

Abstract
Yeast cell lysates produced by mechanical glass bead disruption are widely used in a variety of applications, including for the analysis of native function, e.g. protein–protein interaction, enzyme assays and membrane fractionations. Below, we report a striking case of protein denaturation and aggregation that is induced by this lysis protocol. Most of this analysis focuses on the type 1 casein kinase Yck2, which normally tethers to the plasma membrane through C-terminal palmitoylation. Surprisingly, when cells are subjected to glass bead disruption, non-palmitoylated, cytosolic forms of the kinase denature and aggregate, while membrane-associated forms, whether attached through their native palmitoyl tethers or through a variety of artificial membrane-tethering sequences, are wholly protected from denaturation and aggregation. A wider look at the yeast proteome finds that, while the majority of proteins resist glass bead-induced aggregation, a significant subset does, in fact, succumb to such denaturation. Thus, yeast researchers should be aware of this potential artifact when embarking on biochemical analyses that employ glass bead lysates to look at native protein function. Finally, we demonstrate an experimental utility for glass bead-induced aggregation, using its fine discrimination of membrane-associated from non-associated Yck2 forms to discern fractional palmitoylation states of Yck2 mutants that are partially defective for palmitoylation.
Keywords: protein aggregation, protein denaturation, protein acylation, Yck2, yeast, cell lysis
Introduction
The tough exterior yeast cell wall presents a substantial obstacle for the biochemist, with access to cellular contents gained only through enzymatic cell wall digestion or through mechanical disruption. For enzymatic digestion, cells are incubated with digestive enzymes for a substantial period of time (e.g. 15–60 min) at temperatures in the range of 25–37 °C, i.e. conditions compatible with continued biological activity. Thus, the enzymatic approach is not well suited to analyses where one wants to acquire a quick ‘snapshot’ of intracellular processes. For mechanical disruption, the most widely used approach involves the vigorous agitation of the cells in the cold with small glass beads in the 100–600 μm diameter range. Yeast cell lysates produced by such glass bead disruption are widely used for a variety of applications, including the analysis of native protein function — for enzyme assays, subcellular fractionations and for analysing protein complexes. Below, we report a case where such glass bead disruption of yeast cells leads to a troubling instance of protein aggregation, likely reflecting induced protein denaturation. Given the huge body of literature and the multitude of scientific advances that have relied upon this fundamental yeast method, it is obvious that for most yeast proteins, glass bead lysis-induced protein denaturation and aggregation is not a problem. Indeed, we find only a relatively small minority of yeast proteins to be susceptible to such induced aggregation. Nonetheless, yeast researchers should be alert to this potential artifact.
Most of the analysis below focuses on yeast type 1 casein kinase Yck2. Yck2, together with its close homologue Yck1, provides an essential yeast function (yck1Δ yck2Δ cells are nonviable). The two kinases tether to the cytoplasmic surface of the plasma membrane through dual palmitoylation of the C-terminal Cys–Cys dipeptide (Babu et al., 2004; Roth et al., 2002), where they regulate a variety of cell surface processes, including endocytosis, cell morphogenesis, mRNA localization and nutrient sensing (Abdel-Sater et al., 2004; Feng and Davis, 2000; Hicke et al., 1998; Hwang and Varshavsky, 2008; Moriya and Johnston, 2004; Pal et al., 2008; Panek et al., 1997; Paquin et al., 2007; Robinson et al., 1993; Spielewoy et al., 2004). When palmitoylation is blocked, either through mutation of the C-terminal cysteines or through deletion of the gene encoding their cognate, Golgi-localized palmitoyl-transferase, Akr1, the kinases mislocalize to the cytoplasm (Babu et al., 2004; Roth et al., 2002). The present work starts with a finding of strikingly distinct fractionation phenotypes for palmitoylated Yck2 vs. non-palmitoylated Yck2 when assayed from glass bead-generated lysates. The palmitoylated, wild-type Yck2 behaves like a typical integral membrane protein, fractionating to the membrane pellet, but then to the supernatant with prior detergent treatment. Non-palmitoylated Yck2, we expected, would fractionate as a soluble cytoplasmic protein to the supernatant and thus we were surprised to find it fractionating instead to the pellet, in both the presence and absence of detergent. Additional analyses demonstrated this pellet fractionation to reflect a quantitative segregation of non-palmitoylated Yck2 into a high-molecular-weight protein aggregate.
Our interest in this aggregation of non-palmitoylated Yck2 was piqued by a recent publication connecting impaired palmitoylation to protein aggregation in Huntington’s disease (HD) (Yanai et al., 2006). Huntingtin, the disease protein of HD, is palmitoylated, and decreased huntingtin palmitoylation was shown to result in its increased aggregation. Thus, in light of our Yck2 results, we wondered if this link between impaired palmitoylation and aggregation might be more general. However, as we document below, rather than occurring within the living yeast cell, this aggregation of non-palmitoylated Yck2 occurs instead in vitro, being artifactually induced during the process of glass bead cell disruption, apparently a consequence of mechanical shear forces produced by the vortexing glass beads. Given the widespread use of glass bead lysis within the yeast community, we felt obliged to investigate this artifact more deeply and have found that glass bead-induced aggregation does indeed apply to other, but certainly not all, yeast proteins.
Materials and methods
Strains
Two wild-type yeast strains, namely the MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 strain BY4741 (Brachmann et al., 1998) and the MATa ura3-52 leu2 his3 strain LRB759 (Panek et al., 1997), were used interchangeably in this work, with identical results (LRB759 was used for all experiments except for those reported in Figures 1E, 3, 5 and 6). The akr1Δ strain used was NDY1405 (Roth et al., 2002), which is isogenic to LRB759 except for its unmarked akr1Δ allele.
Figure 1.

Non-palmitoylated Yck2 from glass bead lysates fractionates as a part of a high molecular-weight aggregate. Glass bead lysates prepared from either wild-type or akr1Δ yeast cells, expressing either Yck2(wt) or the palmitoylation-deficient Yck2(SS) mutant, were fractionated by centrifugation. The expressed Yck2 proteins were all N-terminally tagged with the 6× His/FLAG/HA tri-tag, allowing Western blot detection with anti-HA-HRP. (A) Lysates from cells expressing the indicated Yck2 proteins from GAL1 promoter (2 h of expression from CEN/ARS plasmid-borne alleles) were treated with a 30 min, 4 °C membrane solubilizing incubation with 1% Triton X-100 or a parallel mock solubilization (no detergent) to high-speed centrifugation, to yield the supernatant (S) or pellet (P) fractions, which were analysed by anti-HA Western blotting. (B) Glass bead lysates deriving from cells expressing Yck2(SS) from the GAL1 promoter were subjected to 30 min treatments with either 1% Triton X-100, 1 m NaCl, 10 mm DTT, 1% SDS or 6 m urea (incubations were at 4 °C except for SDS and DTT, which were at 23 °C), then fractionation by high-speed centrifugation and, finally, anti-HA Western blotting. (C) Lysates from cells expressing either Yck2(wt) or Yck2(SS) from the GAL1 promoter were treated with 1% Triton X-100 prior to sucrose gradient fractionation. Samples of each gradient fraction were analysed by anti-HA Western blotting. The positions of the endogenous 80S ribosome and added haemoglobin are indicated. (D) Detergent-treated lysates prepared from cells expressing either Yck2(wt) or Yck2(SS) from the YCK2 promoter were analysed as for (A), except that, in addition, samples taken just prior to the centrifugal fractionation were also analysed (Σ)
Figure 3.

Lysis by the mortar–pestle method yields soluble Yck2(SS). Yeast cells, expressing Yck2(wt) or Yck2(SS) from the GAL1 promoter, were lysed by the mortar–pestle method (see Materials and methods), then subsequently treated by Triton X-100 solubilization and centrifugal fractionation, as described for Figure 1A
Figure 5.
