

SUMMARY
Protein palmitoylation, a reversible lipid modification of proteins, is widely used in the nervous system, with dysregulated palmitoylation being implicated in a variety of neurological disorders. Described below is ABE/SILAM, a new proteomic strategy that couples acyl-biotinyl exchange (ABE) purification of palmitoyl-proteins to whole animal stable isotope labeling (SILAM) to provide an accurate tracking of palmitoylation change within rodent disease models. As a first application, we have used ABE/SILAM to look at Huntington disease (HD), profiling palmitoylation change in two HD-relevant, mouse mutants – the transgenic HD model mouse YAC128 and the hypomorphic Hip14-gt mouse, which has sharply reduced expression for HIP14 (Dhhc17), a palmitoyl-transferase implicated in the HD disease process. Rather than mapping to the degenerating neurons themselves, the biggest disease changes instead map to astrocytes and oligodendrocytes, i.e. the supporting glial cells.
Keywords: protein palmitoylation, Huntington disease, neurodegenerative disease, HIP14, palmitoyl-proteome, tandem mass spectrometry, astrocytes, glutamate excitotoxicity, carbonic anhydrase II, glutamine synthetase
INTRODUCTION
Protein palmitoylation, the thioesterification of fatty acyl moieties (often, the saturated 16C palmitate) to selected cysteines, serves both for tethering proteins to membranes and for in-bilayer localizations to membrane microdomains. For many proteins, palmitoylation is reversible, allowing dynamic regulation of protein-membrane localization. Palmitoylation is widely used at the neuronal synapse (Conibear and Davis, 2010; Fukata and Fukata, 2010; Young et al., 2012) where, by modulating protein and receptor content at synaptic membranes, it participates in regulating synaptic plasticity. Perhaps not surprisingly, dysregulated palmitoylation has been implicated as being causal in a number of neurological disorders, notably in schizophrenia and mental retardation, as well as in Huntington and Alzheimer diseases (Young et al., 2012). Below, we describe ABE/SILAM, a new proteomic strategy that profiles and quantifies brain palmitoylation change within rodent models of neurological disease.
ABE/SILAM combines acyl-biotinyl exchange (ABE) purification of palmitoylated proteins (Roth et al., 2006; Wan et al., 2007) with whole animal stable isotope-labeling (SILAM; stable isotope labeling of mammals (McClatchy et al., 2007a; McClatchy et al., 2007b; Savas et al., 2012; Wu et al., 2004)) to enable precise sample-to-sample quantification of palmitoylation change. Our prior application of ABE proteomics to rat brain and rat neuronal culture identified most all of the known neural palmitoyl-proteins, plus over one hundred new palmitoyl-protein candidates (Kang et al., 2008). The ABE neural palmitoyl-proteome, a set of ~300 proteins, includes many well known synaptic regulators – receptors (e.g. AMPA and NMDA glutamate receptors, G protein-coupled receptors), downstream G protein signalers (e.g. H- and N-Ras, Gα subunits), as well as the scaffolding proteins (e.g. PSD-95, SAP97) that organize receptors with downstream signalers within the post-synaptic density. Non-neuronal brain cells, in particular astrocytes and oligodendrocytes, also are well surveyed by this analysis. These glial cells, which make up the majority of cells within the brain, act to support the neuronal circuitry – oligodendrocytes ensheath neuronal axons in insulating myelin membranes, while astrocytes provide nutritional support. The addition of SILAM, we expected, would provide our ABE analysis with an internally-controlled, quantitative capacity that would allow palmitoylation levels on individual palmitoyl-proteins to be comprehensively monitored across the neural palmitoyl-proteome, providing a new, deeper look into the neurological disease process.
As a first ABE/SILAM application, we have targeted Huntington disease (HD), where dysregulated palmitoylation has been suggested to play an important role (Huang et al., 2010; Huang et al., 2011; Singaraja et al., 2011; Yanai et al., 2006). HD is a progressive, dominantly-inherited neurodegenerative disease which begins as a movement disorder due to the early loss of striatal medium spiny neurons. Progression of the neurodegenerative disease to other brain regions leads to cognitive dysfunction and ultimately, to death. The HD causal mutation is an expansion of a polyglutamine string within the N-terminal exon of the disease protein huntingtin (HTT): individuals with polyglutamine tracts of <30 are disease-free, while those with expanded glutamine tracts of >40 succumb to disease. HD typically is characterized by a long disease-free latent period, with overt disease symptoms not becoming apparent until middle age. Mutant HTT (mHTT), i.e. harboring an expanded polyglutamine tract, is aggregation-prone and the appearance of large mHTT aggregates within neurons is a prominent molecular hallmark of progressing disease. While mHTT aggregation likely plays some role in disease pathogenesis, it is now widely thought that the largest aggregates, by sequestering mHTT, are primarily protective (Arrasate et al., 2004; Kuemmerle et al., 1999; Saudou et al., 1998; Slow et al., 2005). Indeed, despite its simple genetics and three decades of research, the process by which mHTT induces neuronal cell death still remains uncertain, with quite a large array of diverse mechanisms suggested to be the central disease driver; these include disordered transcription, disordered post-translational mechanisms, impaired mitochondrial function, impaired vesicular trafficking, impaired protein homeostasis, increased excitotoxicity, and finally impaired protein palmitoylation (Ross and Tabrizi, 2011; Zuccato et al., 2010).
A role for palmitoylation in HD was first suggested by the identification of HIP14 (Zdhhc17) as a 2-hybrid interactor with HTT (Singaraja et al., 2002). HIP14 is one of the 23-member mammalian DHHC protein acyl transferase (PAT) family. These PATs exhibit different, as well as overlapping, specificity for substrate; together, they appear to mediate the bulk of the protein palmitoylation within the cell (Fukata et al., 2004; Greaves and Chamberlain, 2011; Huang et al., 2004; Lobo et al., 2002; Roth et al., 2002; Roth et al., 2006). Recently, a Hip14-deficient mouse was found to show neuroanatomical, behavioral, and physiological disease phenotypes that largely recapitulate those seen in mouse models of HD (Singaraja et al., 2011). The shared disease phenotypes suggested that disease mechanisms also might be shared. More specifically, impaired palmitoylation of HIP14 substrate proteins may drive neuropathology not only in the Hip14-deficient mouse, but also perhaps, in HD. Here, we use ABE/SILAM to profile palmitoylation both in the Hip14-deficient brain and in the YAC128 HD model mouse brain, which expresses a mHTT transgene with a 128 residue glutamine tract. Significant dysregulations are uncovered in both brains. ABE/SILAM profiling of the Hip14-deficient brain identifies a number of proteins showing reduced representations within the ABE palmitoyl-proteome, including several previously identified HIP14 substrates, namely SNAP-25, the cysteine string protein (Dnajc5), and synaptotagmin-1 (Greaves et al., 2010; Greaves et al., 2008; Huang et al., 2009; Huang et al., 2004). More interesting, perhaps is the YAC128 analysis, which highlights a prominent role for glial support cells in the HD disease process, finding brainwide reductions for palmitoylated carbonic anhydrase II (CA II), a glial enzyme involved in the disposable of neuronal CO2 waste, as well as for three central players of the astrocytic glutamate-glutamine pathway, which acts to maintain low neural levels of the excitatory neurotransmitter glutamate, reducing, thereby, the potential for excitotoxicity. Overall, we find ABE/SILAM to be quite an effective tool, able to discern even quite subtle palmitoylation change in the 5–10% range, which should prove useful in analyzing palmitoylation dysregulation in other rodent disease models beyond HD.
RESULTS
ABE/SILAM Analysis
The ABE chemistry substitutes biotin tags for thioester-linked acyl modifications, allowing the palmitoyl-protein contingent of the proteome to be affinity-purified by streptavidin-agarose (Drisdel and Green, 2004; Roth et al., 2006). For ABE, denatured protein extracts are processed through a sequence of three chemical treatments: 1) N-ethylmaleimide-mediated blockade of free thiols, then 2) cleavage of thioester linkages with neutral pH hydroxylamine to uncover acylation site thiols, which then are 3) marked with the thiol-specific biotinylation reagent HPDP-biotin. SILAM applies the powerful quantitative advantages of stable isotope labeling, which have been quite widely employed in cell culture analysis (Oda et al., 1999; Ong et al., 2002), to the whole animal (either mouse or rat) (McClatchy et al., 2007a; McClatchy et al., 2007b; Wu et al., 2004). The analysis below uses mice, wholly labeled with 15N, generated by raising post-weaned mice for 10 weeks on a diet where the nitrogen component is exclusively derived from 15N-labeled Spirulina. Greater than 95% of brain proteins within these reference mice are 15N-labeled (Adibekian et al., 2011).
The first ABE/SILAM step is a 1:1 mixing of brain homogenates from a 14N-test mouse and the 15N-reference mouse. The ABE purification is a multi-step, multi-day protocol (Roth et al., 2006; Wan et al., 2007). The prior homogenate mixing is key to eliminating the experimental error that would otherwise accumulate during the subsequent ABE protocol. Together in the same test tube, 14N- and 15N-proteins are subject to exactly the same experimental forces throughout the ABE purification. Following the ABE purification, the mixed samples are proteolyzed and subjected to MS/MS analysis. Because 14N- and 15N-labeled peptides differ by predictable mass differences, the relative contributions of the 14N- and 15N-animals can be deconvolved by a downstream data analysis (Park et al., 2008). Each identified peptide provides a quantitative data point that compares the amount of protein present within the test animal palmitoyl-proteome to its presence within the reference palmitoyl-proteome (Fig. 1A). Abundant palmitoyl-proteins typically are identified by multiple peptides per MS/MS run, providing thus, multiple, reinforcing measurements.
