregations were common (C & D, arrows). The negative control slides (E: WT mice colon without primary antibody; F: ClC-2−/− mice colon) did not reveal any staining. Bar = 25μM.
ClC-2 Immunolocalization and mRNA expression in Ulcerative Colitis (UC)
To determine the relevance of our experimental findings to ulcerative colitis (UC) in human patients, we examined human colonic biopsies for immunolocalization of ClC-2 as well as ClC-2 mRNA expression by qPCR. In total, we examined six normal and six ulcerative colitis tissues for immunolocalization of ClC-2. In normal colon, surface epithelial cells showed ample staining for ClC-2 (Figure 10A). However, expression of ClC-2 in the surface epithelium of inflamed colon was found to be markedly reduced (Figure 10B & C). Furthermore, qPCR analysis of colonic biopsies from control patients and UC patients revealed significant reductions in ClC-2 mRNA expression in UC patients (Figure 10D). Thus, this study implicates a crucial role for ClC-2 in epithelial homeostasis and epithelial barrier recovery in experimental colitis, and possibly affected patients.
Figure 10.
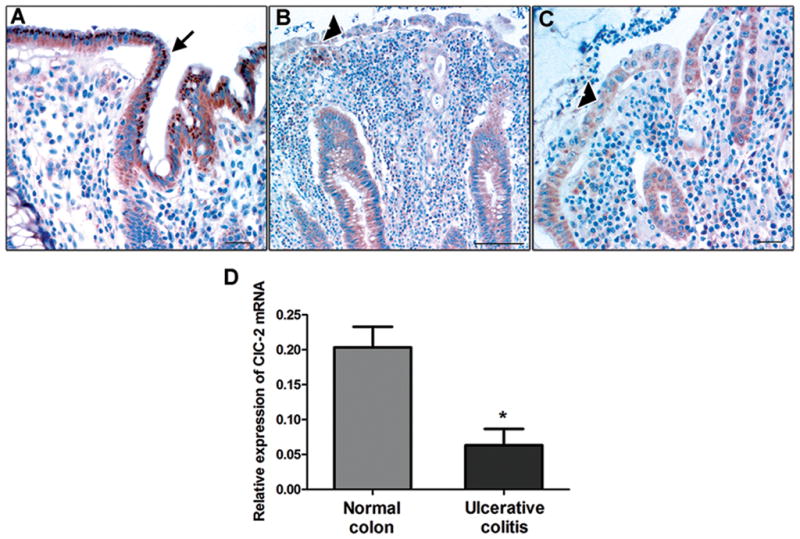
Immunohistochemistry and qPCR for ClC-2 in ulcerative colitis biopsies. A: In unaffected colonic tissue, ample ClC-2 staining was present (brown color, arrow). However, in UC patients, expression of ClC-2 in the colonic surface epithelium was found to be markedly reduced (B and C, arrowhead). Bar = 25μM. The images are representative of 6 normal and 6 UC biopsies. D: In the qPCR analysis, mRNA expression of ClC-2 was found to be significantly reduced in UC colonic biopsies compared to control patients. The ClC-2 mRNA expression is normalized to mRNA expression of GAPDH. (n = 3 in each group, *p < 0.05).
Discussion
The focus of the present study was to investigate the role of ClC-2 in epithelial barrier function and recovery in the event of epithelial injury for which we used a chemical colitis model as well as human colonic biopsies. We found that the severity of DSS colitis is increased and the recovery of colonic barrier function is impaired in the absence of ClC-2. The clinical scores and colonic histological scores of inflammatory changes were significantly higher in ClC-2−/− DSS mice compared to WT DSS mice. We found that the relative loss of TER and increase in mannitol permeability was significantly higher in ClC-2−/− DSS mice. The measurement of TER correlates well with paracellular ion transport and epithelial barrier status in healthy tissues. In inflamed tissues, however, the presence of inflammatory cells and edematous fluid within the lamina propria and the muscular layers can profoundly mask the actual loss of barrier function if solely measured by TER.21 This may explain the fact that the differences in the actual TER after DSS colitis in WT and ClC-2−/− colon in the present studies were not large quantitatively, whereas the severe inflammatory changes and marked increase in the mannitol permeability in ClC-2−/− DSS mice were striking. Furthermore, barrier recovery following withdrawal of DSS, in terms of TER and mannitol permeability, was completely lacking in ClC-2−/− mice. These observations are consistent with our previous reports in which the intestinal barrier recovery was found to be impaired in the absence of ClC-2.11 The persistent loss of the epithelial barrier in ClC-2−/− mice was also accompanied by a lack of resolution of intestinal inflammation, compared to WT mice.
