

Abstract
In recent years, it has become apparent that there exist several roles for respiratory complex II beyond metabolism. These include: (i) succinate signaling, (ii) reactive oxygen species (ROS) generation, (iii) ischemic preconditioning, (iv) various disease states and aging, (v) a role in the function of the mitochondrial ATP-sensitive K+ (mKATP) channel. This review will address the involvement of complex II in each of these areas, with a focus on how complex II regulates or may be involved in the assembly of the mKATP.
Keywords: ATP sensitive potassium channel, mKATP, preconditioning, ischemia, succinate dehydrogenase, diazoxide, mitochondria
1. Introduction
Mitochondrial respiratory complex II/succinate dehydrogenase (SDH)/succinate ubiquinone oxidoreductase (SQR); EC 1.3.5.1 (referred to in this review as complex II) is a 124 kDa protein complex located on the matrix side of the mitochondrial inner membrane [1]. It is comprised of four subunits: SDHA, SDHB, SDHC and SDHD. SDHA, the flavin-adenine dinucleotide (FAD) containing subunit and SDHB, the iron-sulfur cluster containing subunit [2], are anchored to the membrane by the cytochrome b containing membrane proteins SDHC and SDHD. Complex II is a component of both the Krebs cycle and the respiratory chain [3]. Complex II oxidizes the Krebs cycle intermediate succinate, generating fumarate by passing electrons from succinate to FAD. The electrons are passed along the 2Fe-2S, 4Fe-4S and 3Fe-4S centers in SDHB, finally reducing one molecule of ubiquinone (co-enzyme Q10) to ubiquinol (reviewed in [4,5]). A schematic is shown in Figure 1.
Figure 1.

Complex II: Complex II is the succinate dehydrogenase enzyme of the TCA cycle oxidizing succinate to fumarate. The succinate ubiquinone oxidoreductase enzymatic activity transfers electrons from succinate through the FAD moiety of SDHA to three iron sulfur centers of SDHB and finally to Coenzyme Q10 via SDHC/D. Although the enzyme containe a heme iron, its role in electron transfer is uncertain. Complex II acts to increase the pool of reduced Coenzyme Q10 (QH2) in the mitochondrial inner membrane. Reduced Coenzyme Q10 then transfer electrons to complex III.
Unlike respiratory Complexes I or III, complex II does not pump protons across the inner membrane. However, complex II is capable of reducing co-enzyme Q10, which can then be re-oxidized by complex III and thus participate in the proton pumping Q cycle of oxidative phosphorylation (Ox-Phos). It is generally believed that the primary function of complex II is to maintain the reduced state of the integral membrane Q pool [6]. Fully reduced co-enzyme Q10 has been shown to function as an antioxidant, protecting mitochondrial lipids and proteins from damage by constitutively produced reactive oxygen species (ROS) [7].
There exist several roles for complex II beyond metabolism, including succinate signaling, reactive oxygen species (ROS) generation, ischemic preconditioning, various disease states and aging, and in the function of the mitochondrial ATP-sensitive K+ (mKATP) channel. This review will address the involvement of complex II in each of these areas.
2. Complex II inhibition and disease states
2.1 Cancer
Each of the four subunits of complex II is encoded by nuclear DNA. This is a unique feature of complex II compared to the other respiratory chain complexes, which contain additional mitochondrial DNA (mtDNA) encoded subunits. Mutations in each of the four complex II subunits in humans, although rare, have been identified (see Table 1), and result in two general phenotypic classes. Loss of function or nonsense mutations typically produce slow growing or benign (SDHC [8] and SDHD [9]) or highly aggressive (SDHB [10]) tumors of the carotid body, classified as hereditary paragangliomas or pheochromocytomas. In these cases there is typically no detectable SDH activity in tumors [11]. Mutations in the succinate dehydrogenase assembly factor 2 (SDHAF2) gene have also been identified, and produce tumors similar to those caused by SDH-B/C/D mutations [12]. Tumorous phenotypes only manifest after both copies of a particular SDH gene have been lost and thus are classified as tumor suppressor genes [13]. The link between complex II and tumor formation may involve the Warburg effect, wherein defects in Ox-Phos may play a role in driving metabolism more toward glycolysis. The shift in metabolism, which has been reported in SDH-derived pheochromocytomas, is thought to be a an important tumor survival mechanism [14].
Table 1.
Complex II mutant phenotypes associated with cancer and other diseases.
| Subunit | Gene Locus | Phenotype | Malignancy | MIM # | Reference |
|---|---|---|---|---|---|
| SDHA | 5p15.33 [55] | Cardiomyopathy | ? | 613642 | [209] |
| Leigh Syndrome | 256000 | [55,210–212] | |||
| Mitochondrial Cx II deficiency | 252011 | [55,210–212] | |||
| Paraganglioma type 5 | 614165 | [213] | |||
| SDHB | 1p36.13 [214] | Cowden-like syndrome | High | 612359 | [215] |
| GIST | 606764 | [216] | |||
| Paragangliomas and GIST | 606864 | [217,218] | |||
| Paraganglioma type 4 | 115301 | [10,219–223] | |||
| Pheochromocytoma | 171300 | [10,22,224] | |||
| SDHC | 1q23.3 [217] | GIST | Low | 606764 | [216] |
| Paragangliomas and GIST | 606864 | [217] | |||
| Paraganglioma type 3 | 605373 | [8,225] | |||
| SDHD | 11q23.1 [226] | Carciniod Tumors Intestinal | Low | 114900 | [227] |
| Cowden-like syndrome | 612359 | [215] | |||
| Merkel Cell Carcinoma (Somatic) | - | [227] | |||
| Paragangliomas and GIST | 606864 | [217] | |||
| Paraganglioma type 1 | 168000 | [9,221,224,228–232] | |||
| Pheochromocytoma | 171300 | [229,233] | |||
| SDHAF1 | 19q13.12 [234] | Mitochondrial Cx II deficiency | 252011 | [234] | |
| SDHAF2 | 11q12.2 [12] | Paraganglioma type 2 | ? | 601650 | [12,235] |
Legend. GIST, Gastrointestinal stromal turmors (GIST); Cx II, complex II.
The mechanisms through which loss of SDH activity promotes tumor growth via the Warburg effect are debated, and are summarized in Figure 2. The most prominent theories pose that complex II deficiency results in accumulation of metabolic intermediates that signal activation of tumor-promoting pathways. The first theory is that loss of SDH activity leads to an increase in matrix succinate, which freely enters the cytosol through the dicarboxylic acid transporter. Succinate has been shown to act as a cytosolic signaling molecule in the hypoxia-response pathway [15–17], by acting as an inhibitor of prolyl hydroxylases (PHDs) [18]. In this signaling pathway, cells respond to environmental oxygen levels though hypoxia inducible factor (HIF)-1α. In the presence of molecular oxygen HIF-1α is hydroxylated by PHDs and targeted by the E3 ubiquitin ligase Von-Hippel-Lindau (VHL) protein for degradation [19]. In low oxygen, or if PHD is inhibited, HIF-1α migrates to the nucleus where it forms a heterodimeric transcription factor complex with HIF-1β, activating genes that lead to increased glycolysis, angiogenesis, motility and survival [20]. This signaling pathway is generally regarded as pro-tumor survival, enabling cancer cells to survive the low oxygen environment of the tumor. In support of this mechanism as a link between complex II mutations and cancer, increased cytoplasmic succinate levels have been confirmed in SDH–A [21], –B [22], -C and –D [11,23] mutants in association with an activated HIF pathway (see also[24,25]). For further reviews see:[6,26–28].
Figure 2.

Metabolic and cancer related consequences of mutations in the SDH complex: 1) wild-type SDH subunits form a functional complex II. Succinate is oxidized to fumarate keeping cellular succinate levels low. Electrons are transferred to Coenzyme Q10 which can act as an antioxidant maintaining low levels of ROS. 2) Mutations in any of the four SDH subunits inhibit or impair normal election flow. Left: Increased succinate enters the cytosol where it inhibits prolyl hydroxylase (PHD) mediated degradation of HIF-1α, resulting in translocation of HIF-1α to the nucleus and increased transcription of genes for tumor survival. Right: Disrupted electron flow through complex II leads to increased ROS (either directly or via less scavenging by reduced Co-enzyme Q10). ROS can act directly as a mutagenic agent on DNA or as a signaling molecule activating p53. p53 responds to ROS by increasing genes responsible for inhibiting glycolysis and ROS scavenging and decreasing genes involved in stimulating Ox-Phos. Loss of p53 in conjunction with mutations in complex II can result in dysregulation of these genes.
Another key player in cancer, which has been linked to complex II signaling, is the tumor suppressor p53. Mutant SDHB/D backgrounds are associated with decreased levels of p53 and reduced p53 binding to NADH ubiquinone oxidoreductase I [29], further strengthening the link between decreased Ox-Phos and tumorous phenotype. Of interest from a mitochondrial viewpoint, transcriptional targets of p53 include the glycolysis-inhibiting gene, TP53-induced glycolysis regulator (TIGAR), and the Ox-Phos activating gene, synthesis of cytochrome c oxidase 2 (SCO2) [30,31]. Cellular p53 can be activated by increased reactive oxygen species (ROS). When p53 is not bound to the SCO2 gene, SCO2 is active and stimulates Ox-Phos, leading to increased generation of reactive oxygen species (ROS) as a byproduct. If ROS levels rise p53 is activated and translocates to the nucleus where it binds to the SCO2 and TIGAR genes, inactivating and activating transcription, respectively. The activated TIGAR leads to increased production of the ROS scavenger glutathione. Interestingly, both hyper-activation of HIF and reduced p53 activity have been observed in the rare autosomal-dominant conditions Cowden syndrome (CS) and Cowden-like syndrome (CSL). These syndromes are associated with increased risk of tumors and hamartomas. In addition, CS and CSL patients harboring SDHB and SDHD mutations have been identified [29]. This suggests complex II may play a role in other tumors in which both HIF and p53 are implicated.
Complex II has begun to be recognized as a source of ROS [27,32,33]. Thus a second mechanism by which complex II mutations may facilitate cancer is by the generation of damaging levels of ROS. Structural models of bacterial complex II first indicated that the flavin subunit SHDA was capable of generating ROS [34]. Other complex II subunits were later identified as a sources of ROS, specifically SDHB [35–37], -C and –D [38] in yeast, SDHC in C. elegans [39] and mice [40], and SDHB but not SDHA in human cells. In one study the increase in ROS induced by SDHB mutants was found to be capable of stimulating the hypoxia-response by inhibiting PHD [41]. However in another study ROS generation by SHDB was not observed with siRNA knockdown [42] or in human cancer cells exhibiting an SDHB mutation [23]. The concept that complex II is a biologically significant source of ROS generation is relatively new, and the production of ROS by various mutants of complex II subunits therefore requires further analysis for a consensus to be reached.
2.2 Diabetes
ROS and succinate signaling, implicated in cancer formation as a result of complex II mutations, have also been linked to diabetes. Diabetes is classically characterized by an inability to regulate blood glucose levels due to the absence of (type 1) or acquired resistance to (type 2) insulin. In diabetes, mitochondria are subjected to an increase in substrate delivery as a result of prolonged increases in blood glucose levels. This leads to an increased rate of succinate and ROS generation [43–45]. In addition to its role as a signaling molecule in the hypoxia response, succinate has been identified as a ligand for the orphan G protein coupled receptor GPCR91 [46]. This receptor is expressed in the kidney suggesting that increased succinate signaling could contribute to renal hypertension, a disease that is closely linked to diabetes.
