

Abstract
Vigabatrin, a structural analogue of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA), is widely used as initial monotherapy in infantile spasms and add on therapy in partial onset seizures. Vigabatrin is associated with retinal toxicity causing constriction of the visual field. Our aim was to assess what effect add-on antiepileptic drug therapy has on the incidence of retinal toxicity in patients being treated with vigabatrin. Medication dosages, duration of treatment, and electroretinogram results were examined in a single center retrospective study. Retinal toxicity was detected in 18 of 160 patients (11.25%) over a 10-year period. A total of 14 (77%) were in the group treated with additional antiepileptic drugs, the other 4 received vigabatrin as monotherapy. We detected a significantly higher percentage of toxicity in the group of patients treated with vigabatrin and additional antiepileptic drugs. Our numbers were not sufficient to detect which drug or combination of drugs might be associated with higher risk.
Keywords: vigabatrin, toxicity, retina, electroretinogram, infantile spasms
Vigabatrin has been used since 1979 in the treatment of epilepsy. Therefore clinical experience of this drug spans a longer time than most of the newer antiepileptic drugs.1 Vigabatrin is a structural analogue of the inhibitory neurotransmitter gamma-aminobutyric acid, more commonly referred to as GABA, and it produces its antiepileptic effect by irreversibly inhibiting the degradative enzyme GABA transaminase.2–4 This leads to an increase in the levels of GABA in the brain and the retina.5 The levels of GABA are selectively higher in the retina.6
Vigabatrin is used as initial monotherapy in Europe and Canada in patients with infantile spasms and as add-on therapy in patients with partial-onset seizures.7 It was also approved in the United States in August 2009 for treatment of infantile spasms and as an adjunctive agent in the treatment of refractory complex partial seizures in adults.
In the early years of its clinical use, there was concern of its potential neurotoxic effect. Animal studies had shown that doses as low as 30 mg/kg caused intramyelinic vacuolation or edema in specific areas of rodent and dog brains, primarily the hippocampus, cerebellum, and visual pathways.8 These microvacuoles were reversible when vigabatrin was stopped. Delayed conduction times using visual evoked potentials were found to correlate with the onset of vacuolation in dogs, as detected by magnetic resonance imaging (MRI).9 With clinical use, over 500 patients were followed using MRI and visual evoked potentials with no indication that microvacuolation occurs in humans.2 Further studies have described reversibility of MRI defect in infants.10,11 In 1997, Eke et al reported 3 patients receiving a variety of antiepileptic drugs including vigabatrin showed symptomatic constriction of the visual field.11
Electroretinogram abnormalities have been identified in these patients with visual field defined toxicity.12,13 The cone b-wave and 30-Hertz flicker parameters have demonstrated the best correlation with vigabatrin-induced visual field loss, with high sensitivity (100%) and specificity (75%).12 More recently animal studies have reported light dependence as a factor in the development of the retinal toxicity.14,15
The true prevalence of visual field defect in patients receiving vigabatrin is unknown. Figures range from 14% to 92%16,17 with a lower rate of toxicity reported in pediatric patients. In the infant population with infantile spasms, visual field assessment, which requires attention and cooperation, has not been found to be a good measure of toxicity. Instead electroretinogram assessment is the measure used to identify toxicity. The risk factors for developing toxicity are equivocal18; some association has been identified with male gender,19 cumulative dose,20 and duration of therapy.21 The changes are generally believed to be lifelong; however, a recent report shows partial recovery after reduction in dose or discontinuation of therapy.22
Published studies report the incidence of toxicity in patients receiving vigabatrin as add-on therapy. A recent study at our center23 followed the electroretinogram responses in a pediatric population treated with vigabatrin. They reported that the incidence of any vigabatrin-induced defect was 54% (39.5 cases per 100 subject years). The incidence of sustained abnormality was 25% (15.3 cases per 100 subject-years). The group of patients who were treated with vigabatrin and “other antiepileptic agents in combination” showed the earliest onset of abnormality versus the vigabatrin monotherapy group.
