

Abstract
Maintaining correct DNA and histone methylation patterns is essential for the development of all eukaryotes. In Arabidopsis, we identified SHOOT GROWTH1 (SG1), a novel protein involved in the control of gene methylation. SG1 contains both a Bromo-Adjacent Homology (BAH) domain found in several chromatin regulators and an RNA-Recognition Motif (RRM). The sg1 mutations are associated with drastic pleiotropic phenotypes. The mutants degenerate after few generations and are similar to mutants of the histone demethylase INCREASE IN BONSAI METHYLATION1 (IBM1). A methylome analysis of sg1 mutants revealed a large number of gene bodies hypermethylated in the cytosine CHG context, associated with an increase in di-methylation of lysine 9 on histone H3 tail (H3K9me2), an epigenetic mark normally found in silenced transposons. The sg1 phenotype is suppressed by mutations in genes encoding the DNA methyltransferase CHROMOMETHYLASE3 (CMT3) or the histone methyltransferase KRYPTONITE (KYP), indicating that SG1 functions antagonistically to CMT3 or KYP. We further show that the IBM1 transcript is not correctly processed in sg1, and that the functional IBM1 transcript complements sg1. Altogether, our results suggest a function for SG1 in the maintenance of genome integrity by regulating IBM1.
Introduction
Methylation of cytosines and histones are epigenetic modifications found in many eukaryote species, including plants and animals that influence gene expression and transposon mobilization. In contrast to mammals for which DNA methylation occurs predominantly in the CG cytosine context, with the exception of embryonic stem cells [1], plant DNA methylation is found in both symmetrical (CG and CHG where H is A, T or C) and non-symmetrical (CHH) contexts. These three types of cytosine methylations are maintained by different specific pathways in Arabidopsis [2]. The DNA methyltransferases METHYLTRANSFERASE1 (MET1) and CHROMOMETHYLASE3 (CMT3) are required for the maintenance of CG and CHG methylation, respectively [2]. Maintaining CHH methylation depends on both CHROMOMETHYLASE2 (CMT2) [3] and the small RNA-directed DNA methylation (RdDM) pathway, which involves plant specific RNA polymerases (Pol IV and V) and the production of 24-nucleotide small RNAs controlled by the RNA-DEPENDENT RNA POLYMERASE2 (RDR2) together with DICER-LIKE3 (DCL3) [2]. Histones can be methylated in several ways, one of them being at lysine 9 on histone H3 tail (H3K9me), a hallmark of silent chromatin in several organisms, including plants [4]. Methylation at H3K9 can be classified into three different types: mono- di- or tri-methylated, but di-methylation (H3K9me2) is the most common in Arabidopsis. H3K9me2 is catalysed by three histone methyltransferases, KRYPTONITE/SUVH4 (KYP), SUVH5, and SUVH6 [5].
Locus specific CHG and H3K9me2 methylations are intimately connected. Indeed, CMT3 physically interacts with H3K9me2 marks through both its chromo and BAH domains [6] while KYP binds CHG-methylated cytosines through its SRA domain [7]. Altogether CMT3 and KYP participate in a self-reinforcing loop between DNA and histone methylation that is essential for transposons and repeated sequences silencing. Another evidence for an interplay between DNA and histone modifications was the identification of INCREASE IN BONSAI METHYLATION1 (IBM1), a Jumonji C (JmjC) domain protein belonging to the JHDM2 family of H3K9 demethylases that is specific to H3K9 mono- and dimethylation [8]–[10]. IBM1 was identified in a screen for increased methylation at the BONSAI (BNS) locus since cytosines of BNS in the CHG context are highly methylated in ibm1. In both ibm1/cmt3 and ibm1/kyp double mutants, CHG methylation at BNS returns to a WT level, showing that IBM1 counteracts CMT3 and KYP activities at this locus. In fact, epigenome-wide analyses of ibm1 identified thousands of other genes that are ectopically enriched for both CHG and H3K9 methylations in ibm1 mutants [9]. Thus, IBM1 prevents genes from being targeted by the machinery that silences repeated elements and transposons. Additionally, IBM1 acts on components of the RdDM pathway itself through direct targeting of RDR2 and DCL3 genes [11].
Here, we report the identification of a novel player, named SG1, in the regulation of DNA and histone methylations. Initially identified as a major QTL candidate for shoot and root growth in several segregating populations, the sg1 mutant exhibited pleiotropic developmental defects with a higher penetrance in successive generations similarly to previously reported mutants involved in chromatin modifications. SG1 encodes a protein with two putative domains, one of them, a Bromo-Adjacent Homology (BAH) domain being found in several chromatin binding proteins of eukaryotes. A genome-wide methylation analysis in the sg1 mutant background using bisulfite sequencing showed that a large number of gene body sequences were enriched in CHG methylation, similarly to previous reports on ibm1. We also found an increase in H3K9me2 levels within the gene bodies of all CHG hypermethylated candidates tested. We confirmed through a genetic approach that the sg1 mutant phenotype is suppressed by cmt3 or kyp mutations, but not ibm1. We analysed the expression of some targets of SG1 and in particular genes involved in the RdDM pathway, but we found no correlation between an increase in gene body CHG methylation and a lack of function. Finally, we show that the accumulation of full-length functional IBM1 mRNAs is compromised in sg1, which can be complemented by the introduction of a transgene producing the long version of IBM1 transcripts. Altogether, our data reveals a new player in the maintenance of Arabidopsis DNA and histone methylations with an essential role in epigenome stability.
Results
Identification and Characterization of sg1 Mutants
In a previous report we described a quantitative genetic screen to investigate natural genetic variation for shoot growth in Arabidopsis thaliana [12]. Among four significant QTLs mapped in the Bur-0 x Col-0 recombinant inbred line population, one QTL named SHOOT GROWTH1 (SG1) was mapped at the top of chromosome 5 (Figure S1 in File S1). Plants carrying a Col-0 allele at SG1 locus showed drastic shoot growth defects with respect to plants carrying the Bur-0 allele, as confirmed in heterogeneous inbred families (HIF). Fine-mapping analysis using recombinant HIF mapped SG1 to a locus localized in a 8.5 kb interval comprising a single gene, At5g11470. In parallel, we fine-mapped a QTL detected in the Ct-1 x Col-0 RIL population with a major effect on root growth (named ROOT GROWTH1; RG1) down to the same region and gene (Figure S2 in File S1).
A sequence analysis on SG1 did not evidenced obvious DNA polymorphisms common to stock accessions Ct-1, Bur-0 and Col-0, however, different gene model predictions for the SG1 gene were found in the databases (Figure S3 in File S1). Hence, we decided to sequence the SG1 cDNAs from the RILs themselves to exclude any possible mRNA mis-splicing that could account for the phenotype. Surprisingly, we found a single C to T mutation in the SG1 cDNA from plants of the mapping populations carrying Col-0 alleles, compared to the sequences of parental accessions. Analysis of the cloned SG1 cDNA, that matches EuGène prediction [13] with slight variations in regard to TAIRv10 prediction (Figure S3 in File S1), suggested that this sg1-1 mutation identified in the RILs occurs in the sixth exon of the gene (Figure 1A). By analyzing the original seeds of the Col-0 parent used to produce the RILs, we confirmed the presence of the sg1-1 mutation compared to the Col-0 reference sequence. We concluded that sg1-1 is a non-silent spontaneous mutation that appeared in our Col-0 parental line before we developed our RIL sets.
