

Abstract
Objective
To describe the clinical pattern of retinal atrophy in children caused by the anticonvulsant vigabatrin.
Design
An interventional case series report.
Participants
One hundred thirty-eight patients, mainly infants, were evaluated regularly for evidence of possible vigabatrin toxicity in the Eye and Neurology clinics at the Hospital for Sick Children, Toronto.
Method
Sequential clinical and electroretinographic (International Society for Clinical Electrophysiology of Vision standards) evaluations every 6 months.
Main Outcome Measures
Presence of recognizable retinal and optic atrophy in the presence of abnormal electroretinogram (ERG) and other clinical findings.
Results
Three children being treated for seizures with vigabatrin showed definite clinical findings of peripheral retinal nerve fiber layer atrophy, with relative sparing of the central or macular portion of the retina and relative nasal optic nerve atrophic changes. Some macular wrinkling was evident in 1 case. Progressive ERG changes showing decreased responses, especially the 30-Hz flicker response, supported the presence of decreased retinal function.
Conclusions
A recognizable and characteristic form of peripheral retinal atrophy and nasal or “inverse” optic disc atrophy can occur in a small number of children being treated with vigabatrin. The changes in superficial light reflexes of the retina in children facilitate the clinical recognition of nerve fiber layer atrophy. The macula is relatively spared, although superficial retinal light reflexes indicating wrinkling of the innermost retina suggest early macular toxicity as well. Because these changes are accompanied by electrophysiologic evidence of retinal dysfunction, discontinuation of vigabatrin should be strongly considered.
Vigabatrin (Sabril, Aventis Pharma, Laval, Canada) is an effective and popular γ-aminobutyric acid–ergic (GABA-ergic) anticonvulsant used in the management of infantile spasm (West syndrome),1,2 in seizures associated with tuberous sclerosis,3 and in partial seizures of adults as adjunctive therapy.4 It acts by irreversible inhibition of GABA transaminase with the resultant accumulation of the inhibitory neurotransmitter GABA in the brain with even higher concentrations in the retina. Since 1997, it has been recognized that a rather high proportion (30%–40%) of mainly adult patients undergoing vigabatrin therapy have significant deleterious effects develop on peripheral visual fields that are usually, but not always, asymptomatic.5–8 The detection of the toxic effect of vigabatrin in infants and young children has posed a significant clinical challenge because of the inaccuracy of clinical testing of peripheral vision in this age group. There is a lack of defined clinical signs that provide a reliable measure of toxicity. In investigative studies, however, electroretinogram (ERG) results have been correlated with visual field defect.9–12 Specifically, vigabatrin-attributed visual field loss has been associated with evidence of reduced cone b-wave response,9,13 decreased amplitude of the 30-Hz flicker response,12 and abnormalities in photopic and scotopic oscillatory potentials.9–11,13 Demonstration of these ERG abnormalities in retinal function opened up the possibility of using ERG parameters as indicators of visual loss, hopefully in its early stages.
Clinicians have described various retinal appearances mainly in adults with vigabatrin-attributed visual field defects. These included optic disc pallor; hypopigmentation of the peripheral retina, with an easily visible choroidal circulation6; nonspecific optic atrophy; hypopigmented spots in the retina; vascular sheathing14; and surface wrinkling of the retina.9 One case report described a patient who had visual deterioration and blurring develop and showed pronounced bilateral optic atrophy and maculopathy.15 Cases of optic atrophy have been reported in 1 adult16 and in 1 child with steroid-responsive optic neuritis.17 Russell-Eggitt et al18 have reported a series of children with visual field defects caused by the drug, some with optic disc pallor.
Participants and Methods
To monitor possible vigabatrin visual toxicity, the current practice at The Hospital for Sick Children (HSC) is to assess each child with seizure activity before and during drug treatment with neuro-ophthalmologic examination and with an ERG (under sedation in most cases). Electroretinograms are recorded according to the standards of The International Society for Clinical Electrophysiology of Vision (ISCEV).19 Because ERGs in young children who are still developing, ERGs are compared with age-corrected normal values derived from C.R.A.C.K. ERG (The Hospital for Sick Children), a software package that derives age-appropriate ERG data. The complete assessment is repeated at 6-month intervals. Because a variety of ERG parameters (amplitude and implicit time) change during vigabatrin treatment, the current studies are helping to clarify which of these changes represent nontoxic side effects and which correlate with drug toxicity.20–22 Changes in oscillatory potential amplitude result, at least in part, from non-toxic changes.22 Conversely, if the ERG, particularly the 30-Hz flicker response, decreases more than expected from intervisit variability, both the clinical assessment and the ERG are repeated within 3 months. If the reduction is maintained, the treating neurologist is informed of the likelihood of vigabatrin toxicity.