Objective
The purpose of this study is to assess the effect of add-on anti-epileptic drug therapy has on retinal toxicity in patients being treated with vigabatrin.
Methods
This was a single center retrospective observational study. Our study population was identified from the ophthalmology vigabatrin study database, which has been collecting data since 1999 at our institution. Ethical approval was obtained from the hospital’s Research and Ethics board. We examined the register between the period of January 1999 and December 2009. All data obtained were encoded with patient identifiers removed. Medications received, dosages, and durations were reviewed and correlated with electroretinogram results. Definite toxicity was defined as a quantitative reduction in amplitude (using defined normative values) between baseline and follow-up measurements of 30-Hertz flicker response, which was present on at least 2 consecutive occasions. Adequate data were defined as at least 3 visits with completed electroretinogram studies, including B wave amplitudes. Age-specific normative values for the electroretinogram parameters were determined from the large normative dataset in our institution. Age specificity is essential because the infant retina undergoes rapid development during the first months and years of life.
Statistical analysis was performed using commercially available software and a P value of less than .05 was considered statistically significant.
Results
During this 10-year period, we identified 446 children treated with vigabatrin therapy. Data from children 1 month to 18 years of age, who received vigabatrin as monotherapy or add-on therapy, were analyzed. These children received vigabatrin for treatment of infantile spasms, in the infant population, or refractory partial onset seizures in the older children. We identified 247 patients who received vigabatrin and had electroretinogram studies performed to evaluate the development of retinal toxicity. One hundred sixty of these had adequate electroretinogram data to determine if toxicity was present. Seventy-three were treated with vigabatrin monotherapy and 87 with add-on vigabatrin. The add-on vigabatrin group was treated with a combination of 7 medications and between 1 and 4 agents used together. The medications involved were phenobarbital, clonazepam, lamotrigine, valproic acid, adrenocorticotropic hormone, prednisolone, clobazam, topirimate, and carbamazepine. The majority received vigabatrin in combination with 1 other medication.
Strictly defined toxicity (electroretinogram reduction from baseline on at least 2 consecutive occasions) was detected in 18 of the 160 patients (11.25%). Electroretinogram reduction is defined as reduction greater than normal limits. The 95th percentile of normal intervisit variability where is 50% reduction in the electroretinogram flicker response. Of the 160 patients 18, 14 (77%) were in the group of patients being treated with multiple antiepileptic drugs and vigabatrin as add-on therapy (P = .0018). Figure 1 illustrates a normal electroretinogram response on the left compared with 1 showing vigabatrin related retinal toxicity.
Figure 1.
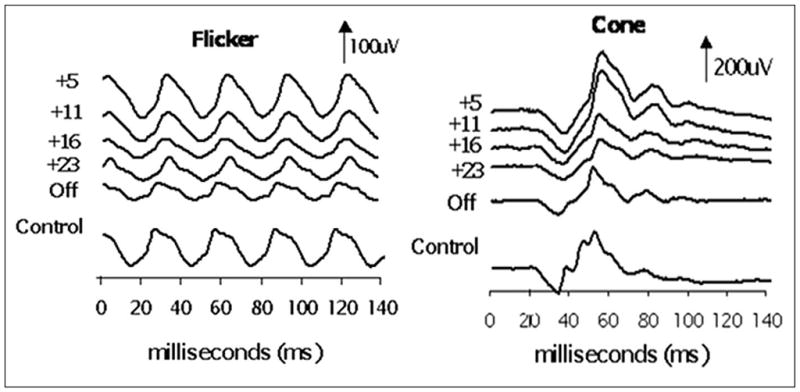
Electroretinogram traces showing flicker response and cone response. The subject is a child who began vigabatrin at 5 months of age and had electroretinogram testing at approximately 6-month intervals during vigabatrin treatment and after vigabatrin treatment (labeled age off). The last waveform (gray electroretinogram trace) is data recorded from a 30-month-old control (with normal vision). Adapted from: Westall CA, Nobile R, Morong S, Buncic JR, Logan WJ, Panton CM. Changes in the electroretinogram resulting from discontinuation of vigabatrin in children. Doc Ophthalmol. 2003;107:299–309.