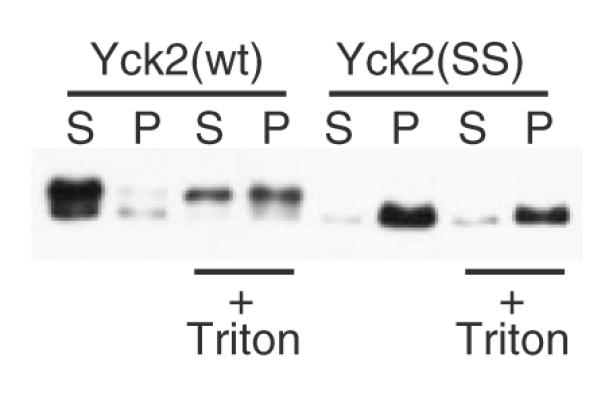
Inclusion of detergent during glass bead disruption reduces the protection from aggregation afforded by membrane association. Glass bead lysates were prepared from yeast cells expressing GAL1-driven Yck2(wt) or Yck2(SS), either by our typical protocol, which adds Triton X-100 subsequent to glass bead disruption, or by an alternative protocol, which included 1% Triton X-100 during the cell disruption step (+ Triton). Subsequent sample processing was as described for Figure 1A, except that no additional detergent was added during the membrane solubilization incubation for the ‘+ Triton’ samples
Figure 6.

Tests of the contribution of elevated temperature to glass bead-induced aggregation. (A) Comparison of the effects of elevated temperature to those of glass bead agitation on the aggregation of both Yck2(SS) (upper panel) and bulk yeast proteins (lower panel). A supernatant fraction, prepared by high-speed centrifugation (no Triton X-100 present) of a mortar–pestle lysate from yeast cells expressing GAL1-driven Yck2(SS), was subjected to a variety of treatments, including either a mock glass bead lysis (glass beads) or 10 min incubations at the indicated temperatures. The samples then were subjected to a second high-speed centrifugation yielding S100 and P100 fractions that were analysed either by anti-HA Western blotting [Yck2(SS), upper panel] or by SDS–PAGE and Coomassie staining (total protein, lower panel). Two proteins that show differential fractionation to the pellet from the glass bead-treated sample, identified by LC–MS/MS as both being translational elongation factors, namely the 116 kDa EF-3 (Yef3) and the 50 kDa EF-1A (Tef1/2), are indicated by asterisks (lower panel). (B) Lysates were prepared from yeast cells expressing GAL1-driven Yck2(wt) or Yck2(SS), either by our standard glass bead lysis protocol (i.e. five 45 s vortexings interspersed with 1 min ice-bath rests) or by a revised protocol intended to minimize heating (45 5 s vortexings interspersed with 1 min slushy ice-bath rests). Subsequent lysate processing and fractionation (i.e. Triton X-100 solubilization and centrifugation) was as described for Figure 1A
Plasmids
Many different plasmids were used in these studies (listed in Table 1). All yeast expression plasmids were based upon the URA3/CEN/ARS vector plasmid pRS316 (Sikorski and Hieter, 1989). Details of plasmid sequence and construction will be furnished upon request.
Table 1.
Plasmids
| Description | Laboratory designation |
Source |
|---|---|---|
| GAL1p-6×His/FLAG/HA/YCK2(wt) on pRS316 | pND1427 | (Roth et al., 2002) |
| GAL1p-6×His/FLAG/HA/YCK2(SS) on pRS316 | pND1446 | (Roth et al., 2002) |
| YCK2p-HA/YCK2(wt) on pRS316 | pND1515 | (Sun et al., 2004) |
| YCK2p-HA/YCK2(SS) on pRS316 | pND1907 | This study |
| T7p-6×His/HA/YCK2(SS) on pET30a (E. coli expression) | pND1397 | (Roth et al., 2002) |
| GAL1p-6×His/FLAG/HA/YCK2(CCIIS) on pRS316 | pND1432 | (Roth et al., 2002) |
| GAL1p-6×His/FLAG/HA/YCK2(SCIIS) on pRS316 | pND1447 | (Roth et al., 2002) |
| GAL1p-3×HA/YCK2(TMD) on pRS316 | pND1131 | This study |
| GAL1p-PSR1(1–20)/HA/YCK2(1–546,SS) on pRS316 | pND2167 | This study |
| GAL1p-PSR1(1–20,Δ2–10)/HA/YCK2(2–546,SS) on pRS316 | pND2189 | This study |
| GAL1p-6×His/HA/YCK1 on pRS316 | pND1345 | This study |
| GAL1p-6×His/HA/YCK1(SS) on pRS316 | pND1475 | This study |
| GAL1p-6×His/FLAG/HA/YCK3 on pRS316 | pND1547 | (Sun et al., 2004) |
| GAL1p-6×His/FLAG/HA/YCK3(ΔCys) on pRS316 | pND1799 | (Sun et al., 2004) |
| GAL1p-6×His/FLAG/HA/RAS2 on pRS316 | pND1463 | (Roth et al., 2006) |
| GAL1p-6×His/FLAG/HA/RAS2(Δ318–322) on pRS316 | pND2127 | This study |
| GAL1p-VAC8/3×HA/FLAG/6×His on pRS316 | pND2038 | This study |
| GAL1p-VAC8(Δ2–7)/3×HA/FLAG/6×His on pRS316 | pND2125 | This study |
| GAL1p-PSR1/3×HA/FLAG/6×His on pRS316 | pND2094 | (Roth et al., 2006) |
| GAL1p-PSR1(Δ2–10)/3×HA/FLAG/6×His on pRS316 | pND2126 | This study |
| GAL1p-6×His/FLAG/HA/LCB4 on pRS316 | pND2256 | This study |
| GAL1p-6×His/FLAG/HA/LCB4(C43S,C46S) on pRS316 | pND2307 | This study |
| GAL1p-6×His/FLAG/HA/YKL047W on pRS316 | pND2095 | (Roth et al., 2006) |
| GAL1p-6×His/FLAG/HA/YPL199C on pRS316 | pND2121 | (Roth et al., 2006) |
| GAL1p-YPL236C/3×HA/FLAG/6×His on pRS316 | pND2200 | (Roth et al., 2006) |
| GAL1p-6×His/FLAG/HA/YCK2(SC) on pRS316 | pND2631 | This study |
| GAL1p-6×His/FLAG/HA/YCK2(CS) on pRS316 | pND2632 | This study |
Plasmids for galactose-induced expression were based upon the GAL1p-6×HIS/FLAG/HA/Yck2(wt) construct pND1427 (Roth et al., 2002). A series of pND1427 derivatives that mutate the Yck2 C-terminal Cys–Cys palmitoyl acceptors to Ser–Ser [Yck2(SS)], to Cys–Cys–Ile–Ile–Ser [Yck2(CCIIS)] and to Ser–Cys–Ile–Ile–Ser [Yck2(SCIIS)] have been previously described (Roth et al., 2002). Additional C-terminal Yck2 mutants, constructed for the current work, include mutation of Cys–Cys to Ser–Cys [Yck2(SC)] and to Cys–Ser [Yck2-(CS)]. The Yck2(TMD) allele replaces the Yck2 C-terminal Cys–Cys with the C-terminal 31 residues of the plasma membrane v-SNARE Snc2 (including the C-terminal Snc2 TMD). Psr1–Yck2(SS) appends the N-terminal 20 residues from dually myristoylated and palmitoylated Psr1 to the N-terminus of Yck2(SS), with the Psr1 sequences separated from the Yck2 sequences by a 3 × HA tag. Psr1(Δ2–10)-Yck2(SS) is identical to Psr1–Yck2-(SS) except for its deletion of coding for Psr1 residues Gly2–Cys10, thus removing the putative Psr1 myristoyl and palmitoyl acceptors. Plasmids for expression from the YCK2 promoter utilized the 623 bp sequence immediately upstream of the YCK2 ORF, i.e. −623 to −1.
