

Abstract
The purpose of this longitudinal study was to identify changes in ERG responses associated with vigabatrin treatment. We accomplished this by recording longitudinally ERGs in children before and during vigabatrin treatment and comparing results between children on vigabatrin monotherapy and those taking additional anticonvulsive medications. Thirty-three children on vigabatrin therapy were tested; the duration between visits was approximately 6 months. Thirteen children were assessed initially before starting vigabatrin therapy and seven were assessed soon after (age range 1.5–126 months, median 6 months). The remaining 13 patients were already on vigabatrin at the time of initial visit (age range 6.5–180 months, median 16 months). ERGs were tested using the standard protocol established by the International Society for Clinical Electrophysiology of Vision, with Burian-Allen bipolar contact-lens electrodes. In addition to standard responses we recorded photopic oscillatory potentials (OPs). All 33 patients were tested longitudinally on at least two occasions and 11 were tested on three occasions. For children whose only anticonvulsive drug was vigabatrin there was a significant curvature (quadratic function, p<0.05) of the predicted cone b-wave amplitude with time; exhibited as increase in b-wave amplitude followed by subsequent decrease. Descriptive data demonstrated the same pattern in the group taking anticonvulsive medications in addition to vigabatrin. In most children the flicker amplitude declined between 6 months and 1 year of vigabatrin treatment. Our data demonstrated that rod responses, which may be abnormal before initiation of vigabatrin, did not change substantially with vigabatrin treatment.
Keywords: ERG, pediatric, retinal toxicity, vigabatrin
Introduction
Vigabatrin (γ-vinyl-GABA) is an antiepileptic drug, proven to be successful in the management of partial seizures and infantile spasms [1–4]. The structure of vigabatrin closely resembles the inhibitory neurotransmitter γ-aminobutyric acid (GABA), found in both the brain and retina. The anticonvulsant effect of vigabatrin is achieved by irreversible inhibition of the enzyme GABA-transaminase used to break down GABA. This results in increased levels of GABA in the brain and in the retina.
There have been ongoing reports linking the use of vigabatrin to visual field defects [5–13]. The pattern of visual field deficit is that of a bilateral concentric constriction [7] and occurs in between 40 and 50% of adult patients on vigabatrin treatment [7, 10, 12]. Wohlrab et al. [14] found a bilateral, concentric visual field constriction in six children on vigabatrin treatment, out of 12 tested. Although 153 pediatric patients had been treated with vigabatrin only 12 had visual field assessment because the very young age or presence of developmental delay precluded assessment in the other children [14]. The incidence of visual field deficits in infants treated with vigabatrin who are under one year of age is unknown [14].
Retinal dysfunction assessed with the electroretinogram (ERG) has also been found in patients taking vigabatrin [5, 8, 11, 12, 15, 16]. The type of retinal dysfunction has been reported to involve rod systems [8], cone systems [12, 15, 17, 18] and oscillatory potential (OP) responses [5, 11, 12, 15, 16, 18]. Daneshvar [8] found patients on vigabatrin with visual field deficits had abnormalities of ERG scotopic responses and Arndt [16] showed that all patients with severe visual field defects showed non-recordable scotopic OP responses. Krauss [15] found that patients with symptomatic visual field loss demonstrated evidence of a reduced cone response b-wave. Decreased amplitude of the 30-Hz flicker response was highly associated with the severity and presence of vigabatrin-attributed visual field deficits [17], whereas vigabatrin-related increase in cone b-wave implicit time was not associated with visual field deficit [19].
Johnson et al. [20] found there was no significant change in ERG results after cessation of vigabatrin, although there were improvements in some responses in several patients with less marked visual field deficit. The questions of whether ERG abnormalities result from vigabatrin treatment or whether other drugs complicate the effect of vigabatrin treatment on the ERG are unknown. To answer these questions we assessed ERGs in children before vigabatrin treatment and longitudinally after the initiation of treatment. We compared also ERG changes between children taking vigabatrin as the only anticonvulsant drug with children taking other anticonvulsant therapies in combination with vigabatrin.
Methods
Subjects
Thirty-three children on vigabatrin treatment, referred for ophthalmological, visual and electrophysiological assessment, were tested longitudinally between September 1998 and August 2000; the duration between visits was approximately 6 months. Thirteen of these children were assessed initially before starting vigabatrin treatment and another seven were assessed soon after. As it was important not to delay initiation of vigabatrin treatment it was not always possible able to conduct the ERG before vigabatrin was started; five were tested within 1 week of initiation of vigabatrin treatment; one was tested within 30 days and one within 60 days of initiation of treatment. Data from these 20 children constituted the baseline ERG response (age range 1.5–126 months, mean 14 months, median 6 months). The remaining 13 children were already on vigabatrin at the time of initial visit (age range 6.5–180 months, mean 36 months, median 16 months). All subjects had at least one follow-up visit with 11/33 having a total of three visits. Patient characteristics, including a description of whether the children were on vigabatrin monotherapy or on a combination of anticonvulsant medications, are shown in Table 1.