A mechanism was recently proposed where succinate regulation/accumulation is a key mediator of insulin release by pancreatic β-cells [47]. Altered mitochondrial function as well as increased ROS production were both implicated as pathogenic products of the diabetic state [48,49]. ROS generation was shown to be increased in insulin deficient rats respiring on complex I or complex II substrates. The increased ROS was largely attributable to increased electron leak from the co-enzyme Q10 binding site of complex III [50], or reverse electron flow through complex II [51].
Recent work has shown that the flavoprotein subunit of complex II (SDHA) is capable of generating physiologically relevant levels of ROS in isolated mitochondria when concentrations of succinate were low (400μM) and the inner-membrane pool of co-enzyme Q10 was mostly in its reduced form (i.e. complex I and III were inhibited). The authors determined that complex II was capable of generating ROS either from forward flow of succinate to fumarate or from the reverse reaction from reduced co-enzyme Q10. It has been demonstrated that complex II was capable of reducing much smaller pools of co-enzyme Q10 than any of the other respiratory complexes, and that complex II was fully active upon reduction of the respiratory chain and in the presence of ATP [6,52]. This suggested that the main function of complex II is maintenance of the reduced co-enzyme Q10 pool. Fully reduced co-enzyme Q10 serves as an antioxidant and helps protect mitochondria from free radicals, whereas the semi-reduced form is a pro-oxidant and is actually a generator of free radicals [53]. Overall, these results suggest that both the succinate signaling and the ROS generating mechanisms, potentially linking complex II dysfunction and diabetes, are not mutually exclusive. For further review see: [5,32,54].
2.3 Neurodegenerative diseases
Rare instances exist where loss of function mutations in SDHA result in paragangliomas, however such mutations more commonly result in a neurodegenerative disease known as Leigh’s syndrome [55]. Leigh’s syndrome develops in early infancy and is characterized by centers of necrotic cells in the brain stem and other regions of the central nervous system. SDH activity has been shown to be reduced by 25–50% relative to normal tissue. Interestingly another common neurodegenerative disease, Huntington’s, has also been linked to dysfunctional SDHA [56]. Huntington’s Disease (HD) is an autosomal dominant neurodegenerative disease that affects the striatum and cerebral cortex. People affected with HD typically show behavior symptoms including loss of coordination, decline of cognitive faculties, dystonia, and the involuntary movement disorder chorea [57]. Patients with HD have shown reduced mitochondrial complex II and III activities [58,59], and decreases in the levels of SDHA and SDHB have been observed in the striatum [60]. Tissue from postmortem HD patents has increased oxidative damage indicative of increased ROS (reviewed in [61]). Additionally systematic treatment of animal models with complex II specific inhibitors 3-nitroproponic acid or malonate cause phenotypes similar to HD, further supporting a link between complex II and HD [62,63].
Finally, another neurological condition in which complex II may play a role is Down syndrome (trisomy 21). It is now recognized that at least some of the robust nature of Down syndrome individuals (e.g., resistance to trauma) may originate from their having higher levels of the cytoprotective gas hydrogen sulfide (H2S), since the H2S generating enzyme cystathionine β synthase is encoded on chromosome 21 [64]. Notably, SDH has been identified as a potential site of inhibition by H2S [65], such that prolonged elevation of H2S may lead to neurodegenerative effects [66] possibly mediated by complex II inhibition.
2.4 Aging
A commonly held theory is that oxidative damage caused in part by ROS from mitochondrial respiration contributes to aging, however this model has been questioned recently, as roles for ROS as protective signaling molecules have been documented [67]. Furthermore, recent evidence in the nematode C. elegans indicates that removal of ROS detoxification enzymes has little effect on worm lifespan [68,69]. Similar results have recently been reported in mammals [70].
A mild decrease in mitochondrial respiration can extend lifespan in many model organisms, including worms [71–73], and this effect is conserved in mice [74,75]. In particular, mutations in complex I and III subunits as well as in the clk-1 gene which encodes a protein involved in ubiquinone synthesis confer longevity [76]. Interestingly, while the ubiquinone product of the CLK-1 protein can transfer electrons between complexes I–III or between complexes II–III, the I–III transfer is specifically inhibited in the mutant [77]. Not coincidentally, complexes I and III are classically thought to be the main sites of ROS production in the respiratory chain, and ROS production is critical for lifespan extension in the mutants [78].
Furthermore, lifespan is sensitive to oxygen concentration and may be HIF dependent [79,80], though other signaling pathways appear to respond to mitochondrial dysfunction as well [78,81–84]. The interface between these pathways may ultimately determine the rate of aging. In concert these results argue that ROS produced by complexes I and III as a result of decreased respiration can act as a signal that promotes longevity.
In dramatic contrast to the life-extending effects of mutations in complex I and III subunits, the mev-1 mutation in the SDHC subunit of C. elegans complex II also results in increased ROS production, but decreases lifespan [85]. This association between complex II dysfunction and reduced lifespan was strengthened by the finding that independent mutation of sdhb-1 (SDHB) resulted in an phenotype that was indistinguishable from mev-1, including increased ROS production [86]. Many aspects of the mev-1 mutant phenotype are recapitulated in Drosophila [87], and introducing the mev-1 mutant into mouse SDHC results in excessive apoptosis, low birth weight and neonatal growth retardation due to ROS overproduction and mitochondrial stress [88]. Although more apoptosis occurs in the mev-1 model through activation of the canonical pathway, there is also premature accumulation of aging biomarkers, suggesting that it is not merely inappropriately timed death occurring, but rather accelerated aging.
There are also unique metabolic signatures that differentiate complex II mutants from those in complexes I and III, and it has been suggested that long-lived mit mutants utilize a novel metabolism normally seen under hypoxic conditions [89]. However, mutations in ucp-4 that decrease succinate-driven complex II respiration suppress shortened lifespan in mev-1 mutants [90], suggesting that it is the physical act of respiring through the mutant complex II that is damaging. Taken together, these results support the idea that ROS production from complex II differs in either quantity or quality from that generated by complexes I and III. For example, one possibility is that the amount of superoxide produced by complex II is much greater than I or III, and merely overwhelms the detoxification processes. Another possibility is that the ROS from complex II is distributed in a way that it is less easily detoxified or more easily creates molecular damage. A third possibility is that the ROS arising from complex II function (or dysfunction) may not be able to elicit adaptive responses through similar signaling pathways as ROS from complexes I and III.
Regardless, there appears to be an evolutionary bias toward protecting complex II function. For example, Drosophila that have been adapted to live under otherwise lethal hypoxia exhibit nearly a two-fold decrease in complex II activity, with only mild changes to the other respiratory complexes and consequently less superoxide leakage occurring during mitochondrial respiration [91]. With respect to mitochondrial disease, for nearly all the other complexes (esp. complexes I and IV), there exist age-dependent correlations for mutations in the complex and in particular the mtDNA encoded subunits, which are acutely subject to mitochondrial ROS. However, complex II is entirely nuclear-encoded, which may be a kind of a safe-haven, removed from the ROS onslaught that the other mtDNA encoded complexes are bombarded with, and protected by nuclear DNA damage repair enzymes. It is intriguing to speculate that this is because the effects of complex II dysfunction are so detrimental.
A recent study on mitochondrial biochemistry in the developing mouse heart reported on a preference for complex II over complex I as the electron entry point in the respiratory chain at embryonic day 9.5, which switches to a preference for complex I by embryonic day 13.5 [92]. A second interesting finding of this paper is that cellular ROS levels rose at the time complex II was the preferred electron entry point [92]. This work provides evidence that complex II is important in early development as well as aging.
3. Ischemia reperfusion (IR) injury and ischemic preconditioning (IPC)
Complex II and ROS are implicated in the acute pathology of ischemia reperfusion (IR) injury and the following sections are focused on the cardiac model. Key hallmarks of IR injury are a loss of oxygen and substrate supply, acidification, mitochondrial Ca2+ overload, loss of adenine nucleotides, permeability pore transition opening, and the overproduction of ROS [93]. One mechanism of protecting the heart from stress is ischemic preconditioning (IPC), whereby brief periods of ischemia prior to a longer ischemic insult provide protection against IR injury [94]. Mitochondria have emerged as a critical component of the signaling mechanisms involved in IPC [95–97]. The precise mechanism of IPC is elusive but involves an array of signaling cascades activated by a mild increase in ROS [98–100]. Many of these signaling events converge on the mitochondrion and the protection of IPC can be mimicked via the administration of pharmacological agents that act on the mitochondrion; in particular, complex II and the mitochondrial ATP-sensitive K+ channel (mKATP) are targets of protective compounds [96].
3.1 complex II and IPC
IPC is known to inhibit mitochondrial electron transport and not surprisingly mimicking this reduction in activity with mitochondrial respiratory inhibitors is cardioprotective [97]. IPC reversibly inhibits complex II [101,102]; however, the mechanism remains unclear. One potential mechanism involves formation of endogenous complex II inhibitors. ROS are an important component in IPC signaling, and under conditions of oxidative stress the complex II inhibitor malonate can be generated [102] via the non-enzymatic decarboxylation reaction between H2O2 and oxaloacetate (OAA) [103]. The competitive inhibitor malonate is also cardioprotective [102,104].
While both malonate and OAA are inhibitors of complex II, malonate-mediated inhibition is more readily reversed. Thus, a malonate-occupied complex II would be desirable at reperfusion [97]. However, the removal of malonate from complex II requires ATP [105], which is not immediately available in early reperfusion. This suggests that malonate removal and reversal of complex II inhibition following ischemia may be a gradual process. As such, malonate may be considered an appropriate inhibitor, since it is easy to remove at the right time. Furthermore, malonate formation from OAA may offer additional benefit since a decrease in the concentration of OAA would prevent rapid re-introduction of acetyl-CoA into the Krebs cycle at reperfusion. Effectively, malonate formation supports a “gradual wake-up” of the respiratory chain from ischemia/reperfusion, thereby providing cardioprotection [97].
In addition to an ROS-mediated mechanism of complex II inhibition during IPC, reactive nitrogen species (RNS) may also participate. For example, nitro-linoleate is generated during IPC, is cardioprotective [106,107] and inhibits complex II [108]. Also, nitroxyl (HNO) inhibits complex II [108,109] and is cardioprotective [108,110]. These compounds react with important thiols in complex II and suggest that complex II may act as a redox sensor. Notably, complex II can also be glutathionylated on the SDHA subunit, resulting in an increase in activity [111]. In IR injury, SDHA becomes deglutathionylated, resulting in impaired activity [111]. It is currently unknown if protective interventions such as IPC are capable of modulating the degree of complex II de-gluathionylation that occurs in subsequent IR injury. If so, this would provide another mechanism of control over complex II during the critical early reperfusion phase.
Finally, while the modulation of complex II by endogenous inhibitors may provide protection, it has recently become apparent that more potent and specific complex II inhibitors such as 3-nitropropionic acid [112,113] and atpenin A5, [104,114] are cardioprotective, opening the door for complex II as a therapeutic target in IR injury.
3.2 complex II and the mitochondrial ATP-sensitive K+ channel (mKATP)
The mechanism of complex II inhibition and how it is protective remains unclear, but is thought to involve the mitochondrial ATP-sensitive potassium channel (mKATP). The mKATP is a central component of IPC-mediated protection, such that channel openers can mimic IPC [115,116] and channel blockers block IPC [117–119]. The mitochondrial K+ cycle plays an established role in regulating mitochondrial volume and function [120,121]. The precise mechanism of protection elicited by the mKATP remains elusive but may involve the regulation of mitochondrial matrix volume and adenine nucleotide status, as well as the prevention of mitochondrial Ca2+ overload and the overproduction of ROS [122,123].
