

Abstract
A role for the flavoprotein NRH:quinone oxidoreductase 2 (NQO2, QR2) in human diseases such as malaria, leukemia and neurodegeneration has been proposed. In order to explore the potential of NQO2 as a therapeutic target, we have developed potent and selective mechanism-based inhibitors centered on the indolequinone pharmacophore. The compounds show remarkable selectivity for NQO2 over the closely related flavoprotein NQO1 with small structural changes defining selectivity. Biochemical studies confirmed mechanism-based inhibition while X-ray crystallography and mass spectrometry revealed the nature of the inhibitor interaction with the protein. These indolequinones represent the first mechanism-based inhibitors of NQO2, and their novel mode of action involving alkylation of the flavin cofactor, provides significant advantages over existing competitive inhibitors in terms of potency and irreversibility, and will open new opportunities to define the role of NQO2 in disease.
Keywords: quinones, redox chemistry, enzymes, inhibitors, protein structure
Introduction
The quinone reductase NRH:quinone oxidoreductase 2 (NQO2, QR2) has been implicated in the etiology of a variety of diseases.[1,2] Thus, the identification of a range of structurally diverse competitive inhibitors of NQO2 such as quercetin, resveratrol, melatonin, chloroquine and imatinib (Figure 1) has generated considerable interest, and focused attention on the role of the enzyme in diseases such as malaria, leukemia, and in neurodegenerative disorders. NQO2 has been identified as a new melatonin binding site (MT3),[3,4] linking the antioxidant effects of the neurohormone with a possible mechanism of action. More recently it has been shown that NQO2 functions as a catechol quinone reductase,[5] can reduce potentially toxic estrogen ortho-quinones,[6] and may have an important role in the regulation of catecholamine oxidation,[5] and hence their implication in the etiology of neurogenerative conditions such as Parkinson’s disease. The possible role of NQO2 in malaria and cancer has been highlighted by the discovery that it is a molecular target for the anti-malarial drug chloroquine,[7,8] and also for the clinically used anti-leukemia drug imatinib.[9,10] The fact that NQO2 might also represent the first identified target for resveratrol,[11] backed up by X-ray crystallographic studies,[12] has added substance to the role of this much discussed chemopreventive agent. However, in order to understand the molecular basis of the relationship between NQO2 activity and its role in disease, and to exploit its potential as a chemotherapeutic target, new chemical biology tools are required in the form of potent and selective mechanism-based inhibitors. In this study we report the first mechanism-based inhibitors of NQO2 and demonstrate that small structural variations dramatically alter the specificity of compounds for either NQO2 or the closely related reductase NQO1 (QR1). We also describe a novel mechanism of inhibition of NQO2 that involves electrophilic attack on the FAD cofactor, rather than the apoprotein as observed for the closely related reductase NQO1.
Figure 1.
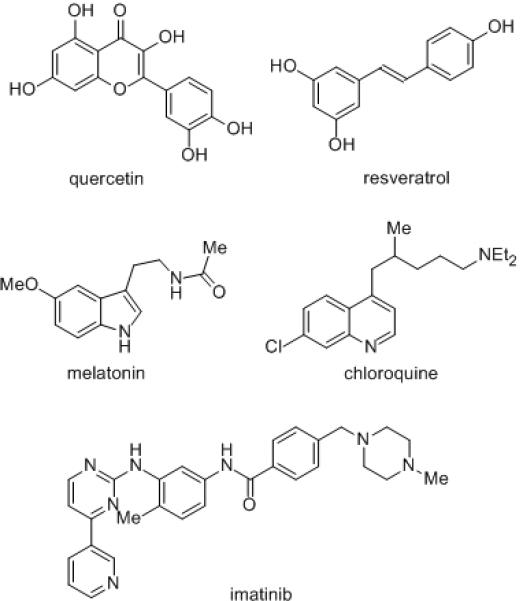
Competitive inhibitors of NQO2.