Table 1.
| Subject | Age start vigabatrin (months) | Type of seizure | Other health problems | Other meds | Age first ERG (months) |
|---|---|---|---|---|---|
| 1. | 6 | GenT/C | TS | Started carbamazepine | 6 |
| 2. | 16 | IS | DD, strab microcephaly | Nitrazepam VGB d/c at 26 months started VPA | 13 |
| 3. | 6 | IS GenT/C |
DD. | Phenobarbital clobazam, lamotrigine | 6 |
| 4. | 7 | IS | DD, nystag, strab lissencephaly | Phenobarbital | 7 |
| 5. | 5 | IS | Trisomy 21 | 5 | |
| 6. | 7 | IS | TS | Phenobarbital | 7 |
| 7. | 1.5 | Tonic | DD | Clobazam, phenobarbital | 1.5 |
| 8. | 7 | IS | DD | 7 | |
| 9. | 126 | CPS GenT/C |
Valproic acid | 126 | |
| 10. | 5 | IS | DD | Phenobarbital | 5 |
| 11. | 6 | IS | 6 | ||
| 12. | 4 | IS | ACC, DD | Phenobarbital lamotrigine ranitidine started ACTH | 4 |
| 13. | 11 | IS | TS, DD | 11 | |
| 14. | 6 | IS | Mild DD | 6 | |
| 15. | 26 | IS | 26 | ||
| 16. | 13 | IS | NF-1, Optic glioma | 13 | |
| 17. | 4 | IS | Lissencephaly | Phenobarbital | 4 |
| 18. | 5 | IS | Lissencephaly | 7 | |
| 19. | 5 | IS. | HC, shunt, IBD | Started ACTH | 10 |
| 20. | 6 | IS | TS | Started clobazam started VPA | 13 |
| 21. | 4 | IS, T M |
DD | Phenobarbital, ACTH | 8 |
| 22. | 13 | CPS GenT/C |
TS, DD | 17 | |
| 23. | 5 | IS | 6 | ||
| 24. | 24 | Focal. | Herpes simplex encephalitis | Nitrazepam, lamotrigine | 42 |
| 25. | 7 | IS | MDF, DD | Clobazam | 19 |
| 26. | 6 | IS, CPS | TS | Carbamazepine | 16 |
| 27. | 4 | IS | DD, hypotonia, HSM, GI tube | Cisapride started ACTH | 8 |
| 28. | 12 | CPS | DW; HC, OA | 36 | |
| 29. | 7 | IS | TS | VGB d/c at 18 months | 11 |
| 30. | 156 | CPS | TS | Valproic acid carbamazepine | 180 |
| 31. | 96 | GenT/C | TS | Valproic acid carbamazepine | 120 |
| 32. | 8 | Partial GenT/C | TS | 46 | |
| 33. | 6 | IS | 6 |
TS, tuberous sclerosis; IS, infantile spasms; CPS, complex partial seizures; genT/C, generalized tonic/clonic seizures; T, tonic seizures; M, myoclonic seizures; DW, Dandy Walker syndrome; DD, developmental delay; OA, optic atrophy; HC = hydrocephalus; IBD, ischemic brain damage; HSM, hepatosplenomegaly; NF-1, neurofibromatosis type 1; MDF, Mild dysmorphic features, enceph, encephalopathy, nystag, nystagmus; strab, strabismus; VPA, valproic acid, when other drugs were started after initiation of vigabatrin therapy this is indicated.
All testing was performed in the Visual Electrophysiology Unit at the Hospital for Sick Children. The research followed the tenets of the Declaration of Helsinki. Subjects 16 years and over as well as parents of subjects under 16 years gave signed consent for their participation in the study. The consent form acknowledged that research procedures were described, any questions answered and that harms and benefits of participating were fully explained. Approval for this study was obtained from the Research Ethics Board at the Hospital For Sick Children.
Subjects were excluded from the study if they had a retinal dystrophy or a known family history of retinal dystrophy, any eye disease associated with abnormal ERG, previous intraocular surgery or systemic disease known to affect the retina. Visual acuity was assessed using preferential looking-based techniques, with Teller acuity cards (Vistech Consultants, Dayton, OH) or Cardiff Acuity test (Keeler, Windsor, UK). Twenty-five patients were assessed with the Teller Acuity Cards (median age at first test, 13 months) and four patients were assessed with Cardiff Acuity test (median age at first test, 25 months).