Despite the central role of the mKATP in protecting the heart, the molecular composition of the channel remains debated (see Figure 3). Similarities in pharmacologic agents and channel characteristics suggest that the mKATP may be related to the well-defined surface KATP channel. However, mKATP specific agents (e.g., diazoxide and 5-HD) which are used to distinguish the channel from the surface KATP [115] suggest a variation in structure. Furthermore, the mKATP specific opener diazoxide inhibits complex II activity, and other complex II inhibitors open mKATP [102,104,104,108,113]. While the relationship between complex II inhibitors and the mKATP has been alluded to, this relationship remains to be fully characterized.
Figure 3.
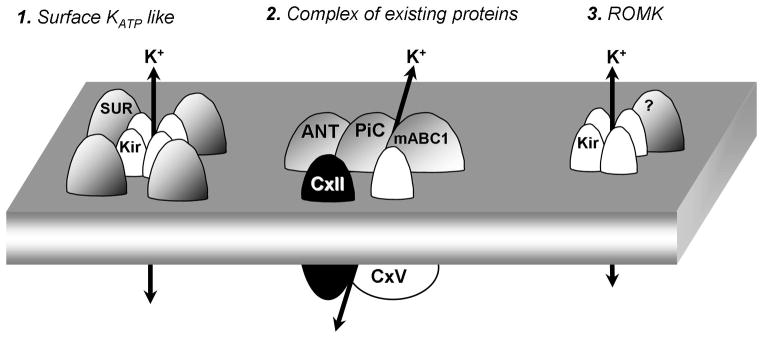
Current models for the mKATP: 1) The mKATP mayo be composed of a Kir/SUR octamer. Evidence is based on the composition of known KATP channels from cell membranes and immunoreactivity of mitochondrial inner membrane fractions to anti-Kir and anti-SUR antibodies. 2) The mKATP may be composed of a super complex of known inner membrane proteins adenine nucleotide transporter, phosphate carrier, mitochondrial ATP binding cassette protein 1, complex II and complex V. This hypothesis is based on detection of mKATP activity in reconstituted proteo-liposomes containing these five proteins [137]. 3) The mKATP is proposed to be composed of a tetramer of the Kir1.1 protein (renoal outer medullary potassium channel, ROMK) [148].
The combination of a lack of a consensus structure and ‘off-target’ effects of channel modulators has led to the hypothesis that the mKATP may be composed of known mitochondrial components such as complex II, rather than canonical KATP subunits. The evidence for each hypothesis is provided in the following sections, with current models for mKATP shown in Figure 3.
3.3 Molecular composition of the mKATP
3.3.1 Canonical Kir6.x/SUR composition?
A canonical surface KATP channel is made of a pore-forming inwardly rectifying K+ channel subunit (Kir6.1 or Kir6.2) coupled with an auxiliary sulfonylurea receptor (SUR1, SUR2A or SUR2B) subunit [124], in an octomeric conformation (Figure 3). Agents which inhibit (e.g., glyburide) or activate (e.g., pinacidil and nicorandil) the surface KATP also affect the mKATP, and following the discovery of ATP-sensitive K+ currents in mitochondria [125] efforts were focused on ascribing a Kir6.x/SURx composition to the channel. However, Kir6.x/SURx are not found in mitochondrial proteome databases [126] and they lack canonical mitochondrial targeting sequences [127]. It has therefore been proposed that other mechanisms that involve structural features such as protein folding [128] or post-translational modification [129] may target these proteins to the mitochondrion.
Previous approaches to identify the mKATP have relied heavily on immunologic techniques and yield a complicated outlook, with reports of Kir6.1 [130–132], Kir6.2 [133], both [134–136] or neither [137] in mitochondria. Moreover two widely used anti-Kir6.1 antibodies have been shown to recognize mitochondrial proteins not related to a Kir subunit [138]. Trafficking of a Kir6.x subunit to the cell surface requires its association with a SURx and suggests that if a Kir6.x subunit exists in the mitochondrion, it would be associated with SURx. Recently, a splice variant of SUR2 was found to localize to mitochondria, however it did not contribute to mKATP activity [139].
The mKATP is central to IPC-mediated protection, and while knockout mice for Kir6.1, Kir6.2, SUR1 and SUR2 [140–142] exist, confounding vascular effects preclude definitive evidence for the role of Kir6.x/SURx in IPC [140,143–145]. A key example is that many of these KATP channel mice exhibit diabetes, which is known to diminish the response to IPC [144]. Previous measurements of mKATP activity relied on flavoprotein fluorescence as an indicator of channel activity and found that the loss of Kir6.1 or Kir6.2 in mice does not affect flavoprotein fluorescence [146,147].
More recent studies suggest an alternative mKATP composition, hypothesizing that a variant of Kir1.1 (the canonical renal outer medullary K+ channel, ROMK) may be the pore-forming subunit of the mKATP [148]. However, reconciliation of several inconsistencies would help to validate this claim. For example, the ATP sensitivity of ROMK is much higher (K1/2 = 2.3 mM) [149] than that measured for the mKATP (1–25 μM) [150,151]. Furthermore, the tricyclic antidepressant drug fluoxetine (Prozac) is known to inhibit both the mKATP (IC50 2.4 μM) and IPC [151], but Kir1.1 is insensitive to fluoxetine [152,153]. Direct measurements of mKATP activity from genetic models (e.g., ROMK knockout mice), which are currently lacking, may be necessary to designate mKATP channel status to Kir1.1. Therefore, while Kir1.1 may indeed be present in mitochondria, its role in IPC remains to be fully elucidated.
The mKATP channel activity is conserved across a range of species and cross-species comparisons may help to identify or rule out certain candidate genes. For example, while the mammalian genome codes for 15 Kir isoforms, the C. elegans Kir family contains only three genes, irk-1, irk-2, and irk-3. Using the power of worm genetics, we recently showed that a triple knockout irk-1/2/3 worm exhibited perfectly normal mKATP activity and preconditioning [154]. These data demonstrate that at least in C. elegans, the mKATP is not derived from the Kir family. While more work is needed to fully elucidate the role of canonical Kir/SUR subunits in the mammalian mitochondrion, these results suggest that non-Kir proteins should also be considered as candidates for the mKATP. In this regard, the lack of consensus regarding off-target effects of KATP agents (described below) has led to the hypothesis that the mKATP is composed of known mitochondrial proteins, including complex II.
3.3.2. Alternative hypothesis: a complex II composition for mKATP?
The notion that the mKATP resembles the surface KATP was largely built on the use of small molecules. Inhibitors and activators of the defined surface KATP had expected effects on isolated mitochondria K+ fluxes. However, most of these molecules have off-target effects on mitochondrial function (reviewed in [155]). For example, glyburide is a sulfonylurea used to treat type 2 diabetes. Via its interaction with the SUR (140–180 kDa), glyburide inhibits KATP channels [156]. Glyburide also prevents the protective effects of mKATP activation. Studies using labeled glyburide found that it binds to a 28 kDa protein in heart [157] and a 64 kDa in brain mitochondria [158]. These weights are significantly smaller than the apparent molecular weight of the surface KATP channel, and they may represent a mitochondrial targeted splice variant [139] or a SUR-independent target. In this regard, glyburide at high doses inhibits respiration [159,160] and molecular modeling demonstrates that it may interact with the adenine nucleotide translocator (ANT) [161]. Work in our laboratory (unpublished) has found that a BODIPY conjugate of glyburide binds to intact complexes I and V on clear-native electrophoresis gels.
Perhaps the most widely used tool related to mKATP studies is diazoxide. While diazoxide is a general KATP opener, it is more potent at opening the mKATP and is considered a mKATP specific agonist [115]. However, diazoxide has off-target mitochondrial effects, such as mitochondrial uncoupling at high concentrations, and inhibition of complex II. It is important to note that while diazoxide does implicate a relationship between complex II and the mKATP, other channel openers such as cromakalim do not affect complex II activity. This suggests that complex II is not the sole component of the channel and may represent an important regulator. Nonetheless, these effects of diazoxide on complex II are interesting when viewed alongside another characteristic of complex II – namely the fact that ATP allosterically activates complex II activity [102,162]. This suggests an inverse relationship between complex II and mKATP activities.
Rather than serving as the mKATP channel alone, it has been proposed that complex II may form a complex with other known mitochondrial proteins, to elicit mKATP channel activity [137]. This complex includes the phosphate carrier, mitochondrial ATP-binding cassette protein-1, the ANT, and respiratory complex V [137]. These proteins were purified as a fraction from mitochondria, and while the fraction contained over twenty proteins identifiable by silver staining, these 5 proteins were the majority components [137]. The purification and reconstitution of these proteins into proteoliposomes reproduced the pharmacologic characteristics of the mKATP channel [137], including the ability of complex II inhibitors to stimulate K+ flux. Thus, it was suggested that these proteins may exist in a super-complex comprising the mKATP channel. There is a precedence for super-complexes in the respiratory chain [163] and evidence for coupling between complex II and complex V deficiencies in humans [164]. It has also been suggested that diazoxide interacts with complex V, facilitating the binding of its endogenous inhibitor to the F(1) subunit [165] and reversibly inhibiting ATP hydrolysis.
A shortcoming of this proposed mKATP super-complex is the lack of an actual K+ transporting protein in its composition [137]. All known K+ channels contain the consensus sequence GYG in their pore region, providing a selectivity filter for potassium. There is some evidence that the ANT can transport K+ [166,167], and molecular modeling suggests that the KATP blockers glyburide and 5-HD can interact with the ANT [168], but ANT does not have a GYG motif. Thus the possibility remains that a low abundance [169] bona fide K+ channel subunit may be present in the super-complex, to confer mKATP activity.
The functional and pharmacologic evidence supporting the hypothesis that complex II is a component or regulator of the mKATP channel is summarized as follows: 1) complex II inhibition is observed in IPC [101,102], 2) mKATP inhibitors stimulate complex II [102,162,170], 3) mKATP openers inhibit complex II [102,104,170,171], 4) complex II inhibitors open mKATP and are cardioprotective in a manner sensitive to mKATP blockade [104,108,113]. The array of structurally distinct complex II inhibitors capable of protecting by opening mKATP range from endogenously generated (e.g., malonate) to potent and specific (e.g., atpenin A5). These inhibitors interact with complex II at different sites and all elicit mKATP activity.
One unique aspect that has arisen from these studies is that, like diazoxide, the amount of inhibitor required for maximal mKATP activity is much lower than that required for the enzymatic inhibition of complex II. For example, atpenin A5 has an IC50 for complex II activity of 10 nM, while 1 nM yields maximal mKATP activity. Taking advantage of the potency of atpenin A5, the relationship between complex II and mKATP activity was quantified, with the finding that only 0.4% of complex II molecules were necessary to elicit maximal mKATP activity [169]. Not only does this account for the low abundance of the channel [172] it also demonstrates that bulk complex II activity is not affected by the presence of the inhibitor. These results imply that the specificity of diazoxide for the mKATP vs. the surface KATP may reside at the level of complex II [169]. Finally, it is notable that all known complex II inhibitors can activate the mKATP channel even when mitochondria are respiring on complex I linked substrates (e.g. pyruvate plus malate). This suggests that complex II does not need to be enzymatically active to participate in the mKATP.
4. What is the endogenous physiologic role of the complex II – mKATP channel interaction?
As discussed in the preceding sections, a role for both complex II and mKATP channels in IPC has been well established. However, these proteins clearly did not evolve for this purpose. Therefore, how do they interact under normal conditions, and what is their baseline/endogenous physiologic role in the absence of a stress signal such as IPC?