Results and Discussion
The two-electron reductase NQO2,[1,2] originally reported in 1961,[13] was “rediscovered” some 30 years later.[14] Not as widely expressed as the related and much better known enzyme NQO1 (QR1),[15-17] NQO2 is assumed to adopt a similar detoxification function, in that it can catalyze the reduction of quinones directly to hydroquinones bypassing the semiquinone intermediate and protecting cells against the potentially toxic effects of quinones. It is a 231-amino-acid homodimer, and like NQO1 is also a flavin-dependent reductase,[18] with a molecule of flavin adenine dinucleotide (FAD) per subunit as a cofactor tightly, but noncovalently, bound to the enzyme. However, unlike NQO1 that can efficiently utilize either NADH or NADPH as reducing cofactors, NQO2 uses N-ribosylnicotinamide (NRH) as a source of reducing equivalents. The structural details of NQO2 and its binding of FAD and simple quinone substrates have been confirmed by protein X-ray crystallography.[19] The natural substrate(s), if any, of NQO2 remain unknown, but this enzyme can metabolically activate a range of quinone substrates including menadione and coenzyme Q0.[1] In addition, NQO2 can also function as a nitroreductase in activating the antitumor drug 5-aziridinyl-2,4-dinitrobenzamide (CB1954).[20,21]
During our long-standing interest in quinone reductases, we have undertaken a detailed chemical biology study on the correlation of quinone structure with rate of metabolism by NQO1 and toxicity towards human tumor cell lines.[22,23] As part of these studies, we developed a specific, and potent mechanism-based inhibitor of NQO1, 5-methoxy-1,2-dimethyl-3-(4-nitrophenoxymethyl)indole-4,7-dione 1a, a compound known as ES936, and fully characterized its interaction with NQO1 using a range of biochemical techniques.[23,24] These studies confirmed a suicide-substrate mechanism in which, following enzymatic reduction, the leaving group (4-nitrophenoxide) in indolequinone 1a is eliminated to generate an electrophilic iminium ion (cf. Step C, Scheme 1),[25-28] which subsequently alkylates the enzyme in the active site, the various stages of which are outlined in Scheme 1. We also demonstrated the therapeutic potential of such compounds by their potent activity against human pancreatic tumor cells.[29,30] In contrast to NQO1, there are no known mechanism-based inhibitors of NQO2. Such inhibitors would be extremely valuable biochemical tools to characterize the role of this enzyme in leukemia, malaria and neurodegenerative disorders.
Scheme 1.
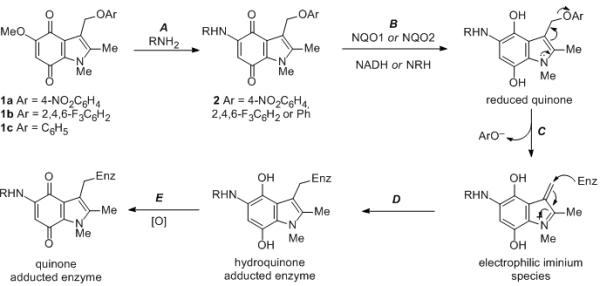
Preparation of indolequinone inhibitors (step A), and proposed mechanism of enzyme inactivation by covalent modification involving enzymatic reduction of quinone (step B), loss of leaving group from 3-indolylmethyl position (step C), and attack by the enzyme (as a nucleophile) on the electrophilic iminium ion to give the hydroquinone adducted enzyme (step D), followed by aerial oxidation to the quinone-enzyme adduct (step E). Compounds 2; a, R = H, Ar = 2,4,6-F3C6H2; b, R = CH3(CH2)4, Ar = 2,4,6-F3C6H2; c, R = H2N(CH2)4, Ar = 4-NO2C6H2; d, R = H2N(CH2)4, Ar = 2,4,6-F3C6H2; e, R = H2N(CH2)8, Ar = 2,4,6-F3C6H2; f, R = Me2N(CH2)3, Ar = 2,4,6-F3C6H2; g, R = Me2N(CH2)2, Ar = 2,4,6-F3C6H2; h, R = Me2N(CH2)3, Ar = 2,4,6-F3C6H2; i, R = Me2N(CH2)3, Ar = 2,4,6-F3C6H2; j, R = Me2N(CH2)3, Ar = C6H5. For compounds 2h and 2i, the 2-methyl group is replaced by H and phenyl respectively.