Figure 1. SG1 encodes a protein with domains involved in chromatin modification.

(A) Top: SG1 gene structure showing UTRs (black boxes), exons (clear boxes), introns (dashed lines). The position of sg1-1 mutation (cytosine at position 2,954 relative to ATG) and sg1-2 or sg1-3 T-DNA insertions are indicated by plain arrows. Bottom: protein structure showing the two putative BAH and RRM domains (black boxes). (B) Analysis of the projected rosette area (pixels) of 15 days-old seedlings grown in vitro. sg1-1(gen2) : progeny of sg1-1; sg1-1(gen3) : progeny of sg1-1(gen2). n: number of individuals analyzed for each genotype. **p<0.01, ***p<0.001, ns: not significant. (C) Left panel: sg1-1 phenotype of a 25 days-old plant (right) compared to WT Col-0 plants. Right panel: sg1-1 floral defects. Note the aberrantly shaped petals, incorrect organs number.
In the original Col-0 accession carrying the sg1-1 mutation at the homozygous state, we observed that the phenotype was not fully penetrant since we identified both plants with no or subtle phenotype in addition to plants with reduced shoot size and a phenotype that increases throughout generations (Figure 1B and Figure S4 in File S1). The shoot growth reduction phenotype was associated with dark green plants and pleiotropic developmental abnormalities including altered leaf morphology (narrow and serrated), and flowers with more petals, some being fused or with an altered morphology (Figure 1C). After at least four generations at the sg1-homozygous state, plants exhibited a terminal phenotype with small deformed leaves associated with strong sterility. We then isolated two T-DNA knockouts (sg1-2 and -3) with insertions localized within the SG1 coding sequence (Figure 1A). Both mutants were similar to sg1-1: the severity of the phenotype varied between individuals and became stronger in successive generations to reach a complete sterility from the 4th–5th generation (Figure S5 in File S1).
SG1 (At5g11470) encodes an unknown protein with two predicted domains: an RNA Recognition Motif (RRM) and a Bromo-Adjacent Homology (BAH) domain (Figure 1A). The sg1-1 mutation results in a Gln to STOP codon, leading to a potentially truncated protein with no RRM domain. While the Arabidopsis genome encodes a unique SG1 protein, we found a total of 21 proteins that contain a BAH domain (Figure S6 in File S1), including well characterized DNA methyltransferases like MET1 and CMT3, pointing towards a function for SG1 in chromatin regulation.
Gene Body CHG Methylation and H3K9 Marks are Impaired in sg1
The pleiotropic and generational nature of the phenotype is reminiscent of those observed in mutants disrupted in chromatin modification pathways such as ibm1 [10], met1 [14] or ddm1 [15]. Thus, we analysed the methylome of sg1-1 mutant through Illumina deep sequencing after bisulfite conversion, in comparison to the Col-0 background. The coverage of sequenced Arabidopsis methylomes was 30X with an error rate of 0.4% (Table S1 in File S2). First, we calculated the percentage of methylation for cytosines in either one of the three contexts CG, CHG or CHH. Genome-wide, we observed no significant differences between Col-0 and sg1-1 for cytosines methylated in CG or CHH, while sg1-1 seemed to contain more methylated CHG than Col-0 (Figure 2A; Table S2 in File S2). This was further confirmed when we calculated the percentages of methylation in 200 bp-windows (with a 50 bp overlap) to scan the whole-genome. The sg1-1 mutant contained a large number of regions enriched in methylated-CHG compared to Col-0, while no such large-scale contrast was observable for cytosines in the CHH or CG contexts (Figure S7 in File S1). To determine whether CHG regions highly represented in sg1-1 corresponded to genes or other parts of the genome, we calculated the percentage of cytosine methylated per gene and we found that many genes of sg1-1 were enriched in CHG methylation compared to Col-0 (Figure 2B). Gene body-methylation in the CG context was similar between Col-0 and sg1-1 (Figure S8A in File S1), while -overall- genes seem to be slightly over-methylated in the CHH context in sg1-1 (Figure S8B in File S1). The average distribution of CHG methylation over genes indicates that genes are more methylated in their core bodies (Figure 2C). We found no systematic contrast in methylated-cytosine content between sg1-1 and Col-0 transposable elements (Figure S9 in File S1).
Figure 2. sg1-1 methylome analysis.
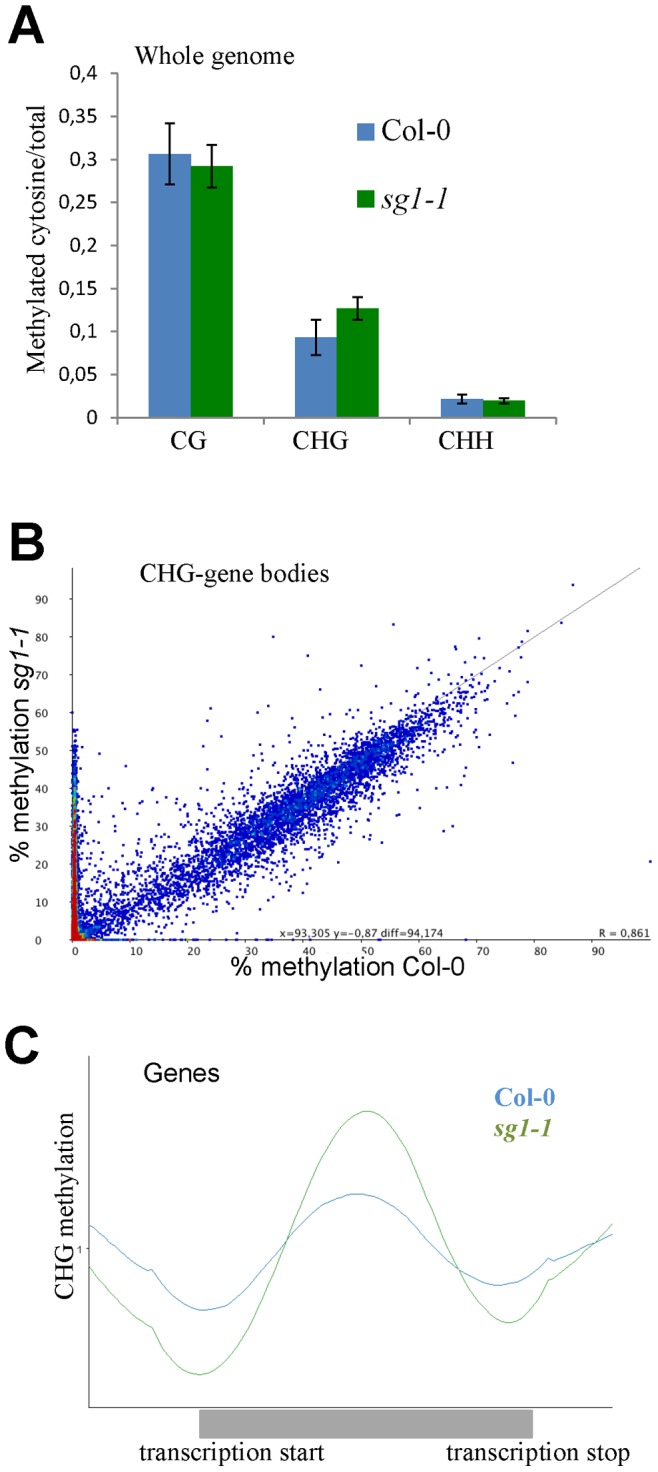
(A) Whole genome analysis of cytosine methylated in all three contexts of methylation (CG, CHG and CHH) in Col-0 and sg1-1 mutants. Error bars indicate the SD between two biological replicates. (B) Scatter plot representing the percentage of CHG methylation calculated for each gene in sg1-1 and Col-0. Hypermethylated genes in sg1-1 are visualized by the dense red area. (C) Graphic representation of the average distribution of CHG methylation in gene bodies.