At time of initial visit for electroretinography 24 children (under 56 months of age) were sedated with oral chloral hydrate (80 mg kg of body weight; maximum single dose of 1 g). The remaining nine children (27%) were not sedated, six toddlers (age range 1.5–13 months) and three children (age range 120–180 months).
ERG technique
Before ERG assessment, both pupils were pharmacologically dilated with 1% cyclopentolate and 2.5% phenylephrine in children and with 0.5% cyclopentolate in infants under 4 months. Each subject was dark-adapted for at least 30 min before recording began. After dark adaptation, the tester, wearing a head-mounted red-lamp (Wratten gelatin filter no. 70, Kodak-Eastman, Rochester, NY), instilled one drop of proparacaine 0.5%. A bipolar Burian-Allen electrode (Hansen Ophthalmic Development Laboratory, Iowa City, IA) of the appropriate size for the individual subject’s eye was placed on the corneal eye surface. An electrode attached to the forehead served as the ground.
ERG rod, maximal (standard flash), oscillatory potentials, cone response and flicker responses were recorded according to ISCEV standards [21] using a Grass PS22 photic stimulator (Grass Instruments, Quincy, MA) and Ganzfeld stimulation (LKC Technologies, Gaithersburg, MD). Neuroscan (Herndon, VA) manufactured the recording equipment and software used. The standard flash intensity was 2 cd·s/m2(Gamma Scientific DR-2000 integrated photometer, San Diego). The rod response was elicited by the standard flash attenuated by 2.6 log units using a neutral density filter mounted on a filter wheel (LKC Technologies).
After dark-adapted responses were recorded, subjects were light-adapted (at 30 cd/m2) for 10 min. The single-flash cone responses (2 cd·s/m2and4 cd·s/m2), followed by 30-Hz flicker (2.0 cd.s/m2) responses were recorded in the presence of an adapting background.
Individual ERG responses were amplified (gain, 2833×; bandpass, 0.3–300 Hz; analogue-to-digital rate, 1000), digitized and saved on a computer disk for subsequent analysis. ERG responses were analyzed off-line. OPs were isolated from the standard flash ERG response and from the cone response by digital-filtering (bandwidth, 100–300 Hz; roll off, 12 dB/octave) and averaging the epochs. The sum of OP amplitude was calculated for both dark and light adapted OPs.
After ERG testing, both direct and indirect ophthalmoscopy was performed through pharmacologically dilated pupils on all patients. Refractive errors were assessed with cycloplegic refraction.
Data analysis
Much of our data were collected from infants while ERG responses were still developing [22]. To avoid confounding effects of age-expected developmental changes in longitudinal measures of ERGs, all data were expressed as relative units to age-expected responses. For amplitude data the log of the age-expected amplitude was subtracted from the log of the individual response; negative values therefore represented reduction in ERG amplitude relative to expected value. For implicit time data, age expected implicit times were subtracted from individual patient values and therefore responses that were delayed relative to expected values were represented by positive values.
Descriptive statistics were used to illustrate changes in ERG responses over time. The visit classification depended on approximate duration of vigabatrin treatment. The baseline visit was defined as the time at initiation of vigabatrin. Subsequent visits were approximately 6 months after baseline testing (range 4–8 months); at 12 months (range 10–14 months); at 18 months (range 16–20 months). Children were separated into two groups. One group was comprised of children who were taking no other anticonvulsant drugs at time of baseline testing or in combination with vigabatrin. The other group was comprised of children who were taking other anticonvulsant medications with vigabatrin or before the initiation of vigabatrin.
A repeated measures analysis of variance was used to establish the change over time of the 30-Hz flicker amplitude, the cone b-wave amplitude and the sum of the oscillatory potential amplitude and to determine if there were any significant group differences over time (vigabatrin only vs. vigabatrin plus other anticonvulsive medication) for any of these outcome measures.