Initially, known inhibitors of NQO1 were screened against human recombinant NQO2 in the presence and absence of the reducing cofactor NRH. Interestingly, potent inhibitors of NQO1 such as indolequinone 1a which are very efficient at inactivating NQO1 in the presence of reducing cofactor NADH (Figure 2, partition ratio of 3.5),[31] did not significantly inhibit the NQO2 enzyme even at 1 μM in the presence or absence of cofactor NRH (Figure 2, partition ratio 135). These data demonstrate that indolequinone 1a at nanomolar concentrations is highly selective as an inhibitor of NQO1 over NQO2. To develop an inhibitor that was highly selective for NQO2 over NQO1, we elected to modify the indolequinone core structure, initially at C-5, taking advantage of the facile displacement of the 5-methoxy group with nucleophiles (step A, Scheme 1). A range of novel compounds was therefore prepared from 1a, and the equally potent NQO1 inhibitor 1b (that bears an alternative phenolic leaving group – 2,4,6-trifluorophenol),[31] and assayed against both the quinone reductase enzymes. Even a relatively simple structural modification – the displacement of the methoxy group in quinone 1b with ammonia – brought about a substantial change in that the resulting quinone 2a inhibited both quinone reductases (Figure 2). Since the catalytic site of NQO2 is larger and more hydrophobic than that of NQO1, we chose to introduce a more lipophilic side chain by reaction of the 5-methoxy quinone 1b with pentylamine, although the resulting compound 2b proved an ineffective inhibitor for both enzymes. However, when a 4-aminobutylamino substituent was introduced by reaction of either the 4-nitro- or the 2,4,6-trifluoro-phenoxy quinones 1a or 1b with 1,4-diaminobutane, the desired selectivity for inhibition of NQO2 over NQO1 was observed. Thus, indolequinone 2c, a compound known as MAC627, was a potent mechanism-based inhibitor of NQO2 (partition ratio 2.0), whilst essentially inactive against NQO1 (Figure 2). Compound 2d exhibited a very similar profile, confirming that, just as with related NQO1 inhibitors, the nature of the leaving group (4-nitrophenol vs. 2,4,6-trifluorophenol in this case) has little effect on inhibition provided that they have similar pKa values.
Figure 2.

Inhibition of recombinant human NQO1 and NQO2 by indolequinones 1a and 2a – 2j. The activities of NQO1 (top panel) and NQO2 (bottom panel) were measured after treatment with indolequinones (1 μM) in the absence (solid bars) and presence (striped bars) of the relevant reducing cofactor (NADH and NRH respectively). Results are the mean ± standard deviation of three separate determinations. [1a* NQO1 inhibition studies with 1a were performed with an inhibitor concentration of 0.1 μM]
With a potential mechanism-based inhibitor of quinone reductase 2 now available, we explored the structural requirements for this selective enzyme inhibition. The advantages of a side chain with a terminal amino group was already clear from comparison of compounds 2b and 2c, although a longer side chain as in the 8-aminooctylamino compound 2e was less satisfactory. Compounds containing shorter three or two carbon substituents terminating in dimethylamino groups, on the other hand, proved potent and selective inhibitors of NQO2 although not of NQO1, as exemplified by indolequinones 2f and 2g. The role of the 2-substituent was also briefly investigated through the 2-unsubstituted and 2-phenyl compounds 2h and 2i. Both were poor inhibitors of NQO1, and whilst 2h showed some effect on NQO2, 2i did not (Figure 2).