We fixed a minimal methylation ratio of two from Col-0 to sg1-1 to classify genes as hyper- (2x increase in sg1-1) or hypo-methylated (2x decrease in sg1-1; see Materials and Methods). Under these criteria, we found less than 100 genes that were hypomethylated in either one of the three contexts, one gene hypermethylated in the CG context, 0 in CHH and over 3,300 in the CHG context (Table S3 in File S2). Genes found to be CHG-hypermethylated were equally distributed among the five Arabidopsis chromosomes (Figure S10 in File S1). We selected six genes from this list to analyze in more details the distribution of the methylated cytosines within gene bodies. We confirmed that all these genes were significantly enriched in CHG-methylated cytosines in sg1-1 mutants compared to Col-0 throughout the gene sequence (Figure 3), while no differences were observable for CG methylated cytosines, and a slight increase for some of them in the CHH contexts (Figure S11 in File S1). Altogether, our results indicate that SG1, very similarly to IBM1, protects certain protein-coding genes from ectopic CHG methylation. Notably, as observed in ibm1-1 mutants [10], CHG and -to some extent- CHH methylation, extend from an adjacent transposon into the BONSAI locus in sg1-1 (Figure 4A).
Figure 3. Gene-body CHG hypermethylation in sg1-1.

Methylation on top (positive values) and bottom strand (negative values) across the coding sequence of the genes indicated is shown. Data are based on methylome results. The corresponding gene models are shown according to TAIR v10.
Figure 4. Methylation analysis at BONSAI in sg1-1.

(A) Methylation in the three cytosine contexts near BNS. The spreading of DNA methylation from the adjacent LINE element (At1g73175) is visible in sg1-1. (B) Histone H3K9me2 accumulation in BNS determined by ChIP analyses. The correspondence of genomic regions tested by ChIP is shown on a gene schematic based on TAIR v10. Error bars are SEM from at least three biological replicates.
Since IBM1 encodes a histone demethylase catalysing the removal of H3K9 marks, a process tightly linked to CHG methylation, we determined H3K9me2 contents by ChIP analyses in the subset of genes that present elevated levels of CHG methylation (Figure 5). Using several couples of oligos along gene bodies, we observed in both sg1-1 and sg1-2 mutant alleles as well as in ibm1-1, a significant enrichment of H3K9me2 localized within the center of genes, and to a lesser extent in UTRs (Figure 5). Similarly, the BNS gene was enriched in H3K9me2, particularly in regions adjacent to the transposon nearby (Figure 4B). Altogether, our results suggest a function for SG1 in the protection of genes from H3K9me2 and CHG DNA methylation, similarly to that described for IBM1.
Figure 5. Gene-body H3K9me2 accumulation in sg1-1 and ibm1 mutants.

H3K9me2 profile across six genes mentioned in Figure 3, in WT Col-0, sg1-1, sg1-2 and ibm1-1 plants. The correspondence of genomic regions tested by ChIP is shown on a gene schematic based on TAIR v10. Error bars are SEM from at least three biological replicates.
Recently, Jacobsen and collaborators revealed the DNA methylome landscapes of several Arabidopsis mutants involved in gene silencing and chromatin remodeling, including components of the methylation pathways [16]. We retrieved the ibm1 and the corresponding WT methylome sequences to identify the genes differently methylated in sg1-1 versus ibm1-6 (SALK_006042). Interestingly, we found that the vast majority of gene bodies differentially CHG methylated in sg1-1 were also targeted by IBM1, but we also found that many genes more CHG methylated in ibm1 were unchanged in sg1-1 (Figure 6). We verified that the CHG methylomes of the WT samples were similar between the two experiments (Figure S12 in File S1).
Figure 6. Comparison of sg1-1 and ibm1-6 CHG methylated genes.

Scatter plot representing the percentage of CHG methylation calculated for each gene in sg1-1 and ibm1-6, according to the data publicly available [16]. Genes solely methylated in ibm1 are visualised by the dense red area.
CMT3 and KYP act Antagonistically to SG1
IBM1 and KYP have opposite effects on H3K9 marks, and this has a direct impact on CMT3 activity and CHG methylation. Thus, IBM1 restricts CMT3 and KYP activities to transposons and repeated sequences, protecting genes from elevated levels of CHG methylation and H3K9me2. To investigate the relationships between SG1, IBM1, KYP and CMT3 we crossed sg1-2 with mutants corresponding to these genes. Both sg1/cmt3 and sg1/kyp double mutants showed a WT phenotype, indicating that either cmt3 or kyp mutations alone can suppress the severe phenotype of sg1 (Figure 7) and ibm1 [8]. On the other hand, the double sg1/ibm1 mutant is phenotypically similar to the sg1 or ibm1 mutants (Figure 7). This suggests a function for SG1 in the protection of genes from H3K9me2 and CHG DNA methylations, together with IBM1, and antagonistically to that of CMT3 and KYP.
Figure 7. sg1 phenotype is suppressed by cmt3 or kyp mutations, but not ibm1.

(A) Pictures of typical 15 days-old seedling corresponding to homozygous double mutants (F4) resulting from the cross between sg1-2 and cmt3-11, kyp (line SALK_069326) or ibm1-1 mutants. Contrast was enhanced for picture visibility. (B) Analysis of the projected rosette area (pixels) of 15 days-old double mutant seedlings (as described in A) grown in vitro. n: number of individuals analyzed for each genotype. ***p<0.001, ns: not significant.
CMT3 is the methyltransferase that maintains CHG methylation and MET1 is involved in the maintenance of CG methylation. To understand whether perturbing one of the other types of methylation in sg1 would have some consequences, we combined the met1 mutation with sg1. In the F2 population, we obtained only three plants fixed for both mutations, on a total of 96 plants (Figure S13A in File S1). The F3 progenies of one of these met1/sg1 double mutants had a strong phenotype (Figure S13B in File S1) and were all sterile. Therefore, suppressing the CG-methylation in both genes and transposons (i.e. met1), while adding silencing marks in the core of genes (i.e. sg1) has a deleterious immediate effect on plant development.
Previous genetic analysis showed that some components of the RdDM pathway, including RDR2, NRPD1A and AGO4, are dispensable for ibm1-induced DNA hypermethylation defects, suggesting that de novo DNA methylation of the IBM1 targets is subject to siRNA-independent regulation [9]. Therefore, we investigated if hypermethylation in sg1 was also siRNA-independent, similarly to what was reported for ibm1. When we combined sg1 with a mutant lacking the AGO4 function that is crucial for RdDM processes [2], we found that the double sg1-2/ago4-2 mutant was phenotypically very similar to sg1-2, suggesting that sg1-induced hypermethylation does not depend on components of RdDM (Figure S14 in File S1).