Results
Representative data describing ERG cone system responses from two children on vigabatrin treatment are shown in Figure 1. Data from four subject visits are shown and data from a control subject (22 months of age) are shown for comparison (Figure 1c). Photopic ERGs: the cone isolated response, photopic oscillatory potentials and the 30-Hz flicker response are shown. Subject 1 (Figure 1a) was diagnosed with tuberous sclerosis at 6 months of age. She was developmentally normally and had a normal birth history. Her first ERG was recorded at 6 months of age before initiation of vigabatrin treatment. At this time, cone responses and OPs were within normal limits for age, although rod and rod-cone b wave amplitudes were below normal (not shown) even though she was on no medication. Her initial dose of vigabatrin was 1250 mg day. After 6 months of vigabatrin treatment (12 months of age) there was an increase in amplitudes, greater than that expected for normal development, of rod, cone a- and b-waves, flicker amplitudes and scotopic and photopic OPs. After 12 months of vigabatrin (18 months of age) scotopic and photopic responses had decreased in amplitude more than would be expected for age determined inter-visit variability (our normal lab age corrected limits of inter-visit variability are 0.09 log units). Scotopic responses remained lower than age-expected limits, photopic OP amplitudes reduced falling below age expected limits although the photopic a- and b- wave responses and flicker response were still within normal limits despite the reduction in amplitude. At 23 months of age (4th visit; 17 months of vigabatrin treatment) there was little change in ERGs from the third visit. By this time she had been given carbamazepine and the vigabatrin was being tapered off. The decrease in cone, flicker and photopic OPs from that recorded at 12 months of age can be seen (Figure 1). The amplitude of the early photopic OP (OPs 2 and 3) were reduced while the amplitude of OP4 was within normal limits. The nomenclature for OP responses is shown in Figure 1 and corresponds to that of Lachapelle [23].
Figure 1.

Sample ERG traces (average responses) for: (a) subject 1, a 6-month-old child before and at approximately 6 month intervals during vigabatrin treatment; (b) subject 5, a 5-month-old child before and at approximately 6-month intervals during vigabatrin treatment; and (c) a 22-month-old child with normal vision taking no medication. (a), (b) length of time on vigabatrin is shown by the numbers at the left of the series of graphs. ERG implicit time is shown in milliseconds. Positive electrical signals are in the upward direction. Zero points are arbitrarily matched. Left traces are cone responses (two repetitions; vertical arrow represents 100 μV). Middle traces are photopic OPs (vertical arrow represents 20 μV). The definition of OPs 2, 3 and 4 are shown. The right hand traces are flicker responses (vertical arrows represent 100 μV). The stimulus flash is at time 0.
Subject 5 (Figure 1b) shows data from a boy with trisomy 21 diagnosed with infantile spasms at 5.5 months of age, who previously had been healthy and had a normal birth history. At the time of the initial ERG (5.5 months) he was on no medication. All ERG responses were within normal limits except for the scotopic OPs, which were non recordable above noise and the photopic OP3 which was also non-recordable. After 6 months on vigabatrin treatment most ERG responses had increased in amplitude with rod–cone a- and b-waves, scotopic OPs and cone response a- and b-wave amplitudes showing increases greater than would be expected for normal development. After 12 months on vigabatrin ERG responses were within normal limits apart from the earlier photopic OPs. After 17 months on vigabatrin treatment there were abnormalities in the early photopic OPs (OP2 and OP3) with OP4 remaining within normal limits. The flicker response had decreased in amplitude.
Descriptive results of ERG responses from ISCEV-recommended stimulus conditions are shown in Figure 2. Each graph is composed of two lines; one describing data from the group of children taking vigabatrin only (vigabatrin monotherapy; black lines) and the other describing data from children taking vigabatrin along with other anticonvulsive medication (gray lines). The error bars describe the standard deviation (mean of four visits). Grey shading describes the upper and lower limits of lab normal control data and contains 95% of responses for each ERG response described. Scotopic ERG amplitudes are described in Figure 2a–d. The mean rod response amplitudes of each group were towards the lower limit of the normal range before vigabatrin treatment and showed no change with length of time on vigabatrin treatment. At baseline and subsequent visits many individual responses fell below the lower limits of normal. The scotopic rod–cone b-wave amplitude data showed a small increase from baseline whereas the rod–cone a-wave amplitude steadily increased from baseline. The mean of the sum of scotopic oscillatory potentials was below the lower limits of normal for baseline and subsequent visits. Photopic responses are described in Figure 2e, f. The cone b-wave and flicker amplitudes show a pattern of initial increase followed by decrease over time. The sum of photopic OPs showed different patterns depending on whether other antiepileptic medications were taken or had been taken previously. Data from children on vigabatrin monotherapy (Figure 2h, black line) were sometimes within normal limits at time of baseline testing (pre-vigabatrin), with the mean amplitude being towards the lower limit of normal. In contrast, data from children on other anticonvulsive treatments in addition to vigabatrin (Figure 2h, grey line) were frequently below normal limits at baseline. The mean amplitude declined over time. There was a trend for the photopic OP2 implicit time data and flicker implicit time to increase with time on vigabatrin treatment (Figure 2k, 2l).
Figure 2.