4.1 A ROS signal?
A key question in the mKATP field is how complex II transmits a signal to induce mitochondrial K+ uptake, and the physiologic relevance of this phenomenon. One proposed mechanism is that, independent from direct complex II ROS generation [173], complex II inhibition may trigger ROS formation by a different part of the respiratory chain (complex III), and this is responsible for subsequent protective signaling [174]. In brief, the reduction state of the co-enzyme Q10 pool can influence complex III ROS production, especially when complex III is inhibited with antimycin [32]. When electrons are supplied via complex II, inhibitors of complex II can stimulate antimycin-induced ROS production in a bell-shaped response, as a consequence of the redox state of the co-enzyme Q10 pool [175,176] (recently reviewed in [177]). Since mild ROS generation is a critical component of IPC, this mechanism was hypothesized to mediate the protective effect of complex II inhibitors and diazoxide, completely independent of any role for the mKATP channel [174]. However, several pieces of evidence suggest this ROS phenomenon is independent of complex II’s role in the mKATP or in IPC. These will now be discussed in detail:
The increase in ROS is dependent on succinate: The increase in ROS with complex II inhibitors was only observed when mitochondria respired on succinate and not present when complex I or complex III were used as electron sources [174,178]. On the contrary, complex II inhibitors can open the mKATP even when mitochondria respire on complex I or complex IV linked substrates [102,104,104,169], or when channel components are reconstituted in proteoliposomes or lipid bilayers [137] (reviewed in [179]).
ROS generation requires the presence of antimycin A: In the absence of antimycin, complex II inhibitors did not increase ROS formation, rather the addition of inhibitors resulted in a decrease in ROS [174,176]. Another study demonstrated a decrease in ROS formation with complex II inhibitors in the presence of succinate, rotenone and myxothiazol [173]. Furthermore, modulation of the co-enzyme Q10 pool redox state by cyanide [176] or loss of cytochrome c does not increase complex III ROS [180]. Thus, the increase in ROS induced by complex II inhibitors appears unique to antimycin. Contrastingly, studies demonstrating mitochondrial K+ flux induced by complex II inhibitors did not have antimycin present [102,104,108,137,151,169,181].
ROS generation is independent of K+, or mKATP inhibitors: A hallmark of mKATP activity is its sensitivity to K+. In this regard, both mitochondrial K+ fluxes and the protective effects of complex II inhibition are K+ sensitive [102,104,108,113,137,151], while antimycin induced ROS generation was insensitive to loss of K+ [174,178]. Similarly, both mitochondrial K+ fluxes and the protective effects of complex II inhibition are sensitive to mKATP channel inhibitors [102,104,108,113,137,151,182], while antimycin-induced ROS generation is not [174,178]. These findings highlight that ROS generation in response to complex II modulation is completely independent from mKATP and mitochondrial K+ flux.
ROS generation via this mechanism requires higher concentrations of complex II inhibitors than required to activate the mKATP channel [174]: Diazoxide at high concentrations (>100μM) may result in uncoupling or activate surface KATP channels. When used at appropriate concentrations (10–30 μM) diazoxide displays mKATP specificity [115]. Recently we demonstrated that a low concentration of the complex II inhibitor atpenin A5 activates mKATP activity in isolated mitochondria and results in mKATP-sensitive cardioprotection [104]. Moreover, this concentration (1 nM) is without effect on bulk complex II enzymatic activity [169]. However, the antimycin-induced ROS generation was only seen at atpenin A5 concentrations (5–50 nM) that would influence complex II activity and subsequently the state of the co-enzyme Q10 pool.
Together, these findings suggest that the mKATP activity and cardioprotection mediated by complex II inhibition are independent of antimycin-induced ROS generation. The currently available evidence does not favor ROS generation as an intermediate signal between complex II, mKATP, and cardioprotection/IPC.
4.2 Energy sensing
The mKATP has been described in humans, rats, mice, plants, amoeba, T. cruzi and C. elegans [125,133,183–186]. Although the primary research interest in the mKATP field is cardioprotection, most of these species do not have a heart or suffer from any cardiac disease, suggesting that the channel may have an evolutionarily conserved physiologic role. If the mKATP is a super-complex of complex II, the phosphate carrier, mitochondrial ATP-binding cassette protein-1, ANT, and complex V, this may suggest a relationship between mitochondrial K+ fluxes and bioenergetics. This view is compatible with the energy-sensing capabilities of the canonical surface KATP channel. While other metabolic sensing pathways such as mTOR [187], SIRT3 [188] and AMPK [189] may involve changes in complex II [190–192], they integrate more long term changes in metabolic state. In contrast, the complex II/mKATP system may respond more rapidly to energetic status. Since regulation of metabolism is known to be a strategy for protecting the heart against ischemia [97,193], any component which can regulate metabolism or the energetic status of the mitochondrion (e.g. mKATP) may also be a critical component of a cell’s cardioprotective machinery.
The complex II/mKATP complex is hypothesized herein to exert control over metabolism in addition to “classical” respiratory control (i.e., state 3 / state 4), to meet the cell’s energy demand. These mechanisms are shown in Figure 4. First, the introduction of electrons into the respiratory chain is competitive, such that the oxidation of succinate at complex II inhibits the oxidation of NADH at complex I [52]. Since complex II does not pump protons, the H+/e− stoichiometry is lower when electrons enter at complex II. Thus, the ratio of electron entry between complex II vs. I can lead to changes in the overall efficiency of Ox-Phos. This becomes an important mechanism of regulating metabolism, when coupled with the fact that complex II is activated by ATP. Under conditions of high ATP, complex II is active, thus inhibiting complex I, and lowering H+/e−, leading to inefficient Ox-Phos. Alternatively, when ATP is low, complex II activity is also low, so more electrons enter at complex I, and H+/e− rises to meet the demand for more ATP [52].
Figure 4.

Complex II/mKATP interaction in energy sensing: “Classical” respiratory coupling is shown in the center, in which mitochondria transition between quiescent (state 4) and phosphorylating (state 3) conditions depending on availability of ADP. Additional levels of control are shown in outer circuits, including: (i) ATP is a complex II activator, so low ATP leads to preferred electron entry via complex I, which in turn increases the efficiency/stoichiometry of Ox-Phos (H+/e− ratio), increasing ATP synthesis (right side). The reverse is true when ATP is high (left side). (ii) Low ATP dis-inhibits (opens) the mKATP channel, leading to K+ and water entry and swelling of the matrix (right side). This leads to greater contact between the inner and outer membranes (IM/OM), enhancing the creatine kinase shuttle and ATP availability for the cell. When ATP is low, the cycle reverses, as ATP closes the mKATP channel, the matrix shrinks, and IM/OM contact decreases, lowering ATP transport to the cytosol (left side).
In a second novel mechanism of control, regulation of mitochondrial matrix volume by the mKATP channel may serve as a coupling link between ATP levels and the activity of the Ox-Phos machinery. Mitochondrial K+ uptake is followed by osmotically obliged water, resulting in matrix swelling [120]. Matrix volume controls efficient energy transfer (via the creatine kinase (CK) shuttle system), and controls the activity of matrix enzymes necessary for ATP production, and is critical to the regulation of energy metabolism [121,172,194–200]. As such, when ATP levels are high, the mKATP is inhibited, the matrix shrinks, and the resulting loss of inner/outer membrane contacts decreases the efficiency of the CK shuttle system. In contrast, low ATP levels facilitate K+ influx via mKATP, leading to matrix swelling, which enhances inner/outer membrane contact and stimulates efficient shuttling of high energy phosphates.
As depicted in Figure 4, the combination of the influence of K+ on mitochondrial volume and metabolism, with complex II activity and energy sensing capabilities, suggest that these phenomena may be components of a concerted mechanism to modulate Ox-Phos to meet energy demands. This proposed physiologic role of the mKATP also makes the channel an important component of how the cell responds to pathologic conditions (e.g., IR injury).
4.3 Mitochondrial Fission/Fusion?
Recently however, an intriguing link has been proposed between a super-complex type mKATP channel (see section 3.3.2) and Charcot-Marie-Tooth disease type 2A [181]. Typically, this disease is associated with mutations in mitofusin 2 (MFN2), which is an important regulator of both mitochondrial fusion [201] and membrane potential [202]. Notably, the study found that a mouse model carrying an MFN2 mutation had a defect in both complex II (40% reduction) and complex V (30% reduction) [181]. Interestingly, the lost enzymatic activities were reversed by the mKATP inhibitor 5-HD. Furthermore, the phenotype could be mimicked by the mKATP channel opener diazoxide [181]. What remains unclear, is how a mutation in MFN2 (an outer membrane protein with the bulk of its structure facing the cytosol) communicates with complex II and V in the inner membrane. However, this study highlighted a role for complex II (and complex V) and the mKATP in disease. Further studies are needed to investigate the link between mitochondrial K+ flux and fission/fusion.
5. Conclusions and Future Directions
Complex II inhibition plays a dual role in biology, being both a destructive force in many disease states and also a protective mediator in others. Complex II “the destroyer” is capable of generating ROS in a manner that may be different in quantity or quality from complexes I and III. The genes encoding complex II are protected in the nucleus and may represent a mechanism to protect the cell from devastating ROS production resulting from mutated complex II. Complex II “the protector” connects mitochondrial volume regulation and metabolism via the mKATP channel. Despite a role for mKATP in both physiological and pathological conditions, the underlying molecular structure of this channel and why it is evolutionarily conserved remains unknown. Since the phenomenon of the mKATP has been described in a range of organisms from plants to mammals, comparative genomics may yield promising candidates, as was recently applied to the identification of the mitochondrial calcium uniporter [203,204]. However, if the mKATP is composed of known mitochondrial components (e.g. complex II) this approach may not be successful. Pharmacologic evidence has suggested that the channel is molecularly related to the surface KATP, and while a significant pharmacologic overlap made this hypothesis attractive, model organism genomics has shown that a Kir subunit is not necessary for mKATP channel activity or protection against stress [154]. On the other hand, complex II (and other components in the super-complex) are implicated not only in energy sensing but also mitochondrial K+ fluxes.
Despite recent reports [148], the quest for the precise molecular identity of the mKATP and the role of complex II in regulating it continue. In particular, although knockout mice for the mKATP candidate gene ROMK are available, these mice exhibit renal pathology [205] which could the interpretation of experiments on cardioprotection. Clearly, a cardiac-specific ROMK−/− mouse would be a useful tool to elucidate the role of ROMK in cardioprotection. Furthermore, newly discovered inhibitors or activators of ROMK and other members of the inward rectifying K+ channel family [206] will be useful tools, and (in the case of activators) potential therapeutics.
Although the complete loss of complex II in mammalian models is lethal [207], the generation of inducible tissue-specific knockout models may provide the tools necessary to address the role of complex II in cytoprotection. In such models, an acute loss of complex II activity in adulthood would be necessary, to avoid the contribution of developmental and metabolic affects associated with long term loss of function. Alternatively, microRNA regulation of complex II (e.g., by miR-210 [208]) may be a promising avenue to influence complex II levels, and is worthy of pursuit not only from a mechanistic angle, but also due to the increasing interest in miRNAs as therapeutic targets.
Finally, although complex II inhibitors are prevalent (Table 2), there is currently only one allosteric activator of complex II activity, namely ATP. Clearly such activators could have both therapeutic and investigational value, and the search for more such molecules should be encouraged.
Table 2.