FIGURE 1.

Example data for flottilin-1 (Flot1) from the ABE/SILAM analysis of palmitoylation in Hip14-gt and WT littermate brains. (A) Example chromatograms are shown for two Flot1 peptides from a 14N/15N-WT sample run (at left) and from a 14N/15N-Hip14-gt sample run (at right). Each chromatogram compares the abundance of the 14N-test peptide (red trace) deriving from either the WT littermate (at left) or Hip14-gt animal (at right) to the corresponding 15N-reference peptide (black trace). The ratio of the peak volumes for the test and reference peptides is reported below. (B) Graphical summary of the ABE/SILAM peptide data for Flot1, for the eight MS/MS runs that comprise the Hip14-gt analysis. For the two test genotypes analyzed, i.e. Hip14-gt and WT littermate, four mixed 14N/15N-ABE samples were prepared from three mice (m1, m2, and m3) plus one technical replicate (m1-t2). 14N/15N-peak ratios are reported for each Flot1 peptide, identified from each MS/MS run are reported. At right, the data aggregated from the four component MS/MS runs is shown with the median value and standard deviation indicated (***, P = 4 × 10−12). See related Figure S1.
Hip14-gt Brain Palmitoyl-Profile
We first used ABE/SILAM to analyze brain palmitoylation change within the recently characterized Hip14-deicient mouse, which is homozygous for a gene-trapped allele of HIP14 (Hip14-gt) (Singaraja et al., 2011). HIP14 protein expression is reduced by >90% in Hip14-gt mouse brain (Singaraja et al., 2011). Our comparison of Hip14-gt mouse brain palmitoylation to isogenic, wild-type (WT) littermates relied on the analysis of eight 14N/15N, ABE-purified samples, with four samples (three biological replicates plus one technical replicate) being analyzed for each of the two genotypes (i.e. Hip14-gt and isogenic WT). The Hip14-gt and WT littermate mice were raised on typical mouse chow and thus are, in essence, 14N-labeled. Rather than being the control, the 15N-mouse component of each sample instead provides an invariant reference palmitoyl-proteome, allowing the amount of each purified 14N-palmitoyl-protein to be measured against a reference level of 15N-palmitoyl-protein. The comparison of interest, i.e. between Hip14-gt and WT littermates, is assessed by a ratio of ratios approach that compares averaged 14N/15N-ratios for each protein for the two test mice. Note that in this triangulated scheme, the 15N-reference mouse need not be of the same age or strain background as the Hip14-gt and WT test mice.
Figure 1B reports peptide datapoints mapping to the protein flotillin-1 (Flot1) from each of the eight component MS/MS runs, comprising the Hip14-gt/WT analysis. The individual peptide datapoints although showing some scatter, do normally distribute around median values (Fig. 1B). These median values clearly differ for the Hip14-gt and the WT sample runs, indicating a reduced presence for palmitoylated Flot1 in Hip14-gt relative to WT brain. Aggregation of like datasets (Fig. 1B, at right), leads us to conclude that palmitoylated Flot1 is reduced by 36% in Hip14-gt brain relative to WT brain. Indeed, Flot1 is one of the proteins showing the greatest and most significant change in Hip14-gt brain.
The above-noted scatter for the peptide ratio data (Fig. 1B) is not a consequence of the extensive ABE sample processing, since a similar degree of datapoint variance is also seen for minimally processed 14N/15N samples, generated by a simple mixing of 14N- and 15N-brain homogenates, i.e. no ABE purification (Fig. S1). In order to overcome uncertainty due to this scatter, we have focused our analysis on the most abundant proteins within the ABE palmitoyl-proteome, i.e. proteins like Flot1 (Fig. 1B) that are identified on average by five or more quantifiable peptides per MS/MS run (see Experimental Procedures). For the Hip14-gt/WT analysis, this consists of the set of ~300 relatively abundant palmitoyl-proteins (Table S1). The vast majority of these proteins do not show substantial change in Hip14-gt versus WT brain, however, 19 proteins do show significant change that is >10% in magnitude (Table 1 and Fig. S2). Indeed, in addition to Flot1, we find that the Flot1 paralog, Flot2, also is 36% reduced in Hip14-gt brain. Flot2 is sufficiently diverged from Flot1 (50% identity) to be identified here fully independently, i.e. by a non-overlapping set of peptides. The two flotillins share roles in endocytosis and in plasma membrane microdomain formation (Cremona et al., 2011; Frick et al., 2007; Gkantiragas et al., 2001; Glebov et al., 2006; Morrow et al., 2002).
Table 1. Palmitoyl-proteome change in Hip14-gt versus WT brain.
| Proteomics | |||||
|---|---|---|---|---|---|
| Gene Symbol | Description | Change | Ref | ABE | click |
| Down in HIP14-gt: | |||||
| Flot1 | Flotillin 1; endocytosis; plasma membrane microdomain formation | −36%*** | (1) | + | + |
| Flot2 | Flotillin 2; endocytosis; plasma membrane microdomain formation | −36%*** | (2) | + | + |
| Arhgap21 | GAP for CDC42 | −30%** | + | ||
| Ank2 | Ankyrin 2; links integral membrane proteins to cytoskeleton; membrane domain role. | −22%** | |||
| Syn2 | Synapsin 2; associates with cytosolic face of synaptic vesicles | −18%*** | |||
| Gnal | Gα(olf); Gα olfactory type; stimulates adenylyl cyclase | −17%*** | (3) | + | |
| Sept8 | Septin 8; GTPase that, together with other septins, forms filamentous, cytoskeletal structures that demarcate polarized plasma membrane structures; neuronal polarity | −13%** | + | ||
| Camkv | CAM kinase-like vesicle-associated | −11%** | + | ||
| Fasn | Fatty acid synthase (ABE detection may reflect thioester utilization by acyl-carrier function). | −10%*** | + | + | |
| Cnp | Cyclic nucleotide phosphodiesterase; highly concentrated in myelin sheaths. | −10%*** | (4) | + | + |
| Gdi2 | Guanosine diphosphate dissociation inhibitor; Rab GDI beta. | −10%*** | + | ||
| Up in Hip14-gt: | |||||
| Pde2a | Cyclic nucleotide phosphodiesterase, cGMP-activated. | +25%*** | (5) | + | |
| Glul | GS; glutamine synthetase | +12%*** | + | ||
| Lower significance: | |||||
| Add1 | Adducin 1; α-adducin; heterodimer with γ-adducin; membrane-cytoskeleton associated; promotes assembly of actin/spectrin network; binds calmodulin. | −19%* | |||
| Slc44a1 | CTL1; choline transporter-like 1; multi-pass membrane protein; component of mitochondrial outer membrane; may be involved in myelin synthesis. | −15%* | + | ||
| Rap2a | Small RAS family G protein; regulates dendrite extension and arborization; farnesylated and palmitoylated. | −14%* | (6) | + | + |
| Mtap1b | Microtubule-associated protein; role in neurite outgrowth. | −13%* | + | ||
| Gpd2 | Glycerol-3—phosphate dehydrogenase 2; mitochondrial inner membrane localization. | −11%* | |||
| Pfkl | Phosphofructokinase, liver. | −10%* | |||
The proteins showing significant change (* P < 0.05; ** P < 0.01; *** P < 0.001) of greater than 10% in the Hip14-gt brain palmitoyl-proteome relative to the WT brain palmitoyl-proteome are listed. The three rightmost columns provide the published evidence supporting palmitoylation documentation for each protein, including either a specific reference (see below), or a “+” indication that the protein was identified as likely palmitoylated in prior ABE- or click-based proteomic analyses (see Table S1 for the specific references to these proteomic studies and for the expanded ABE/SILAM data set). See Figure S2 for graphical depictions of the peptide data for the proteins reported within this table.
References:
The ABE/SILAM data also has been analyzed for the putative HIP14 substrates that have been identified by prior reports (Table 1), either by in vitro palmitoylation assays (i.e. utilizing purified substrate protein and HIP14 enzyme) or by enhanced palmitoylation in cells, co-transfected together with HIP14 and the putative substrate protein (Huang et al., 2009; Huang et al., 2004; Ohyama et al., 2007). Six of these seven proteins are well covered by our ABE/SILAM analysis (Table 2 and Fig. S2). Three, namely synaptotagmin-1 (Syt1), cysteine string protein (CSP; Dnajc5), and SNAP-25, show small, but significant reductions within Hip14-gt brain context of 7, 8, and 5%, respectively. For Syt1 and CSP, the high significance of these changes (P = 5 × 10−5 and 5 × 10−4, respectively) provides an indication of the power of this approach in discerning even quite subtle change (Fig. S2).