Further analysis of the patients with retinal toxicity on electroretinogram found no difference between those in the vigabatrin monotherapy group and those with additional anti-epileptic drugs in terms of their total dose of Vigabatrin, maximum dose of vigabatrin received, age at onset of vigabatrin therapy, duration of vigabatrin therapy, and number of electroretinograms performed and found no statistical difference between the groups. We examined the add-on therapy group to assess whether number of antiepileptic drugs was associated with an increased toxicity and found it was not. We examined the add-on antiepileptic drug looking at what agent was used and there was no difference between the group who developed toxicity and those who did not. Similarly there was no statistical difference whether the medication exhibited GABAergic mechanism of action or not in relation to the development of toxicity.
Discussion
Much debate surrounds the potential risk factors for development of and mechanisms of development of retinal toxicity in patients receiving vigabatrin. The true incidence is uncertain.
The mechanism of vigabatrin-induced retinal toxicity is generally presumed to be GABA mediated. An interesting study by Kinirons at al24 looked at whether there may be a genetic predisposition among the patients who develop this toxicity. They examined 6 candidate genes; however, they failed to identify a replicable association.
Another study14 examines the possibility of light-dependent retinotoxicity in these patients. In this animal study, using albino rats, it was shown that vigabatrin’s mechanism of toxicity is light-dependent rather than GABA-dependent. Thus, this may explain the difference in incidence of toxicity according to climate/geographic location. Another study also examined light dependence in the development of vigabatrin-related retinal toxicity and found similar results examining both electroretinogram amplitude and retinal layer.15 Most studies reporting toxicity involve patients receiving vigabatrin as add on therapy not monotherapy.11,25–29 Therefore, one may propose a possible additive or cumulative effect of additional antiepileptic drugs (whether GABAergic or not) in this evolution of toxicity.
Kalviainen et al examined the incidence of visual field defects in patients receiving vigabatrin monotherapy versus carbamazepine.30 They reported an incidence of 40% with some visual field defect in their group treated with vigabatrin monotherapy and none in the group receiving carbamazepine. Of the 40%, 9% had severe defect identified using perimetry. They reported a typical consistent concentric pattern of defect detected by perimetry in all patients. They concluded that, even if patients are on multiple antiepileptic drugs, the pattern of visual field defect can only be explained by a single common factor, namely vigabatrin therapy.
Vigabatrin causes bilateral concentric visual field constriction11 which is more pronounced nasally than temporally.31 The pattern of visual field constriction can vary from nasally predominant to a pattern of more central involvement and thus is not always a pathognomonic pattern.32 Associated with the visual field loss are changes in the retinal nerve fiber layer.33,34
In our study of 160 patients, we detected an overall incidence of 11.25% of visual field defect. This is likely due to the strictly defined criteria for definite abnormality. We detected a significantly higher percentage of toxicity in the group of patients treated with vigabatrin and additional antiepileptic drugs. This increased toxicity may represent a cumulative effect of other GABAergic antiepileptic drugs acting in conjunction with vigabatrin to produce additive toxicity. It has been postulated that valproic acid, by causing an increased retinal concentration of GABA, may lead to increased toxicity. Alternatively other antiepileptic drugs may have retinotoxic effects by an independent mechanism. Carbamazepine has been shown to induce electroretinogram changes. It causes a reduction in amplitude of another measured parameter (b amplitude) in the photopic electroretinogram.12 Our numbers with toxicity were small, and the variety of additional antiepileptic drugs meant we were unable to draw any conclusions on whether specific antiepileptic drugs may be associated with an increased risk. Another study limitation was that none of our patients had formal visual field perimetry performed. For many, this decision was due to patient age or developmental ability and it was believed that most would not be able to comply with formal visual field testing.