In the TJ analyses, we found that occludin was prominantly redistributed from low density detergent insoluble fractions to high density detergent soluble fractions in the ClC-2−/− DSS colon. Loss of epithelial barrier function has been shown to be associated with loss of occludin localization and expression in several studies.22–25 Alternatively, total expression of claudin-2, a pore forming TJ protein was found to be up regulated in ClC-2−/− DSS colon, compared to WT DSS colon. The increased expression of claudin-2 was also found to be present within detergent insoluble low density sucrose fractions of colonic tissue of DSS ClC-2−/− mice (data not shown). Upregulation of claudin-2 has been reported in animal models of intestinal inflammation,26 IBD,27,28 and colonic cancer.29 However, it is not clear if increased expression of claudin-2 in ClC-2−/− DSS colon is the primary determinant of the defect in barrier function.
Previously, we have demonstrated that ClC-2 modulates barrier function via intracellular caveolar movement of occludin.9 In this study, the displacement of occludin to the caveolin-1 containing detergent soluble fractions was particularly noticeable in ClC-2−/− DSS colon compared to WT DSS colon. Endocytosis and recycling of TJ proteins in response to physiological and pathological stimuli occurs via different pathways.30,31 The importance of caveolar-mediated movement of occludin in the epithelial barrier has been demonstrated in our previous study 9 as well as studies by other investigators.32,33 The link between ClC-2 and caveolae in the maintenance of the epithelial barrier was also evident in the present study when caveolar disruption during DSS treatment of ClC-2 shRNA cells caused acute and persistent loss of TER, unlike similarly treated control shRNA cells.
As reported in our previous study, the tighly confined intestinal epithelial tight junctions and the narrowed paracellular space of ClC-2−/− mice are altered in such a way that the baseline permeability is paradoxically reduced, as compared to WT counterparts.10 In the present study, we found that the colonic barrier in ClC-2−/− mice was altered in a similar way; this observation is consistent with another recent study of Catalan et al.5 Moreover, knockdown of ClC-2 in human intestinal Caco-2 cells in this study resulted in a similar modification in the paracellular barrier. In spite of reduced basal permeability, the effect of DSS treatment was severe in the colon of ClC-2−/− mice as well as ClC-2 knockdown Caco-2 cells, as compared to ClC-2+/+ mice and ClC-2+/+ Caco-2 cells, respectively. Furthermore, unlike barrier recovery in WT mice following withdrawal of DSS, the ClC-2−/− colon showed continued loss of TER and increased mannitol permeability. Additionally, in the cell culture model, control but not ClC-2 knockdown cells showed barrier recovery after DSS-MβCD treatment. Therefore, the present study indicates that DSS treatment causes distinct changes in the TJ in the absence of ClC-2, leading to augmented inflammatory responses and impaired barrier recovery. Overall, loss of a chloride channel at the level of the TJ seems to ‘tighten’ the paracellular space, but other actions of ClC-2, most importantly trafficking of the TJ protein occludin, are crucial to rapidly repair the TJ barrier.
Consistent with the higher histological scores and the inflammatory changes in the ClC-2−/− DSS colon, we found that the mRNA levels of the pro-inflammatory cytokines TNF-α and IL-1β were significantly increased in these tissues. The importance of cytokines in IBD as well as experimental colitis models has been underscored in several studies (see review34). We found that the ClC-2 knockdown Caco-2 cells had increased loss of TER following treatment with TNF-α and IL-1β, as compared to similarily treated control shRNA cells. TNF-α–induced increases in TJ permeability have been shown to be dependent on caveolin-1-mediated occludin endocytosis in an in vivo study.32 Lipid rafts and caveolin-1 have been shown to be involved in intracellular signaling in response to IL-1β35 and TNF-α,36 and association of ClC-2 with caveolin-19 in lipid rafts might be relevant to the organization of ligand-receptor complexes in the cell membrane.
Lastly, we were able to immunolocalize ClC-2 in the murine colon and human colonic biopsies from UC patients. We found that in DSS colitis tissue, localization of ClC-2 was disrupted and mostly intracytoplasmic compared to membrane distribution in control murine colon. In human biopsies, loss of ClC-2 expression in colonic epithelium was clearly evident in UC inflamed regions. Furthermore, the qPCR analysis revealed significant reductions in mRNA expression of ClC-2 in UC colonic biopsies. The exact association between inflammation and loss of expression of ClC-2 requires further investigation.