Levels of mRNAs in sg1, Methylation and Consequences on Gene Function
One of the most intriguing questions regarding the methylation marks that appear in gene bodies of sg1 or ibm1 mutants is whether they have direct consequences on gene expression, and ultimately on the function of these affected genes. Recently, IBM1 was suggested to act indirectly on the RdDM pathway by targeting both RDR2 and DCL3, reducing their expression, correlating with a final up-regulation of some RdDM-targets [11]. We analyzed the H3K9me2 enrichment at RDR2 and DCL3 locus, by ChIP analysis. We found that, like ibm1, sg1 mutants contain higher levels of H3K9me2 and cytosines methylated in the CHG context at both locus compared to the Col-0 WT plants (Figure 8A); this correlated with more subtle modifications of CHH cytosine methylation but not with CG methylation (Figure S15 in File S1). We confirmed that both RDR2 and DCL3 gene expression is reduced by half in ibm1 and we found similar results for the two sg1 alleles tested (Figure 8B). We also found other members of the RdDM pathway with a CHG-methylation status affected in sg1, including NRPD1A, one of the polIV subunits (Table S4 in File S2). However, even if some of the RdDM key players are impacted by sg1-induced methylation defects (at CHG, CHH and H3K9 levels), the functional consequences on the RdDM pathway activity remain to be determined.
Figure 8. sg1 and key players of the RdDM pathway.

(A) H3K9me2 profile across RDR2 and DCL3 genes in WT Col-0, sg1-1, sg1-2 and ibm1-1 plants. The correspondence of genomic regions tested by ChIP is shown on a gene schematic based on TAIR v10; CHG methylation is presented below. Error bars are SEM from at least three biological replicates. (B) Transcript accumulation of RDR2 and DCL3 in sg1 and ibm1 determined by qPCR analyses. Error bars are SEM from three replicates.
We tested the expression of several other targets having gene bodies enriched in both H3K9me2 and CHG, and found genes (At4g00450 and At5g35210, see Figures 3 and 5) whose expression was not modified between sg1-1 and WT plants (Figure S16 in File S1). We concluded that sg1-induced methylation defects are not necessarily correlated with changes in the final quantity of transcripts detected, or that sg1 may induce other changes such as transcript processing alteration that were not detected in our gene expression assays.
Since sg1 and ibm1 phenotypes are very similar, both molecularly and morphologically, we thought that the function of IBM1 could be compromised in sg1 mutants. The IBM1 gene encodes two different transcripts and only the longest one (IBM1-L) is functional and can complement an ibm1 mutant [17]. We first tested whether the sg1 phenotype could be the result of IBM1 transcript misregulation. To this end we examined by qRT-PCR the proportion of the IBM1-L mRNA product relative to total IBM1 mRNA product (as defined by the amount of transcript upstream of the large intron heterochromatic region). Interestingly, we found a stronger reduction in the sg1-1/WT mRNA ratio corresponding to the IBM1-L product relative to the total amount (Figure S17 in File S1). This suggested that a significant fraction of the mRNA produced at the IBM1 locus in a sg1 mutant may correspond to the short, non-functional IBM1-S product. We then transformed sg1 mutants with a transgene carrying a full-length functional IBM1-L cDNA construct [17]. Both ibm1-1 and sg1-1 were complemented by IBM1-L, indicating that the phenotype of sg1 mutants most likely results from the compromised function of IBM1 in sg1 (Figure S18 in File S1).
Discussion
SG1 and IBM1 act in the Same Pathway
In an effort to map molecular determinants quantitatively controlling growth, we identified a new gene, SG1 (At5g11470), coding a protein with domains found in chromatin regulators. We identified the sg1-1 allele corresponding to a recent spontaneous point mutation in the SG1 coding sequence from the specific Col-0 parent line used to build our mapping population. We also isolated two additional T-DNA alleles (sg1-2 and -3) allowing us to determine that sg1-related phenotypes became apparent from the second homozygous generation. All sg1 mutants present similar phenotypes appearing stochastically, namely growth defects, abnormally shaped organs (including flowers and leaves) and ultimately leading to sterility. The penetrance and severity of the phenotype increase across generations, similar to phenotypic defects previously reported for ibm1 [10]. Since IBM1 is controlling H3K9 methylation, with consequences on the levels of cytosines methylated in the CHG context, we performed a high-throughput methylome sequencing of sg1-1 mutants, showing that gene bodies are CHG-hypermethylated in the mutant background (Figure 2B and C) while transposable elements are not affected (Figure S9 in File S1). This was correlated with an enrichment of the heterochromatin mark H3K9me2 at gene bodies of all CHG hypermethylated candidates tested in our study (Figures 3, 4, 5 and 8A). These results strongly suggest that SG1 is working closely to IBM1, indeed, similar enrichments within gene-bodies have been reported in an ibm1 mutant context [9]. Altogether, these observations suggested a function for SG1 in the maintenance of genome integrity, likely at the chromatin regulation level, together with IBM1. Recently, two other sg1 alleles were identified, namely ibm2 and asi1 [18]–[19]. In agreement with our hypothesis for SG1 function, the authors showed that SG1/IBM2/ASI1 is required for the processing of long transcripts over intronic heterochromatic marks. Given the presence of an intragenic heterochromatin region in the large intron of IBM1, this conclusion is supported by findings that IBM1 full-length transcript was decreased in the sg1 mutant compared to the short transcript (Figure S17 in File S1) and that a functional IBM1 transcript complement sg1 (Figure S18 in File S1). SG1/IBM2/ASI1 might indeed function by promoting the use of distal polyadenylation sites over proximal ones located in large introns [18]–[19].
SG1 contains two predicted domains, one of them, the BAH domain, being found in various chromatin regulating factors. In plants, one of the two BAH domains of MET1 for instance can physically interact with the C-terminal region of the histone deacetylase HDA6, [20]. Interestingly, BAH domains can also directly recognize histone marks. A component of the Origin of Replication Complex-1 (ORC1) can bind in a specific way H4K20me2 marks that are enriched at replication origins in mammalians [21]. In maize, the homolog of CMT3 binds H3K9me2 marks via a dual recognition through both the BAH domain and the chromodomain of the protein [6]. Therefore SG1 may be recruited to the chromatin via its BAH domain. In addition, SG1 contains a RRM motif shared by a large number of RNA-binding proteins that could also mediate the targeting of SG1 to particular loci. Accordingly, Wang et al. showed in vitro that SG1/IBM2/ASI1 has RNA-binding properties, and that SG1/IBM2/ASI1 is enriched at intronic heterochromatin regions [19]. The authors proposed a model in which SG1/IBM2/ASI1 is binding to intronic heterochromatin regions and nascent pre-mRNA via its BAH domain and RRM motif respectively, but whether SG1/IBM2/ASI1 BAH domain is directly involved in heterochromatin recruitment remains to be determined.