The testing of other yeast palmitoyl proteins (Figure 7) relied on GAL1-driven expression of epitope-tagged constructs carried on pRS316. For these constructs the epitope tags were positioned at either the N- or C-terminus (depending on the protein’s putative lipidation site location), as previously described (Roth et al., 2006).
Figure 7.

Testing other yeast palmitoyl-proteins for glass bead-induced aggregation. The indicated yeast palmitoyl proteins expressed from the GAL1 promoter both in their native lipidated state, as well as in a mutant, non-lipidated state, were assessed for potential aggregation in the glass bead lysis protocol described for Figure 1A. For most of these proteins (viz. Yck2, Yck1, Yck3, Ras2, Vac8, Psr1 and Lcb4), the wild-type, lipidated form was compared to a mutant version with the lipidation sites specifically ablated. These mutations were: for Yck2, C545S,C546S [i.e. Yck2(SS)]; for Yck1, C537S, C538S [i.e. Yck1(SS)]; for Yck3, Δ517-524 [truncation removing the eight C-terminal residues, including the seven putative palmitoyl-accepting cysteines (Sun et al., 2004); Yck3(ΔCys)]; for Ras2, Δ318–322 [truncation removing the five C-terminal residues, i.e. the dually palmitoylated and prenylated pentapeptide CCIIS (Deschenes and Broach, 1987), i.e. Ras2(Δ318–322)]; for Vac8, Δ2–7 [an in-frame deletion of Gly2 through Cys7; removes the myristoylation site as well as the three cysteinyl palmitoyl-acceptors (Peng et al., 2006; Subramanian et al., 2006), i.e. Vac8(Δ2–7)]; for Lcb4, C43S, C46S mutation of the two palmitoyl acceptors (Kihara et al., 2005) [i.e. Lcb4(C43S, C46S)]. In addition, for some of these palmitoyl proteins known to depend on Akr1 for their palmitoylation (viz. Lcb4, Ykl047w, Ypl199c and Ypl236c) (Kihara et al., 2005; Roth et al., 2006), the akr1Δ cell context was used to assess fractionation of the lipidation-minus form
For the Escherichia coli expression of Yck2(SS), a pET30a vector (EMD Biosciences) for high-level, T7 polymerase-driven expression was used.
Yeast cultures
Plasmid-transformed yeast were inoculated from selective plates, into either YP-Raf medium (1% yeast extract, 2% peptone, 2% raffinose) or YPD (1% yeast extract, 2% peptone, 2% glucose) for overnight log-phase growth. The next day, following appropriate culture dilution and an additional 2 h period of log-phase growth, cells were either directly harvested (cultures in YPD) or subjected to a 2 h galactose-induction period (addition of 2% galactose to YP-Raf cultures) prior to the collection of 4 × 108 cells (i.e. 20 A600 units) for cell lysis and fractionation.
Glass bead cell lysis, membrane solubilization and centrifugal fractionation
Cell pellets were resuspended in 0.3 ml ice-cold lysis buffer (LB; 150 mm NaCl, 50 mm Tris/Cl, 5 mm EDTA, pH 7.4) containing 2×PI (1×PI: 1 mm PMSF and 0.25 μg/ml each of antipain, leupeptin, pepstatin and chymostatin) and then transferred to 2 ml centrifuge tubes (Thermo-Fisher 02-681-344) that contained a 200 μl dry volume of 212–300 mm acid-washed glass beads (Sigma-Aldrich, G1277). The cells were then disrupted by five 45 s blasts of vigorous vortexing, interspersed with 2 min rests of the tube on ice (to minimize heating). Following the final vortexing, the beads were allowed to settle and the supernatant was decanted to a fresh tube. The glass beads were then washed by resuspension into an additional 0.2 ml LB containing 1×PI, with this supernatant being pooled together with the first. To solubilize cellular membranes, 400 μl of this lysate was adjusted to 1% Triton X-100 (Ana-trace APX100) and incubated for 30 min at 4 °C with rotation. Triton X-100 was omitted from this incubation when unsolubilized membranes were to be collected (Figure 1A). Prior to the high-speed, fractionating centrifugation, lysates were first subjected to a low-speed spin (30 s at 3000 × g) to remove unbroken cells. This supernatant then was diluted to 2.7 ml with LB containing 1×PI and 1% Triton X-100 and centrifuged at 200 000 × g for 30 min, yielding S100 and P100 fractions for analysis. Equal portions of the S100, P100 and the total sample (taken just prior to centrifugation) were subjected to SDS–PAGE and then Western blotting with anti-HA-HRP (Roche). Some experiments (Figures 1D, 4C, 8C, D) relied on a lower-speed spin (20 min, 15 000 × g) to fractionate the supernatant from the pellet.
Figure 4.

Membrane association affords protection from glass bead-induced denaturation. (A) Schematics for ectopically-tethered Yck2 mutants. (B) Mutant Yck2 protein intracellular localization analysed by anti-HA immunofluorescent analysis. The indicated Yck2 proteins were analysed following a 2 h period of GAL1-driven expression. (C) Fractionation of mutants in the glass bead lysis–centrifugation protocol. Cells expressing the indicated mutants were lysed, subjected to Triton X-100 solubilization, then fractionated as described for Figure 1A. Also tested was the lipidation-minus control version of Psr1–Yck2(SS), i.e. Psr1(Δ2-10)-Yck2(SS), which deletes Psr1 residues Gly2 through Cys10 (i.e. Δ2-10), a segment which includes the acceptor sites for both myristoylation and palmitoylation
Figure 8.
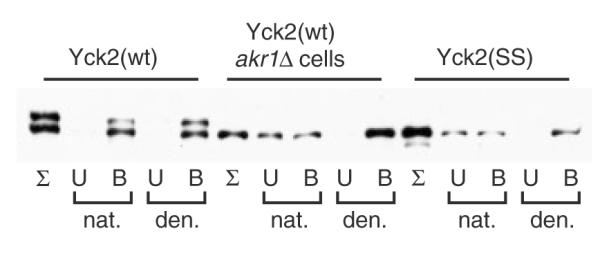
Yck2 immunoprecipitation from glass bead lysates. Glass bead lysates were prepared and Triton X-100-solubilized from cells expressing GAL1-driven Yck2(wt) or Yck2(SS), as described for Figure 1A. The detergent-solubilized lysates were then divided, with half diluted directly into the IP reaction with anti-FLAG agarose (‘nat.’) and with the other half being subjected to denaturation prior to the anti-FLAG IP (‘den.’). Following the IP incubation, equivalent portions of the fractions of the eluted (B, bound), the unbound fraction (U) and the initial input sample (Σ) were analysed by anti-HA Western blotting
For sucrose gradient analyses, Triton X-100-solubilized supernatants were layered onto a 4.5 ml 15–50% sucrose gradient prepared in 1% Triton X-100, 150 mm Tris/Cl, pH 7.5, with a 0.5 ml 80% sucrose cushion in the same buffer. Gradients were centrifuged at 4 °C in a swinging-bucket rotor for 2 h at 140 000 × g and 300 μl fractions were collected from the tube bottom. Absorbance of each fraction was read at both 260 and 415 nm to identify the gradient positions for both endogenous 80S ribosomes and the 50 μg bovine haemoglobin, which was added to the sample prior to centrifugation.
Mortar–pestle lysis
Cells (5 × 109) collected from log-phase cultures were resuspended into 0.5 ml LB containing 2×PI, then quick frozen as droplets into liquid nitrogen. As previously described (Roth et al., 2002), the cells were lysed by grinding in a cold mortar and pestle under liquid nitrogen. The lysate then was subjected to Triton X-100 membrane solubilization and centrifugal fractionation, as described above.