Evidence that the observed selective inhibition of NQO2 is mechanism-based (suicide substrate) involving irreversible modification of the protein was supported by the requirement for the presence of the reducing cofactor NRH for inhibition (Figure 2) and the observation that the inhibition of NQO2 by indolequinones was both concentration- and time-dependent (data not shown).[32] In addition, the lack of inhibition by compounds such as the phenoxy derivative 2j that contain a poor leaving group at the 3-position and which less easily generate iminium ions in the active site of the enzyme (cf. step C, Scheme 1), provides further evidence supporting mechanism-based inhibition of the enzyme.
In order to elucidate the details of the binding of our new inhibitors to the NQO2 protein, indolequinone 2c was co-crystallized with the enzyme, and the structure of the resulting complex solved by X-ray crystallography. Inspection of the initial electron density maps for the NQO2-indolequinone complex clearly showed the presence of the inhibitor within both NQO2 active sites, where the position of the indolequinone ring was clearly visible in a parallel stacked alignment above the isoalloxazine ring of the FAD cofactor. However the electron density for the two conformationally flexible side chains was less well defined. A number of alternative orientations of the inhibitor were therefore explored during refinement, and the preferred orientation shows the aminobutylamino side chain of the inhibitor oriented into a space in the active site. This is clearly seen in the surface model (Figure 3a). This orientation allows a hydrogen bond between the primary amine of the aminobutylamino side chain and the Asp117 main chain carbonyl oxygen, and also positions the quinone moiety of the inhibitor above the N5 of the isoalloxazine (Figure 3b), suggesting this to be a productive binding mode for hydride transfer from FADH2 during reduction of the quinone.
Figure 3.
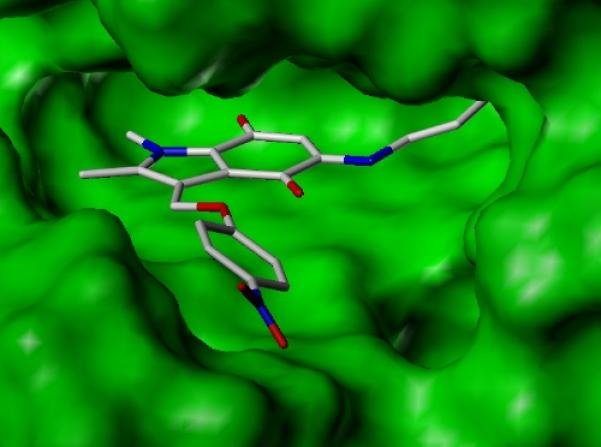

X-Ray crystallographic studies of indolequinone 2c bound to NQO2. a. Surface model of active site showing the indolequinone ring positioned above the planar surface of the FAD isoalloxazine ring, and with the aminobutylamino side chain pointing into the active site. b. Diagram showing quinone ring stacked above the FAD, and interactions to nearby residues (2c with carbon atoms colored in white, NQO2 residues and FAD with carbon atoms colored in green); (PDB ID 3O73).
In keeping with other published crystallographic structures of NQO2 complexes, the ligand 2c is positioned to allow favorable π–π stacking interactions between the planar indolequinone ring and the planar FAD isoalloxazine ring,[12,19-21] with the aromatic residues Phe126 and Phe178 positioned above the inhibitor ring. The role of hydrogen bonds in ligand binding within the NQO2 active site has also been observed previously, in particular Asn161 is implicated in hydrogen bond formation in both the resveratrol and CB1954 complexes.[12,21] In this case the hydrogen bond between inhibitor 2c and Asp117 occurs in the opposite side of the active site pocket. It is of interest that in a number of NQO2 crystallographic structures, including that of the enzyme with no ligand bound,[19] and the complex structures with resveratrol[12] and CB1954,[21] a number of water molecules are located in this region of the active site, in close proximity to Asp117.
Additional evidence for the non-covalent binding of the intact inhibitor 2c to NQO2 under non-reducing conditions was also obtained by nano-electrospray mass spectrometry (ESIMS). A mass spectrum of the enzyme:indolequinone complex showed the presence of species with the inhibitor occupying one or both of the active sites in the dimeric protein (see Supplementary Information). In addition, it was also possible to estimate the apparent Kd for binding of the intact inhibitor by titrating 2c (1 to 20 μM) into a constant concentration of NQO2 (5 μM) and detecting ligand binding by ESI-MS. From the binding curve (see Supplementary Information) it was possible to estimate the Kd for non-covalent binding of one and two ligands to the active site(s) of the protein as Kd1 = 71 μM and Kd2 = 19 μM.