By comparing our methylome data with the one published recently for ibm1 mutants [16], we observed that most IBM1 target genes are also affected in sg1 mutants, while IBM1 has additional targets (Figure 6). However, this does not necessarily mean that IBM1 has a broader range of target genes compared to SG1. This observation might be the consequence of a generational effect since the phenotypes of sg1 and ibm1 mutants become more deleterious after several generations. We suspect that the ibm1 mutant used in the recent study of Jacobsen and coworkers [16] had been accumulating gene methylation for more generations than the sg1 mutant used in our methylome analysis. In regions where both ibm1 and sg1 mutations affect the same genes, we observed that the CHG methylation in sg1 seems to be more localized to the center of gene bodies. Contrariwise, the CHG methylation in the ibm1 mutant, while being also localized to gene bodies, seems to be more extended toward the UTRs (Figure S19 in File S1). It is tempting to speculate that generations after generations, the methylation that first invade specifically certain regions of the genes slowly spread to adjacent regions in plants lacking IBM1 or SG1 function. We also detected genes that are methylated in ibm1 while being almost free of CHG methylation in the sg1 background (Figure S20 in File S1). Since the phenotype is strongly correlated with the number of generations, it is possible that secondary targets, not impacted in the first generations, are gradually becoming methylated in the progenies, increasing the deleterious effects of the ibm1 mutation. We currently do not know how methylation acts on mRNA accumulation, but the lethal effects observed after several generations indicate that the constant and possible increasing invasion of the methylation have strong consequences on plant development. To further confirm these hypotheses, methylome analyses of both sg1 and ibm1 mutants will have to be compared for the same generations.
Interconnection between Different Methylation Contexts of Gene Bodies
We have shown that the maintenance DNA methyltransferase CMT3 or the H3K9 methyltransferase KYP are epistatic to SG1. Both cmt3 or kyp mutations suppress the phenotype of sg1, similarly to what was reported for ibm1 and suggesting that SG1 functions antagonistically to CMT3 or KYP. In addition to KYP, two other methyltransferases are found in Arabidopsis (SUVH5 and 6), but since the suppression of KYP can restore the WT phenotype in a sg1 background, it is likely that ectopic H3K9me2 marks deposition in gene bodies of sg1 mostly involves KYP. The double ibm1/sg1 mutant displayed similar phenotypes to a single sg1 or ibm1 mutant, confirming that IBM1 and SG1 act in the same pathway. Altogether, the interplay between the self-reinforcing CMT3/KYP loop and SG1/IBM1 maintains the Arabidopsis epigenome integrity to ensure correct expression of genes. How SG1 functions to prevent CMT3 and KYP activities at a given locus remains unclear, as well as the initial starting signal that first promotes their activities in genes. But the self-reinforcing loop and the subsequent spreading of the methylation generations after generations seem to be key elements to explain the increase invasion of genes by silencing marks. Recently, DDM1, encoding a chromatin remodeling factor, was shown to restrict CMT3/KYP activities at BNS and many other locus. The methylation at BNS in all cytosine contexts in a ddm1 background depends on KYP and CMT3 since the corresponding mutants individually suppress this methylation [22]. In ddm1, residual methylation from the adjacent LINE element spreads to BNS after several generations, and through the repeated action of both CMT3 and KYP, exactly as in sg1 or ibm1 mutants.
The activity of IBM1 seems to be more prone to target actively transcribed genes [9] and the functions of IBM1 and SG1 are essential to keep gene bodies free of CHG methylation. Paradoxically, these genes are not exempt of methylation in the WT, but in a CG-context (Figures S11 and S15 in File S1). The presence of CG-methylation in actively transcribed genes has been described for years, with a role still undetermined at present. Several hypotheses are being examined to discover possible functions for that methylation, some converging towards a potential role in regulating splicing [23]. SG1 could possibly target CG-methylated genes in WT plants, and such targeting could rely on active transcription, or CG-methylation of gene bodies, or both, by direct binding of SG1 to proteins involved in these processes. Intriguingly, even if methylation invades some genes in sg1 or ibm1, it does not prevent their transcription, or at least it does not change the level of transcripts detected by qPCR which is only tested through small transcript sections of 200 bp. The links between transcription or splicing and methylation (CG, CHG or H3K9me2) remains to be investigated further but sg1 mutants could be appropriate candidates to clarify that point in plants.
Another intriguing observation is that certain CHG-enriched gene bodies of sg1 seem to be also slightly enriched in CHH methylation, compared to the WT (Figures S11 and S14 in File S1); an observation also true for ibm1 mutant plants. Part of the CHH methylation depends on the RdDM pathway and the production of sRNAs, but the ectopic CHH methylation in sg1 is unlikely due to a stimulation of the RdDM pathway and a production of sRNAs from genic regions: first, ago4/sg1 double mutants are phenotypically similar to sg1, indicating that disrupting the RdDM pathway has limited consequences on sg1 plants and, second, it is unlikely that sg1 mutants and WT plants have genome-wide dissimilar contents of sRNAs, since ibm1 and WT plants have not [11]. However, recent evidences point toward the crucial role of CMT2 in maintaining the CHH methylation that is independent on RdDM [3]. The authors hypothesized that CMT2 might bind H3K9me2 marks, exactly like CMT3, to target DNA and methylate cytosines in the CHH context. Genes enriched in H3K9me2 methylation are then also probably targeted by CMT2 in sg1 and ibm1, explaining their enrichment in CHH. H3K9me2 marks could therefore be linked to two contexts of methylated cytosines, CHH and CHG. Further work is required to shed the light on the mechanisms of interplay between SG1 and IBM1, as well as to understand the functional consequences of genic non-CG methylation.
Materials and Methods
Plant Material and Growth Conditions
A. thaliana RILs and HIFs were obtained from the INRA Versailles collection (http://publiclines.versailles.inra.fr/) and described before [24]–[25]. sg1 T-DNA mutants were obtained from the Arabidopsis stock center: sg1-2 is SAIL_310B06 and sg1-3 corresponds to GK_045A07. In addition, the following Arabidopsis mutants were used: ago4-2 [26], cmt3-11 [27], kyp (SALK_069326) [28], met1-1 [14], and ibm1-1 [10]. Projected area analyses of leaves were performed as previously described [12].
Whole-genome Bisulfite Sequencing and Analyses
Col-0 and sg1-1 homozygous seeds were grown in long day conditions for 25 days in culture rooms and 5 to 25 µg of genomic DNA were extracted using a CTAB phenol-chloroform extraction protocol followed by RNAse treatment (Qiagen). Bisulfite treatment, library preparation and whole genome sequencing were performed at BGI (Shenzhen, China) using Illumina’s recommendations and HiSeq technology for paired-end 91 bp reads. We sequenced the methylome of two biological replicates for the WT Col-0 and two for sg1-1. The sequencing results are described in Table S1 in File S2.
For sequencing analysis low-quality reads (q <30) were discarded and clean reads were mapped to the Col-0 Arabidopsis thaliana TAIR 10 reference genome using the Bowtie and Bismark softwares with options ‘-q -n 3–non directional’. Subsequent analyses were done with SeqMonk (Babraham Bioinformatics - SeqMonk Mapped Sequence Analysis Tool by Simon Andrews). Only cytosines covered by at least 5 unique reads were retained. For genome-wide analyses, we defined windows of 200 bp overlapping by 50 bp intervals to calculate the percentages of methylation along the genome. To determine differentially methylated genes, a Fisher’s exact test was performed comparing methylated and unmethylated counts. We then corrected nominal P-values and estimated the corresponding q-values to retain genes with q-values <0.05. For ibm1-6 and the corresponding WT Col-0, we retrieved the raw data from NCBI (GEO query: GSE39901) to align it as described for the data obtained in the present study.