Table 2. ABE/SILAM data for proteins identified by prior work to be likely HIP14 palmitoylation substrates.
| Gene Symbol | Description | Hip14-gt Change | YAC128 Change |
|---|---|---|---|
| Dnajc5 | Cysteine string protein (CSP); may be involved in Ca2+-dependent neurotransmitter release | −8%*** | −7% (ns) |
| Syt1 | Synaptotagmin 1; synaptic vesicle integral membrane protein; may be Ca2+ sensor for vesicle release. | −6%*** | −1% (ns) |
| Snap25 | SNAP-25; neuronal SNARE protein that acts together with Vamp2 and Stx1 to effect synaptic vesicle release. | −5%* | −7%* |
| Dlg4 | PSD-95; SAP-90; MAGK scaffolding protein that clusters NMDA glutamate receptors at synapse. | −5% (ns) | −13%* |
| Gad2 | GAD-65; glutamate decarboxylase 2; GABA neurotransmitter biosynthesis; tethers to GABA-containing synaptic vesicles. | +2% (ns) | +2% (ns) |
| Gap43 | GAP-43; may play a role in neurite outgrowth and nerve regeneration. | +7% (ns) | +12% (ns) |
| Htt | Huntingtin; disease protein of HD. | not detected | not detected |
| Lck | Lymphocyte-specific protein tyrosine kinase | not detected | not detected |
The change detected in Hip14-gt brain palmitoyl-proteome relative to WT brain palmitoyl-proteome is reported (Significance levels: * P < 0.05; ** P < 0.01; *** P < 0.001). See Figure S2 for graphical depictions of the peptide-level data for the proteins of this table.
Overall, the palmitoylation change detected in Hip14-gt brain is less pronounced than anticipated. The two best hits, Flot1 and Flot2, are reduced by only 36% (Table 1). Furthermore, in contrast to our prior analysis of palmitoylation in yeast strains knocked out for the different DHHC PATs (Roth et al., 2006), no protein within our abundant palmitoyl-protein set is found to be fully lost from the Hip14-gt palmitoyl-proteome, suggesting that none of these proteins is fully reliant upon HIP14 for palmitoylation. In part, this relatively mild palmitoylation defect likely reflects the well-documented specificity overlaps among the DHHC PATs (Fukata et al., 2004; Greaves et al., 2010; Hou et al., 2009; Huang et al., 2009; Roth et al., 2006). Furthermore, the Hip14-gt allele may allow some leaky expression of wild-type HIP14 gene product. Although the gene-trap moiety, inserted within HIP14 intron-5, is designed for efficient gene product truncation, any residual, low efficiency splicing around the gene trap moiety will yield wild-type enzyme, potentially dampening the magnitude of the Hip14-gt palmitoylation defect. Prior immunoblot analysis indicated HIP14 expression to be reduced in Hip14-gt brain by >90% (Singaraja et al., 2011). Irrespective of whether HIP14 protein is reduced by 90% or by 99.9% in Hip14-gt brain, this level of reduction clearly is associated with relatively severe consequences, as evidenced by the HD-like phenotypes documented for this mouse (Singaraja et al., 2011).
While Hip14-gt palmitoyl-proteome change is dominated by reductions, two proteins, glutamine synthetase (GS; Glul) and the phosphodiesterase Pde2a, show sharply increased levels within the Hip14-gt palmitoyl-proteome (Table 1; Fig. S2). Increases could result from a compensative up-regulation of other PATs, or alternatively, could simply be a secondary feature of the Hip14-gt disease process (Singaraja et al., 2011), i.e. palmitoylation and/or expression changes, secondary to the Hip14-gt-induced neural injury. Indeed, as is further delineated below, palmitoyl-proteome change also can be a consequence of altered expression. Changes in the transcription or the turnover for a particular palmitoyl-protein obviously will impact the amount of protein available for purification into the ABE palmitoyl-proteome. Palmitoylation and expression-level change may also be linked in some instances, with impaired palmitoylation resulting in mislocalization and consequently, increased turnover (Valdez-Taubas and Pelham, 2005). A recent proteomic analysis, which assessed palmitoylation in cells knocked down for the DHHC PAT DHHC5, found that the vast majority of proteins identified as showing reduced palmitoylation, also showed reduced expression levels (Li et al., 2012). To assess the impact of expression-level change on the reduced Flot1 and Flot2 levels measured by ABE/SILAM, we have used immunoblot-based quantification to analyze and compare levels of these two proteins in whole brain homogenates from WT and Hip14-gt mice (Fig. 2A). Significant expression-level reductions of 43% and 28% are seen for Flot1 and Flot2, respectively, well in line with the 36% reductions found for both by ABE/SILAM (Table 1). This same immunoblot analysis showed 18% increased expression in Hip14-gt for GS (Fig. 2A). Though not reaching our significance threshold, the upward trend for GS in this immunoblot analysis is nonetheless again in line with the highly significant 12% increase seen by ABE/SILAM (Table 1). Thus, the palmitoyl-proteome change seen for Flot1, Flot2, and GS, appears to be well correlated with expression-level change. However, as noted above, cause and effect are not easily parsed. Reduced expression obviously will reduce the amount of palmitoyl-protein that is available to be ABE-purified. However, reduced palmitoylation, through effects on subcellular localization, also may impact protein turnover. Indeed, in addition to the reduced expression levels found for Flot1 and Flot2 (Fig. 2A), a more focused analysis also finds reduced levels of palmitoylation per protein in Hip14-gt versus WT brain homogenates (Fig. S3A).
FIGURE 2.

Flot1 and Flot2 expression levels and DHHC PAT specificities. (A) Quantitative immunoblotting was used to analyze expression-level change in Hip14-gt and WT brains for carbonic anhydrase II (CA II), flotillin 1 (Flot1), flotillin 2 (Flot2), and glutamine synthetase (GS). Whole brain homogenates from Hip14-gt and WT animals were analyzed by immunoblotting with specific antibodies. SNAP-25 was used as the normalization control. Example immunoblots are shown at left and quantified results represented as means +/− SD are shown at right (n = 3, * P = 0.01, ** P = 0.003). (B) Analysis of HIP14- and DHHC5-mediated palmitoylation of Flot1 and Flot2 in yeast. Yeast cells were co-transformed with two plasmids, a plasmid that drives constitutive expression of a DHHC PAT (HIP14, DHHC5, or empty vector control) and a plasmid for GAL1 promoter-inducible expression of the test substrate protein (SNAP-25, Flot1, or Flot2). Palmitoylation was assessed using a click chemistry-based approach in the expressing yeast cells were metabolically labeled with the alkynated palmitate analog ODYA (see Experimental Procedures). Substrate and enzyme proteins, anti-FLAG immune precipitated from protein extracts, were click-reacted with azido-Alexa647 allowing fluorographic detection. Anti-HA immunoblotting was used to monitor the levels of these proteins both within the immune precipitation (at left) and within the initial protein extracts (at right). See related Figure S3.
Interestingly, reduced Flot1 and Flot2 palmitoylation also was highlighted in a recent proteomic analysis of palmitoylation in neuronal stem cells, deficient for DHHC5 PAT (Li et al., 2012). Furthermore, co-transfection of DHHC5 with Flot2 into COS cells served to up-regulate Flot2 palmitoylation, suggesting that Flot2 likely is a DHHC5 substrate (Li et al., 2012). To test if the flotillins might also be HIP14 substrates, we have used the yeast cell as a context for co-expression of the mammalian flotillins with mammalian HIP14 (Fig. 2B). Support for the validity of this yeast approach is provided by a recent report, which assessed palmitoylation for several mammalian palmitoyl-proteins, co-expressed with a wide panel of the mammalian PATs in yeast (Ohno et al., 2012). For our analysis, in addition to looking at Flot1 and Flot2, we also examined SNAP-25, a well-documented HIP14 substrate (Greaves et al., 2009; Huang et al., 2009; Huang et al., 2004). SNAP-25, we find, is palmitoylated in HIP14-expressing cells, but not in DHHC5-expressing cells, nor in naive yeast cells, not expressing any mammalian PATs (Fig. 2B). In contrast, Flot1 and Flot2 are palmitoylated only in the DHHC5-expressing cells, not in the HIP14-expressing cells. Thus, at least in yeast, HIP14 is active against SNAP-25, but not the flotillins, while both flotillins are recognized by DHHC5. Therefore, Flot1 and Flot2 appear not to be bona fide HIP14 substrates. The strong reductions seen for these two proteins in the Hip14-gt palmitoyl-proteome likely is a secondary manifestation of the Hip14-gt disease process. As DHHC5 is regulated by a rapid degradative turnover mechanism (Li et al., 2012), we entertained the possibility that reduced flotillin palmitoylation might be a consequence of reduced DHHC5 levels in the Hip14-gt brain. However, our ABE/SILAM analysis does not reveal any major change in palmitoylated DHHC5 levels in Hip14-gt versus WT brain (Fig. S3B).