Despite the detailed structural information available from X-ray crystallography of the NQO2 – indolequinone 2c complex, it was unclear how an electrophilic center generated by loss of 4-nitrophenol might alkylate the protein since there are no nucleophilic amino acid residues nearby. This is in direct contrast to our previous studies on the NQO1 – indolequinone 1a complex where structural biology played a key role in identifying nucleophilic tyrosine residues as the most likely site of covalent enzyme modification by the inhibitor.[24] Therefore we considered an alternative possibility for the inactivation of NQO2 by indolequinone 2c involving the formation of an adduct on the flavin coenzyme rather than the apoprotein, particularly since our structural studies confirm the close proximity of the inhibitor to the flavin. The inactivation of flavoproteins by mechanism-based inhibitors (suicide substrates) by covalent modification of the flavin coenzyme is known,[33,34] although the proposed molecular mechanisms for such inhibitors involve nucleophilic attack at C-4a, C-6 or N-5 of the isoalloxazine ring or radical addition to C-4a or N-5.[35-38] However, in our case the mechanism-based inhibitors generate an electrophile rather than a nucleophile, and therefore we sought experimental evidence for the formation of an FAD – indole adduct using the potent NQO2 inhibitor MAC627 2c. Purified rhNQO2 was incubated with 2c in the absence or presence of NRH after which the enzyme was immediately examined by MS. The ESI-MS spectrum of native NQO2 exhibited a mass shift of 276 amu following incubation with 2c (Figure 4a). Denaturation of the protein revealed no modification to the polypeptide of NQO2, but an ion at m/z 1061, corresponding to FAD+276 amu was observed (Figure 4b). A mass of 276 amu is consistent with the indole-hydroquinone electrophile generated from 2c (A, Scheme 2). Quadrupole isolation and MS/MS fragmentation of m/z 1061 showed product ions at m/z 786 (equivalent to [M+H]+ of FAD, and thus the loss of 275), and m/z 714 (due to the loss of AMP from m/z 1061) (Figure 4c). Notably, this latter fragment ion locates the 276 amu adduct to the flavin region of FAD (B, Scheme 2).
Figure 4.



Mass spectrometric analysis of NQO2 – indolequinone 2c interactions. Electrospray ionization mass spectra following incubation of NQO2 with IQ inhibitor in the presence of NRH showing: a NQO2 under native conditions (measured dimer mass 53332 amu) with an additional mass of 276 amu due to adduct formation; b NQO2 under denaturing conditions revealing covalent modification of FAD with the inhibitor residue of mass 276 amu (FAD+276 seen at m/z 1061.2) and no modification of the polypeptide (measured monomer mass 25834 amu); and c an MS/MS spectrum of 1061.2 localising the inhibitor residue to the FMN portion of FAD.
Scheme 2.