In summary, we have shown that the severity of DSS-induced murine colitis is increased and the recovery after DSS colitis is hampered in the absence of the chloride channel ClC-2. The DSS colitis in ClC-2 knockout mice was found to be characterized by increased colonic permeability, distinct changes in the TJ barrier, marked histopathological changes, and increased levels of TNFα and IL-1β mRNA. Increased intestinal permeability due to a defective intestinal epithelial TJ barrier is known to be an important pathogenic factor contributing to the development and progression of intestinal inflammation.37–41 Although the mechanistic link between ClC-2 and colonic inflammation needs to be investigated further, ClC-2-mediated TJ barrier function was found to be critical for tissue homeostasis as well as predisposition to severity of injury and barrier recovery in the event of epithelial injury. The potential of screening patients for genetic defects in ClC-2 or improvement of therapeutic efforts using ClC-2 agonists in inflammatory bowel disease will be further investigated.
Acknowledgments
The authors thank the Laboratory of Electron and Light Microscopy (LAELOM), Laboratory Animal Resources (LAR), and Monica Mattmuller, Histopathology Laboratory, North Carolina State University for their excellent technical assistance.
Funding: This work was funded by the Center for Comparative Medicine & Translational Research (P.N., A.B.); R01-DK054452 (S.P); the Center for Gastrointestginal Biology and Disease histology core (P30 DK34987); and a Crohn’s and Colitis Foundation of America Research Fellowship Award (N.M.)
Footnotes
Conflicts of interest: The authors disclose no conflicts.
References
- 1.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- 3.Niemeyer MI, Yusef YR, Cornejo I, et al. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol Genomics. 2004;19:74–83. doi: 10.1152/physiolgenomics.00070.2004. [DOI] [PubMed] [Google Scholar]
- 4.Bosl MR, Stein V, Hubner C, et al. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(−) channel disruption. EMBO J. 2001;20:1289–1299. doi: 10.1093/emboj/20.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Catalan MA, Flores CA, Gonzalez-Begne M, et al. Severe Defects in Absorptive Ion Transport in Distal Colons of Mice That Lack ClC-2 Channels. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gyomorey K, Yeger H, Ackerley C, et al. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am J Physiol Cell Physiol. 2000;279:C1787–94. doi: 10.1152/ajpcell.2000.279.6.C1787. [DOI] [PubMed] [Google Scholar]
- 7.Kirk KL. Chloride channels and tight junctions. Focus on “Expression of the chloride channel ClC-2 in the murine small intestine epithelium”. Am J Physiol Cell Physiol. 2000;279:C1675–6. doi: 10.1152/ajpcell.2000.279.6.C1675. [DOI] [PubMed] [Google Scholar]
- 8.Moeser AJ, Haskell MM, Shifflett DE, et al. ClC-2 chloride secretion mediates prostaglandin-induced recovery of barrier function in ischemia-injured porcine ileum. Gastroenterology. 2004;127:802–815. doi: 10.1053/j.gastro.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Nighot PK, Blikslager AT. Chloride channel ClC-2 modulates tight junction barrier function via intracellular trafficking of occludin. Am J Physiol Cell Physiol. 2012;302:C178–87. doi: 10.1152/ajpcell.00072.2011. [DOI] [PubMed] [Google Scholar]
- 10.Nighot PK, Blikslager AT. ClC-2 regulates mucosal barrier function associated with structural changes to the villus and epithelial tight junction. Am J Physiol Gastrointest Liver Physiol. 2010;299:G449–56. doi: 10.1152/ajpgi.00520.2009. [DOI] [PubMed] [Google Scholar]
- 11.Nighot PK, Moeser AJ, Ryan KA, et al. ClC-2 is required for rapid restoration of epithelial tight junctions in ischemic-injured murine jejunum. Exp Cell Res. 2009;315:110–118. doi: 10.1016/j.yexcr.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mruk DD, Lau AS, Conway AM. Crosstalk between Rab GTPases and cell junctions. Contraception. 2005;72:280–290. doi: 10.1016/j.contraception.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Yu D, Turner JR. Stimulus-induced reorganization of tight junction structure: the role of membrane traffic. Biochim Biophys Acta. 2008;1778:709–716. doi: 10.1016/j.bbamem.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barrett KE. Building better bugs to deliver biologics in intestinal inflammation. Gut. 2010;59:427–428. doi: 10.1136/gut.2009.195016. [DOI] [PubMed] [Google Scholar]
- 16.Nehrke K, Arreola J, Nguyen HV, et al. Loss of hyperpolarization-activated Cl(−) current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem. 2002;277:23604–23611. doi: 10.1074/jbc.M202900200. [DOI] [PubMed] [Google Scholar]