Complex II modulators.
| Reagent | Conc. | Effects | mKATP effect |
|---|---|---|---|
| ATP | mM | Activate [102,162] | Inhibit [102,182,198] |
| Atpenin A5 | nM | Inhibit [104,114,173,174] | Activate [104] |
| Diazoxide | μM | Inhibit [171] | Activate [102,115,182,198] |
| HNO | μM | Inhibit [108,109] | Activate [108] |
| LNO2 | μM | Inhibit [108] | Activate [108] |
| Malonate | mM | Inhibit [137] | Activate [102,104,137,181] |
| 3-nitropropionate | mM | Inhibit [236,237] | Activate [113,137] |
| Oxaloacetate | μM | Inhibit [52,102,238,239] | |
| Harzianopyridone | nM | Inhibit [114] | |
| carboxin | μM | Inhibit [114] | |
| HQNO | μM | Inhibit [114] | |
| TTFA | μM | Inhibit [114] |
Legend. HQNO, 2-heptyl-4-hydroxyquinoline N-oxide; TTFA, 4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedione
Acknowledgments
This work was supported by an American Heart Association Founder’s Affiliate Postdoctoral Fellowship award 11POST7290028 (to A.P.W.), and by US National Institutes of Health grants HL071158 (to P.S.B.), GM087483 (to P.S.B. & K.W.N.).
References
- 1.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 2.Hagerhall C. Succinate: quinone oxidoreductases. Variations on a conserved theme. Biochim Biophys Acta. 1997;1320:107–141. doi: 10.1016/s0005-2728(97)00019-4. [DOI] [PubMed] [Google Scholar]
- 3.Scheffler IE. Molecular genetics of succinate:quinone oxidoreductase in eukaryotes. Prog Nucleic Acid Res Mol Biol. 1998;60:267–315. doi: 10.1016/s0079-6603(08)60895-8. [DOI] [PubMed] [Google Scholar]
- 4.Cecchini G. Function and structure of complex II of the respiratory chain. Annu Rev Biochem. 2003;72:77–109. doi: 10.1146/annurev.biochem.72.121801.161700. [DOI] [PubMed] [Google Scholar]
- 5.Lenaz G, Genova ML. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid Redox Signal. 2010;12:961–1008. doi: 10.1089/ars.2009.2704. [DOI] [PubMed] [Google Scholar]
- 6.Rustin P, Munnich A, Rotig A. Succinate dehydrogenase and human diseases: new insights into a well-known enzyme. Eur J Hum Genet. 2002;10:289–291. doi: 10.1038/sj.ejhg.5200793. [DOI] [PubMed] [Google Scholar]
- 7.Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 8.Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 9.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der MA, Taschner PE, Rubinstein WS, Myers EN, Richard CW, III, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 10.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, Rotig A, Jeunemaitre X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001;69:1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baysal BE, Rubinstein WS, Taschner PE. Phenotypic dichotomy in mitochondrial complex II genetic disorders. J Mol Med (Berl) 2001;79:495–503. doi: 10.1007/s001090100267. [DOI] [PubMed] [Google Scholar]
- 14.Favier J, Briere JJ, Burnichon N, Riviere J, Vescovo L, Benit P, Giscos-Douriez I, De RA, Bertherat J, Badoual C, Tissier F, Amar L, Libe R, Plouin PF, Jeunemaitre X, Rustin P, Gimenez-Roqueplo AP. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4:e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holme E. A kinetic study of thymine 7-hydroxylase from neurospora crassa. Biochemistry. 1975;14:4999–5003. doi: 10.1021/bi00693a033. [DOI] [PubMed] [Google Scholar]
- 16.Myllyla R, Tuderman L, Kivirikko KI. Mechanism of the prolyl hydroxylase reaction. 2. Kinetic analysis of the reaction sequence. Eur J Biochem. 1977;80:349–357. doi: 10.1111/j.1432-1033.1977.tb11889.x. [DOI] [PubMed] [Google Scholar]
- 17.MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27:3282–3289. doi: 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schofield CJ, Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr Opin Struct Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 19.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 20.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Briere JJ, Favier J, Benit P, El GV, Lorenzato A, Rabier D, Di Renzo MF, Gimenez-Roqueplo AP, Rustin P. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet. 2005;14:3263–3269. doi: 10.1093/hmg/ddi359. [DOI] [PubMed] [Google Scholar]
- 22.Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Kerlan V, Plouin PF, Rotig A, Jeunemaitre X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab. 2002;87:4771–4774. doi: 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- 23.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 24.Baysal BE. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003;14:453–459. doi: 10.1016/j.tem.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Pollard PJ, Wortham NC, Tomlinson IP. The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Ann Med. 2003;35:632–639. doi: 10.1080/07853890310018458. [DOI] [PubMed] [Google Scholar]
- 26.Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med. 2011;17:641–649. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ralph SJ, Moreno-Sanchez R, Neuzil J, Rodriguez-Enriquez S. Inhibitors of succinate: quinone reductase/Complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm Res. 2011;28:2695–2730. doi: 10.1007/s11095-011-0566-7. [DOI] [PubMed] [Google Scholar]
- 28.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Ni Y, He X, Chen J, Moline J, Mester J, Orloff MS, Ringel MD, Eng C. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum Mol Genet. 2012;21:300–310. doi: 10.1093/hmg/ddr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–3999. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bensaad K, Vousden KH. p53: new roles in metabolism. Trends Cell Biol. 2007;17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paddenberg R, Ishaq B, Goldenberg A, Faulhammer P, Rose F, Weissmann N, Braun-Dullaeus RC, Kummer W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol. 2003;284:L710–L719. doi: 10.1152/ajplung.00149.2002. [DOI] [PubMed] [Google Scholar]
- 34.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 35.Smith EH, Janknecht R, Maher LJ., III Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 36.Goffrini P, Ercolino T, Panizza E, Giache V, Cavone L, Chiarugi A, Dima V, Ferrero I, Mannelli M. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Hum Mol Genet. 2009;18:1860–1868. doi: 10.1093/hmg/ddp102. [DOI] [PubMed] [Google Scholar]
- 37.Szeto SS, Reinke SN, Sykes BD, Lemire BD. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J Biol Chem. 2007;282:27518–27526. doi: 10.1074/jbc.M700601200. [DOI] [PubMed] [Google Scholar]
- 38.Szeto SS, Reinke SN, Sykes BD, Lemire BD. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J Biol Chem. 2007;282:27518–27526. doi: 10.1074/jbc.M700601200. [DOI] [PubMed] [Google Scholar]
- 39.Adachi H, Fujiwara Y, Ishii N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. J Gerontol A Biol Sci Med Sci. 1998;53:B240–B244. doi: 10.1093/gerona/53a.4.b240. [DOI] [PubMed] [Google Scholar]
- 40.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005;65:203–209. [PubMed] [Google Scholar]
- 41.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cervera AM, Apostolova N, Crespo FL, Mata M, McCreath KJ. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Res. 2008;68:4058–4067. doi: 10.1158/0008-5472.CAN-07-5580. [DOI] [PubMed] [Google Scholar]
- 43.Brownlee M. A radical explanation for glucose-induced beta cell dysfunction. J Clin Invest. 2003;112:1788–1790. doi: 10.1172/JCI20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53(Suppl 1):S110–S118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 45.Herlein JA, Fink BD, Sivitz WI. Superoxide production by mitochondria of insulin-sensitive tissues: mechanistic differences and effect of early diabetes. Metabolism. 2010;59:247–257. doi: 10.1016/j.metabol.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 47.Fahien LA, MacDonald MJ. The succinate mechanism of insulin release. Diabetes. 2002;51:2669–2676. doi: 10.2337/diabetes.51.9.2669. [DOI] [PubMed] [Google Scholar]
- 48.Park TS, Yamashita H, Blaner WS, Goldberg IJ. Lipids in the heart: a source of fuel and a source of toxins. Curr Opin Lipidol. 2007;18:277–282. doi: 10.1097/MOL.0b013e32814a57db. [DOI] [PubMed] [Google Scholar]
- 49.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herlein JA, Fink BD, Henry DM, Yorek MA, Teesch LM, Sivitz WI. Mitochondrial superoxide and coenzyme Q in insulin-deficient rats: increased electron leak. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1616–R1624. doi: 10.1152/ajpregu.00395.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gutman M. Modulation of Mitochondrial Succinate-Dehydrogenase Activity, Mechanism and Function. Molecular and Cellular Biochemistry. 1978;20:41–60. doi: 10.1007/BF00229453. [DOI] [PubMed] [Google Scholar]
- 53.Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 54.Rustin P, Rotig A. Inborn errors of complex II--unusual human mitochondrial diseases. Biochim Biophys Acta. 2002;1553:117–122. doi: 10.1016/s0005-2728(01)00228-6. [DOI] [PubMed] [Google Scholar]
- 55.Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rotig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- 56.Pandey M, Mohanakumar KP, Usha R. Mitochondrial functional alterations in relation to pathophysiology of Huntington’s disease. J Bioenerg Biomembr. 2010;42:217–226. doi: 10.1007/s10863-010-9288-5. [DOI] [PubMed] [Google Scholar]
- 57.Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 58.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 59.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 60.Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johri A, Beal MF. Antioxidants in Huntington’s disease. Biochim Biophys Acta. 2012;1822:664–674. doi: 10.1016/j.bbadis.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 63.Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci Biobehav Rev. 1997;21:289–293. doi: 10.1016/s0149-7634(96)00027-9. [DOI] [PubMed] [Google Scholar]
- 64.Kamoun P, Belardinelli MC, Chabli A, Lallouchi K, Chadefaux-Vekemans B. Endogenous hydrogen sulfide overproduction in Down syndrome. Am J Med Genet A. 2003;116A:310–311. doi: 10.1002/ajmg.a.10847. [DOI] [PubMed] [Google Scholar]
- 65.Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, Lillie LE. Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats. Toxicol Appl Pharmacol. 1990;103:482–490. doi: 10.1016/0041-008x(90)90321-k. [DOI] [PubMed] [Google Scholar]
- 66.Kamoun P. Mental retardation in Down syndrome: a hydrogen sulfide hpothesis. Med Hypotheses. 2001;57:389–392. doi: 10.1054/mehy.2001.1377. [DOI] [PubMed] [Google Scholar]
- 67.Gems D, Doonan R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle. 2009;8:1681–1687. doi: 10.4161/cc.8.11.8595. [DOI] [PubMed] [Google Scholar]
- 68.Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22:3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cabreiro F, Ackerman D, Doonan R, Araiz C, Back P, Papp D, Braeckman BP, Gems D. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med. 2011;51:1575–1582. doi: 10.1016/j.freeradbiomed.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, Thorpe SR, Baynes JW, Epstein C, Richardson A, Van RH. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci. 2009;64:1212–1220. doi: 10.1093/gerona/glp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lakowski B, Hekimi S. Determination of life-span in Caenorhabditis elegans by four clock genes. Science. 1996;272:1010–1013. doi: 10.1126/science.272.5264.1010. [DOI] [PubMed] [Google Scholar]