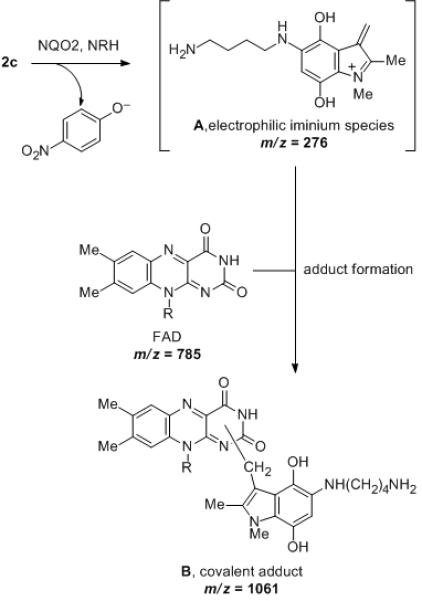
Proposed mechanism for covalent modification of the FAD coenzyme during mechanism-based inhibition of NQO2 by indolequinone 2c. (R = ribityl adenosine diphosphate)
The above mass spectrometric data are completely consistent with the formation of a FAD – indole-hydroquinone adduct by covalent modification of the coenzyme. Hence the mechanism must involve nucleophilic attack by FAD on the electrophilic iminium species (A, Scheme 2) generated upon reductive activation of the indolequinone and subsequent loss of 4-nitrophenoxide from the hydroquinone (Scheme 2). There are several possible nucleophilic sites in FAD (N-1, N-3, N-5, the ribitol hydroxyl groups or the adenine group), but of these, the adenine is ruled out by the mass spectrometric data, and therefore the isoalloxazine ring nitrogens (most probably N-1 given the relative electron density in the FAD ring[39]) seem most likely given their proximity to the reactive center in the inhibitor as revealed by our structural studies (Scheme 2). We are not aware of previous reports of mechanism-based inactivation of flavoproteins as a result of alkylation of the flavin cofactor by electrophiles, and therefore our data suggest a novel mode of enzyme inhibition.
In summary, we have developed a series of indolequinones that are potent mechanism-based inhibitors of quinone reductases and demonstrate high selectivity for NQO2 over NQO1 as a result of small structural changes. Using X-ray crystallography and mass spectrometry, we have established the nature of binding of inhibitor to the protein and our data suggest an unprecedented mode of flavoprotein inhibition by electrophilic covalent adduction of FAD. We believe that these potent inhibitors provide a useful chemical biology tool to investigate the potential of NQO2 as a molecular therapeutic target in disease.
Experimental Section
General methods, experimental protocols and spectroscopic characterization data for all inhibitors are given in the Supporting Information. The data for indolequinone 2c (MAC627) are given below. Full details of the biochemistry, protein crystallography, and mass spectrometry are also given in the Supporting Information.
5-(4-Aminobutyl)amino-1,2-dimethyl-3-(4-nitrophenoxy)methylindole-4,7-dione 2c
5-Methoxy-1,2-dimethyl-3-(4-nitrophenoxy)methylindole-4,7-dione 1a (30 mg, 0.08 mmol) in acetonitrile (1 mL) was treated with 1,4-diaminobutane (85 μL, 0.84 mmol). Following workup, the residue was purified by chromatography using alumina, eluting with 95% dichloromethane/methanol to give the title compound (11 mg, 30%) as a purple solid; mp 155-157 °C; (Found: M + H+, 413.1816. C21H24N4O5 + H+ requires 413.1819); λmax (acetonitrile)/nm 241 (log ε 4.15), 311 (4.19); νmax (CHCl3)/cm−1 3379, 2996, 1596, 1504, 1462, 1343; δH (400 MHz; CDCl3) 8.20 (2 H, d, J 8.0, ArH), 7.06 (2 H, d, J 8.0, ArH), 6.04 (1 H, bs, NH), 5.34 (2 H, s, CH2), 5.16 (1 H, s, H-6), 3.94 (3 H, s, NMe), 3.14 (2 H, ~q, J 6.6, CH2), 2.76 (2 H, t, J 6.6, CH2), 2.29 (3 H, s, 2-Me), 1.73 (2 H, quin, J 7.3, CH2), 1.55 (2 H, quin, J 7.3, CH2); δC (100 MHz; CDCl3) 179.0 (C), 178.8 (C), 163.8 (C), 148.1 (C), 141.5 (C), 136.2 (C), 131.4 (C), 126.0 (CH), 119.3 (C), 114.8 (CH), 114.6 (C), 96.7 (C), 61.3 (CH2), 42.7 (CH2), 41.6 (CH2), 32.4 (Me), 30.9 (CH2), 25.6 (CH2), 9.8 (Me); m/z (ESI) 435 (MNa+, 21%), 413 (MH+, 61), 274 (100).
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health National Cancer Institute [Grant R01-CA114441] (to D.R.) and the University of Nottingham.