ABE/SILAM Analysis of the HD Mouse YAC128
We have also analyzed YAC128, a well-characterized mouse model of HD that expresses a full-length human mHTT transgene containing a 128 residue-long poly-glutamine string (Slow et al., 2003). As with the human disease, the YAC128 mouse shows a disease-free latent period, with frank disease becoming evident in terms of striatal atrophy and associated motor defects at ~10–12 months of age. For our ABE/SILAM analysis, 12 month-old YAC128 mice were compared to their WT littermates. Our analysis again focuses on the abundant palmitoyl-protein set (Table S2), with the 19 proteins showing significant change of >10% being listed in Table 3. Prominent within this list are a number of glial-specific proteins, including: carbonic anhydrase II (CA II; Car2), which is present mainly in astrocytes and oligodendrocytes; two myelin-associated oligodendrocyte proteins, the phosphodiesterase CNP and the transmembrane protein Mog; as well as three well known components of the astrocyte glutamate-glutamine pathway, namely glutamine synthetase (GS; Glul) and the two glutamate transporters GLT-1 (Slc1a2) and GLAST (Slc1a3). GLT-1, GLAST, and GS act together to limit brain levels of the excitatory neurotransmitter glutamate, limiting thus, potential excitotoxicity. GLT-1 and GLAST localize to the astrocytic plasma membrane where they act to clear glutamate from excitatory synapses to the astrocyte cytoplasm where it is converted into the less toxic glutamine by GS. Excitotoxicity is implicated as a key driver of neuronal cell death in many neurodegenerative disorders, including HD (Lau and Tymianski, 2010; Raymond et al., 2011). Reduced function of any of these three proteins (GLT-1, GLAST, or GS) would be expected to enhance the potential for excitotoxicity. CA II, the protein with the most prominent YAC128 reduction (Table 2), also likely plays an important role in maintaining the healthy brain environment. CA II, the most abundant brain carbonic anhydrase isoform, accelerates the equilibrium of CO2 and H2O with HCO3− and H+ and thus helps to maintain the proper brain acid-base balance, by ridding the brain of the excess CO2 waste produced by the high rate of neuronal respiration (Deitmer, 2002; Deitmer and Rose, 2010).
Table 3.
Palmitoyl-proteome change in YAC128 relative to WT littermate brain.
| proteomics | |||||
|---|---|---|---|---|---|
| Gene Symbol | Description | Change | Ref | ABE | click |
| Down in YAC128: | |||||
| Car2 | CA II; carbonic anhydrase II; mediates the interconversion of CO2 and H2O to H+ and HCO3−; localized in brain mainly to oligodendrocytes and astrocytes. | −42%*** | + | ||
| Glul | GS; glutamine synthetase; converts glutamate to glutamine; role implicated in excitotoxicity; localized in brain mainly to astrocytes. | −29%*** | + | ||
| Pde10a | cAMP and cGMP 3′,5′-cyclic phosphodiesterase 10A, cAMP-inhibited; localized mainly to striatum. | −27%*** | (1) | + | |
| Vdac3 | Voltage-dependent anion-selective channel protein 3; mitochondrial outer membrane porin. | −17%*** | + | + | |
| Uqcrc1 | Cytochrome b-c1 complex protein 1, mitochondrial | −16%** | + | ||
| Cnp | CNP II; 2′,3′-cyclic-nucleotide 3′-phosphodiesterase2; localizes to oligodendrocyte myelin membranes | −14%*** | (2) | + | + |
| Atp2b1 | PMCA1; plasma membrane calcium-transporting ATPase 1. | −14%*** | + | ||
| Atp2b4 | PMCA4; plasma membrane calcium-transporting ATPase 4. | −11%** | + | + | |
| Slc1a2 | GLT-1; EAAT2; glial high affinity glutamate transporter; astrocyte protein; mediates retrieval of synaptic glutamate; implicated role in excitotoxicity. | −11%** | (3) | + | |
| Up in YAC128: | |||||
| Gnas | Gαs; Gα heterotrimeric G protein component; stimulates adenylyl cyclase. | +10%*** | (4) | + | + |
| Lower Significance: | |||||
| Igsf8 | EWI-2; PGRL; CD316; immunoglobulin superfamily member; interacts with tetraspannins to regulate cell migration. | −27%* | (5) | + | + |
| Hadhb | Trifunctional enzyme (hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase), subunit beta, mitochondrial. | −24%* | + | + | |
| Slc1a3 | GLAST; EAAT1; glial high affinity glutamate transporter; astrocyte protein; mediates retrieval of synaptic glutamate; implicated role in excitotoxicity. | −14%* | + | ||
| Dlg4 | PSD-95; SAP-90; MAGK scaffolding protein that clusters NMDA glutamate receptors at synapse. | −13%* | (6) | + | + |
| Mog | Myelin oligodendrocyte glycoprotein; IgG superfamily; type 1 TM protein localizing to outer layer of myelin sheath; auto-antigen in multiple sclerosis; causal mutation in familial form of narcolepsy. | −13%* | + | ||
| Gria3 | GluR3; GluA3; AMPA glutamate receptor subunit. | −10%* | (7) | + | |
| Arvcf | Armadillo repeat protein deleted in velo-cardio-facial syndrome homolog. | +33%* | + | ||
| Uba1 | Ubiquitin-like modifier-activating enzyme 1; ubiquitin E1 enzyme; likely detected by ABE due to its transient thioester linkage to ubiquitin. | +17%* | + | + | |
| Arhgap21 | GAP for CDC42. | +11%* | + | ||
Data is reported for the proteins that show significant change (* P < 0.05; ** P < 0.01; *** P < 0.001) of greater than 10% in the YAC128 palmitoyl-proteome relative to the WT palmitoyl-proteome. The three rightmost columns provide the published evidence supporting palmitoylation documentation for each protein, including either a specific reference (see below), or a “+” indication that the protein was identified as likely palmitoylated in prior ABE- or click-based proteomic analyses (see Table S2 for references to the relevant proteomic studies and for the expanded ABE/SILAM dataset). Proteins that are expressed predominantly in either oligodendrocytes or astrocytes are underlined.
References:
Although ABE/SILAM detection infers palmitoylation, neither CA II nor GS have yet been definitively reported to be palmitoylated. As a test of this, we have assessed the hydroxylamine-dependence of their ABE purification (Fig. 3A). Hydroxylamine (HAm), with its cleavage of the acyl-cysteine thioester linkage, plays a central role in ABE. Indeed, parallel, minus-HAm reactions are often employed in ABE proteomic analyses to help distinguish palmitoyl-proteins from contaminant proteins that may purify non-specifically; bona fide palmitoyl-proteins show HAm-dependent purification (Kang et al., 2008; Roth et al., 2006; Wan et al., 2007; Yang et al., 2010). Using the acyl-RAC simplification of the ABE protocol (Forrester et al., 2011), CA II and GS both show strong, HAm-dependent purifications indicative of palmitoylation (Fig. 3A). Additional support for GS and CA II palmitoylation can be gleaned from the published literature: GS was detected in our large-scale ABE analysis of rat brain palmitoylation (Kang et al., 2008), while CA II was identified as a palmitoyl-protein in a click chemistry-based proteomic analysis in mouse brain (Li et al., 2012). In theory, both enzymes could have their functionality altered by palmitoylation. Both CA II and GS have been suggested to participate in membrane-localized metabolons, with import/export efficiency being enhanced by the direct association of the enzyme with the relevant plasma membrane transporter (Moraes and Reithmeier, 2012). Palmitoylation may help these enzymes gain access to their cognate membrane-localized transporters. For GS, an association with GLT-1 and GLAST glutamate transporters would allow for efficient conversion of glutamate into glutamine as it is brought into the astrocytes (Moraes and Reithmeier, 2012). While an interaction of GS with these transporters remains undocumented, some small support for this speculation comes from a co-localization for GS and GLAST at the surface of retinal Muller glial cells (Derouiche and Rauen, 1995). The CA II metabolon stands on firmer ground, with CA II documented to associate with several different plasma membrane bicarbonate and proton transporters (Li et al., 2002; Loiselle et al., 2004; Sterling et al., 2002; Vince and Reithmeier, 1998). Localization of such CA II metabolons to the astrocytic membrane sites that juxtapose with the cerebral vasculature (i.e. the astrocytic foot) could facilitate the coupled excretion of respiratory waste products from the brain.
FIGURE 3.
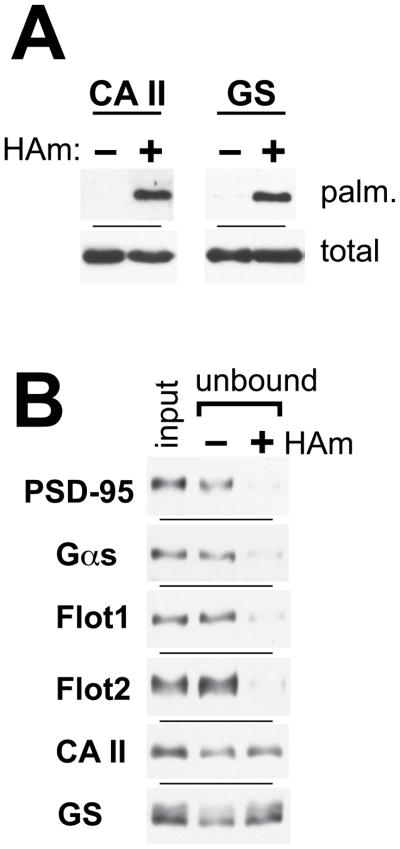
Analysis of carbonic anhydrase II (CA II) and glutamine synthetase (GS) palmitoylation. (A) CA II and GS both show hydroxylamine-dependent ABE purification indicative of palmitoylation. WT mouse brain homogenates were processed through parallel acyl-RAC purifications, either in the presence or absence of hydroxylamine (HAm). The two purified samples were blotted with antibodies specific to the two proteins (“palm.”). (B) Fractional palmitoylation was assessed by thiol-Sepharose-mediated depletion of indicated proteins, following acyl-RAC work-up of WT whole brain homogenates. As for panel A, homogenates were processed through parallel plus- and minus-HAm protocols, with the portion of the samples that failed to bind to the thiol-Sepharose (unbound) being compared to the starting homogenate (total) by immunoblotting with the indicated specific antibodies. See related Figure S4.