The search for potential protective agents against vigabatrin-induced retinal toxicity is under way, with one recent publication suggesting taurine as a potential protective agent.15 Taurine deficiency is known to produce retinal toxicity in animal studies.15 Light exposure in the setting of taurine deficiency potentiates this photoreceptor degeneration.35 Jammoul et al performed randomized study of rats treated with vigabatrin and found that a low level of taurine was highly correlated with both the electroretinogram amplitudes and cone densities in their animals. They supplemented a group of their vigabatrin-treated rats with taurine and found that their electroretinogram amplitude was greater than those treated with vigabatrin without supplementation. They retrospectively evaluated 6 infants who were treated with vigabatrin for infantile spasms and found that 5 of the 6 had low serum levels of taurine. Recent animal studies suggest that taurine may have antiseizure effects.36,37
Conclusion
The prevalence of toxicity detected by electroretinogram was 11.25% in this pediatric cohort. We plan to review our patients with electroretinogram toxicity and assess visual field perimetry where possible to confirm the extent of their visual field defect. We detected a significantly higher percentage of toxicity in the group of children treated with vigabatrin and additional antiepileptic drugs. Our sample size did not provide sufficient statistical power to detect which antiepileptic drugs or combination of antiepileptic drugs that might be associated with higher risk, although we postulated a GABAergic effect may be likely. Further studies are indicated to assess the potential additive deleterious effects of other antiepileptic drugs in causing retinal toxicity. Vigabatrin may not be acting alone in patients who develop toxicity, and the identification of the underlying etiology of their seizures may also impact on development of retinal toxicity.
Footnotes
Author Contributions
First draft of the article was written by B. McCoy, and contribution was received from all co-authors in editing and completing the manuscript for submission.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
Ethical Approval
Ethical approval was obtained from the hospital research and ethics board.
Reprints and permission: sagepub.com/journalsPermissions.nav
Financial Disclosure/Funding
The authors received no financial support for the research and/or authorship of this article.
References
- 1.French JA. Vigabatrin. Epilepsia. 1999;40(Suppl 5):S11–S16. doi: 10.1111/j.1528-1157.1999.tb00914.x. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Menachem E. Vigabatrin. Epilepsia. 1995;36(Suppl 2):S95–S104. doi: 10.1111/j.1528-1157.1995.tb06003.x. [DOI] [PubMed] [Google Scholar]
- 3.Mumford JP, Cannon DJ. Vigabatrin. Epilepsia. 1994;35(Suppl 5):S25–S28. doi: 10.1111/j.1528-1157.1994.tb05962.x. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Menachem E, Persson LI, Scheter PJ, et al. The effect of different vigabatrin treatment regimes on CSF biochemistry and seizure control in epileptic patients. Br J Clin Pharmacol. 1989;27(Suppl 1):79S–85S. doi: 10.1111/j.1365-2125.1989.tb03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petroff OAC, Rothman DL, Behar KL, Collins BA, Mattson RH. Human brain GABA levels rise rapidly after initiation of vigabatrin therapy. Neurology. 1996;47:1567–1571. doi: 10.1212/wnl.47.6.1567. [DOI] [PubMed] [Google Scholar]