- 72.Dillin A, Hsu AL, rantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 73.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 74.Hughes BG, Hekimi S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PLoS One. 2011;6:e26116. doi: 10.1371/journal.pone.0026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dell’agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 76.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 77.Yang YY, Vasta V, Hahn S, Gangoiti JA, Opheim E, Sedensky MM, Morgan PG. The role of DMQ(9) in the long-lived mutant clk-1. Mech Ageing Dev. 2011;132:331–339. doi: 10.1016/j.mad.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010;9:433–447. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- 79.Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science. 2009;324:1196–1198. doi: 10.1126/science.1173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walter L, Baruah A, Chang HW, Pace HM, Lee SS. The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol. 2011;9:e1001084. doi: 10.1371/journal.pbio.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baker BM, Nargund AM, Sun T, Haynes CM. Protective Coupling of Mitochondrial Function and Protein Synthesis via the eIF2alpha Kinase GCN-2. PLoS Genet. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- 86.Huang J, Lemire BD. Mutations in the C. elegans succinate dehydrogenase iron-sulfur subunit promote superoxide generation and premature aging. J Mol Biol. 2009;387:559–569. doi: 10.1016/j.jmb.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 87.Tsuda M, Sugiura T, Ishii T, Ishii N, Aigaki T. A mev-1-like dominant-negative SdhC increases oxidative stress and reduces lifespan in Drosophila. Biochem Biophys Res Commun. 2007;363:342–346. doi: 10.1016/j.bbrc.2007.08.168. [DOI] [PubMed] [Google Scholar]
- 88.Ishii T, Miyazawa M, Onodera A, Yasuda K, Kawabe N, Kirinashizawa M, Yoshimura S, Maruyama N, Hartman PS, Ishii N. Mitochondrial reactive oxygen species generation by the SDHC V69E mutation causes low birth weight and neonatal growth retardation. Mitochondrion. 2011;11:155–165. doi: 10.1016/j.mito.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 89.Butler JA, Ventura N, Johnson TE, Rea SL. Long-lived mitochondrial (Mit) mutants of Caenorhabditis elegans utilize a novel metabolism. FASEB J. 2010;24:4977–4988. doi: 10.1096/fj.10-162941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pfeiffer M, Kayzer EB, Yang X, Abramson E, Kenaston MA, Lago CU, Lo HH, Sedensky MM, Lunceford A, Clarke CF, Wu SJ, McLeod C, Finkel T, Morgan PG, Mills EM. Caenorhabditis elegans UCP4 protein controls complex II-mediated oxidative phosphorylation through succinate transport. J Biol Chem. 2011;286:37712–37720. doi: 10.1074/jbc.M111.271452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ali SS, Hsiao M, Zhao HW, Dugan LL, Haddad GG, Zhou D. Hypoxia-adaptation involves mitochondrial metabolic depression and decreased ROS leakage. PLoS One. 2012;7:e36801. doi: 10.1371/journal.pone.0036801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu SS, Porter GA., Jr The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev Cell. 2011;21:469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 95.Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 97.Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–810. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- 99.Valen G, Starkopf J, Takeshima S, Kullisaar T, Vihalemm T, Kengsepp AT, Lowbeer C, Vaage J, Zilmer M. Preconditioning with hydrogen peroxide (H2O2) or ischemia in H2O2-induced cardiac dysfunction. Free Radic Res. 1998;29:235–245. doi: 10.1080/10715769800300271. [DOI] [PubMed] [Google Scholar]
- 100.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 101.Pasdois P, Beauvoit B, Tariosse L, Vinassa B, Bonoron-Adele S, Santos PD. MitoK(ATP)-dependent changes in mitochondrial volume and in complex II activity during ischemic and pharmacological preconditioning of Langendorff-perfused rat heart. J Bioenerg Biomembr. 2006;38:101–112. doi: 10.1007/s10863-006-9016-3. [DOI] [PubMed] [Google Scholar]
- 102.Wojtovich AP, Brookes PS. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: Implications for ischemic preconditioning. Biochim Biophys Acta. 2008;1777:882–889. doi: 10.1016/j.bbabio.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fedotcheva NI, Sokolov AP, Kondrashova MN. Nonezymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med. 2006;41:56–64. doi: 10.1016/j.freeradbiomed.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 104.Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim YS. Malonate metabolism: Biochemistry, molecular biology, physiology, and industrial application. Journal of Biochemistry and Molecular Biology. 2002;35:443–451. doi: 10.5483/bmbrep.2002.35.5.443. [DOI] [PubMed] [Google Scholar]
- 106.Nadtochiy SM, Baker PR, Freeman BA, Brookes PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovasc Res. 2009;82:333–340. doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schopfer FJ, Batthyany C, Baker PR, Bonacci G, Cole MP, Rudolph V, Groeger AL, Rudolph TK, Nadtochiy S, Brookes PS, Freeman BA. Detection and quantification of protein adduction by electrophilic fatty acids: mitochondrial generation of fatty acid nitroalkene derivatives. Free Radic Biol Med. 2009;46:1250–1259. doi: 10.1016/j.freeradbiomed.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Queliconi BB, Wojtovich AP, Nadtochiy SM, Kowaltowski AJ, Brookes PS. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys Acta. 2011;1813:1309–1315. doi: 10.1016/j.bbamcr.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shiva S, Crawford JH, Ramachandran A, Ceaser EK, Hillson T, Brookes PS, Patel RP, Darley-Usmar VM. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem J. 2004;379:359–366. doi: 10.1042/BJ20031758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 111.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial Complex II in the Post-ischemic Heart: Oxidative Injury and the Role of Protein S-glutathionylation. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 112.Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, Ludolph AC, Dirnagl U, Hugon J. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning”. J Cereb Blood Flow Metab. 1997;17:257–264. doi: 10.1097/00004647-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 113.Ockaili RA, Bhargava P, Kukreja RC. Chemical preconditioning with 3-nitropropionic acid in hearts: role of mitochondrial K(ATP) channel. Am J Physiol Heart Circ Physiol. 2001;280:H2406–H2411. doi: 10.1152/ajpheart.2001.280.5.H2406. [DOI] [PubMed] [Google Scholar]
- 114.Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, Miyoshi H, Osanai A, Kita K, Omura S. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase) Proc Natl Acad Sci U S A. 2003;100:473–477. doi: 10.1073/pnas.0237315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 116.Mizumura T, Nithipatikom K, Gross GJ. Bimakalim, an ATP-sensitive potassium channel opener, mimics the effects of ischemic preconditioning to reduce infarct size, adenosine release, and neutrophil function in dogs. Circulation. 1995;92:1236–1245. doi: 10.1161/01.cir.92.5.1236. [DOI] [PubMed] [Google Scholar]
- 117.Auchampach JA, Grover GJ, Gross GJ. Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovasc Res. 1992;26:1054–1062. doi: 10.1093/cvr/26.11.1054. [DOI] [PubMed] [Google Scholar]
- 118.Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res. 1992;70:223–233. doi: 10.1161/01.res.70.2.223. [DOI] [PubMed] [Google Scholar]
- 119.Hide EJ, Thiemermann C. Limitation of myocardial infarct size in the rabbit by ischaemic preconditioning is abolished by sodium 5-hydroxydecanoate. Cardiovasc Res. 1996;31:941–946. [PubMed] [Google Scholar]
- 120.Garlid KD, Paucek P. Mitochondrial potassium transport: the K(+) cycle. Biochim Biophys Acta. 2003;1606:23–41. doi: 10.1016/s0005-2728(03)00108-7. [DOI] [PubMed] [Google Scholar]
- 121.Halestrap AP. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim Biophys Acta. 1989;973:355–382. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- 122.Costa AD, Quinlan CL, Andrukhiv A, West IC, Jaburek M, Garlid KD. The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am J Physiol Heart Circ Physiol. 2006;290:H406–H415. doi: 10.1152/ajpheart.00794.2005. [DOI] [PubMed] [Google Scholar]
- 123.da Silva MM, Sartori A, Belisle E, Kowaltowski AJ. Ischemic preconditioning inhibits mitochondrial respiration, increases H2O2 release, and enhances K+ transport. Am J Physiol Heart Circ Physiol. 2003;285:H154–H162. doi: 10.1152/ajpheart.00955.2002. [DOI] [PubMed] [Google Scholar]
- 124.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 125.Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- 126.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guda C, Guda P, Fahy E, Subramaniam S. MITOPRED: a web server for the prediction of mitochondrial proteins. Nucleic Acids Res. 2004;32:W372–W374. doi: 10.1093/nar/gkh374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Balss J, Papatheodorou P, Mehmel M, Baumeister D, Hertel B, Delaroque N, Chatelain FC, Minor DL, Jr, Van Etten JL, Rassow J, Moroni A, Thiel G. Transmembrane domain length of viral K+ channels is a signal for mitochondria targeting. Proc Natl Acad Sci U S A. 2008;105:12313–12318. doi: 10.1073/pnas.0805709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Anandatheerthavarada HK, Biswas G, Mullick J, Sepuri NB, Otvos L, Pain D, Avadhani NG. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic AMP-dependent phosphorylation at ser128. EMBO J. 1999;18:5494–5504. doi: 10.1093/emboj/18.20.5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Suzuki M, Kotake K, Fujikura K, Inagaki N, Suzuki T, Gonoi T, Seino S, Takata K. Kir6.1: a possible subunit of ATP-sensitive K+ channels in mitochondria. Biochem Biophys Res Commun. 1997;241:693–697. doi: 10.1006/bbrc.1997.7891. [DOI] [PubMed] [Google Scholar]
- 131.Zhou M, Tanaka O, Sekiguchi M, Sakabe K, Anzai M, Izumida I, Inoue T, Kawahara K, Abe H. Localization of the ATP-sensitive potassium channel subunit (Kir6. 1/uK(ATP)-1) in rat brain. Brain Res Mol Brain Res. 1999;74:15–25. doi: 10.1016/s0169-328x(99)00232-6. [DOI] [PubMed] [Google Scholar]
- 132.Zhou M, He HJ, Suzuki R, Tanaka O, Sekiguchi M, Yasuoka Y, Kawahara K, Itoh H, Abe H. Expression of ATP sensitive K+ channel subunit Kir6.1 in rat kidney. Eur J Histochem. 2007;51:43–51. [PubMed] [Google Scholar]
- 133.Jiang MT, Ljubkovic M, Nakae Y, Shi Y, Kwok WM, Stowe DF, Bosnjak ZJ. Characterization of human cardiac mitochondrial ATP-sensitive potassium channel and its regulation by phorbol ester in vitro. Am J Physiol Heart Circ Physiol. 2006;290:H1770–H1776. doi: 10.1152/ajpheart.01084.2005. [DOI] [PubMed] [Google Scholar]
- 134.Singh H, Hudman D, Lawrence CL, Rainbow RD, Lodwick D, Norman RI. Distribution of Kir6.0 and SUR2 ATP-sensitive potassium channel subunits in isolated ventricular myocytes. J Mol Cell Cardiol. 2003;35:445–459. doi: 10.1016/s0022-2828(03)00041-5. [DOI] [PubMed] [Google Scholar]
- 135.Lacza Z, Snipes JA, Miller AW, Szabo C, Grover G, Busija DW. Heart mitochondria contain functional ATP-dependent K+ channels. J Mol Cell Cardiol. 2003;35:1339–1347. doi: 10.1016/s0022-2828(03)00249-9. [DOI] [PubMed] [Google Scholar]