CA II and GS are generally appreciated to be soluble enzymes, which reside in the cytoplasm. Palmitoylation may apply to only a minor, membrane-targeted sub-population. The current palmitoylation detection methodologies do not provide information regarding palmitoylation stoichiometry and thus it is generally unclear if 90 or only 0.1% of the protein population is being modified. Here, to investigate this issue, we have used a modified acyl-RAC protocol (Forrester et al., 2011). Following the NEM-mediated thiol blockade and subsequent HAm-mediated thioester cleavage steps, the fraction of the protein population that is available for thiopropyl-Sepharose-mediated pull-down is assessed. The palmitoylated sub-population should bind to the resin and thus be removed, while the non-palmitoylated sub-population should remain unbound. We have examined CA II and GS, along with the two flottilins, as well as PSD-95 and Gαs (Fig. 3B). Flot1, Flot2, PSD-95, and Gαs all are efficiently captured, with 70–90% of each population being depleted in the plus-HAm condition, but not in the minus-HAm control condition. Thus, in WT brain, these proteins appear to be mainly palmitoylated. These results with the two “control” proteins, i.e. PSD-95 and Gαs, are significant, since both proteins are well known to undergo dynamic palmitoylation, cycling between palmitoylated and de-palmitoylated states (Conibear and Davis, 2010), with essentially nothing being known about their basal palmitoylation status in vivo. This analysis indicates that the palmitoylated forms of these two proteins predominate. In contrast to the high palmitoylation stoichiometries seen for four of the six proteins tested, CA II and GS both are quite poorly depleted by the thiol-Sepharose resin (Fig. 3B), indicating that palmitoylation likely applies only to a minor population of these two proteins (<10%). Using the assumption that both the capture by, and elution from, the thiol-Sepharose affinity resin is 100% efficient, we estimate a lower limit for the palmitoylation of CA II and GS of ~0.5% (Fig. S4), indicating that ~0.5–10% of these two protein populations are palmitoylated. This relatively low palmitoylation level may yet prove critical for targeting sub-populations of these enzymes to membranes, perhaps facilitating interaction with relevant membrane transporters.
The 42 and 29% decreases for CA II and GS detected by ABE/SILAM in YAC128 brain (Table 3), indicate sharp reductions for the palmitoylated sub-populations of these two enzymes. To test if reduced levels of the palmitoyl-forms, might reflect broader expression-level changes, whole brain homogenates from YAC128 and WT littermate mice were subjected to quantitative immunoblot analysis (Fig. 4A). Indeed, CA II and GS both show reduced expression levels in YAC128 homogenates that are roughly consistent with decreases seen by ABE/SILAM: CA II shows a significant 35% reduction (P = 0.017), while the 28% reduction detected for GS (P = 0.13) fell short of our significance threshold. Additional support for sharp and significant expression-level reductions for these two proteins is provided both by a whole proteome SILAM analysis of WT and YAC128 brain homogenates (data not shown), and by immunoblot analysis of dissected WT and YAC128 brain regions (Fig. 4B). In light of the low palmitoylation stoichiometries detected for both proteins (Fig. 3B), the sharply reduced expression levels measured here suggest that the large disease-induced changes detected by ABE/SILAM for CA II and GS likely are fundamentally the result of disordered expression, not disordered palmitoylation.
FIGURE 4.
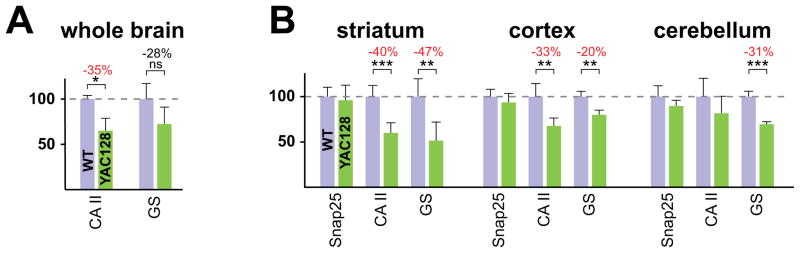
Carbonic anhydrase II (CA II) and glutamine synthetase (GS) expression level changes (A) Expression-level analysis of CA II and GS in 12 month-old YAC128 and WT littermate whole brain homogenates. An immunoblot analysis identical to that in Fig. 1C was employed (WT, n=2; YAC128, n=4). (B) Brain distribution of CA II and GS expression reductions. Homogenates from striatum, cortex and cerebellum dissected from five 15 month-old YAC128 and WT littermate brains were subjected to quantitative immunoblot analysis. Results are depicted as means +/− SD. Significance: * P < 0.05, ** P < 0.01, *** P < 0.001.
In human HD and in the YAC128 model, neuronal cell death is first evident in the striatum, with progression then to cortex and hippocampus and then finally, to the rest of the brain (Slow et al., 2003). To analyze the distribution of GS and CA II expression-level changes within the brain, we applied our quantitative immunoblot analysis to homogenates of striatum, cortex, and cerebellum dissected from YAC128 and WT littermate mice (Fig. 4B) at 15 months, a relatively late stage in the YAC128 disease process (Slow et al., 2003). Consistent with the spreading pathology typically seen at this stage, significant reductions for CA II and GS are seen in all three sub-regions, with the most extreme and significant reductions focused within the striatum (Fig. 4B).
Comparison of Hip14-gt and YAC128 Palmitoyl-Profiles
In light of the hypothesis that HIP14 dysfunction plays a causal role in HD pathogenesis (Singaraja et al., 2011), we were interested to compare the palmitoylation changes documented here for Hip14-gt and YAC128 brain. Do the two mice show similar palmitoylation derangements? For this, the 13 top Hip14-gt hits (Table 1) were assessed for correlated change in the YAC128 brain (Fig. 5A) and in a reciprocal analysis, the 10 top YAC128 hits (Table 3) were assessed for correlated Hip14-gt change (Fig. 5B). Overall, palmitoylation derangements for the two mice are not well correlated. Indeed, some proteins, e.g. Flot2, Arhgap21, Ank2, Gdi2, Gαs, and most notably GS, show significant change that is opposite in sign in the two mutant palmitoyl-proteomes. For instance, GS shows a highly significant 29% decrease in the YAC128 palmitoyl-proteome (P = 4 × 10−5), while showing significant 12% increase in the Hip14-gt palmitoyl-proteome (P = 3 × 10−4). Correlated change is however seen for some proteins, notably for CA II, Cnp, and Pde10a. It will be interesting to see if the proteins within this correlated subset play some driving role in the shared Hip14-gt/YAC128 pathologies (Singaraja et al., 2011). In addition, we have examined the Hip14-gt and YAC128 ABE/SILAM data for eight proteins previously identified as HIP14 substrates (Table 2). Some of these, notably CSP, SNAP-25, and PSD-95, do show subtle downward trends in both Hip14-gt and YAC128 brains, consistent with our prior report, which found HIP14 PAT activity to be reduced in YAC128 brain (Singaraja et al., 2011). Thus, while HIP14 dysfunction likely plays an important part in HD pathogenesis (Huang et al., 2011; Singaraja et al., 2011; Yanai et al., 2006), the absence of a strong correlation between Hip14-gt and YAC128 palmitoyl-proteome change suggests that HIP14 dysfunction is unlikely to be the sole disease driver.
FIGURE 5.

Correlating the Hip14-gt and YAC128 palmitoylation profiles. (A) Hip14-gt: YAC128 comparison. The thirteen proteins identified with the most significant ABE/SILAM change in Hip14-gt relative to WT littermates (from Table 1; red bars) were analyzed for change in YAC128 brain relative to its WT littermates (blue bars are indicative of significant change, while light blue bars indicate non-significant change). Significance levels: *P < 0.05, **P < 0.01, ***P < 0.001. Proteins are denoted by gene symbol, except for CA II (Car2; carbonic anhydrase II) and GS (Glul; glutamine synthetase). (B) YAC128:Hip14-gt comparison. The ten proteins identified with the most significant ABE/SILAM change in YAC128 (Table 2; blue bars) were analyzed for change in Hip14-gt (red bars for significant change, while change denoted by light red bars does not pass the significance threshold). Note that the change reported for each protein is change in mutant ABE palmitoyl-proteome levels relative to that derived from isogenic, age-matched WT littermates.