All testing was performed in the Visual Electrophysiology Unit at HSC.
To date, 138 children have had ERGs recorded as part of this practice: 79 ERG recordings were conducted before vigabatrin was started (baseline recordings). Of these, 51 have received baseline and follow-up ERGs; the remainder (28) were eventually not prescribed the drug or were lost to follow-up. Twenty-five children have received longitudinal follow-up without comparative baseline measurements, and the remaining 34 children had a single ERG while on vigabatrin treatment. The research followed the tenets of the Declaration of Helsinki. Subjects 16 years and older and parents or guardians of subjects younger than 16 years gave signed consent for their participation in the study. The consent form acknowledged that research procedures were described, any questions answered, and that harms and benefits of participating were fully explained. Approval for this study was obtained from the Research Ethics Board at HSC.22
Results
During this protocol, 3 cases of a specific, identifiable, vigabatrin-associated peripheral retinal atrophy with secondary optic nerve atrophy in children have been identified and selected for this communication. The authors report the characteristic pattern of atrophy that is found in association with the peripheral field loss caused by this drug in young children. In 1 case, ERGs were recorded sequentially, and results show dramatic parallel deterioration. This clinically characteristic atrophic change seen at the optic nerve head is named “inverse” optic atrophy to emphasize its difference from and contrast with the usual macular and temporal optic nerve atrophy pattern seen in the more common optic neuropathies. This pattern of atrophy also supports the notion of diffuse, but differential, involvement of peripheral and central retinal cells and the ganglion cell layer. The nasal optic nerve pallor seen is secondary to damage to the retinal nerve fiber layer ganglion cells and axons, the change being most obvious in the peripheral retina.
Case 1
A 10-year-old girl was referred to the neuro-ophthalmology clinic in 1999 because of peripheral visual field loss noted over the previous year. She had been treated for seizures that began in her home country of Greece in the form of sudden and unexpected onset of shaking spells and falling to the ground lasting approximately 10 minutes with only partial loss of awareness. Control with carbamazepine alone was insufficient, but improved with the addition of vigabatrin in March of 1995 at a dosage of 2500 mg/day, which continued for the next 4 years. Her visual acuity was 20/20 in each eye unaided, and color vision was normal tested with Hardy–Rand–Rittler plates. Goldmann perimetry showed generally and significantly constricted peripheral fields to approximately the central 30° to 40° in both eyes (Fig 1). This peripheral field loss has remained unchanged on 3 subsequent tests over a 12-month period. Her fundi showed an inverse type of optic atrophy, with loss of retinal nerve fibers diffusely throughout the retina, with relative sparing of the macular areas and the temporal aspect of the disc in both eyes (Figs 2, 3). The maculae were not completely normal, however, showing slight diffuse irregular atrophy of the nerve fiber layer (Fig 4). These changes were symmetric and evident in photographic studies of the retinae. Once the abnormal visual and ocular abnormality was identified, the vigabatrin dosage was decreased gradually and discontinued. The initial ERG performed while she was taking vigabatrin showed little abnormality during the weaning phase and little change when reassessed 1 year later. No baseline previgabatrin ERG was available for comparison. The follow-up ERG performed 2 years later showed slight reduction in the 30-Hz flicker response compared with the initial study (19% reduction), although all the parameters tested remained within accepted normal limits. In August 2000, visual evoked potentials (VEPs) were recorded with Power Diva sweep VEP software23,24 to a stimulus changing constantly in contrast or spatial frequency (bar width). The results demonstrated that visual acuity and contrast sensitivity were completely within normal limits. The funduscopic picture and visual field assessed by perimetry remained unchanged in follow-up over 2 years.
Figure 1.

Goldmann perimetry field examination shows severely constricted visual fields with sloping borders in both eyes in case 1, demonstrating a typical toxic effect after 4 years of vigabatrin treatment.
Figure 2.
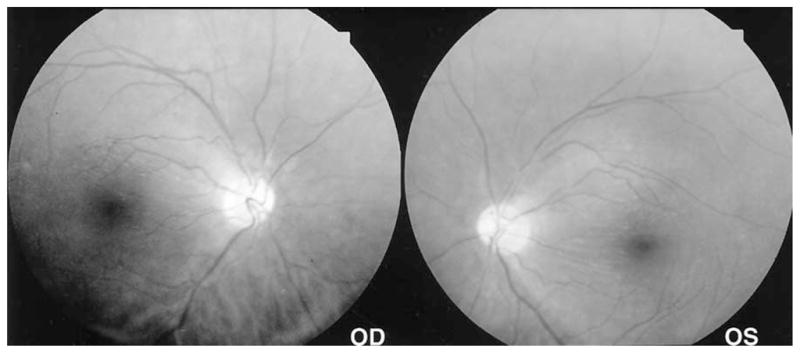
Standard 50° photographs of the fundi in case 1 showing a more panoramic view, emphasizing the contrast of the darker, more granular-appearing peripheral fundus and the more opalescent-appearing macula. A well-defined visible border separates the macula just central to the temporal vascular arcades from the surrounding atrophic retina. The nasal peripapillary nerve fiber layer is typically atrophic.