- 136.Lacza Z, Snipes JA, Kis B, Szabo C, Grover G, Busija DW. Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Res. 2003;994:27–36. doi: 10.1016/j.brainres.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 137.Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci U S A. 2004;101:11880–11885. doi: 10.1073/pnas.0401703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Foster DB, Rucker JJ, Marban E. Is Kir6.1 a subunit of mitoK(ATP)? Biochem Biophys Res Commun. 2008;366:649–656. doi: 10.1016/j.bbrc.2007.11.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ye B, Kroboth SL, Pu JL, Sims JJ, Aggarwal NT, McNally EM, Makielski JC, Shi NQ. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ Res. 2009;105:1083–1093. doi: 10.1161/CIRCRESAHA.109.195040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, Koseki H, Iwanaga T, Nakaya H, Seino S. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- 141.Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, Berggren PO, Magnuson MA. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277:37176–37183. doi: 10.1074/jbc.M206757200. [DOI] [PubMed] [Google Scholar]
- 142.Chutkow WA, Samuel V, Hansen PA, Pu J, Valdivia CR, Makielski JC, Burant CF. Disruption of Sur2-containing K(ATP) channels enhances insulin-stimulated glucose uptake in skeletal muscle. Proc Natl Acad Sci U S A. 2001;98:11760–11764. doi: 10.1073/pnas.201390398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chutkow WA, Pu J, Wheeler MT, Wada T, Makielski JC, Burant CF, McNally EM. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 K(ATP) channels. J Clin Invest. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kersten JR, Toller WG, Gross ER, Pagel PS, Warltier DC. Diabetes abolishes ischemic preconditioning: role of glucose, insulin, and osmolality. Am J Physiol Heart Circ Physiol. 2000;278:H1218–H1224. doi: 10.1152/ajpheart.2000.278.4.H1218. [DOI] [PubMed] [Google Scholar]
- 145.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A. 1998;95:10402–10406. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Seino S, Marban E, Nakaya H. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest. 2002;109:509–516. doi: 10.1172/JCI14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Seharaseyon J, Ohler A, Sasaki N, Fraser H, Sato T, Johns DC, O’Rourke B, Marban E. Molecular composition of mitochondrial ATP-sensitive potassium channels probed by viral Kir gene transfer. J Mol Cell Cardiol. 2000;32:1923–1930. doi: 10.1006/jmcc.2000.1226. [DOI] [PubMed] [Google Scholar]
- 148.Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, Garlid KD, O’Rourke B. The Mitochondrial ROMK Channel is a Molecular Component of MitoKATP. Circ Res. 2012;111:446–454. doi: 10.1161/CIRCRESAHA.112.266445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McNicholas CM, Yang Y, Giebisch G, Hebert SC. Molecular site for nucleotide binding on an ATP-sensitive renal K+ channel (ROMK2) Am J Physiol. 1996;271:F275–F285. doi: 10.1152/ajprenal.1996.271.2.F275. [DOI] [PubMed] [Google Scholar]
- 150.Mironova GD, Negoda AE, Marinov BS, Paucek P, Costa AD, Grigoriev SM, Skarga YY, Garlid KD. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR) J Biol Chem. 2004;279:32562–32568. doi: 10.1074/jbc.M401115200. [DOI] [PubMed] [Google Scholar]
- 151.Wojtovich AP, Williams DM, Karcz MK, Lopes CM, Gray DA, Nehrke KW, Brookes PS. A Novel Mitochondrial KATP Channel Assay. Circ Res. 2010;106:1190–1196. doi: 10.1161/CIRCRESAHA.109.215400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac) Br J Pharmacol. 2003;138:1119–1128. doi: 10.1038/sj.bjp.0705172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kobayashi T, Washiyama K, Ikeda K. Modulators of G protein-activated inwardly rectifying K+ channels: potentially therapeutic agents for addictive drug users. Ann N Y Acad Sci. 2004;1025:590–594. doi: 10.1196/annals.1316.073. [DOI] [PubMed] [Google Scholar]
- 154.Wojtovich AP, DiStefano P, Sherman T, Brookes PS, Nehrke K. Mitochondrial ATP-sensitive potassium channel activity and hypoxic preconditioning are independent of an inwardly rectifying potassium channel subunit in Caenorhabditis elegans. FEBS Lett. 2012;586:428–434. doi: 10.1016/j.febslet.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Szewczyk A, Kajma A, Malinska D, Wrzosek A, Bednarczyk P, Zablocka B, Dolowy K. Pharmacology of mitochondrial potassium channels: dark side of the field. FEBS Lett. 2010;584:2063–2069. doi: 10.1016/j.febslet.2010.02.048. [DOI] [PubMed] [Google Scholar]
- 156.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 157.Szewczyk A, Wójcik G, Lobanov NA, Nalecz MJ. The Mitochondrial Sulfonylurea Receptor: Identification and Characterization. Biochemical and Biophysical Research Communications. 1997;230:611–615. doi: 10.1006/bbrc.1996.6023. [DOI] [PubMed] [Google Scholar]
- 158.Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J Biol Chem. 2001;276:33369–33374. doi: 10.1074/jbc.M103320200. [DOI] [PubMed] [Google Scholar]
- 159.Jaburek M, Yarov-Yarovoy V, Paucek P, Garlid KD. State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J Biol Chem. 1998;273:13578–13582. [PubMed] [Google Scholar]
- 160.Szewczyk A, Pikula S, Wojcik G, Nalecz MJ. Glibenclamide inhibits mitochondrial K+ and Na+ uniports induced by magnesium depletion. Int J Biochem Cell Biol. 1996;28:863–871. doi: 10.1016/1357-2725(96)00040-4. [DOI] [PubMed] [Google Scholar]
- 161.Ziemys A, Toleikis A, Kopustinskiene DM. Molecular modelling of K(ATP) channel blockers-ADP/ ATP carrier interactions. Syst Biol (Stevenage) 2006;153:390–393. doi: 10.1049/ip-syb:20060007. [DOI] [PubMed] [Google Scholar]
- 162.Gutman M, Kearney EB, Singer TP. Control of succinate dehydrogenase in mitochondria. Biochemistry. 1971;10:4763–4770. doi: 10.1021/bi00801a025. [DOI] [PubMed] [Google Scholar]
- 163.Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Briere JJ, Favier J, El GV, Djouadi F, Benit P, Gimenez AP, Rustin P. Succinate dehydrogenase deficiency in human. Cell Mol Life Sci. 2005;62:2317–2324. doi: 10.1007/s00018-005-5237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Comelli M, Metelli G, Mavelli I. Downmodulation of mitochondrial F0F1 ATP synthase by diazoxide in cardiac myoblasts: a dual effect of the drug. Am J Physiol Heart Circ Physiol. 2007;292:H820–H829. doi: 10.1152/ajpheart.00366.2006. [DOI] [PubMed] [Google Scholar]
- 166.Kopustinskiene DM, Toleikis A, Saris NE. Adenine nucleotide translocase mediates the K(ATP)-channel-openers-induced proton and potassium flux to the mitochondrial matrix. J Bioenerg Biomembr. 2003;35:141–148. doi: 10.1023/a:1023746103401. [DOI] [PubMed] [Google Scholar]
- 167.Panov A, Filippova S, Lyakhovich V. Adenine nucleotide translocase as a site of regulation by ADP of the rat liver mitochondria permeability to H+ and K+ ions. Arch Biochem Biophys. 1980;199:420–426. doi: 10.1016/0003-9861(80)90298-2. [DOI] [PubMed] [Google Scholar]
- 168.Ziemys A, Toleikis A, Kopustinskiene DM. Molecular modelling of K(ATP) channel blockers-ADP/ ATP carrier interactions. Syst Biol (Stevenage) 2006;153:390–393. doi: 10.1049/ip-syb:20060007. [DOI] [PubMed] [Google Scholar]
- 169.Wojtovich AP, Nehrke KW, Brookes PS. The mitochondrial complex II and ATP-sensitive potassium channel interaction: quantitation of the channel in heart mitochondria. Acta Biochim Pol. 2010;57:431–434. [PMC free article] [PubMed] [Google Scholar]
- 170.Dzeja PP, Bast P, Ozcan C, Valverde A, Holmuhamedov EL, Van Wylen DG, Terzic A. Targeting nucleotide-requiring enzymes: implications for diazoxide-induced cardioprotection. Am J Physiol Heart Circ Physiol. 2003;284:H1048–H1056. doi: 10.1152/ajpheart.00847.2002. [DOI] [PubMed] [Google Scholar]
- 171.Schafer G, Wegener C, Portenhauser R, Bojanovski D. Diazoxide, an inhibitor of succinate oxidation. Biochem Pharmacol. 1969;18:2678–2681. [PubMed] [Google Scholar]
- 172.Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K(+) channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H649–H657. doi: 10.1152/ajpheart.2001.280.2.H649. [DOI] [PubMed] [Google Scholar]
- 173.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012 doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Drose S, Bleier L, Brandt U. A common mechanism links differently acting complex II inhibitors to cardioprotection: modulation of mitochondrial reactive oxygen species production. Mol Pharmacol. 2011;79:814–822. doi: 10.1124/mol.110.070342. [DOI] [PubMed] [Google Scholar]
- 175.Ksenzenko M, Konstantinov AA, Khomutov GB, Tikhonov AN, Ruuge EK. Effect of electron transfer inhibitors on superoxide generation in the cytochrome bc1 site of the mitochondrial respiratory chain. FEBS Lett. 1983;155:19–24. doi: 10.1016/0014-5793(83)80200-2. [DOI] [PubMed] [Google Scholar]
- 176.Ksenzenko M, Konstantinov AA, Khomutov GB, Tikhonov AN, Ruuge EK. Relationships between the effects of redox potential, alpha-thenoyltrifluoroacetone and malonate on O(2) and H2O2 generation by submitochondrial particles in the presence of succinate and antimycin. FEBS Lett. 1984;175:105–108. doi: 10.1016/0014-5793(84)80579-7. [DOI] [PubMed] [Google Scholar]
- 177.Malinska D, Mirandola SR, Kunz WS. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010;584:2043–2048. doi: 10.1016/j.febslet.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 178.Drose S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim Biophys Acta. 2009;1790:558–565. doi: 10.1016/j.bbagen.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 179.Ardehali H, O’Rourke B. Mitochondrial K(ATP) channels in cell survival and death. J Mol Cell Cardiol. 2005;39:7–16. doi: 10.1016/j.yjmcc.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 181.Guillet V, Gueguen N, Cartoni R, Chevrollier A, Desquiret V, Angebault C, mati-Bonneau P, Procaccio V, Bonneau D, Martinou JC, Reynier P. Bioenergetic defect associated with mKATP channel opening in a mouse model carrying a mitofusin 2 mutation. FASEB J. 2011;25:1618–1627. doi: 10.1096/fj.10-173609. [DOI] [PubMed] [Google Scholar]
- 182.Facundo HT, Fornazari M, Kowaltowski AJ. Tissue protection mediated by mitochondrial K+ channels. Biochim Biophys Acta. 2006;1762:202–212. doi: 10.1016/j.bbadis.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 183.Costa ADT, Krieger MA. Evidence for an ATP-sensitive K+ channel in mitoplasts isolated from Trypanosoma cruzi and Crithidia fasciculata. International Journal for Parasitology. 2009;39:955–961. doi: 10.1016/j.ijpara.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kicinska A, Swida A, Bednarczyk P, Koszela-Piotrowska I, Choma K, Dolowy K, Szewczyk A, Jarmuszkiewicz W. ATP-sensitive potassium channel in mitochondria of the eukaryotic microorganism Acanthamoeba castellanii. J Biol Chem. 2007;282:17433–17441. doi: 10.1074/jbc.M701496200. [DOI] [PubMed] [Google Scholar]
- 185.Wojtovich AP, Burwell LS, Sherman TA, Nehrke KW, Brookes PS. The C. elegans mitochondrial K+(ATP) channel: a potential target for preconditioning. Biochem Biophys Res Commun. 2008;376:625–628. doi: 10.1016/j.bbrc.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pastore D, Stoppelli MC, Di FN, Passarella S. The existence of the K(+) channel in plant mitochondria. J Biol Chem. 1999;274:26683–26690. doi: 10.1074/jbc.274.38.26683. [DOI] [PubMed] [Google Scholar]