Yck2 Production from E. coli
Log-phase 50 ml cultures of Rosetta DE3 strain E. coli cells (Merck), transformed by pET30a-derived Yck2(SS) expression plasmids, were induced for 2 h with 1 mm IPTG. The harvested cells were resuspended into 1 ml 50 mm Tris, 100 mm NaCl, 1 mm DTT, pH 8.0, then incubated with chicken egg white lysozyme (Sigma-Aldrich) at 1 mg/ml for 20 min on ice, and then lysed with five 20 s blasts (interspersed with 1 min ice rests) with a Fisher Scientific 60 Sonic Dismembrator set at power level 3.5. The lysates then were centrifuged at 15 000 × g for 10 min at 4 °C to yield pellet and supernatant fractions.
Indirect immunofluorescent localizations
Cells were grown and processed as described previously (Sun et al., 2004). Images were acquired using a Zeiss Axiovert-200 equipped with a Perkin-Elmer Ultraview ERS (spinning disk laser confocal).
LC–MS/MS protein identification
Following glass bead treatment, proteins were separated by SDS–PAGE and stained. Bands showing differential fractionation to the centrifugal pellet were excised. Peptides were prepared by a stepwise, in-gel process of reduction, alkylation and trypsinization. LC–MS/MS analysis utilized an LTQ-XL mass spectrometer (Thermo Scientific), with MS2 spectra being searched by Mascot (Matrix Science) simultaneously against yeast forward and reverse protein sequences. The protein with the greatest number of associated spectra per protein size was considered to be the major component of the excised gel band. The EF-3 identification was based on 254 spectral identifications (the next most abundant protein was associated with 48 identifications). The EF-1A identification was associated with 346 spectral identifications (132 for the next most abundant protein).
Immunoprecipitation (IP)
Glass bead lysates were Triton X-100-solubilized, then clarified of unbroken cells by low-speed centrifugation, as described above. Equal lysate aliquots then were processed through either ‘native’ or ‘denatured’ IP protocols. For the native IP, the lysate was diluted to 1 ml with LB containing 1×PI, 0.2% Triton X-100 and 0.1% SDS. Samples then were subjected to a 30 min, 4 °C pre-incubation end-over-end rotation, followed by 1 min 15 000 × g centrifugation to remove particulates, and finally a 16 h, 4 °C binding incubation with 20 μl anti-FLAG-agarose (Sigma-Aldrich). The antibody beads were then subjected to four 1 ml washes and then elution into 50 μl 8 M urea, 5% SDS, 40 mm Tris, 0.1 mm EDTA, 0.4 mg/ml bromophenol blue, 1% β-mercaptoethanol, pH 6.8 (10 min at 37 °C). Samples for Western analysis were taken at different stages of the IP reaction: (a) just prior to the anti-FLAG-agarose addition (the ‘total’ samples); (b) the first supernatant following the anti-FLAG-agarose incubation (the ‘unbound’ fraction); and (c) the eluted fraction (‘bound’). Equal portions of each sample were loaded onto SDS–PAGE for anti-HA Western analysis. The denatured IPs were processed identically to the native IPs, except for inclusion of a denaturation step, prior to the IP. For this denaturation, lysate proteins were collected by chloroform–methanol precipitation (Wessel and Flugge, 1984), then dissolved into 50 ml 2% SDS, 8 m urea, 50 mm Tris, 1 mm DTT, pH 7.4, for a 10 min incubation at 37 °C. The denatured protein samples then were diluted 20-fold into 1 ml LB with 1×PI, 0.2% Triton X-100 and 0.1% SDS, with further processing as described for the native IPs.
Protein palmitoylation
Protein palmitoylation was detected using the scaled-down version of the acyl–biotinyl exchange (ABE) protocol (Politis et al., 2005; Roth et al., 2006). In brief, denatured protein extracts, prepared from cells expressing 6×His/FLAG/HA-tagged Yck2 proteins from the GAL1 promoter (2 h expression period), were subjected to the ABE chemical modification steps: (a) blockade of free thiols with N -ethylmaleimide; (b) cleavage of thioester-linked acyl modifications with neutral pH hydroxylamine; (c) marking of the newly uncovered cysteinyl thiols with the thiol-specific biotinylation reagent HPDP–biotin (Thermo Scientific). Finally, the different Yck2 proteins were purified from the biotinylated extracts via anti-FLAG-agarose IP, then subjected to Western blotting with both anti-biotin–HRP (Sigma-Aldrich) to detect palmitoylation and with anti-HA–HRP (Roche) to assess Yck2 expression levels.
Results
Non-palmitoylated Yck2 fractionates to a high molecular-weight aggregate or complex
To assess the effects of palmitoylation on Yck2 membrane fractionation, glass bead lysates were prepared from cells expressing either wild-type Yck2 or a non-palmitoylated Yck2. Wild-type Yck2 fractionates like an integral membrane protein, fractionating to the P100 membrane pellet, then to the supernatant (S100) when membranes are dissolved with Triton X-100 prior to centrifugation (Figure 1A). For non-palmitoylated Yck2, produced either by mutation of the two C-terminal, palmitoyl-accepting cysteines to serine [Yck2(SS)] or by expression of wild-type Yck2 in the akr1Δ cell context (deleted for the cognate Yck2 palmitoyl-transferase Akr1), our expectation, based on prior fluorescence-based localization analyses (Babu et al., 2004; Feng and Davis, 2000; Roth et al., 2002; Vancura et al., 1994), was for non-palmitoylated kinase to fractionate like a soluble cytoplasmic protein, i.e. to the supernatant. Instead, we found it fractionating mainly to the P100 pellet, irrespective of whether lysates were pretreated by detergent (Figure 1A).
The differing mobilities seen for the palmitoylated and non-palmitoylated Yck2 forms (Figure 1A) result from differential phosphorylation (see Figure 2A) (Roth et al., 2002). This is an autophosphorylation which depends on both high-level Yck2 expression (e.g. GAL1 promoter-driven expression) and Yck2 being properly localized to the plasma membrane (I. Papanayotou, A. Roth and N. Davis, unpublished data). Furthermore, it can occur in trans, e.g. over-produced, plasma membrane-localized Yck2 can phosphorylate kinase-inactivated mutant versions of Yck2 that are otherwise normally localized to the cell surface (I. Papanayotou, A. Roth and N. Davis, unpublished data). While the gel mobility shift is directly the consequence of phosphorylation, the requirement for surface localization means that this modification is also dependent on palmitoylation. Thus, the non-palmitoylated Yck2(SS) does not show the upper, hyperphosphorylated band (Figure 1A).
Figure 2.
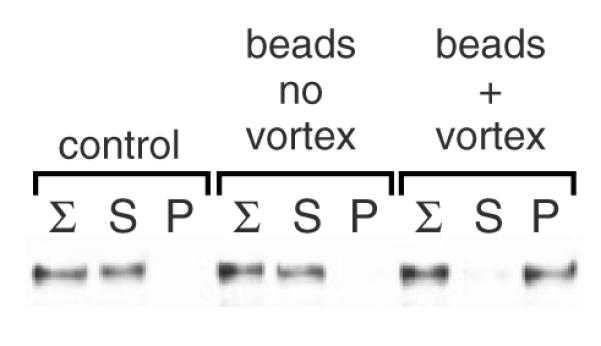
Soluble, E. coli-produced Yck2 is converted to insolubility by the mock glass bead lysis protocol. A supernatant fraction, prepared from Yck2(SS)-overproducing E. coli cells, was subjected to a mock-up of the yeast glass bead lysis protocol (beads + vortex) or, alternatively, either to simple incubation with the glass beads (beads, no vortex) or to no treatment (control). The samples then were treated by the Triton X-100 incubation and centrifugal fractionation described for Figure 1A
As is apparent here (Figure 1A), we also often find non-palmitoylated Yck2 forms, i.e. either Yck2(SS) or Yck2(wt) in the akr1Δ cell context, to be expressed at lower levels than the fully palmitoylated, wild-type Yck2. We suspect that this lowered expression likely reflects that the non-palmitoylated, mislocalized kinase is subject to increased degradative turnover.