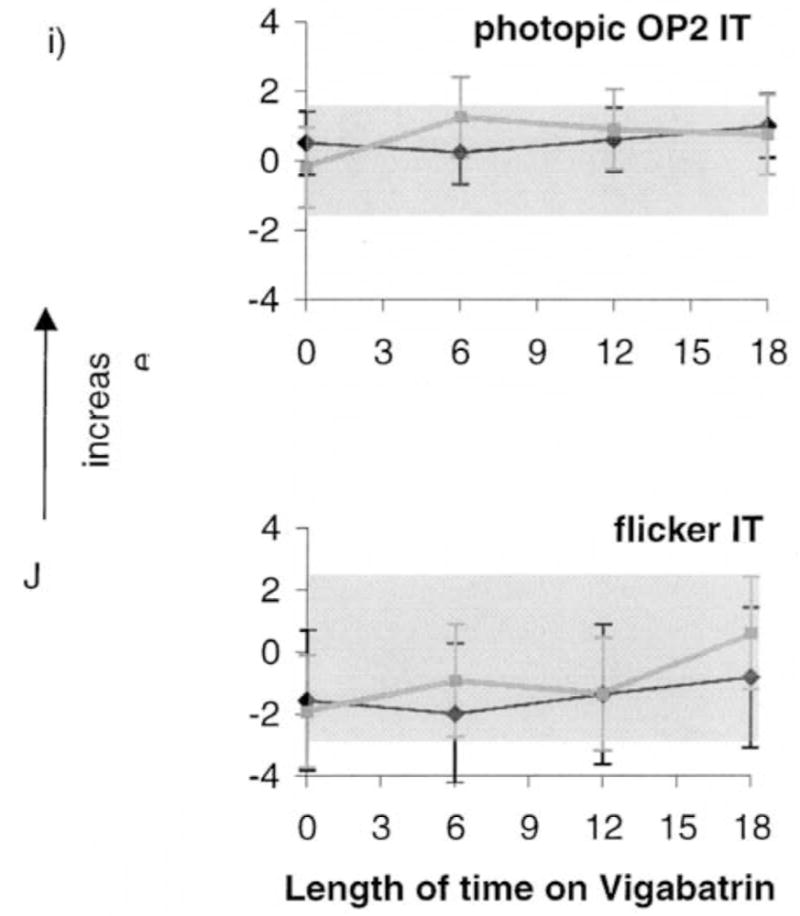
Figures 2a–d. Mean ERG responses from all children in the longitudinal vigabatrin study who received ERGs less than 18 months after initiation of vigabatrin. Log relative amplitudes are plotted against length of time on vigabatrin (months). Black lines represent data on vigabatrin treatment only, gray lines are data from children on vigabatrin treatment and additional anticonvulsive treatment. The error bars represent the standard deviation (mean of four visits). Shaded areas represent lab normal ranges containing 95% of control data. (a) Rod b-wave amplitude; (b) Mixed rod–cone a-wave amplitude; (c) Mixed rode–cone b-wave amplitude; (d) Scotopic sum of OPs.
Figures 2e–h. (e) cone a-wave amplitude responses, (f) cone b-wave amplitude responses (g) flicker amplitude data (h) photopic sum of OPs.
Figures 2i–j. (i) photopic OP2 implicit time, (j) flicker implicit time from all children in the longitudinal study. Vertical arrows signify that the upward direction reflects increase in both amplitude and implicit time data.
A statistical description of changes over time and interaction between children in the two drug groups was accomplished by ANOVA. For the group on vigabatrin monotherapy, there was a significant time effect for the cone b-wave-amplitude quadratic function (Figure 3). The prediction showed significant curvature (p=<0.05) giving relevance to the pattern of initial increase and subsequent decrease in cone b-wave amplitude. Analyses involving other ERG responses showed no significant change of predicted value over time. Although the baseline for the group on vigabatrin monotherapy was higher than for the group taking additional anticonvulsive drugs there were no significant differences between the two groups.
Figure 3.

Terms predicted by the most appropriate curve fit; linear or quadratic plots plotted over time for (a) cone b-wave amplitude data, (b) flicker amplitude data and (c) sum of photopic amplitude. Relative amplitudes are plotted against length of time on vigabatrin (months). Predicted curves from the group taking vigabatrin only (solid line) are shown with predicted curves from the group taking vigabatrin and other anticonvulsive medications (broken line).
The results of visual acuity testing showed that on the initial visit 13 children had visual acuity scores within normal limits for age, 13 children demonstrated reduced visual acuity scores, three children did not respond to testing and four children were not tested. On the final visit two of the children with initial acuity score within normal limits showed a subsequent decrease in visual acuity. One child initially with subnormal visual acuity showed improvement with subsequent visual acuity scores falling within normal limits.