- 17.Wirtz S, Neufert C, Weigmann B, et al. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 18.Moeser AJ, Nighot PK, Ryan KA, et al. Prostaglandin-mediated inhibition of Na+/H+ exchanger isoform 2 stimulates recovery of barrier function in ischemia-injured intestine. Am J Physiol Gastrointest Liver Physiol. 2006;291:G885–94. doi: 10.1152/ajpgi.00380.2005. [DOI] [PubMed] [Google Scholar]
- 19.Li Q, Zhang Q, Zhang M, et al. Effect of n-3 polyunsaturated fatty acids on membrane microdomain localization of tight junction proteins in experimental colitis. FEBS J. 2008;275:411–420. doi: 10.1111/j.1742-4658.2007.06210.x. [DOI] [PubMed] [Google Scholar]
- 20.Clayburgh DR, Musch MW, Leitges M, et al. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–2694. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitz H, Barmeyer C, Fromm M, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116:301–309. doi: 10.1016/s0016-5085(99)70126-5. [DOI] [PubMed] [Google Scholar]
- 22.Al-Sadi R, Khatib K, Guo S, et al. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1054–64. doi: 10.1152/ajpgi.00055.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCarthy KM, Skare IB, Stankewich MC, et al. Occludin is a functional component of the tight junction. J Cell Sci. 1996;109 ( Pt 9):2287–2298. doi: 10.1242/jcs.109.9.2287. [DOI] [PubMed] [Google Scholar]
- 24.Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu AS, McCarthy KM, Francis SA, et al. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol. 2005;288:C1231–41. doi: 10.1152/ajpcell.00581.2004. [DOI] [PubMed] [Google Scholar]
- 26.Ridyard AE, Brown JK, Rhind SM, et al. Apical junction complex protein expression in the canine colon: differential expression of claudin-2 in the colonic mucosa in dogs with idiopathic colitis. J Histochem Cytochem. 2007;55:1049–1058. doi: 10.1369/jhc.7A7211.2007. [DOI] [PubMed] [Google Scholar]
- 27.Oshima T, Miwa H, Joh T. Changes in the expression of claudins in active ulcerative colitis. J Gastroenterol Hepatol. 2008;23 (Suppl 2):S146–50. doi: 10.1111/j.1440-1746.2008.05405.x. [DOI] [PubMed] [Google Scholar]
- 28.Weber CR, Nalle SC, Tretiakova M, et al. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88:1110–1120. doi: 10.1038/labinvest.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinugasa T, Huo Q, Higashi D, et al. Selective up-regulation of claudin-1 and claudin-2 in colorectal cancer. Anticancer Res. 2007;27:3729–3734. [PubMed] [Google Scholar]
- 30.Utech M, Mennigen R, Bruewer M. Endocytosis and recycling of tight junction proteins in inflammation. J Biomed Biotechnol. 2010;2010:484987. doi: 10.1155/2010/484987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hopkins AM, Walsh SV, Verkade P, et al. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci. 2003;116:725–742. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- 32.Marchiando AM, Shen L, Graham WV, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Itallie CM, Fanning AS, Holmes J, et al. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci. 2010;123:2844–2852. doi: 10.1242/jcs.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouguen G, Chevaux JB, Peyrin-Biroulet L. Recent advances in cytokines: therapeutic implications for inflammatory bowel diseases. World J Gastroenterol. 2011;17:547–556. doi: 10.3748/wjg.v17.i5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oakley FD, Smith RL, Engelhardt JF. Lipid rafts and caveolin-1 coordinate interleukin-1beta (IL-1beta)-dependent activation of NFkappaB by controlling endocytosis of Nox2 and IL-1beta receptor 1 from the plasma membrane. J Biol Chem. 2009;284:33255–33264. doi: 10.1074/jbc.M109.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Alessio A, Kluger MS, Li JH, et al. Targeting of tumor necrosis factor receptor 1 to low density plasma membrane domains in human endothelial cells. J Biol Chem. 2010;285:23868–23879. doi: 10.1074/jbc.M110.122853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma TY, Anderson JM. Tight junction and intestinal barrier. In: Johnson LR, editor. Textbook of Gastrointestinal Physiology. Elsevier Health Sciences; Philadelphia, PA: 2006. pp. 1559–1594. [Google Scholar]
- 38.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 39.Arrieta MC, Madsen K, Doyle J, et al. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wyatt J, Vogelsang H, Hubl W, et al. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–1439. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 41.Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol. 2000;35:1163–1169. doi: 10.1080/003655200750056637. [DOI] [PubMed] [Google Scholar]