DISCUSSION
ABE/SILAM
ABE/SILAM builds upon our prior ABE/spectral count-based MS approaches that were used, for instance, to map the yeast palmitoyl-proteins with their modifying DHHC PATs (Roth et al., 2006). The SILAM addition substantially improves quantification: rather than the 2- to 5-fold differences, discernable by spectral count-based methods (Roth et al., 2006), ABE/SILAM allows significant change in the 5–10% range to be reliably detected. While ABE/SILAM should prove useful for analyzing other diseases where dysregulated palmitoylation is implicated (Cheng et al., 2009; Mansouri et al., 2005; Mizumaru et al., 2009; Mukai et al., 2008; Raymond et al., 2007; Tarpey et al., 2009; Vetrivel et al., 2009), it also may prove valuable even when palmitoylation is not directly involved. For instance, we have seen that the proteins showing the biggest palmitoyl-proteomic changes, e.g. Flot1 and Flot2 in Hip14-gt brain, and CA II and GS in YAC128 brain, also show correlated expression-level change. In theory, this change also could have been detected by a whole proteome SILAM analysis, omitting the cumbersome ABE purification steps. A big advantage of including the ABE purification, however, is that it focuses the analysis on just a limited set of functionally important proteins. MS-based proteomic analyses tend to be dominated by the most abundant sample proteins, with abundant housekeeping functions often obscuring interesting regulatory proteins, typically present at lower stoichiometries. By purifying the palmitoyl-proteome, ABE focuses the analysis on a much smaller set of proteins, with an outsized regulatory role. Indeed, the neural palmitoyl-proteome includes many of the well-known synaptic regulators (e.g. receptors, G proteins, scaffolds, and SNAREs), many of the ion channels that serve to initiate and propagate the nerve impulse, as well as many of the transmembrane proteins, mediating the cell-cell adhesions that wire the neuronal circuitry (Kang et al., 2008). Glial cell regulators, as the present work highlights, also are well represented. Overall, ABE/SILAM provides a look at neurological disease that is both detailed and broad.
YAC128 Palmitoyl-Proteome Change
The two proteins showing the most pronounced change in the YAC128 palmitoyl-proteome are CA II and GS, being reduced by 42% and 29%, respectively (Table 3). Both also show correlated expression-level reductions (Fig. 4A). As only small subpopulations of both enzymes appear to be palmitoylated (Fig. 3B), it is likely that expression-level change and not dysregulated palmitoylation is mainly responsible for the reduced ABE/SILAM detection. Indeed, reduced expression for both proteins has been previously documented in profiling analysis of other HD mouse models (Deschepper et al., 2011; Hodges et al., 2008; Kuhn et al., 2007; Luthi-Carter et al., 2000). Neither enzyme has yet been well studied within the HD disease process context. Both could well play important roles. As discussed below, reduced GS activity would be expected to elevate brain glutamate levels, enhancing excitotoxicity potential. CA II is the most abundant brain carbonic anhydrase, being predominantly expressed in oligodendrocytes and astrocytes, where it likely participates in the excretion of the large amounts of CO2 waste produced by the high neuronal respiratory rate. CO2 diffuses across out of neurons and into glial cells, where it is trapped by its CA II-mediated conversion into the ionic HCO3−, allowing for vectorial excretion to the circulatory system, likely by astrocytic bicarbonate transporters. Proper CO2 removal, obviously, is important to avoid toxic acidification. In humans, CA II-deficiency results in marble brain syndrome, a mental retardation that is part of wider syndrome that includes non-neural symptoms, such as osteopetrosis and impaired kidney function (Sly et al., 1983). The substantial brainwide reduction in CA-II, reported here, could easily play a major role in the HD disease process.
The GLT-1 (Slc1a2) glutamate transporter has been previously identified as a potential player in HD pathogenesis, showing reduced function and expression both in mouse HD models and in human HD (Arzberger et al., 1997; Behrens et al., 2002; Estrada-Sanchez et al., 2009; Hassel et al., 2008; Huang et al., 2010; Lievens et al., 2001; Nicniocaill et al., 2001). Indeed, we have previously reported reduced palmitoylation and transporter function for GLT-1 in YAC128 brain (Huang et al., 2010). Here, in addition to GLT-1, we also find significant reductions for two other key players in the astrocyte’s glutamate retrieval mechanism, these being GLAST (Slc1a3), a second astrocyte glutamate transporter that also participates in glutamate re-uptake from the synapse, and the astrocytic enzyme GS, which acts downstream of the transporters to convert the internalized glutamate into glutamine. Reduced function of any of these proteins would be expected to contribute to excitotoxic stress. The co-reduction of all three together, as we find here in YAC128 brain, provides strong support for excitotoxic models of HD pathogenesis.
Glial-Specific Change – Cause or Effect?
What is to be drawn from the preponderance of astrocytic and oligodendrocytic proteins among our top YAC128 hits (Table 3)? Distinguishing the causal change that drives the disease process from change that may be induced in response to the initial injury generally is difficult in profiling analyses like the present one. Both types of change may be therapeutically relevant. Interruption of the causal chain provides an obvious therapeutic strategy. On the other hand, secondary change often is part of the body’s natural ameliorative response. Thus, inducing or augmenting these secondary changes presents a second therapeutic path. The glial change identified here could well be secondary. Indeed, reactive gliosis, a process that involves proliferation, migration, and reprogramming of astrocytes, is a well-known neural response to brain injury (Allaman et al., 2011). Reactive gliosis can be induced either by trauma or by neurodegenerative injury and is well documented in HD (Vonsattel et al., 1985). However, down-regulation of the glutamate-glutamine pathway as is reported here, generally is not part of the gliotic response (Sofroniew, 2009). Nevertheless, such changes could well be part of the HD-specific response to mHTT-induced injury. Alternatively, some of the identified glial change could be causal. HD is a neurodegenerative disease and its disease symptomology is best understood as resulting from neuronal cell death. Thus, over the years, it has made most sense to look within the neuron for the disease origins. However, neuronal cell death can also be the result of reduced glial support. Interestingly, transgenic mice, engineered to express mHTT exclusively within astrocytes, recapitulate key HD pathologies (Bradford et al., 2009), while mice engineered to express mHTT in discrete neuronal populations, including within striatal medium spiny neurons (MSNs), surprisingly, do not manifest HD-like phenotypes (Gu et al., 2007; Gu et al., 2005). Thus, impaired glial cell support could well be a major disease driver. In conclusion, our analysis lends further credence to models that see mHTT-induced glial dysfunction as being a major contributor to HD pathogenesis.
EXPERIMENTAL PROCEDURES
ABE/SILAM Proteomic Analysis
The two ABE/SILAM analyses of this report compared brain palmitoyl-proteomes for 3 month-old Hip14-gt mice to their WT littermates (non-transgenic siblings) and 12 month-old YAC128 mice to their WT littermates. These mutant and WT mice were raised on standard mouse chow and thus are, in effect, 14N-labeled. To generate these comparisons, palmitoylation in the individual mutant and WT test mouse brain were first compared to palmitoylation in the 15N-labeled reference mouse brain, these mice being wild-type C57Bl/6 mice raised on a diet for which the protein component was comprised exclusively of pelletized, 15N-labeled Spirulina algae. For these first level comparisons, palmitoyl-proteins were prepared by ABE purification from a 1:1 mixing of whole brain homogenates from the individual mutant and WT14N-test mice each with 15N-reference mouse homogenate. Details of the ABE protocol, which effects the exchange of thioester-linked acyl moieties for biotin tags and then purifies the tagged palmitoyl-proteins via streptavidin-agarose binding, are provided in the Supplemental Experimental Procedures. These ABE-purified, N14/N15-samples were then prepared for mass spectrometry by TCEP reduction, iodoacetamide alkylation, and overnight trypsinization. Samples were analyzed by MuDPIT (LC/LC/MS/MS) on a LTQ Orbitrap XL instrument (Thermo Finnigan, Palo Alto, CA) (for details, see Supplemental Experimental Procedures). Tandem mass spectra were searched against a combined light (14N) and heavy (15N) versions of the EBI-IPI mouse protein database (EBI-IPI_mouse_01-01-09), with each identified peptide being subsequently matched to either its heavy or light counterpart peptide by Census, a software suite designed for quantitative analysis of MS/MS data from stable isotope-labeled samples (Park et al., 2008). In brief, for each identified 14N- or 15N-peptide, Census constructs a chromatogram from the MS1 data and then, based on the predicted 14N- and 15N-peptide mass differences, finds the MS1 peak for the counterpart peptide. A comparison of 14N- and 15N-peak volumes yields a 14N/15N-ratio for each identified peptide, essentially a datapoint reporting on the relative abundance of the associated palmitoyl-protein in test and reference brain. Abundant palmitoyl-proteins typically are identified by multiple peptides per MS/MS run, thus, multiple datapoints. As described above (Results section), our analysis has largely focused on the ~300 palmitoyl-proteins, identified, on average, by five or more quantifiable peptides per MS/MS run. The comparison of interest, the comparison of individual palmitoyl-protein abundance levels within the mutant palmitoyl-proteome to levels within the isogenic WT palmitoyl-proteomes, is then derived via a ratio of ratios of approach, which compares the median 14N/15N-peptide ratio from mixed 14N/15N-mutant samples to the median ratio derived from the WT sample. For the Hip14-gt analysis, a total of eight samples were analyzed, four 14N/15N samples analyzed each for Hip14-gt and for WT littermate mice, corresponding to three individual mice plus one technical replicate for each genotype. The YAC128 relied on the analysis of four YAC128 brains and two WT littermate brains.
Data sharing
RAW files and complete parameter files will be publically available at http://fields.scripps.edu/published/ABE_SILAM upon publication.