Discussion
Previous investigators have found cone system dysfunction in patients taking vigabatrin [15, 17]. The significant finding in the current study was that for children on vigabatrin monotherapy the predicted cone b-wave amplitude change over time was quadratic; demonstrating an increase followed by a subsequent decrease. Vigabatrin associated increase in b-wave amplitude has been described previously [24, 25]. In these studies patients with partial epilepsy who were treated with carbamazepine or phenytoin showed decreased b-wave and oscillatory potential amplitudes when compared with controls. After the administration of vigabatrin, b-wave amplitudes increased [24, 25]. In the current study three of the children taking other anti-convulsive therapies were taking carbamazepine, two of these were also taking valproic acid; another 13 were taking other anticonvulsant drugs during the time of testing. A variety of antiepileptic drugs have been attributed to different effects on the ERG; decrease in ERG b-wave amplitude [24, 25], increase in b-wave amplitude, reduction in a-wave latency and amplitude, increase in duration of the b-wave, [26]. In some cases other antiepileptic drugs might have masked the effects from vigabatrin explaining the lack of significant change in data over time.
Following exposure of retinae to γ-vinyl-GABA (vigabatrin) [27] animal models demonstrated bi-phasic changes in amacrine cell GABA levels. Amacrine cells and displaced amacrine cells accumulated vigabatrin and exposure was followed by a decrease and subsequent increase of GABA levels [27]. Animal models have shown that a decrease in GABA levels was associated with increased b-wave amplitude [28] and an increase in GABA resulted in decreased ERG b-wave amplitude [28, 29].
Changing GABA levels may contribute to non-toxic changes in b-wave amplitude in humans; perhaps an initial reduction in GABA is responsible for the preliminary enhancement of the b-wave, however it is not clear why the enhancement is apparent 6 months after initiation of vigabatrin therapy.
Human studies involving an adult population have shown an association between reductions in the 30-Hz flicker response and visual field deficits [15,17]. In the current study only two children demonstrated flicker amplitude outside normal limits, although four children demonstrated marked reduction in flicker amplitude with length of time on vigabatrin. The difference between results from the current study and adult studies may be related to the cumulative dosage of vigabatrin. Children had been taking vigabatrin in the current study for a duration of 6 months (n=5), 12 months (n=18) or 18 months (n=10). This was less than the time of vigabatrin use in other studies, e.g., 18–24 months [15] and 31–78 months [20].
The flicker response depends on input from the outer retinal cones and the bipolar cells as well as inner retinal mechanisms. Vigabatrin toxicity may involve one or a combination of these sites. Of relevance might be the pattern of change in the light-adapted oscillatory potentials. We find deficits in early (OP2 and OP3), rather than in the later OP4. Cone responses are processed through a dual system of pathways involving on and off- centred bipolar cells [30], OP2 and OP3 are elicited by the onset of the light stimulus while OP4 is elicited by the offset of the light. This implies that ON rather than OFF mechanisms are preferentially compromised in children on vigabatrin medication. Mooney [31] describes the effect of GABA on the proximal negative response, a transient response from the amacrine cells of the inner retina. This response was markedly reduced in the presence of high GABA concentrations with the off-response being more resistant to changes than the on response.
There have been reports of other types of ERG defects in patients on vigabatrin therapy. Our results do not contradict findings of [8] scotopic effects. We found that scotopic responses are abnormal in some children before the initiation of vigabatrin treatment and therefore deficits in scotopic systems and oscillatory potentials may be associated with mechanisms related to the underlying neurological problem. Scotopic deficits as well as some photopic anomalies have been found in people with brain anomalies [32] and in children with developmental delays [33].
Visual acuity results were subnormal in 50% of children at their first visit and this percentage of subnormal responses was similar for subsequent visits. One child had optic atrophy, another had nystagmus, others had cortical visual loss and therefore it is not unexpected that central visual function was impaired in some children. It is also probable that the presence of developmental delay in many children contributed to apparent visual deficit [34].
In summary, this study provides important data on longitudinal changes in ERG responses from onset of vigabatrin treatment to 18 months after initiation of treatment. Scotopic responses and dark adapted OPs were often abnormal before initiation of vigabatrin treatment and did not change with treatment. In most subjects the flicker amplitude declined after 6 months of vigabatrin treatment. The cone response b-wave showed an initial increase in amplitude followed by a decrease in amplitude after 6 months of vigabatrin treatment. Further studies are under way currently to identify if either or both of these change patterns are associated with subsequent visual deficits.
Acknowledgments
We are grateful to the research institute of The Hospital for Sick Children for support of this study (seed grant 79025), to Rita Buffa for help with data collection and to Ajmal Kahn for help with deriving ERG normal ranges according to age.