- 187.Baltzer C, Tiefenbock SK, Frei C. Mitochondria in response to nutrients and nutrient-sensitive pathways. Mitochondrion. 2010;10:589–597. doi: 10.1016/j.mito.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 188.Finley LW, Haigis MC. Metabolic regulation by SIRT3: implications for tumorigenesis. Trends Mol Med. 2012 doi: 10.1016/j.molmed.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. 2009;8:604–606. doi: 10.1111/j.1474-9726.2009.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Finley LW, Haas W, squiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One. 2011;6:e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dos Santos P, Kowaltowski AJ, Laclau MN, Seetharaman S, Paucek P, Boudina S, Thambo JB, Tariosse L, Garlid KD. Mechanisms by which opening the mitochondrial ATP- sensitive K(+) channel protects the ischemic heart. Am J Physiol Heart Circ Physiol. 2002;283:H284–H295. doi: 10.1152/ajpheart.00034.2002. [DOI] [PubMed] [Google Scholar]
- 195.Alberici LC, Oliveira HC, Patricio PR, Kowaltowski AJ, Vercesi AE. Hyperlipidemic mice present enhanced catabolism and higher mitochondrial ATP-sensitive K+ channel activity. Gastroenterology. 2006;131:1228–1234. doi: 10.1053/j.gastro.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 196.Halestrap AP. The regulation of the oxidation of fatty acids and other substrates in rat heart mitochondria by changes in the matrix volume induced by osmotic strength, valinomycin and Ca2+ Biochem J. 1987;244:159–164. doi: 10.1042/bj2440159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Guzun R, Timohhina N, Tepp K, Gonzalez-Granillo M, Shevchuk I, Chekulayev V, Kuznetsov AV, Kaambre T, Saks VA. Systems bioenergetics of creatine kinase networks: physiological roles of creatine and phosphocreatine in regulation of cardiac cell function. Amino Acids. 2011;40:1333–1348. doi: 10.1007/s00726-011-0854-x. [DOI] [PubMed] [Google Scholar]
- 198.Garlid KD, Dos SP, Xie ZJ, Costa AD, Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim Biophys Acta. 2003;1606:1–21. doi: 10.1016/s0005-2728(03)00109-9. [DOI] [PubMed] [Google Scholar]
- 199.Laclau MN, Boudina S, Thambo JB, Tariosse L, Gouverneur G, Bonoron-Adele S, Saks VA, Garlid KD, Dos SP. Cardioprotection by ischemic preconditioning preserves mitochondrial function and functional coupling between adenine nucleotide translocase and creatine kinase. J Mol Cell Cardiol. 2001;33:947–956. doi: 10.1006/jmcc.2001.1357. [DOI] [PubMed] [Google Scholar]
- 200.Rouslin W, Broge CW, Guerrieri F, Capozza G. ATPase activity, IF1 content, and proton conductivity of ESMP from control and ischemic slow and fast heart-rate hearts. J Bioenerg Biomembr. 1995;27:459–466. doi: 10.1007/BF02110008. [DOI] [PubMed] [Google Scholar]
- 201.Cartoni R, Martinou JC. Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol. 2009;218:268–273. doi: 10.1016/j.expneurol.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 202.Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 203.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.De SD, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lorenz JN, Baird NR, Judd LM, Noonan WT, Andringa A, Doetschman T, Manning PA, Liu LH, Miller ML, Shull GE. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome. J Biol Chem. 2002;277:37871–37880. doi: 10.1074/jbc.M205627200. [DOI] [PubMed] [Google Scholar]
- 206.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, Fallen K, Lindsley CW, Weaver CD, Denton JS. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol. 2009;76:1094–1103. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bayley JP, van MI, Hogendoorn PC, Cornelisse CJ, van der WA, Prins FA, Teppema L, Dahan A, Devilee P, Taschner PE. Sdhd and SDHD/H19 knockout mice do not develop paraganglioma or pheochromocytoma. PLoS One. 2009;4:e7987. doi: 10.1371/journal.pone.0007987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Puissegur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, Maurin T, Lebrigand K, Cardinaud B, Hofman V, Fourre S, Magnone V, Ricci JE, Pouyssegur J, Gounon P, Hofman P, Barbry P, Mari B. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011;18:465–478. doi: 10.1038/cdd.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Levitas A, Muhammad E, Harel G, Saada A, Caspi VC, Manor E, Beck JC, Sheffield V, Parvari R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet. 2010;18:1160–1165. doi: 10.1038/ejhg.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pagnamenta AT, Hargreaves IP, Duncan AJ, Taanman JW, Heales SJ, Land JM, Bitner-Glindzicz M, Leonard JV, Rahman S. Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol Genet Metab. 2006;89:214–221. doi: 10.1016/j.ymgme.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 211.Parfait B, Chretien D, Rotig A, Marsac C, Munnich A, Rustin P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Genet. 2000;106:236–243. doi: 10.1007/s004390051033. [DOI] [PubMed] [Google Scholar]
- 212.Van CR, Seneca S, Smet J, Van HR, Gerlo E, Devreese B, Van BJ, Leroy JG, De ML, Lissens W. Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. Am J Med Genet A. 2003;120A:13–18. doi: 10.1002/ajmg.a.10202. [DOI] [PubMed] [Google Scholar]
- 213.Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo AP. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Leckschat S, Ream-Robinson D, Scheffler IE. The gene for the iron sulfur protein of succinate dehydrogenase (SDH-IP) maps to human chromosome 1p35–36.1. Somat Cell Mol Genet. 1993;19:505–511. doi: 10.1007/BF01233256. [DOI] [PubMed] [Google Scholar]
- 215.Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, O’Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–1056. doi: 10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- 218.Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E, Chompret A, Ellison JW, Briere JJ, Rustin P, Gimenez-Roqueplo AP, Eng C, Carney JA, Stratakis CA. Clinical molecular genetics of patients with the Carney-Stratakis syndrome germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 219.Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, Pacak K. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 220.Cascon A, Montero-Conde C, Ruiz-Llorente S, Mercadillo F, Leton R, Rodriguez-Antona C, Martinez-Delgado B, Delgado M, Diez A, Rovira A, Diaz JA, Robledo M. Gross SDHB deletions in patients with paraganglioma detected by multiplex PCR: a possible hot spot? Genes Chromosomes Cancer. 2006;45:213–219. doi: 10.1002/gcc.20283. [DOI] [PubMed] [Google Scholar]
- 221.Lima J, Feijao T, Ferreira da SA, Pereira-Castro I, Fernandez-Ballester G, Maximo V, Herrero A, Serrano L, Sobrinho-Simoes M, Garcia-Rostan G. High frequency of germline succinate dehydrogenase mutations in sporadic cervical paragangliomas in northern Spain: mitochondrial succinate dehydrogenase structure-function relationships and clinical-pathological correlations. J Clin Endocrinol Metab. 2007;92:4853–4864. doi: 10.1210/jc.2007-0640. [DOI] [PubMed] [Google Scholar]
- 222.Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Young AL, Baysal BE, Deb A, Young WF., Jr Familial malignant catecholamine-secreting paraganglioma with prolonged survival associated with mutation in the succinate dehydrogenase B gene. J Clin Endocrinol Metab. 2002;87:4101–4105. doi: 10.1210/jc.2002-020312. [DOI] [PubMed] [Google Scholar]
- 224.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 225.Baysal BE, Willett-Brozick JE, Filho PA, Lawrence EC, Myers EN, Ferrell RE. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J Med Genet. 2004;41:703–709. doi: 10.1136/jmg.2004.019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hirawake H, Taniwaki M, Tamura A, Kojima S, Kita K. Cytochrome b in human complex II (succinate-ubiquinone oxidoreductase): cDNA cloning of the components in liver mitochondria and chromosome assignment of the genes for the large (SDHC) and small (SDHD) subunits to 1q21 and 11q23. Cytogenet Cell Genet. 1997;79:132–138. doi: 10.1159/000134700. [DOI] [PubMed] [Google Scholar]
- 227.Kytola S, Nord B, Elder EE, Carling T, Kjellman M, Cedermark B, Juhlin C, Hoog A, Isola J, Larsson C. Alterations of the SDHD gene locus in midgut carcinoids, Merkel cell carcinomas, pheochromocytomas, and abdominal paragangliomas. Genes Chromosomes Cancer. 2002;34:325–332. doi: 10.1002/gcc.10081. [DOI] [PubMed] [Google Scholar]
- 228.Badenhop RF, Cherian S, Lord RS, Baysal BE, Taschner PE, Schofield PR. Novel mutations in the SDHD gene in pedigrees with familial carotid body paraganglioma and sensorineural hearing loss. Genes Chromosomes Cancer. 2001;31:255–263. doi: 10.1002/gcc.1142. [DOI] [PubMed] [Google Scholar]
- 229.Cascon A, Ruiz-Llorente S, Cebrian A, Telleria D, Rivero JC, Diez JJ, Lopez-Ibarra PJ, Jaunsolo MA, Benitez J, Robledo M. Identification of novel SDHD mutations in patients with phaeochromocytoma and/or paraganglioma. Eur J Hum Genet. 2002;10:457–461. doi: 10.1038/sj.ejhg.5200829. [DOI] [PubMed] [Google Scholar]
- 230.Hensen EF, Jordanova ES, van MI, Hogendoorn PC, Taschner PE, van Der Mey AG, Devilee P, Cornelisse CJ. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004;23:4076–4083. doi: 10.1038/sj.onc.1207591. [DOI] [PubMed] [Google Scholar]
- 231.McWhinney SR, Pilarski RT, Forrester SR, Schneider MC, Sarquis MM, Dias EP, Eng C. Large germline deletions of mitochondrial complex II subunits SDHB and SDHD in hereditary paraganglioma. J Clin Endocrinol Metab. 2004;89:5694–5699. doi: 10.1210/jc.2004-0769. [DOI] [PubMed] [Google Scholar]
- 232.Taschner PE, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Brocker-Vriends AH, van Der Mey AG, van Ommen GJ, Cornelisse CJ, Devilee P. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer. 2001;31:274–281. doi: 10.1002/gcc.1144. [DOI] [PubMed] [Google Scholar]
- 233.Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res. 2000;60:6822–6825. [PubMed] [Google Scholar]
- 234.Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H, D’Adamo P, Gasparini P, Strom TM, Prokisch H, Invernizzi F, Ferrero I, Zeviani M. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41:654–656. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- 235.Hensen EF, van DN, Jansen JC, Corssmit EP, Tops CM, Romijn JA, Vriends AH, van Der Mey AG, Cornelisse CJ, Devilee P, Bayley JP. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin Genet. 2012;81:284–288. doi: 10.1111/j.1399-0004.2011.01653.x. [DOI] [PubMed] [Google Scholar]
- 236.Alston TA, Mela LHJ. Bright, 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci U S A. 1977;74:3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Coles CJ, Edmondson DE, Singer TP. Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem. 1979;254:5161–5167. [PubMed] [Google Scholar]
- 238.Dervartanian DV, Veeger C. Studies on succinate dehydrogenase. I. Spectral properties of the purified enzyme and formation of enzyme-competitive inhibitor complexes. Biochim Biophys Acta. 1964;92:233–247. [PubMed] [Google Scholar]
- 239.Kearney EB, Ackrell BA, Mayr M. Tightly bound oxalacetate and the activation of succinate dehydrogenase. Biochem Biophys Res Commun. 1972;49:1115–1121. doi: 10.1016/0006-291x(72)90328-2. [DOI] [PubMed] [Google Scholar]