To examine the nature of the forces responsible for the Yck2(SS) pellet fractionation, lysates from Yck2(SS)-expressing cells were treated to various chemical treatments prior to the centrifugal fractionation (Figure 1B). Treatments with Triton X-100, NaCl or DTT had no effect, i.e. Yck2(SS) continued to fractionate to the pellet. However, the two strong protein denaturants, SDS and urea, released Yck2(SS) to the supernatant. To crudely size this Yck2(SS) complex or aggregate, Triton X-100-treated glass bead lysates from Yck2(wt)- or Yck2(SS)-expressing cells were subjected to sucrose gradient analysis (Figure 1C). Yck2(wt), as would be expected following detergent dissolution of membranes, fractionates ostensibly as a soluble protein to the gradient top. In contrast, Yck2(SS) is seen to distribute throughout the gradient with the bulk of the protein concentrating towards the gradient bottom, indicating its incorporation into an extremely large protein aggregate.
The above analyses (Figures 1A–C) all relied on Yck2 expression from the strong and inducible GAL1 promoter, affording approximately 10-fold overexpression relative to the endogenous Yck2 expression level. Since aggregation is often promoted at high expression levels, the fractionation behaviour of Yck2(SS) was also examined at the normal Yck2 expression level, i.e. from the YCK2 promoter (Figure 1D). We found fractionation to be unchanged: Yck2(wt) is solubilized into the supernatant, while Yck2(SS) continues to fractionate to the pellet. At this native level of expression, note the absence of the slowly migrated, hyperphosphorylated species that was seen above for GAL1-driven Yck2(wt) (Figure 1A, C).
Aggregation of E. coli-produced Yck2 in mock glass bead lysis protocol
Prior examinations of non-palmitoylated Yck2, either Yck2(SS) or Yck2(wt), in the akr1Δ context, by either GFP-tagging or indirect immunofluorescence, found no obvious visual evidence of aggregation [i.e. no intracellular puncta were seen (Babu et al., 2002, 2004; Feng and Davis, 2000; Roth et al., 2002; Vancura et al., 1994)]. Instead, non-palmitoylated Yck2 was found to distribute throughout the cytoplasm and nucleus, being excluded from the membrane-enclosed vacuole (e.g. see Figures 4B, 9B). Furthermore, when Yck2 or Yck2(SS) was artificially expressed in E. coli cells, these being cells not thought to utilize protein palmitoylation, we found that a substantial proportion of the Yck2 protein, even when massively over-produced, was both soluble and active (data not shown), indicating that palmitoylation is not intrinsically required for proper Yck2 folding. Together, these two findings, these being the lack of visual evidence of non-palmitoylated Yck2 aggregation seen within the yeast cell as well as non-palmitoylated Yck2’s ability to properly fold within the foreign E. coli context, raised concerns that, rather than occurring in vivo, non-palmitoylated Yck2 aggregation instead might be occurring in vitro during the cell lysis protocol. To test this possibility, a supernatant fraction derived from E. coli overexpressing Yck2(SS) was subjected to conditions replicating our yeast glass bead lysis protocol. To our dismay, this iterative vortexing with glass beads did quite efficiently convert the soluble Yck2(SS) into an insoluble, pellet-fractionating form (Figure 2). As controls, the E. coli supernatant was subjected either to just the mock Triton X-100 membrane solubilization step prior to the centrifugal fractionation (Figure 2, ‘control’) or, alternatively, incubated with the glass beads with no vortexing prior to detergent treatment and centrifugation (Figure 2, ‘beads no vortex’). Neither treatment produced insoluble Yck2(SS). We conclude, therefore, that pellet fractionation can be artifactually induced as part of the glass bead lysis protocol. The finding is that simple incubation with the beads is not sufficient to induce pellet fractionation: thus, denaturation is not being catalysed on the surface of the glass beads, neither is the denaturation induced by a chemical washing off of the beads.
Figure 9.

Use of the glass bead aggregation to analyse the fractional palmitoylation of singly-lipidated Yck2 mutants. (A) Palmitoylation of the singly-lipidated Yck2 mutants. Protein extracts prepared from cells expressing (2 h GAL1-driven expression) either wild-type Yck2 or the indicated mutant versions were analysed for palmitoylation, using the ABE method (see Materials and methods). Palmitoylation is assessed as the intensity of the anti-biotin Western blot signal relative to signal derived from immuno-precipitated Yck2 protein (anti-HA). (B) The subcellular localization of the indicated Yck2 mutant proteins was analysed by anti-HA indirect immunofluorescence, as described for Figure 4B. (C) Glass bead fractionation. Cells expressing the indicated Yck2 mutant proteins from the GAL1 promoter were subjected to glass bead lysis/fractionation as described for Figure 1A. (D) Palmitoylation of supernatant- vs. pellet-fractionated populations. Following the glass bead lysis and centrifugal fractionation described for Figure 9C, supernatant- and pellet-fractionated proteins were subjected to ABE palmitoylation analysis
More gentle cell lysis protocol yields soluble Yck2(SS)
We also have used a different yeast cell lysis protocol to assess the solubility of the non-palmitoylated Yck2 produced within the yeast cell. For this, cells were quickly frozen in liquid nitrogen, then ground, also in the presence of liquid nitrogen, with a mortar and pestle. Further processing of the resulting lysate was essentially as for the glass bead method, i.e. detergent extraction, then a low-speed centrifugation to remove unbroken cells and finally, the high-speed centrifugation. With these conditions, in contrast to what was seen with glass bead lysis, palmitoylated Yck2(wt) and non-palmitoylated Yck2(SS) were both found to fractionate to the supernatant (Figure 3). Thus, this gentler cell lysis protocol yields soluble Yck2(SS), supporting the conclusion that the Yck2(SS) insolubility seen with glass bead lysis (Figure 1) results from protein aggregation artifactually induced by the agitation with glass beads. Note again that Yck2(wt), over-expressed here from the GAL1 promoter, presents here as a doublet, with the slower-migrating band, not seen for cytoplasmic Yck2(SS), corresponding to the hyperphosphorylated form.
Protection from glass bead-induced aggregation by membrane association
What accounts for the striking differential susceptibility of palmitoylated and non-palmitoylated Yck2 to glass bead-induced aggregation? Perhaps the palmitoyl modifications help to maintain native Yck2 structure, making the protein more resistant to denaturation. Alternatively, perhaps membrane association somehow confers resistance. We tested the ability of different ectopic membrane tethering sequences to substitute for the wild-type C-terminal Cys-Cys palmitoylation motif in averting the glass bead-induced aggregation. Three different Yck2 forms were constructed (Figure 4A). Yck2(CCIIS) replaced the Cys–Cys dipeptide with the Ras2 C-terminal peptide –CCIIS. Like Ras2, Yck2(CCIIS) was efficiently farnesylated and palmitoylated (Babu et al., 2002, 2004; Roth et al., 2002). Yck2(TMD) replaced the C-terminal Cys-Cys dipeptide with the C-terminal transmembrane domain (TMD) from the plasma membrane v-SNARE Snc2. Psr1–Yck2 fused a 20 residue-long myristoylation–palmitoylation motif from the plasma membrane phosphatase Psr1 to the N-terminus of Yck2(SS). Like Yck2(wt), all three of the mutant Yck2 proteins localized within the cell, predominantly to the plasma membrane (Figure 4B), indicating avid membrane tethering. Finally, all three ectopic membrane tethers also provided complete protection from glass bead-induced aggregation: like Yck2(wt), all three fractionated to the centrifugal supernatant fraction from Triton X-100-solubilized glass bead lysates (Figure 4C). In contrast, the Psr1(Δ2–10)–Yck2-(SS) control construct, deleted for the Psr1 myristoyl and palmitoyl acceptors, fractionated, like Yck2(SS), to the pellet. Thus, attachment to the membrane appears to be the key to averting glass bead-induced aggregation. One plausible explanation is that membrane-associated proteins may be protected by their incorporation into the vesicles that are produced through the glass bead disruption of cellular membranes. Enclosure within vesicles might allow Yck2 to survive the denaturing actions of the glass beads. With the subsequent Triton X-100 treatment, these proteins then would be released to the supernatant as soluble proteins.