Footnotes
Competing Financial Interests CJM, DR, and DS are scientific cofounders and stockholders in QGenta Inc., which holds an option to license molecules described in this article.
Supporting Information for this article is available on the WWW under http://.........
References
- [1].Vella F, Gilles FA, Delagrange P, Boutin JA. Biochem. Pharmacol. 2005;71:1. doi: 10.1016/j.bcp.2005.09.019. [DOI] [PubMed] [Google Scholar]
- [2].Ferry G, Hecht S, Berger S, Moulharat N, Coge F, Guillaumet G, Leclerc V, Yous S, Delagrange P, Boutin JA. Chem.-Biol. Interact. 2010;186:103. doi: 10.1016/j.cbi.2010.04.006. [DOI] [PubMed] [Google Scholar]
- [3].Mailliet F, Ferry G, Vella F, Berger S, Coge F, Chomarat P, Mallet C, Guenin SP, Guillaumet G, Viaud-Massuard MC, Yous S, Delagrange P, Boutin JA. Biochem. Pharmacol. 2005;71:74. doi: 10.1016/j.bcp.2005.09.030. [DOI] [PubMed] [Google Scholar]
- [4].Calamini B, Santarsiero BD, Boutin JA, Mesecar AD. Biochem. J. 2008;413:81. doi: 10.1042/BJ20071373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fu Y, Buryanovskyy L, Zhang Z. J. Biol. Chem. 2008;283:23829. doi: 10.1074/jbc.M801371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gaikwad NW, Yang L, Rogan EG, Cavalieri EL. Free Radic. Biol. Med. 2009;46:253. doi: 10.1016/j.freeradbiomed.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, Foley M, Haystead TAJ. Mol. Pharmacol. 2002;62:1364. doi: 10.1124/mol.62.6.1364. [DOI] [PubMed] [Google Scholar]
- [8].Kwiek JJ, Haystead TAJ, Rudolph J. Biochemistry. 2004;43:4538. doi: 10.1021/bi035923w. [DOI] [PubMed] [Google Scholar]
- [9].Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Nat. Biotechnol. 2007;25:1035. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- [10].Winger JA, Hantschel O, Superti-Furga G, Kuriyan J. BMC Structural Biology. 2009;9:7. doi: 10.1186/1472-6807-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang ZR, Hsieh TC, Zhang ZT, Ma YL, Wu JM. Biochem. Biophys. Res. Commun. 2004;323:743. doi: 10.1016/j.bbrc.2004.08.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Buryanovskyy L, Fu Y, Boyd M, Ma YL, Hsieh TC, Wu JM, Zhang ZT. Biochemistry. 2004;43:11417. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liao S, Williamsashman HG. Biochem. Biophys. Res. Commun. 1961;4:208. doi: 10.1016/0006-291x(61)90272-8. [DOI] [PubMed] [Google Scholar]
- [14].Jaiswal AK, Burnett P, Adesnik M, McBride OW. Biochemistry. 1990;29:1899. doi: 10.1021/bi00459a034. [DOI] [PubMed] [Google Scholar]
- [15].Ernster L. Chem. Scripta. 1987;27A:1. [Google Scholar]
- [16].Bianchet MA, Faig M, Amzel LM. Method Enzymol. 2004;382:144. doi: 10.1016/S0076-6879(04)82009-3. [DOI] [PubMed] [Google Scholar]
- [17].Dinkova-Kostova AT, Talalay P. Arch. Biochem. Biophys. 2010;501:116. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Deller S, Macheroux P, Sollner S. Cellular and Molecular Life Sciences. 2008;65:141. doi: 10.1007/s00018-007-7300-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Foster CE, Bianchet MA, Talalay P, Zhao QJ, Amzel LM. Biochemistry. 1999;38:9881. doi: 10.1021/bi990799v. [DOI] [PubMed] [Google Scholar]