Antibodies and Immunoblot Quantification
Analysis of Substrate Specificity in Yeast
For this analysis, which assessed the palmitoylation activity of two mammalian DHHC PATs, HIP14 and DHHC5, against several potential mammalian substrates, namely Snap25, Flot1, and Flot2, cDNAs both for the PATs and for the substrate proteins were subcloned as epitope-tagged constructs into appropriate yeast expression vectors, allowing their co-expression in yeast cells. Substrate protein palmitoylation was assessed via a click-labeling protocol (Charron et al., 2009; Roth et al., 2011), for which cultured, co-expressing yeast cells were metabolically labeled with the alkynated palmitate analog, 17-octadecynoic acid (ODYA; Cayman Chemical Corp.; Ann Arbor, MI). Subsequently, extracts were prepared, allowing PAT and substrate proteins to be purified by immune-precipitation and then click-reacted with Alexa Fluor 647 Azide (Life Technologies; Grand Island, NY). Palmitoylation then was assessed by SDS-PAGE followed by fluorography. Details of both the plasmid constructions and the click labeling are provided within the Supplemental Experimental Procedures.
Acyl-RAC-Based Analysis and Palmitoylation Stoichiometry
As detailed within the Supplemental Experimental Procedures, a modified acyl-RAC protocol (Forrester et al., 2011) was used both to confirm palmitoylation several proteins identified by the ABE/SILAM analysis and to examine fractional palmitoylation status for selected individual proteins. The starting points for both analyses were whole brain homogenates deriving from 3 month-old wild-type mice, i.e. the f1 progeny from a C3H x C57Bl/6 cross.
Supplementary Material
Highlights.
ABE/SILAM proteomically tracks brain palmitoylation change.
ABE/SILAM provides a broad and accurate survey of the palmitoyl-proteome.
Disease-induced changes in a mouse model of Huntington Disease were analyzed.
Major hits include carbonic anhydrase-II and regulators of glutamatergic excitotoxicity.
SIGNIFICANCE.
ABE/SILAM tracks palmitoylation change within the whole animal, providing broad and accurate looks, able to discern subtle change in the 5–10% range. With the emerging connections that now link dysregulated palmitoylation to neurological disease and cancer, this approach should help identify the key palmitoylation-dependent targets and pathways that drive the disease process. Furthermore, as drugs are developed to modify disease course through inhibition either of the individual palmitoyl transferases that add these modifications or the thioesterases that remove them, ABE/SILAM will provide a way of assessing drug actions in vivo. Here, the first ABE/SILAM applications to mouse models of HD highlight both the power and limitations of this approach. Intriguingly, much of the change identified in the HD model mouse YAC128 maps to glial-specific palmitoyl-proteins, with major reductions seen for carbonic anhydrase-II, an enzyme which helps rid the brain of its otherwise toxic CO2 waste, and also for three well-known regulators of glutamatergic excitotoxicity, these being glutamine synthetase and the GLT-1 and GLAST glutamate transporters.
Acknowledgments
We thank Robin (Sung Kyu) Park for his expert help with the Census program. Grant support for this work was provided by the NIH: R01GM65525 (N.G.D.), R01MH067880 (J.R.Y.), P41-GM103533 (J.R.Y.), F32-AG039127 (J.N.S.), and by the Canadian Institutes of Health Research grant: (CIHR) GPG-102165. Fellowship support for S. Sanders was provided from both the CIHR and the Michael Smith Foundation for Health Research. M. Hayden is a Killam University Professor and holds a Canada Research Chair in Human Genetics and Molecular Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat Chem Biol. 2011;7:469–478. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal HC, Sprinkle TJ, Agrawal D. 2′,3′-cyclic nucleotide-3′-phosphodiesterase in the central nervous system is fatty-acylated by thioester linkage. J Biol Chem. 1990;265:11849–11853. [PubMed] [Google Scholar]
- Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease--an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Beranger F, Tavitian A, de Gunzburg J. Post-translational processing and subcellular localization of the Ras-related Rap2 protein. Oncogene. 1991;6:1835–1842. [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106:22480–22485. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- Charych EI, Jiang LX, Lo F, Sullivan K, Brandon NJ. Interplay of palmitoylation and phosphorylation in the trafficking and localization of phosphodiesterase 10A: implications for the treatment of schizophrenia. J Neurosci. 2010;30:9027–9037. doi: 10.1523/JNEUROSCI.1635-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Vetrivel KS, Drisdel RC, Meckler X, Gong P, Leem JY, Li T, Carter M, Chen Y, Nguyen P, Iwatsubo T, Tomita T, Wong PC, Green WN, Kounnas MZ, Thinakaran G. S-palmitoylation of gamma-secretase subunits nicastrin and APH-1. J Biol Chem. 2009;284:1373–1384. doi: 10.1074/jbc.M806380200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conibear E, Davis NG. Palmitoylation and depalmitoylation dynamics at a glance. J Cell Sci. 2010;123:4007–4010. doi: 10.1242/jcs.059287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona ML, Matthies HJ, Pau K, Bowton E, Speed N, Lute BJ, Anderson M, Sen N, Robertson SD, Vaughan RA, Rothman JE, Galli A, Javitch JA, Yamamoto A. Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat Neurosci. 2011;14:469–477. doi: 10.1038/nn.2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitmer JW. A role for CO(2) and bicarbonate transporters in metabolic exchanges in the brain. J Neurochem. 2002;80:721–726. doi: 10.1046/j.0022-3042.2002.00765.x. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Rose CR. Ion changes and signalling in perisynaptic glia. Brain Res Rev. 2010;63:113–129. doi: 10.1016/j.brainresrev.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Derouiche A, Rauen T. Coincidence of L-glutamate/L-aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: evidence for coupling of GLAST and GS in transmitter clearance. J Neurosci Res. 1995;42:131–143. doi: 10.1002/jnr.490420115. [DOI] [PubMed] [Google Scholar]
- Deschepper M, Hoogendoorn B, Brooks S, Dunnett SB, Jones L. Proteomic changes in the brains of Huntington’s disease mouse models reflect pathology and implicate mitochondrial changes. Brain Res Bull. 2011 doi: 10.1016/j.brainresbull.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Green WN. Labeling and quantifying sites of protein palmitoylation. Biotechniques. 2004;36:276–285. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- Estrada-Sanchez AM, Montiel T, Segovia J, Massieu L. Glutamate toxicity in the striatum of the R6/2 Huntington’s disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol Dis. 2009;34:78–86. doi: 10.1016/j.nbd.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PJ. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res. 2011;52:393–398. doi: 10.1194/jlr.D011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick M, Bright NA, Riento K, Bray A, Merrified C, Nichols BJ. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr Biol. 2007;17:1151–1156. doi: 10.1016/j.cub.2007.05.078. [DOI] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Gkantiragas I, Brugger B, Stuven E, Kaloyanova D, Li XY, Lohr K, Lottspeich F, Wieland FT, Helms JB. Sphingomyelin-enriched microdomains at the Golgi complex. Mol Biol Cell. 2001;12:1819–1833. doi: 10.1091/mbc.12.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8:46–54. doi: 10.1038/ncb1342. [DOI] [PubMed] [Google Scholar]
- Greaves J, Chamberlain LH. DHHC palmitoyl transferases: substrate interactions and (patho)physiology. Trends Biochem Sci. 2011;36:245–253. doi: 10.1016/j.tibs.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Greaves J, Gorleku OA, Salaun C, Chamberlain LH. Palmitoylation of the SNAP25 protein family: specificity and regulation by DHHC palmitoyl transferases. J Biol Chem. 2010;285:24629–24638. doi: 10.1074/jbc.M110.119289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Prescott GR, Fukata Y, Fukata M, Salaun C, Chamberlain LH. The hydrophobic cysteine-rich domain of SNAP25 couples with downstream residues to mediate membrane interactions and recognition by DHHC palmitoyl transferases. Mol Biol Cell. 2009;20:1845–1854. doi: 10.1091/mbc.E08-09-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Salaun C, Fukata Y, Fukata M, Chamberlain LH. Palmitoylation and membrane interactions of the neuroprotective chaperone cysteine-string protein. J Biol Chem. 2008;283:25014–25026. doi: 10.1074/jbc.M802140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Andre VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Hassel B, Tessler S, Faull RL, Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem Res. 2008;33:232–237. doi: 10.1007/s11064-007-9463-1. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rumbaugh G, Huganir RL. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron. 2005;47:709–723. doi: 10.1016/j.neuron.2005.06.035. [DOI] [PubMed] [Google Scholar]
- He B, Zhang YH, Richardson MM, Zhang JS, Rubinstein E, Zhang XA. Differential functions of phospholipid binding and palmitoylation of tumour suppressor EWI2/PGRL. The Biochemical journal. 2011;437:399–411. doi: 10.1042/BJ20101381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Hughes G, Brooks S, Elliston L, Holmans P, Dunnett SB, Jones L. Brain gene expression correlates with changes in behavior in the R6/1 mouse model of Huntington’s disease. Genes Brain Behav. 2008;7:288–299. doi: 10.1111/j.1601-183X.2007.00350.x. [DOI] [PubMed] [Google Scholar]