To test the protective role of membrane association, Triton X-100 was included during the glass bead lysis step (Figure 5). The inclusion of detergent caused a significant portion of Yck2(wt) to now fractionate to the pellet, indicating that it had been made susceptible to the denaturing actions of the beads. Presumably, the portion of the Yck2(wt) protein population that did get converted to insolubility was the fraction that was released from vesicles during the glass bead agitation step.
Temperature vs. mechanical denaturation
Glass bead-induced aggregation presumably results from protein denaturation. Denaturation could result from increased temperatures produced by the frictional actions of the vortexing glass beads or, alternatively, directly from mechanical shear forces generated by the whirring glass beads. Heat production in glass bead lysis protocols is typically minimized by resting lysates on ice between vortexing blasts. Nonetheless, some temperature elevation is unavoidable. To test the role of temperature in our aggregation phenomenon, a soluble Yck2(SS)-containing supernatant fraction from a high-speed centrifugation of a mortar–pestle lysate was subjected to either the mock glass bead lysis protocol or, alternatively, to 10 min incubations at different elevated temperatures (30 °C, 40 °C or 65 °C). Following these treatments, a second, high-speed centrifugation was used to yield supernatant and pellet fractions for analysis (Figure 6A, upper panel). As expected, the mock glass bead lysis efficiently converted Yck2(SS) into a pelletfractionating aggregate. The effects of temperature, by contrast, were less impressive: a minor amount of Yck2(SS) aggregation was apparent following the 10 min, 40 °C incubation, while incubation at high temperature (65 °C), not surprisingly, resulted in a total conversion of Yck2(SS) into its aggregate form. Neither temperature (40 °C or 65 °C) is likely to be attained during our glass bead lysis protocol; thus, we suspect that the denaturation of non-palmitoylated Yck2 was instead the product of the mechanical actions of the vortexing glass beads. Both the vortexing and the glass beads were required for Yck2(SS) aggregation, since in mock glass bead lysis experiments like those of Figure 6A (upper panel), where the soluble protein extracts were subjected to vortexing in the absence of glass beads, no aggregation was seen (data not shown).
As an additional test of the potential role of thermal denaturation in the generation of the Yck2(SS) glass bead aggregates, the glass bead protocol was altered to further minimize heating (Figure 6B). In addition to our typical protocol, in which cells are glass bead-disrupted with five 45 s vortexing blasts interspersed with 1 min ice rests, the cell disruption was divided into 45 5 s blasts, interspersed by 1 min rests in a slush ice bath. Even with this new protocol, with heating further minimized, Yck2(SS) continued to fractionate to the pellet, indicating again that the mechanical forces generated by the vortexing glass beads play the predominant role in the Yck2(SS) denaturation that leads to aggregation.
Susceptibility of other yeast proteins to glass bead-induced aggregation
The above experiment, testing the effects of temperature and glass beading on Yck2(SS) solubility (Figure 6A, upper panel), also tracked the behaviour of total yeast cytosolic protein by subjecting the total centrifugal fractions to SDS–PAGE and Coomassie staining (Figure 6A, lower panel). Looking at the full retinue of soluble yeast proteins, we found that, while the majority of proteins resisted glass bead-induced aggregation, a few of these highly-abundant yeast proteins quite clearly were converted to insolubility. The two protein species most prominently showing differential fractionation to the pellet were identified by LC–MS/MS as the 50 kDa translation elongation factor EF-1α (the identical proteins Tef1 and Tef2) and the 116 kDa translation elongation factor 3 (Yef3) (indicated by asterisks in the lower panel of Figure 6A). Interestingly, Tef1/2 and Yef3, which are not homologous, do interact and collaborate in the feed and removal of amino acyl-tRNAs to and from the ribosome as each amino acid is added to the growing polypeptide. The identification of these two proteins, we feel, is largely a consequence of their high abundance within the yeast cell. Proteins of lower abundance that might also be converted to insolubility would not be identified by this analysis; identifications were limited to just those proteins showing differential fractionation as visualized by Coomassie SDS–PAGE (i.e. highly abundant yeast proteins). Nonetheless, it is worth noting that, like Yck2, Tef1/2 and Yef3 both bind and hydrolyse nucleotide triphosphates — Tef1/2 is a G protein and Yef3 an ATPase.
We also have tested a number of other yeast palmitoyl proteins for glass bead-induced aggregation (Figure 7). Consistent with the analysis above (Figure 6A, lower panel), only a minority of the tested proteins showed evidence of aggregation. Proteins showing similar glass bead-induced aggregation behaviour to Yck2 included, not surprisingly, the two palmitoylated Yck2 paralogues, Yck1 and Yck3 (Figure 7): like Yck2, mutant, non-palmitoylated versions of Yck1 and Yck3 aggregated following glass bead lysis, while the palmitoylated, wild-type forms of these two proteins were fully protected from the induced aggregation, presumably a consequence of their membrane associations. Likewise, similar behaviour was found for the yeast sphingosine kinase Lcb4: aggregation was induced for a non-palmitoylated Lcb4 mutant [Lcb4(C43S,C46S); (Kihara et al., 2005)], as well as when expressed in akr1Δ cells [where palmitoylation is substantially reduced (Kihara et al., 2005)], but not for the wild-type palmitoylated form (Figure 7). However, the bulk of the palmitoyl proteins tested, viz. Ras2, Psr1, Vac8, Ykl047w, Ypl199c and Ypl236c, showed little evidence for induced aggregation when tested as either lipidated or non-lipidated proteins (Figure 7). Thus, the differential susceptibility to mechanical denaturation seen for Yck2 and the handful of other non-palmitoylated kinases tested does not broadly extrapolate to non-lipidated versions of other lipidated proteins.
Glass bead lysates in IPs
Glass bead lysates are frequently used for coimmunoprecipitation (co-IP) analysis to identify or confirm protein–protein interactions. Thus, we have examined the IP behaviour of Yck2 non-palmitoylated and palmitoylated forms from glass bead lysates under both ‘native’ and ‘denatured’ conditions. As the immuno-absorbent, anti-FLAG-agarose, which reacts with the FLAG epitope of 6×His/FLAG/HA tri-tag marking the N-termini of both Yck2(wt) and Yck2(SS), was used. For the ‘native’ condition, our protocol paralleled that used for the above centrifugal fractionations; the glass bead lysates were subjected to Triton X-100 solubilization and then applied to the anti-FLAG-agarose. For the ‘denatured’ condition, total protein, precipitated from the lysates, was dissolved into a small amount of SDS-containing buffer at 37 °C for 10 min prior to its dilution into the IP-compatible buffer. Binding to anti-FLAG-agarose was assessed through anti-HA Western blotting of both the bound and unbound fractions. Under the denaturing condition, both palmitoylated and non-palmitoylated Yck2 proteins showed efficient binding to the anti-FLAG-agarose (Figure 8). In contrast, for the native IP, only palmitoylated Yck2(wt) showed efficient binding. The two non-palmitoylated forms, either Yck2(SS) or Yck2(wt) expressed in the PAT-deficient akr1Δ context, showed only a partial binding (Figure 8). Equivalent results were seen when the same experiment was performed using Ni-agarose (binds to the N-terminal 6×His sequence) as affinity absorbent (data not shown). We additionally found that non-palmitoylated forms of the Lcb4 sphingosine kinase, i.e. Lcb4(C43S, C46S) or Lcb4(wt) expressed in the akr1Δ cell context, also fail to bind to immuno-absorbent when assessed under ‘native’ conditions from glass bead lysates (data not shown). The failure of these proteins to be properly immunoprecipitated from glass bead lysates presumably reflects the occlusion of the epitope tags within the aggregate. Thus, for some proteins, glass bead-induced aggregation may also confound analysis of protein–protein interaction via co-IP analysis.