Chromatin Immunoprecipitation (ChIP) and Real-time Quantitative PCR (qPCR) Analyses
ChIP assays were performed as previously described [29] on the aerial parts of 25 days-old seedlings using anti-histone H3 dimethyl lysine 9 antibody from Abcam (Catalog No. ab1220) and anti-histone H3 antibody from Abcam (Catalog No. ab1791). All ChIP experiments were quantified by qPCR using primers listed in Table S5 in File S2. SHOOT MERISTEMLESS (STM) was used as the internal control for the ChIP experiments. Data are represented as the ratio of (H3K9me2 gene/H3 gene)/(H3K9me2 STM/H3 STM). Experiments were repeated in at least three biological replicates.
Gene Expression Analysis
Total RNA was isolated from the aerial parts of 15 days-old seedlings using the RNeasy Plant Mini kit (Qiagen) followed by a DNAse treatment (Fermentas). RT-PCR was performed on 500 ng of total RNAs with the M-MLV reverse transcriptase (Fermentas) and cDNAs were diluted 10 times. 5 µl were used for qRT-PCRs using a CFX96 real-time PCR machine (BioRad) with a SYBR solution (Eurogentec) using the primers listed in Table S5 in File S2. Expression levels were normalized against the Arabidopsis UBC21 gene (At5g25760). Experiments were repeated in three biological replicates.
Supporting Information
Contains: Figure S1: SG1 fine mapping. (A) QTL mapping for shoot size in the Bur-0 x Col-0 RIL set identifies a major QTL on chromosome 5 [12]. (B) Fine-mapping of the major-effect locus to 8 kb around At5g11470 on chromosome 5. Figure S2: RG1 fine mapping in the Ct-1 x Col-0 maps to SG1. QTL mapping for root growth (total root length) in the Ct-1 x Col-0 RIL set (A) reveals RG1, a QTL confirmed in vitro 10 days after germination (days after germination) (B) and in the greenhouse 20 days after germination (C). Fine-mapping and sequencing revealed that sg1-1 mutation is responsible for this QTL. Figure S3: SG1 gene models. SG1 gene models according to TAIR v10, EuGène and our cDNA sequencing data is shown with UTRs (black boxes), exons (clear boxes) and introns (dashed lines). Left, 5′ end; right, 3′ end. Figure S4: sg1 phenotype at the flowering stage. Picture of a tray of 40 days-old WT Col-0 plants (left) and sg1-1 plants (middle, right) derived from a parent exhibiting (a) or not (b) a phenotype. The flowering delay can be seen for sg1-1 (a). Figure S5: sg1-2 and sg1-3 mutants. (A) Phenotype of plants homozygous for sg1-2 T-DNA (bottom) and WT Col-0 (top) in two successive generations after fixation at the homozygous state. (B) Analysis of the projected rosette area (pixels) of 15 days-old seedlings grown in vitro. sg1-2(gen2): second homozygous generation. Each following generation (gen3, gen4 and gen5) descends from the previous one. ***p<0.001, ns: not significant. (C) Picture of a mature sg1-2 plant. The arrow points one sterile bud that failed to develop into a silique. The frame shows an enlargement of a rosette leaf showing the over-serrated shape. (D) Segregating plants (left) originating from a unique sg1-3 homozygous parent illustrate the stochasticity of the sg1 phenotype compared to segregating plants from a unique WT Col-0 parent (right). Figure S6: Proteins containing Bromo adjacent homology (BAH) domain (IPR001025) in Arabidopsis. The different domains (according to the InterPro database) are indicated: Agenet (IPR008395), ATPase AAA core (IPR003959), Znf PHD-finger (IPR013083), Znf PHD = finger (IPR019787), Chromodomain (IPR023780), TFIIS centre (IPR003618), TFIIS/elonginA/CRSP70 N (IPR017923), C5 DNA meth (IPR022702) and RRM RNP (IPR012677). Figure S7: Comparison of the percentages of methylation in 200 bp-windows throughout the genome of sg1-1 and Col-0. Each 200 bp-window is represented by a dot. Windows were designed to overlap by 50 bp and only cytosine position covered by at least 5 reads were considered in the calculation. Reads that were not matching at unique locations were discarded. The percentage of methylation in CG (A) and CHH (C) contexts are similar between Col-0 and sg1-1, while CHG methylated regions are over-represented in sg1-1 (B). Figure S8: Scatter plots comparing the percentages of methylation in gene bodies of sg1-1 and Col-0. Each gene according to the TAIR10 annotations is represented by a dot. Only cytosine position covered by at least 5 reads were considered in the calculation. Reads that were not mapping to unique locations were discarded. The percentage of methylation in CG (A) are similar between Col-0 and sg1, while CHH-methylated genes seem to be slightly more methylated in sg1-1 (B). Figure S9: Comparison of the percentages of methylation in transposable elements (TEs) in sg1-1 and Col-0. Each transposable element according to the TAIR10 annotations is represented by a plot. Only cytosine position covered by at least 5 reads were considered in the calculation. Reads that were not matching at unique locations were discarded. The percentage of methylation in CG (A), CHG (B) and CHH (C) are similar between Col-0 and sg1-1. Figure S10: Genomic distribution of the CHG differentially-methylated genes identified in sg1-1 . Only genes that present a 2x increase in CHG methylation are represented (see Mat&Met). One hypermethylated gene corresponds to a black line drawn on one of the five chromosomes of Arabidopsis. Gene-poor centromeres are indicated by a ‘c’. The Chromosome Map Tool of TAIR (www.arabidopsis.org) was used to map the different genes on chromosomes. Figure S11: Gene-body methylation in sg1-1 . Data is based on methylome results. CG, CHG and CHH methylations are shown on top (positive values) and bottom strand (negative values) across the genes indicated. The corresponding gene models are shown according to TAIR v10. Figure S12: Comparison of data samples for sg1-1 and ibm1-6 methylome analyses. (A) Distribution of the percentages of CHG methylation of genes in different samples: sg1-1, ibm1-6, the WT Col-0 used in this study and one of the three Col-0 samples (sample #3) used by Jacobsen and colleagues [16]. (B) Scatter plots comparing the percentages of methylation in gene bodies of the Col-0 WT used in this study and one of the three Col-0 samples (sample #3) used by Jacobsen and colleagues [16]. Each gene according to the TAIR10 annotations is represented by a dot. The percentage of methylation in CHG are similar between the two samples. Figure S13: Phenotype of the double met1-1/sg1-2 mutant. (A) Genotyped F2 plants from the met1-1/sg1-2 cross. We found only three plants fixed for the two mutations (circled in red). (B) F3 plants (descending from a F2-genotyped met1/sg1 plant shown in A) were grown in the greenhouse and pictured after three weeks. Figure S14: Phenotype of the double ago4-2 / sg1-2 mutant. (A) Phenotype of the sg1-2/ago4-2 mutant grown in greenhouse. (B) Analysis of the projected rosette area (pixels) of 15 days-old double mutant seedlings grown in vitro. n: number of individuals analyzed for each genotype. ***p<0.001, ns: not significant. Figure S15: Gene-body methylation of RDR2 and DCL3 in sg1-1 . Data is based on methylome results. CG, CHG and CHH methylations are shown on top (positive values) and bottom strand (negative values) across the genes indicated. The corresponding gene models are shown according to TAIR v10. Figure S16: qRT-PCR expression analysis of At4g00450 and At5g35210. Error bars are SEM from three technical replicates. Figure S17: qRT-PCR expression analysis of IBM1 , expressed as a percentage of mRNA expression in sg1 relative to WT. A gene model representation shows the two mRNA regions assayed, either upstream or downstream of the heterochromatin region located in the large IBM1 intron. Error bars are SEM from three technical replicates. Figure S18: Complementation of sg1-1 by IBM1. ibm1-1 and sg1-1 were transformed by a construct carrying a functional IBM1 cDNA [17]. T2 plants were selected on kanamycin, transferred to the greenhouse and pictured after three weeks of growth. Figure S19: Examples of genes differentially methylated in sg1-1 and ibm1 . Graphic representation is based on the publicly available data for ibm1-6 and the corresponding Col-0 methylome [16]. The genome annotation is based on TAIR v10. The red box indicates the genes differentially methylated. Figure S20: Examples of genes CHG-methylated in ibm1 but not sg1-1 . Graphic representation is based on the publicly available data for ibm1-6 and the corresponding Col-0 methylome [16]. The genome annotation is based on TAIR v10. The red box indicates the genes differentially methylated.