- 6.Sills G, Patsalos PN, Butler E, et al. Concentration related pharmacodynamic studies with vigabatrin and tiagabine in rat brain and eye [abstract] Epilepsia. 1999;40(Suppl 2):132. [Google Scholar]
- 7.Aicardi J, Mumford JP, Dumas C, Woods S. Vigabatrin as initial therapy for infantile spasms: a European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia. 1996;37:638–642. doi: 10.1111/j.1528-1157.1996.tb00627.x. [DOI] [PubMed] [Google Scholar]
- 8.Graham D. Neuropathology of vigabatrin. Br J Clin Pharmacol. 1989;27(Suppl 1):43S–45S. doi: 10.1111/j.1365-2125.1989.tb03460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sussman NM, Weiss KI, Schroeder CE. Vigabatrin: effect on in vivo and ex vivo magnetic resonance imaging of dog brains (Abstract) Epilepsia. 1991;31(Suppl 1):13. [Google Scholar]
- 10.Dracopoulos A, Widjaja E, Raybaud C, Westall CA, Snead OC., 3rd Vigabatrin associated reversible MRI signal changes in patients with infantile spasms. Epilepsia. 2010;51:1297–1304. doi: 10.1111/j.1528-1167.2010.02564.x. [DOI] [PubMed] [Google Scholar]
- 11.Eke T, Talbot JF, Lawdon MC. Severe persistent visual field constriction associated with vigabatrin. BMJ. 1997;31:180–181. doi: 10.1136/bmj.314.7075.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding GF, Wild JM, Robertson KA, Rietbrock S, Martinez C. Separating the retinal electrophysiologic effects of vigabatrin: treatment versus field loss. Neurology. 2000;55:347–352. doi: 10.1212/wnl.55.3.347. [DOI] [PubMed] [Google Scholar]
- 13.Ponjavic V, Anreasson S. Multifocal ERG and full field ERG in patients on long-term vigabatrin medication. Doc Ophthalmol. 2001;102:63–72. doi: 10.1023/a:1017589301855. [DOI] [PubMed] [Google Scholar]
- 14.Izumi Y, Ishikawa W, Benz AM, et al. Acute vigabatrin retino-toxicity in albino rats depends on light but not GABA. Epilepsia. 2004;45:1043–1048. doi: 10.1111/j.0013-9580.2004.01004.x. [DOI] [PubMed] [Google Scholar]
- 15.Jammoul F, Wang Q, Nabbout R, et al. Taurine deficiency is a cause of vigabatrin-induced retinal phototoxicity. Ann Neurol. 2009;65:98–107. doi: 10.1002/ana.21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefan HJ, Bernati K, Knoor HL. Visual field constriction and anti-epileptic drug treatment. Neurol Psychiatr Brain Res. 2000;7:185–190. [Google Scholar]
- 17.Besch DA, Kurtenbach E, Apfelstedt-Sylla B, et al. Visual field constriction and electrophysiological changes associated with vigabatrin. Doc Ophthalmol. 2002;104:151–170. doi: 10.1023/a:1014644307518. [DOI] [PubMed] [Google Scholar]
- 18.Wild JM, Robson CR, Jones A, Cunliffe IA, Smith PE. Detecting vigabatrin toxicity by imaging the retinal nerve fibre layer. Invest Ophthalmol Vis Sci. 2006;47:917–924. doi: 10.1167/iovs.05-0854. [DOI] [PubMed] [Google Scholar]
- 19.Wild JM, Martinez C, Reinshagen G, Harding GF. Characteristics of a unique visual field defect attributed to vigabatrin. Epilepsia. 1999;40:1784–1794. doi: 10.1111/j.1528-1157.1999.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 20.Miller NR, Johnson MA, Paul SR, et al. Visual dysfunction in patients receiving vigabatrin. Neurology. 1999;53:2082–2087. doi: 10.1212/wnl.53.9.2082. [DOI] [PubMed] [Google Scholar]
- 21.Nousiainen I, Mantyjarvi M, Kalviainen R. No reversion in vigabatrin associated visual field defects. Ophthalmology. 2001;57:1916–1917. doi: 10.1212/wnl.57.10.1916. [DOI] [PubMed] [Google Scholar]