Glass bead-induced aggregation provides insights about fractional Yck2 palmitoylation states
Prior work has shown that the individual mutation of the two Yck2 C-terminal cysteines, i.e. Yck2(SC) and Yck2(CS), impairs Yck2 localization and function (Babu et al., 2004), suggesting that normally, both cysteines are palmitoylated. We confirmed this here, using acyl–biotinyl exchange (ABE) chemistry to follow palmitoylation. We found that Yck2(SC) and Yck2(CS) were both palmitoylated: as one might expect, both showed palmitoylation levels that were somewhat lower than those seen for wild-type Yck2(CC), which retained both palmitoyl-acceptors (Figure 9A). This analysis also included Yck2(CCIIS), which was expected to be modified by just a single palmitoyl moiety as well as Yck2(SCIIS), which lacks the palmitoyl-accepting cysteine and was thus expected to be only farnesylated, not palmitoylated (Figure 9A). An examination of the cellular localization of these mutants found that Yck2(CCIIS), like wild-type Yck2(CC), localized to the cell surface, while Yck2(SCIIS) showed reduced surface localization balanced by increased localization to intracellular membranes (Figure 9B). The difference in membrane avidities provided by one vs. two lipid tethers is considerable. Consequently, singly-lipidated proteins rapidly equilibrated onto and off of membranes, precluding their capacity to exploit classic vesicular transport mechanisms for localization. The result was that singly-lipidated proteins were seen to localize randomly to a variety of cellular membranes (Rocks et al., 2005; Shahinian and Silvius, 1995). Nonetheless, the localizations seen for singly-palmitoylated Yck2(SC) and Yck(CS) were quite different from that observed for singly-lipidated Yck2(SCIIS) (Figure 9B). Yck2(SC) and Yck(CS) both showed little evidence of any membrane association, with only small amounts detectable at surface and intracellular membranes, and with the bulk of both proteins localizing diffusely through the cytoplasm, with a presentation not grossly dissimilar to that seen for cytoplasmic Yck2(SS).
Analysis of these mutants in our glass bead lysis/fractionation protocol showed the typical behaviours for Yck2(CC) and Yck2(SS) (Figure 9C): following detergent solubilization, Yck2(CC) fractionated to the supernatant, while Yck2(SS) fractionated to the pellet. Similar to Yck2(CC), Yck2(CCIIS) and Yck2(SCIIS) both fractionated mainly to the supernatant, a finding consistent with the predominant membrane localizations seen for these proteins by immunofluorescence (Figure 9B). Interestingly, Yck2(SC) and Yck2(CS) both showed intermediate phenotypes, showing fractionations divided between both the supernatant and the pellet (Figure 9C). The different gel mobilities of the different Yck2 mutants (Figure 9A and 9C), a reflection of their differing phosphorylation and thereby of their different localization to the plasma membrane, was also well correlated: Yck2(CC) and Yck2(CCIIS), which both localized primarily to the surface, showed the heaviest phosphorylations (i.e. slowest mobility), while Yck2(SCIIS), which is only partially surface-localized (Figure 9B), showed an intermediate mobility shift. In contrast, Yck2(SC) and Yck2(CS) both looked quite under-phosphorylated, showing mobilities that were essentially indistinguishable from the cytoplasmic Yck2(SS). These Yck2(CS) and Yck2(SC) results — the intermediate fractionation, the reduced phosphorylation and the largely cytoplasmic localizations — together indicated inefficient membrane tethering, with membrane associations substantially reduced relative to the singly-lipidated Yck2(SCIIS).
What explains the less efficient tethering of Yck2(SC) and Yck2(CS), relative to Yck2(SCIIS)? It is not that the Yck2(SCIIS) farnesyl lipid provides stronger membrane tethering than do the Yck2(SC) and Yck(CS) palmitates — the opposite is true; membrane avidity of palmitoyl tethers is greater than that of farnesyl tethers (Shahinian and Silvius, 1995). One plausible explanation, however, is that Yck2(SC) and Yck2(CS) palmitoylation is inefficient. Maybe substantial subpopulations of these two proteins fail to receive any palmitoylation. To investigate this, the membrane-associated and cytosolic subpopulations were fractionated from one another via our glass bead lysis/fractionation protocol. Then, palmitoylation on the two sub-populations was assessed by ABE. This analysis clearly showed that these two fractionated subpopulations of Yck2(CS) and Yck2(SC) did, in fact, differ considerably in their palmitoylation, with substantially more palmitoylation seen for supernatant-fractionated protein than for the pellet-fractionated protein (Figure 9D). Thus, for both mutants, the membrane-associated population was palmitoylated and the non-membrane-associated population, which accounted for a substantial fraction (>50%) of the total for both mutants being largely not palmitoylated. Thus, both the SC and CS mutations were substantially more disruptive to palmitoylation than had been previously appreciated (Babu et al., 2004); the removal of either one of the two palmitoyl-acceptor sites substantially impaired palmitoylation at the remaining cysteine. Therefore, the fractionation provided by this artifactual aggregation has led to an important new conclusion regarding the Yck2 recognition elements that specify its palmitoylation.
Discussion
Here we have described a troubling artifact that can confuse yeast analyses that rely on glass bead lysis — some proteins, when subjected to this mode of cell lysis, denature and aggregate, potentially confounding analyses of native function. While this induced aggregation appears to apply to just a subset of yeast proteins, the overall scope of the problem — whether susceptibility to aggregation applies to 1% or to 10% of the yeast proteome — was left open by our analysis. Our attempt at addressing this issue proteomically demonstrated susceptibility for two additional proteins, Tef1/2 and Yef3 (Figure 6A, lower panel). However, this analysis was quite limited in that it allowed susceptibility to be tested only for the most highly abundant yeast proteins. Thus, for the vast majority of the proteins comprising the yeast proteome, susceptibility remains unprobed. Nonetheless, an extrapolation from this limited, abundant-protein test set would suggest that, for the vast majority of yeast proteins, glass bead-induced denaturation is likely not a major concern. Glass bead lysis is, and hopefully will continue to be, an extremely valuable and convenient tool for the yeast researcher. Nevertheless, we felt it important that the yeast researcher be made aware of this potential artifact.
What distinguishes the yeast proteins that are susceptible to glass bead-induced aggregation? Our results suggest that the mechanical shear forces produced by the vortexing glass beads likely cause the denaturation driving this aggregation. The proteins identified herein as undergoing this glass bead-induced aggregation, viz. Yck2, its paralogues Yck1 and Yck3, the sphingosine kinase Lcb4 and the two translation elongation factors identified (Tef1/2 and Yef3), may all be proteins that are particularly labile structurally. Kinases, protein kinases as well as lipid kinases, may be particularly susceptible to forces of mechanical denaturation. Interestingly, a deletion analysis of the Yck2 sequences that contribute to aggregation identified the kinase domain to be the main driver of Yck2 aggregation: mutants retaining the kinase domain were susceptible to glass bead-induced aggregation, while mutants deleted for this domain were not (B. Sun and N. Davis, unpublished data).
The strikingly different behaviour of palmitoylated and non-palmitoylated Yck2 in our glass bead lysis/fractionation protocol is curious. Protection from aggregation, we have shown, requires an association of Yck2 with membranes (Figures 4, 5). As explanation, we suggest that with the glass bead disruption of membranes, membrane-tethered Yck2 becomes encapsulated within newly formed vesicles that confer protection from the whirring glass beads. The discrete nature of this protection, wherein almost all of membrane-associated Yck2 and virtually none of cytosolic Yck2 is protected, suggests that all of the mechanically-generated vesicles are of one orientation, orientated so that the cytosolic, Yck2-harbouring membrane face is orientated towards the vesicle interior. While this is possible, perhaps a more plausible explanation is that major default structure produced in these mechanical disruptions is instead multilamellar vesicles (MLVs), structures in which multiple layers of membranes are enclosed within membranes. MLVs tend to be the default membrane structure produced by a variety of means, with unilamellar vesicle production typically requiring specialized methodologies (Matsuoka and Schekman, 2000). The membranes-within-membranes MLV structure minimizes the amount of membrane surface that is exposed to the external milieu.