References
- 1.Fois A, Buoni S, Bartolo R, Marco V, Mostardini R. Vigabatrin treatment in children. Child’s Nerv Syst. 1994;10:244–48. doi: 10.1007/BF00301162. [DOI] [PubMed] [Google Scholar]
- 2.Kwong L. Vigabatrin as first line therapy in infantile spasms: review of seven patients. J Paediatr Child Health. 1997;33(2):121–4. doi: 10.1111/j.1440-1754.1997.tb01013.x. [DOI] [PubMed] [Google Scholar]
- 3.Uldall P, Alving J, Gram L, Beck S. Vigabatrin in pediatric epilepsy – an open study. J Child Neurol. 1991;6:2, S38–2S44. [PubMed] [Google Scholar]
- 4.Uldall P, Alving J, Gram L, Hogenhaven H. Vigabatrin in childhood epilepsy: a 5-year follow-up study. Neuropediatrics. 1995;26:253–56. doi: 10.1055/s-2007-979766. [DOI] [PubMed] [Google Scholar]
- 5.Eke T, Talbot JF, Lawden MC. Severe persistent visual field constriction associated with vigabatrin. B Med J. 1997;314:180–181. doi: 10.1136/bmj.314.7075.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawden MC, Eke T, Degg C, Harding GF, Wild JM. Visual field defects associated with vigabatrin therapy [see comments] J Neurol Neurosurg Psychiatry. 1999;67(6):716–22. doi: 10.1136/jnnp.67.6.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wild JM, Martinez C, Reinshagen G, Harding GF. Characteristics of a unique visual field defect attributed to vigabatrin. Epilepsia. 1999;40(12):1784–94. doi: 10.1111/j.1528-1157.1999.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 8.Daneshvar H, Racette L, Coupland SG, Kertes PJ, Guberman A, Zackon D. Symptomatic and asymptomatic visual loss in patients taking vigabatrin. Ophthalmology. 1999;106(9):1792–8. doi: 10.1016/S0161-6420(99)90345-7. [DOI] [PubMed] [Google Scholar]
- 9.Kalviainen R, Nousiainen I, Mantyjarvi M, Riekkinen P. Initial vigabatrin monotherapy is associated with increased risk of visual field constriction; a comparative follow-up study with patients on initial carbamazepine monotherapy and healthy controls. Epilepsia. 1998;39:72. [Google Scholar]
- 10.Kalviainen R, Nousiainen I, Mantyjarvi M, Nikoskelainen E, Partanen J, Partanen K, et al. Vigabatrin, a gabaergic antiepileptic drug, causes concentric visual field defects. Neurology. 1999;53(5):922–6. doi: 10.1212/wnl.53.5.922. [DOI] [PubMed] [Google Scholar]
- 11.Ruether K, Pung T, Kellner U, Schmitz B, Hartmann C, Seeliger M. Electrophysiologic evaluation of a patient with peripheral visual field contraction associated with vigabatrin [letter] Arch Ophthalmol. 1998;116(6):817–9. [PubMed] [Google Scholar]
- 12.Miller NR, Johnson MA, Paul SR, Girkin CA, Perry JD, Endres M, et al. Visual dysfunction in patients receiving vigabatrin: clinical and electrophysiologic findings. Neurology. 1999;53(9):2082–7. doi: 10.1212/wnl.53.9.2082. [DOI] [PubMed] [Google Scholar]
- 13.Gross-Tsur V, Banin E, Shahar E, Shalev RS, Lahat E. Visual impairment in children with epilepsy treated with vigabatrin. Ann Neurol. 2000;48(1):60–4. [PubMed] [Google Scholar]
- 14.Wohlrab G, Boltshauser E, Schmitt B, Schriever S, Landau K. Visual field constriction is not limited to children treated with vigabatrin. Neuropediatrics. 1999;30(3):130–2. doi: 10.1055/s-2007-973477. [DOI] [PubMed] [Google Scholar]
- 15.Krauss GL, Johnson MA, Miller NR. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings [see comments] Neurology. 1998;50(3):614–8. doi: 10.1212/wnl.50.3.614. [DOI] [PubMed] [Google Scholar]
- 16.Arndt CF, Derambure P, Defoort-Dhellemmes S, Hache JC. Outer retinal dysfunction in patients treated with vigabatrin. Neurology. 1999;52(6):1201–5. doi: 10.1212/wnl.52.6.1201. [DOI] [PubMed] [Google Scholar]
- 17.Harding GF, Wild JM, Robertson KA, Rietbrock S, Martinez C. Separating the retinal electrophysiologic effects of vigabatrin: treatment versus field loss [In Process Citation] Neurology. 2000;55(3):347–52. doi: 10.1212/wnl.55.3.347. [DOI] [PubMed] [Google Scholar]