- 22.Fledelius HC. Vigabatrin-associated visual field constriction in a longitudinal series: reversibility suggested after drug withdrawal. Acta Ophthalmol Scand. 2003;81:41–46. doi: 10.1034/j.1600-0420.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 23.Westall CA, Buncic JR, Donner EJ, et al. Understanding the Vigabatrin-related Visual Field Defect: Infants. Poster presented at annual meeting of AES; 2007. [Google Scholar]
- 24.Kinirons P, Cavalleri GL, O’Rourke D, et al. Vigabatrin Retinopathy in an Irish cohort: lack of correlation with dose. Epilepsia. 2006;47:311–317. doi: 10.1111/j.1528-1167.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 25.Wong IC, Mawer GE, Sander JW. Severe persistent visual constriction associated with vigabatrin. Reaction may be dose dependent. BMJ. 1997;314:1693–1694. [PMC free article] [PubMed] [Google Scholar]
- 26.Blackwell N, Hayllar J, Kelly G. Severe persistent visual costrication associated with vigabatrin. Patients taking vigabatrin should have regular visual field testing [letter] BMJ. 1997;314:1694. [PMC free article] [PubMed] [Google Scholar]
- 27.Harding GF. Severe persistent visual constriction associated with vigabatrin. Four possible explanations exist [letter] BMJ. 1997;314:1694. [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer G, Scollo-Lavizzari G, Jallon P, et al. Vigabatrin-associated bilateral concentric visual field defects in four patients. Epilepsia. 1997;38(Suppl 8):179. [Google Scholar]
- 29.Mackenzie R, Klistorner A. Severe persistent visual constriction associated with vigabatrin. Asymptomatic as well as symptomatic defects occur with vigabatrin [letter] BMJ. 1998;316:232. [PMC free article] [PubMed] [Google Scholar]
- 30.Kalviainen R, Nousiainen I, Mantyjarvi M, et al. Vigabatrin, a gabaergic antiepileptic drug, causes concentric visual field defects. Neurology. 1999;53:922–926. doi: 10.1212/wnl.53.5.922. [DOI] [PubMed] [Google Scholar]
- 31.Lawden MC, Eke T, Degg C, et al. Visual field defects associated with vigabatrin therapy. J Neurol Neurosurg Psychiatry. 1999;67:716–722. doi: 10.1136/jnnp.67.6.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wild JM, Martinez C, Reinshagen G, Harding GF. Characteristics of a unique visual field defect attributed to vigabatrin. Epilepsia. 1999;40:1784–1794. doi: 10.1111/j.1528-1157.1999.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 33.Buncic JR, Westall CA, Panton CM, Munn JR, MacKeen LD, Logan WJ. Characteristic retinal atrophy with secondary “inverse” optic atrophy identifies vigabatrin toxicity in children. Ophthalmology. 2004;111:1935–1942. doi: 10.1016/j.ophtha.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawthom C, Smith PE, Wild JM. Nasal retinal nerve fiber layer attenuation: a biomarker for vigabatrin toxicity. Ophthalmology. 2009;116:565–571. doi: 10.1016/j.ophtha.2008.09.047. [DOI] [PubMed] [Google Scholar]
- 35.Rapp LM, Thum LA, Anderson RE. Synergism between environmental lighting and taurine depletion in causing photoreceptor cell degeneration. Exp Eye Res. 1988;46:229–238. doi: 10.1016/s0014-4835(88)80080-0. [DOI] [PubMed] [Google Scholar]
- 36.El Idrissi A, Messing J, Scalia J, Tranker E. Prevention of epileptic seizures by taurine. Adv Exp Med Biol. 2003;526:515–525. doi: 10.1007/978-1-4615-0077-3_62. [DOI] [PubMed] [Google Scholar]
- 37.Gupta RC, Win T, Bittner S. Taurine analogues: a new class of therapeutics: retrospect and prospects. Curr Med Chem. 2005;12:2021–2039. doi: 10.2174/0929867054546582. [DOI] [PubMed] [Google Scholar]