(PDF)
Contains: Table S1. Sequencing statistics. Table S2. Bisulfite sequencing statistics. Data were obtained by the bismark methylation extractor software. Table S3. List of genes differentially CHG enriched by at least a factor 2 in sg1-1 compared to Col-0. The ratios between the number of reads containing methylated cytosines and the total number of reads per gene is indicated. The data correspond to the mean from two different biological replicates for both Col-0 and sg1-1. Hyper- and hypomethylated genes are listed. Genes selected to analyze both CHG methylation and H3K9 enrichment within genes bodies (Figures 3 and 5) are indicated in red. Table S4. Members of the RdDM pathway with a CHG-methylation status affected in sg1-1. Table S5. List of oligos used in this study.
(XLSX)
Acknowledgments
We thank Hidetoshi Saze for providing seeds of ibm1-1. We thank Olivier Mathieu for the IBM1-L construct [17]. We thank Astrid Agorio for critical reading of the manuscript.
Funding Statement
VC was supported by the BAP INRA division and a Marie Curie European Reintegration Grant (FP7-PERG08-GA-2010-277068) to NB and OL. The authors acknowledge funding from ANR (Project 11-JSV7-0013) to NB. DV was supported by funding from the European Commission Framework Programme 6, Integrated Project ‘AGRON-OMICS’/LSHG-CT-2006-037704 to OL. EK was supported by funding from ANR grant ‘ARABRAS’/ANR-06-ERAPG-003-01 to OL. FC was supported by funding from the European Commission Framework Programme 7, ERC Starting Grant ‘DECODE’/ERC-2009-StG-243359 to OL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, et al. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Rev. Genet. 11: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, et al. (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huff JT, Zilberman D (2012) Regulation of biological accuracy, precision, and memory by plant chromatin organization. Curr. Opin. Genet. Dev. 22: 132–138. [DOI] [PubMed] [Google Scholar]
- 5. Ebbs ML, Bender J (2006) Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell 18: 1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Du J, Zhong X, Bernatavichute YV, Stroud H, Feng S, et al. (2012) Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151: 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johnson LM, Bostick M, Zhang X, Kraft E, Henderson I, et al. (2007) The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. : CB 17: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Inagaki S, Miura-Kamio A, Nakamura Y, Lu F, Cui X, et al. (2010) Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J. 1 29: 3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miura A, Nakamura M, Inagaki S, Kobayashi A, Saze H, et al. (2009) An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J. 28: 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saze H, Shiraishi A, Miura A, Kakutani T (2008) Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science 319: 462–465. [DOI] [PubMed] [Google Scholar]
- 11. Fan D, Dai Y, Wang X, Wang Z, He H, et al. (2012) IBM1, a JmjC domain-containing histone demethylase, is involved in the regulation of RNA-directed DNA methylation through the epigenetic control of RDR2 and DCL3 expression in Arabidopsis. Nucl. Acids Res. 40: 8905–8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vlad D, Rappaport F, Simon M, Loudet O (2010) Gene transposition causing natural variation for growth in Arabidopsis thaliana. PLoS Genet. 6: e1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiex T, Moisan A, Rouzé P (2001) EuGene: An Eucaryotic Gene Finder that combines several sources of evidence. Comp. Biol. LNCS 2066: LNCS 2066. pp. 111–125.
- 14. Kankel MW, Ramsey DE, Stokes TL, Flowers SK, Haag JR, et al. (2003) Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163: 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vongs A, Kakutani T, Martienssen RA, Richards EJ (1993) Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928. [DOI] [PubMed] [Google Scholar]
- 16. Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152: 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rigal M, Kevei Z, Pélissier T, Mathieu O (2012) DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J. 31(13): 2981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saze H, Kitayama J, Takashima K, Miura S, Harukawa Y, et al. (2013) Mechanism for full-length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nat Commun. 4: 2301. [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Duan CG, Tang K, Wang B, Zhang H, et al. (2013) RNA-binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin-containing gene IBM1. Proc Natl Acad Sci U S A. 110(38): 15467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X, Yu CW, Duan J, Luo M, Wang K, et al. (2012) HDA6 directly interacts with DNA methyltransferase MET1 and maintains transposable element silencing in Arabidopsis. Plant Physiol. 158: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, et al. (2012) The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484: 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sasaki T, Kobayashi A, Saze H, Kakutani T (2012) RNAi-independent de novo DNA methylation revealed in Arabidopsis mutants of chromatin remodeling gene DDM1. Plant J. 70: 750–758. [DOI] [PubMed] [Google Scholar]
- 23. Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Rev. Genet. 13: 484–492. [DOI] [PubMed] [Google Scholar]
- 24. Loudet O, Gaudon V, Trubuil A, Daniel-Vedele F (2005) Quantitative trait loci controlling root growth and architecture in Arabidopsis thaliana confirmed by heterogeneous inbred family. TAG 110: 742–753. [DOI] [PubMed] [Google Scholar]
- 25. Simon M, Loudet O, Durand S, Berard A, Brunel D, et al. (2008) Quantitative trait loci mapping in five new large recombinant inbred line populations of Arabidopsis thaliana genotyped with consensus single-nucleotide polymorphism markers. Genetics 178: 2253–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agorio A, Vera P (2007) ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 19: 3778–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, et al. (2001) Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292: 2077–2080. [DOI] [PubMed] [Google Scholar]
- 28. Mathieu O, Probst AV, Paszkowski J (2005) Distinct regulation of histone H3 methylation at lysines 27 and 9 by CpG methylation in Arabidopsis. EMBO J. l 24: 2783–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Angel A, Song J, Dean C, Howard M (2011) A Polycomb-based switch underlying quantitative epigenetic memory. Nature 476: 105–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains: Figure S1: SG1 fine mapping. (A) QTL mapping for shoot size in the Bur-0 x Col-0 RIL set identifies a major QTL on chromosome 5 [12]. (B) Fine-mapping of the major-effect locus to 8 kb around At5g11470 on chromosome 5. Figure S2: RG1 fine mapping in the Ct-1 x Col-0 maps to SG1. QTL mapping for root growth (total root length) in the Ct-1 x Col-0 RIL set (A) reveals RG1, a QTL confirmed in vitro 10 days after germination (days after germination) (B) and in the greenhouse 20 days after germination (C). Fine-mapping and sequencing revealed that sg1-1 mutation is responsible for this QTL. Figure S3: SG1 gene models. SG1 gene models according to TAIR v10, EuGène and our cDNA sequencing data is shown with