Finally, we show that glass bead-induced aggregation, with its exquisite discrimination of Yck2-soluble from membrane-bound forms, can be of some experimental utility, allowing us to discern the fractional associations of different Yck2 mutants with membranes. This is of particular value when examining palmitoylation. Palmitoylation labelling methods, whether through metabolic labelling with [3H]-palmitate or through the in vitro chemical exchange of biotin moieties at palmitoylation sites (i.e. ABE), do not well discern the fraction of the total protein population that is palmitoylated; it is generally unclear for a protein that is found to be palmitoylated, whether 100% or 1% of the protein population is modified by the attached lipid. For instance, palmitoylation analysis of Yck2(SC) and Yck2(CS) showed that both labelled to somewhat lower levels than did wild-type Yck2(CC) (Figure 9A); the level of this reduction seemed roughly in line with expectations for proteins having one vs. two attached palmitates. However, coupling this ABE analysis to glass bead lysis/fractionation revealed that a substantial proportion of both the Yck2(SC) and Yck2(CS) protein populations (>50%), resisted palmitoylation altogether. Apparently, the bulk of these two mutants fail to be recognized by their cognate palmitoyl-transferase Akr1, thus providing an important new insight regarding the nature of the Yck2 palmitoylation signal — efficient recognition of the Yck2 substrate depends on both C-terminal cysteines being present. This novel approach for discriminating membrane-bound from unbound Yck2 may find additional utility in future analyses of the Yck2 palmitoylation signal, and also for following the kinetics of Yck2 palmitoylation and membrane-association in vivo. It is presently unclear whether this fractionation approach might apply more widely beyond Yck2. In any case, despite this minor utility, our main goal with this report was to alert yeast colleagues to the troubling possibility of aggregation when employing glass bead lysis.
Acknowledgements
This work was supported by NIH Grant No. GM65525. We thank Paul Stemmer and Joseph Caruso at the Karmanos Cancer Institute proteomics core (NIH ES06639) for the mass spectral analysis.
References
- Abdel-Sater F, El Bakkoury M, Urrestarazu A. Amino acid signaling in yeast: casein kinase I and the Ssy5 endoprotease are key determinants of endoproteolytic activation of the membrane-bound Stp1 transcription factor. Mol Cell Biol. 2004;24:9771–9785. doi: 10.1128/MCB.24.22.9771-9785.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu P, Bryan JD, Panek HR, et al. Plasma membrane localization of the Yck2p yeast casein kinase 1 isoform requires the C-terminal extension and secretory pathway function. J Cell Sci. 2002;115:4957–4968. doi: 10.1242/jcs.00203. [DOI] [PubMed] [Google Scholar]
- Babu P, Deschenes RJ, Robinson LC. Akr1p-dependent palmitoylation of Yck2p yeast casein kinase 1 is necessary and sufficient for plasma membrane targeting. J Biol Chem. 2004;279:27138–27147. doi: 10.1074/jbc.M403071200. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Deschenes RJ, Broach JR. Fatty acylation is important but not essential for Saccharomyces cerevisiae RAS function. Mol Cell Biol. 1987;7:2344–2351. doi: 10.1128/mcb.7.7.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Davis NG. Akr1p and the type I casein kinases act prior to the ubiquitination step of yeast endocytosis: Akr1p is required for kinase localization to the plasma membrane. Mol Cell Biol. 2000;20:5350–5359. doi: 10.1128/mcb.20.14.5350-5359.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Zanolari B, Riezman H. Cytoplasmic tail phosphorylation of the α-factor receptor is required for its ubiquitination and internalization. J Cell Biol. 1998;141:349–358. doi: 10.1083/jcb.141.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Varshavsky A. Regulation of peptide import through phosphorylation of Ubr1, the ubiquitin ligase of the N-end rule pathway. Proc Natl Acad Sci USA. 2008;105:19188–19193. doi: 10.1073/pnas.0808891105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Kurotsu F, Sano T, et al. Long-chain base kinase Lcb4 Is anchored to the membrane through its palmitoylation by Akr1. Mol Cell Biol. 2005;25:9189–9197. doi: 10.1128/MCB.25.21.9189-9197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka K, Schekman R. The use of liposomes to study COPII- and COPI-coated vesicle formation and membrane protein sorting. Methods. 2000;20:417–428. doi: 10.1006/meth.2000.0955. [DOI] [PubMed] [Google Scholar]
- Moriya H, Johnston M. Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proc Natl Acad Sci USA. 2004;101:1572–1577. doi: 10.1073/pnas.0305901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal G, Paraz MT, Kellogg DR. Regulation of Mih1/Cdc25 by protein phosphatase 2A and casein kinase 1. J Cell Biol. 2008;180:931–945. doi: 10.1083/jcb.200711014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panek HR, Stepp JD, Engle HM, et al. Suppressors of YCK-encoded yeast casein kinase 1 deficiency define the four subunits of a novel clathrin AP-like complex. EMBO J. 1997;16:4194–4204. doi: 10.1093/emboj/16.14.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin N, Menade M, Poirier G, et al. Local activation of yeast ASH1 mRNA translation through phosphorylation of Khd1p by the casein kinase Yck1p. Mol Cell. 2007;26:795–809. doi: 10.1016/j.molcel.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Peng Y, Tang F, Weisman LS. Palmitoylation plays a role in targeting Vac8p to specific membrane subdomains. Traffic. 2006;7:1378–1387. doi: 10.1111/j.1600-0854.2006.00472.x. [DOI] [PubMed] [Google Scholar]
- Politis EG, Roth AF, Davis NG. Transmembrane topology of the protein palmitoyl transferase Akr1. J Biol Chem. 2005;280:10156–10163. doi: 10.1074/jbc.M411946200. [DOI] [PubMed] [Google Scholar]
- Robinson LC, Menold MM, Garrett S, Culbertson MR. Casein kinase I-like protein kinases encoded by YCK1 and YCK2 are required for yeast morphogenesis. Mol Cell Biol. 1993;13:2870–2881. doi: 10.1128/mcb.13.5.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocks O, Peyker A, Kahms M, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- Roth AF, Feng Y, Chen L, Davis NG. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol. 2002;159:23–28. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, et al. Global analysis of protein palmitoylation in yeast. Cell. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian S, Silvius JR. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry. 1995;34:3813–3822. doi: 10.1021/bi00011a039. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielewoy N, Flick K, Kalashnikova TI, et al. Regulation and recognition of SCFGrr1 targets in the glucose and amino acid signaling pathways. Mol Cell Biol. 2004;24:8994–9005. doi: 10.1128/MCB.24.20.8994-9005.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K, Dietrich LE, Hou H, et al. Palmitoylation determines the function of Vac8 at the yeast vacuole. J Cell Sci. 2006;119:2477–2485. doi: 10.1242/jcs.02972. [DOI] [PubMed] [Google Scholar]
- Sun B, Chen L, Cao W, et al. The yeast casein kinase Yck3p is palmitoylated, then sorted to the vacuolar membrane with AP-3-dependent recognition of a YXXPhi adaptin sorting signal. Mol Biol Cell. 2004;15:1397–1406. doi: 10.1091/mbc.E03-09-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancura A, Sessler A, Leichus B, Kuret J. A prenylation motif is required for plasma membrane localization and biochemical function of casein kinase I in budding yeast. J Biol Chem. 1994;269:19271–19278. [PubMed] [Google Scholar]
- Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Yanai A, Huang K, Kang R, et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]