- 18.Westall CA, Smith K, Logan WJ, Buncic JR, Panton CM, Khan A. Longitudinal investigation of ERGs in children on vigabatrin therapy. Invest Ophthal; ARVO meeting; 2000; May 2000; Fort Lauderdale, Florida. 2000. p. S35. [Google Scholar]
- 19.Harding GF, Robertson KA, Edson AS, Barnes P, Wild J. Visual electrophysiological effect of a GABA transaminase blocker. Doc Ophthalmol. 1999;97(2):179–88. doi: 10.1023/a:1002045223358. [DOI] [PubMed] [Google Scholar]
- 20.Johnson MA, Krauss GL, Miller NR, Medura M, Paul SR. Visual function loss from vigabatrin: effect of stopping the drug. Neurology. 2000;55(1):40–5. doi: 10.1212/wnl.55.1.40. [DOI] [PubMed] [Google Scholar]
- 21.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update) Doc Ophthalmol. 1999;97:143–56. doi: 10.1023/a:1002016531591. [DOI] [PubMed] [Google Scholar]
- 22.Westall CA, Panton CM, Levin AV. Time course for maturation of electroretinogram response, from infancy to adulthood. Doc Ophthalmol. 1999;96:355–79. doi: 10.1023/a:1001856911730. [DOI] [PubMed] [Google Scholar]
- 23.Lachapelle P. The human suprathreshold photopic oscillatory potentials: method of analysis and clinical application. Doc Ophthalmol. 1994;88(1):1–25. doi: 10.1007/BF01203698. [DOI] [PubMed] [Google Scholar]
- 24.Bayer A, Zrenner E, Reid S, Schmidt D. Effects of anticonvulsant drugs in retinal function. Psychophysical and electrophysiological findings in patients with epilepsy. Invest Ophthalmol Vis Sci. 1990;31:S427. Abstract. [Google Scholar]
- 25.Harding GFA, Jones LA, Tipper VJ, Betts TA, Mumford JP. Electroretinogram, pattern electroretinogram and visual evoked potential assessment in patients receiving vigabatrin. Eplilepsia. 1995;36 (Suppl):108. Abstract. [Google Scholar]
- 26.Sannita WG. Neuropsychiatric drug effects on the visual nervous system. In: Hecken-lively JRA, editor. Principles and Practice of Clinical Electrophysiology of Vision. St Louis: Mosby Year Book, Inc; 1991. pp. 167–73. [Google Scholar]
- 27.Pow DV, Baldridge W, Crook DK. Activity-dependent transport of GABA analogues into specific cell types demonstrated at high resolution using a novel immunocytochemical strategy. Neuroscience. 1996;73(4):1129–43. doi: 10.1016/0306-4522(96)00097-8. [DOI] [PubMed] [Google Scholar]
- 28.De Vries GW, Friedman AH. GABA, picrotoxin and retinal sensitivity. Brain Res. 1978;148(2):530–5. doi: 10.1016/0006-8993(78)90743-6. [DOI] [PubMed] [Google Scholar]
- 29.Gottlob I, Wundsch L, Pflug R. Possible role of amacrine cells in the generation of the mammalian ERG b-wave. Doc Ophthalmol. 1985;61(1):55–63. doi: 10.1007/BF00143216. [DOI] [PubMed] [Google Scholar]
- 30.Watchtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998;17(4):485–521. doi: 10.1016/s1350-9462(98)00006-8. [DOI] [PubMed] [Google Scholar]
- 31.Mooney R. GABA-mediated control of transient signals in the inner retina. Brain Res. 1978;145:97–115. doi: 10.1016/0006-8993(78)90799-0. [DOI] [PubMed] [Google Scholar]
- 32.Cibis GW, Fitzgerald KM. Optic nerve hypoplasia in association with brain anomalies and an abnormal electroretinogram. Doc Ophthalmol. 1994;86(1):11–22. doi: 10.1007/BF01224624. [DOI] [PubMed] [Google Scholar]
- 33.Fulton AB, Hansen RM. Electroretinography: application to clinical studies of infants. J Pediatr Ophthalmol Strabismus. 1985;22:251–55. doi: 10.3928/0191-3913-19851101-12. [DOI] [PubMed] [Google Scholar]
- 34.Westall CA, Ainsworth JR, Buncic JR. Which ocular and neurological conditions cause disparate results in visual acuity scores recorded with VEP and teller acuity cards? J AAPOS. 4(5):295–301. doi: 10.1067/mpa.2000.107898. [DOI] [PubMed] [Google Scholar]