UTRs (black boxes), exons (clear boxes) and introns (dashed lines). Left, 5′ end; right, 3′ end. Figure S4: sg1 phenotype at the flowering stage. Picture of a tray of 40 days-old WT Col-0 plants (left) and sg1-1 plants (middle, right) derived from a parent exhibiting (a) or not (b) a phenotype. The flowering delay can be seen for sg1-1 (a). Figure S5: sg1-2 and sg1-3 mutants. (A) Phenotype of plants homozygous for sg1-2 T-DNA (bottom) and WT Col-0 (top) in two successive generations after fixation at the homozygous state. (B) Analysis of the projected rosette area (pixels) of 15 days-old seedlings grown in vitro. sg1-2(gen2): second homozygous generation. Each following generation (gen3, gen4 and gen5) descends from the previous one. ***p<0.001, ns: not significant. (C) Picture of a mature sg1-2 plant. The arrow points one sterile bud that failed to develop into a silique. The frame shows an enlargement of a rosette leaf showing the over-serrated shape. (D) Segregating plants (left) originating from a unique sg1-3 homozygous parent illustrate the stochasticity of the sg1 phenotype compared to segregating plants from a unique WT Col-0 parent (right). Figure S6: Proteins containing Bromo adjacent homology (BAH) domain (IPR001025) in Arabidopsis. The different domains (according to the InterPro database) are indicated: Agenet (IPR008395), ATPase AAA core (IPR003959), Znf PHD-finger (IPR013083), Znf PHD = finger (IPR019787), Chromodomain (IPR023780), TFIIS centre (IPR003618), TFIIS/elonginA/CRSP70 N (IPR017923), C5 DNA meth (IPR022702) and RRM RNP (IPR012677). Figure S7: Comparison of the percentages of methylation in 200 bp-windows throughout the genome of sg1-1 and Col-0. Each 200 bp-window is represented by a dot. Windows were designed to overlap by 50 bp and only cytosine position covered by at least 5 reads were considered in the calculation. Reads that were not matching at unique locations were discarded. The percentage of methylation in CG (A) and CHH (C) contexts are similar between Col-0 and sg1-1, while CHG methylated regions are over-represented in sg1-1 (B). Figure S8: Scatter plots comparing the percentages of methylation in gene bodies of sg1-1 and Col-0. Each gene according to the TAIR10 annotations is represented by a dot. Only cytosine position covered by at least 5 reads were considered in the calculation. Reads that were not mapping to unique locations were discarded. The percentage of methylation in CG (A) are similar between Col-0 and sg1, while CHH-methylated genes seem to be slightly more methylated in sg1-1 (B). Figure S9: Comparison of the percentages of methylation in transposable elements (TEs) in sg1-1 and Col-0. Each transposable element according to the TAIR10 annotations is represented by a plot. Only cytosine position covered by at least 5 reads were considered in the calculation. Reads that were not matching at unique locations were discarded. The percentage of methylation in CG (A), CHG (B) and CHH (C) are similar between Col-0 and sg1-1. Figure S10: Genomic distribution of the CHG differentially-methylated genes identified in sg1-1 . Only genes that present a 2x increase in CHG methylation are represented (see Mat&Met). One hypermethylated gene corresponds to a black line drawn on one of the five chromosomes of Arabidopsis. Gene-poor centromeres are indicated by a ‘c’. The Chromosome Map Tool of TAIR (www.arabidopsis.org) was used to map the different genes on chromosomes. Figure S11: Gene-body methylation in sg1-1 . Data is based on methylome results. CG, CHG and CHH methylations are shown on top (positive values) and bottom strand (negative values) across the genes indicated. The corresponding gene models are shown according to TAIR v10. Figure S12: Comparison of data samples for sg1-1 and ibm1-6 methylome analyses. (A) Distribution of the percentages of CHG methylation of genes in different samples: sg1-1, ibm1-6, the WT Col-0 used in this study and one of the three Col-0 samples (sample #3) used by Jacobsen and colleagues [16]. (B) Scatter plots comparing the percentages of methylation in gene bodies of the Col-0 WT used in this study and one of the three Col-0 samples (sample #3) used by Jacobsen and colleagues [16]. Each gene according to the TAIR10 annotations is represented by a dot. The percentage of methylation in CHG are similar between the two samples. Figure S13: Phenotype of the double met1-1/sg1-2 mutant. (A) Genotyped F2 plants from the met1-1/sg1-2 cross. We found only three plants fixed for the two mutations (circled in red). (B) F3 plants (descending from a F2-genotyped met1/sg1 plant shown in A) were grown in the greenhouse and pictured after three weeks. Figure S14: Phenotype of the double ago4-2 / sg1-2 mutant. (A) Phenotype of the sg1-2/ago4-2 mutant grown in greenhouse. (B) Analysis of the projected rosette area (pixels) of 15 days-old double mutant seedlings grown in vitro. n: number of individuals analyzed for each genotype. ***p<0.001, ns: not significant. Figure S15: Gene-body methylation of RDR2 and DCL3 in sg1-1 . Data is based on methylome results. CG, CHG and CHH methylations are shown on top (positive values) and bottom strand (negative values) across the genes indicated. The corresponding gene models are shown according to TAIR v10. Figure S16: qRT-PCR expression analysis of At4g00450 and At5g35210. Error bars are SEM from three technical replicates. Figure S17: qRT-PCR expression analysis of IBM1 , expressed as a percentage of mRNA expression in sg1 relative to WT. A gene model representation shows the two mRNA regions assayed, either upstream or downstream of the heterochromatin region located in the large IBM1 intron. Error bars are SEM from three technical replicates. Figure S18: Complementation of sg1-1 by IBM1. ibm1-1 and sg1-1 were transformed by a construct carrying a functional IBM1 cDNA [17]. T2 plants were selected on kanamycin, transferred to the greenhouse and pictured after three weeks of growth. Figure S19: Examples of genes differentially methylated in sg1-1 and ibm1 . Graphic representation is based on the publicly available data for ibm1-6 and the corresponding Col-0 methylome [16]. The genome annotation is based on TAIR v10. The red box indicates the genes differentially methylated. Figure S20: Examples of genes CHG-methylated in ibm1 but not sg1-1 . Graphic representation is based on the publicly available data for ibm1-6 and the corresponding Col-0 methylome [16]. The genome annotation is based on TAIR v10. The red box indicates the genes differentially methylated.
(PDF)
Contains: Table S1. Sequencing statistics. Table S2. Bisulfite sequencing statistics. Data were obtained by the bismark methylation extractor software. Table S3. List of genes differentially CHG enriched by at least a factor 2 in sg1-1 compared to Col-0. The ratios between the number of reads containing methylated cytosines and the total number of reads per gene is indicated. The data correspond to the mean from two different biological replicates for both Col-0 and sg1-1. Hyper- and hypomethylated genes are listed. Genes selected to analyze both CHG methylation and H3K9 enrichment within genes bodies (Figures 3 and 5) are indicated in red. Table S4. Members of the RdDM pathway with a CHG-methylation status affected in sg1-1. Table S5. List of oligos used in this study.
(XLSX)