- [20].Fu Y, Buryanovskyy L, Zhang ZT. Biochem. Biophys. Res. Commun. 2005;336:332. doi: 10.1016/j.bbrc.2005.08.081. [DOI] [PubMed] [Google Scholar]
- [21].AbuKhader M, Heap J, De Matteis C, Kellam B, Doughty SW, Minton N, Paoli M. J. Med. Chem. 2005;48:7714. doi: 10.1021/jm050730n. [DOI] [PubMed] [Google Scholar]
- [22].Beall HD, Winski S, Swann E, Hudnott AR, Cotterill AS, O’Sullivan N, Green SJ, Bien R, Siegel D, Ross D, Moody CJ. J. Med. Chem. 1998;41:4755. doi: 10.1021/jm980328r. [DOI] [PubMed] [Google Scholar]
- [23].Winski SL, Swann E, Hargreaves RHJ, Dehn DL, Butler J, Moody CJ, Ross D. Biochem. Pharmacol. 2001;61:1509. doi: 10.1016/s0006-2952(01)00631-1. [DOI] [PubMed] [Google Scholar]
- [24].Winski SL, Faig M, Bianchet MA, Siegel D, Swann E, Fung K, Duncan MW, Moody CJ, Amzel M, Ross D. Biochemistry. 2001;40:15135. doi: 10.1021/bi011324i. [DOI] [PubMed] [Google Scholar]
- [25].For other loss of leaving groups from indolequinones upon reduction, see refs 25-28: Hernick M, Borch RF. J. Med. Chem. 2003;46:148. doi: 10.1021/jm0203229.
- [26].Ferrer S, Naughton DP, Threadgill MD. Tetrahedron. 2003;59:3445. [Google Scholar]
- [27].Tanabe K, Makimura Y, Tachi Y, Imagawa-Sato A, Nishimoto S. Bioorg. Med. Chem. Lett. 2005;15:2321. doi: 10.1016/j.bmcl.2005.03.013. [DOI] [PubMed] [Google Scholar]
- [28].Zhang Z, Tanabe K, Hatta H, Nishimoto S. Org. Biomol. Chem. 2005;3:1905. doi: 10.1039/b502813b. [DOI] [PubMed] [Google Scholar]
- [29].Reigan P, Colucci MA, Siegel D, Chilloux A, Moody CJ, Ross D. Biochemistry. 2007;46:5941. doi: 10.1021/bi700008y. [DOI] [PubMed] [Google Scholar]
- [30].Yan C, Shieh B, Reigan P, Zhang Z, Colucci MA, Chilloux A, Newsome JJ, Siegel D, Chan D, Moody CJ, Ross D. Mol. Pharmacol. 2009;76:163. doi: 10.1124/mol.109.055855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Colucci MA, Reigan P, Siegel D, Chilloux A, Ross D, Moody CJ. J. Med. Chem. 2007;50:5780. doi: 10.1021/jm070396q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Siegel D, Yan C, Dufour M, Shieh B, Reigan P, Moody CJ, Ross D. Proc. Amer. Assoc. Cancer Res. 2010;51:5471. [Google Scholar]
- [33].Abeles RH, Maycock AL. Acc. Chem. Res. 1976;9:313. [Google Scholar]
- [34].Walsh C. Acc. Chem. Res. 1980;13:148. [Google Scholar]
- [35].Ghisla S, Olson ST, Massey V, Lhoste JM. Biochemistry. 1979;18:4733. doi: 10.1021/bi00588a038. [DOI] [PubMed] [Google Scholar]
- [36].McCann AE, Sampson NS. J. Am. Chem. Soc. 2000;122:35. [Google Scholar]
- [37].Chen ZW, Zhao GH, Martinovic S, Jorns MS, Mathews FS. Biochemistry. 2005;44:15444. doi: 10.1021/bi0515422. [DOI] [PubMed] [Google Scholar]
- [38].Mitchell DJ, Nikolic D, Rivera E, Sablin SO, Choi S, van Breemen RB, Singer TP, Silverman RB. Biochemistry. 2001;40:5447. doi: 10.1021/bi010388q. [DOI] [PubMed] [Google Scholar]
- [39].Cavelier G, Amzel LM. Proteins. 2001;43:420. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.