- Hou H, John Peter AT, Meiringer C, Subramanian K, Ungermann C. Analysis of DHHC acyltransferases implies overlapping substrate specificity and a two-step reaction mechanism. Traffic. 2009;10:1061–1073. doi: 10.1111/j.1600-0854.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- Huang K, Kang MH, Askew C, Kang R, Sanders SS, Wan J, Davis NG, Hayden MR. Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2010;40:207–215. doi: 10.1016/j.nbd.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Huang K, Sanders S, Singaraja R, Orban P, Cijsouw T, Arstikaitis P, Yanai A, Hayden MR, El-Husseini A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Sanders SS, Kang R, Carroll JB, Sutton L, Wan J, Singaraja R, Young FB, Liu L, El-Husseini A, Davis NG, Hayden MR. Wild-type HTT modulates the enzymatic activity of the neuronal palmitoyl transferase HIP14. Hum Mol Genet. 2011;20:3356–3365. doi: 10.1093/hmg/ddr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, Hayden MR, El-Husseini A. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR, 3rd, Davis NG, El-Husseini A. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin R, Shelbourne P, Delorenzi M, Augood SJ, Faull RL, Olson JM, Bates GP, Jones L, Luthi-Carter R. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Li X, Alvarez B, Casey JR, Reithmeier RA, Fliegel L. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem. 2002;277:36085–36091. doi: 10.1074/jbc.M111952200. [DOI] [PubMed] [Google Scholar]
- Li Y, Martin BR, Cravatt BF, Hofmann SL. DHHC5 protein palmitoylates flotillin-2 and is rapidly degraded on induction of neuronal differentiation in cultured cells. J Biol Chem. 2012;287:523–530. doi: 10.1074/jbc.M111.306183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Linder ME, Middleton P, Hepler JR, Taussig R, Gilman AG, Mumby SM. Lipid modifications of G proteins: alpha subunits are palmitoylated. Proc Natl Acad Sci U S A. 1993;90:3675–3679. doi: 10.1073/pnas.90.8.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo S, Greentree WK, Linder ME, Deschenes RJ. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2002;277:41268–41273. doi: 10.1074/jbc.M206573200. [DOI] [PubMed] [Google Scholar]
- Loiselle FB, Morgan PE, Alvarez BV, Casey JR. Regulation of the human NBC3 Na+/HCO3− cotransporter by carbonic anhydrase II and PKA. Am J Physiol Cell Physiol. 2004;286:C1423–1433. doi: 10.1152/ajpcell.00382.2003. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Mansouri MR, Marklund L, Gustavsson P, Davey E, Carlsson B, Larsson C, White I, Gustavson KH, Dahl N. Loss of ZDHHC15 expression in a woman with a balanced translocation t(X;15)(q13.3;cen) and severe mental retardation. Eur J Hum Genet. 2005;13:970–977. doi: 10.1038/sj.ejhg.5201445. [DOI] [PubMed] [Google Scholar]
- McClatchy DB, Dong MQ, Wu CC, Venable JD, Yates JR., 3rd 15N metabolic labeling of mammalian tissue with slow protein turnover. J Proteome Res. 2007a;6:2005–2010. doi: 10.1021/pr060599n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchy DB, Liao L, Park SK, Venable JD, Yates JR. Quantification of the synaptosomal proteome of the rat cerebellum during post-natal development. Genome Res. 2007b;17:1378–1388. doi: 10.1101/gr.6375007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumaru C, Saito Y, Ishikawa T, Yoshida T, Yamamoto T, Nakaya T, Suzuki T. Suppression of APP-containing vesicle trafficking and production of beta-amyloid by AID/DHHC-12 protein. J Neurochem. 2009;111:1213–1224. doi: 10.1111/j.1471-4159.2009.06399.x. [DOI] [PubMed] [Google Scholar]
- Moraes TF, Reithmeier RA. Membrane transport metabolons. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamem.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Morrow IC, Rea S, Martin S, Prior IA, Prohaska R, Hancock JF, James DE, Parton RG. Flotillin-1/reggie-2 traffics to surface raft domains via a novel golgi-independent pathway. Identification of a novel membrane targeting domain and a role for palmitoylation. J Biol Chem. 2002;277:48834–48841. doi: 10.1074/jbc.M209082200. [DOI] [PubMed] [Google Scholar]
- Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos JA. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann-Giesen C, Falkenbach B, Beicht P, Claasen S, Luers G, Stuermer CA, Herzog V, Tikkanen R. Membrane and raft association of reggie-1/flotillin-2: role of myristoylation, palmitoylation and oligomerization and induction of filopodia by overexpression. The Biochemical journal. 2004;378:509–518. doi: 10.1042/BJ20031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicniocaill B, Haraldsson B, Hansson O, O’Connor WT, Brundin P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in R6/1 Huntington transgenic mice. Eur J Neurosci. 2001;13:206–210. doi: 10.1046/j.0953-816x.2000.01379.x. [DOI] [PubMed] [Google Scholar]
- Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Kashio A, Ogata R, Ishitomi A, Yamazaki Y, Kihara A. Analysis of substrate specificity of human DHHC protein acyltransferases using a yeast expression system. Mol Biol Cell. 2012;23:4543–4551. doi: 10.1091/mbc.E12-05-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama T, Verstreken P, Ly CV, Rosenmund T, Rajan A, Tien AC, Haueter C, Schulze KL, Bellen HJ. Huntingtin-interacting protein 14, a palmitoyl transferase required for exocytosis and targeting of CSP to synaptic vesicles. J Cell Biol. 2007;179:1481–1496. doi: 10.1083/jcb.200710061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, Yates JR., 3rd A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods. 2008;5:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond FL, Tarpey PS, Edkins S, Tofts C, O’Meara S, Teague J, Butler A, Stevens C, Barthorpe S, Buck G, Cole J, Dicks E, Gray K, Halliday K, Hills K, Hinton J, Jones D, Menzies A, Perry J, Raine K, Shepherd R, Small A, Varian J, Widaa S, Mallya U, Moon J, Luo Y, Shaw M, Boyle J, Kerr B, Turner G, Quarrell O, Cole T, Easton DF, Wooster R, Bobrow M, Schwartz CE, Gecz J, Stratton MR, Futreal PA. Mutations in ZDHHC9, which encodes a palmitoyltransferase of NRAS and HRAS, cause X-linked mental retardation associated with a Marfanoid habitus. Am J Hum Genet. 2007;80:982–987. doi: 10.1086/513609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA, Andre VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–273. doi: 10.1016/j.neuroscience.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- Roth AF, Feng Y, Chen L, Davis NG. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol. 2002;159:23–28. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Papanayotou I, Davis NG. The yeast kinase Yck2 has a tripartite palmitoylation signal. Mol Biol Cell. 2011;22:2702–2715. doi: 10.1091/mbc.E11-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, 3rd, Davis NG. Global analysis of protein palmitoylation in yeast. Cell. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm C, Zoidl G, Koesling D, Russwurm M. Dual acylation of PDE2A splice variant 3: targeting to synaptic membranes. J Biol Chem. 2009;284:25782–25790. doi: 10.1074/jbc.M109.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR, 3rd, Hetzer MW. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942. doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, Yanai A, Gutekunst CA, Leavitt BR, Yi H, Fichter K, Gan L, McCutcheon K, Chopra V, Michel J, Hersch SM, Ikeda JE, Hayden MR. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- Singaraja RR, Huang K, Sanders SS, Milnerwood AJ, Hines R, Lerch JP, Franciosi S, Drisdel RC, Vaid K, Young FB, Doty C, Wan J, Bissada N, Henkelman RM, Green WN, Davis NG, Raymond LA, Hayden MR. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 2011;20:3899–3909. doi: 10.1093/hmg/ddr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci U S A. 1983;80:2752–2756. doi: 10.1073/pnas.80.9.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling D, Brown NJ, Supuran CT, Casey JR. The functional and physical relationship between the DRA bicarbonate transporter and carbonic anhydrase II. Am J Physiol Cell Physiol. 2002;283:C1522–1529. doi: 10.1152/ajpcell.00115.2002. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O’Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, de Brouwer AP, van Bokhoven H, Van Esch H, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topinka JR, Bredt DS. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron. 1998;20:125–134. doi: 10.1016/s0896-6273(00)80440-7. [DOI] [PubMed] [Google Scholar]
- Valdez-Taubas J, Pelham H. Swf1-dependent palmitoylation of the SNARE Tlg1 prevents its ubiquitination and degradation. EMBO J. 2005;24:2524–2532. doi: 10.1038/sj.emboj.7600724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Meckler X, Chen Y, Nguyen PD, Seidah NG, Vassar R, Wong PC, Fukata M, Kounnas MZ, Thinakaran G. Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J Biol Chem. 2009;284:3793–3803. doi: 10.1074/jbc.M808920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince JW, Reithmeier RA. Carbonic anhydrase II binds to the carboxyl terminus of human band 3, the erythrocyte C1−/HCO3− exchanger. J Biol Chem. 1998;273:28430–28437. doi: 10.1074/jbc.273.43.28430. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nature protocols. 2007;2:1573–1584. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., 3rd Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem. 2004;76:4951–4959. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, Hayden MR. Palmitoylation of huntingtin by HIP14is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010;9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young FB, Butland SL, Sanders SS, Sutton LM, Hayden MR. Putting proteins in their place: palmitoylation in Huntington disease and other neuropsychiatric diseases. Prog Neurobiol. 2012;97:220–238. doi: 10.1016/j.pneurobio.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.