Figure 3.
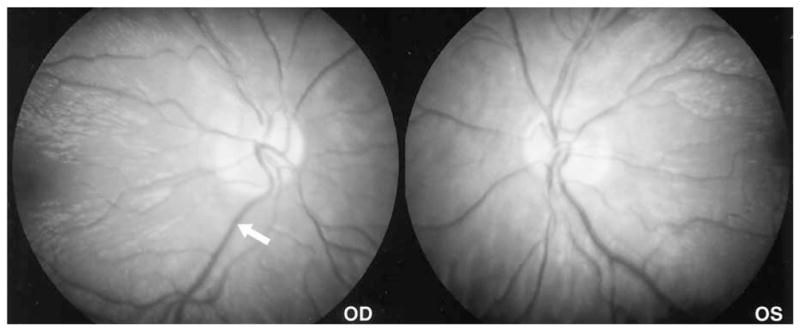
Genesis portable fundus camera (Kowa Genesis, Tokyo, Japan) 30° views provide more obvious contrast of the differently affected retinal areas, showing loss of the peripapillary nerve fiber layer and relative sparing of the temporal area. The white arrow indicates the obvious demarcation border between the differently affected parts of the retina in the posterior pole.
Figure 4.
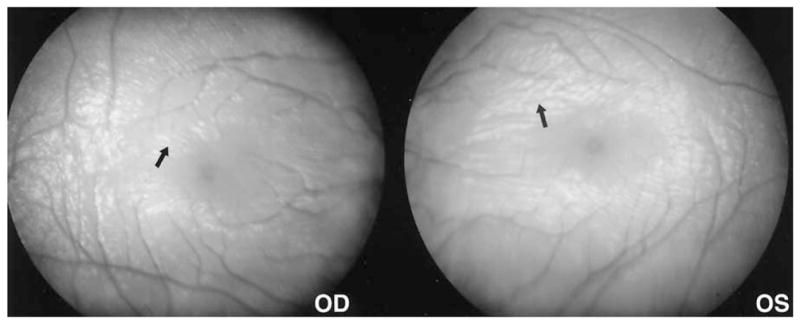
Genesis portable fundus camera (Kowa Genesis, Tokyo, Japan) 30° photos of the maculae in case 1 demonstrate the wrinkled appearance (arrow) of the superficial retinal nerve fiber layer as demonstrated by the irregular white light reflexes, suggesting a lesser and perhaps variable effect on the macular retina. The macular surface reflexes emphasize horizontal wrinkling centrally, more radially oriented wrinkles peripheral to this, and a generally decreased nerve fiber layer as seen by the light reflexes settling between the more exposed vessels.
Case 2
A 13-year-old autistic, delayed, nonverbal boy with mild microcephaly and seizure disorder was reviewed in the neuro-ophthalmology clinic. He had been treated for generalized tonic clonic seizures lasting several minutes since the age of 6. Control of his seizures was difficult even with 3 anticonvulsants (carbamazepine [Tegretol, Novartis Pharmaceuticals Canada Inc., Dorval, Canada], phenobarbital, and lorazepam [Ativan, Wyeth Pharmaceuticals, St. Laurent, Canada]). Vigabatrin (1000 mg per day) was added to carbamazepine and phenobarbital after 4 months in 1994, 6 years before his ophthalmic referral. His seizure control improved, the phenobarbital was discontinued, and control was well maintained with the 2 other drugs. His ophthalmic examination was described as normal. A transient pattern VEP recorded in February 1998 was normal and consistent with good acuity and normal optic nerve function. An ERG performed under general anesthesia was abnormal. Mixed rod–cone responses were delayed and mildly reduced in amplitude, although cone responses were within normal limits. The ERG flicker response was not recorded at this time because of technical difficulties. According to his mother, he was not having any visual symptoms that she could identify at this time. Repeat ERG 6 months later showed the mixed rod–cone responses to be at the lower level of normal, and the cone responses showed no change over time. A transient VEP recorded in December 1999 was completely normal, again supporting intact optic nerve function.
He did not attend the Eye Clinic again until June 2001 (18 months later), when his mother described that he was bumping into things occasionally. He seemed not to see where he was walking, and his lower field seemed to be problematic. On examination, he tended to stare straight ahead and made few exploratory eccentric movements of his eyes either spontaneously or in response to confrontation visual field testing. He showed no refixational eye movements toward objects placed in his binocular peripheral fields in any quadrant. His fixation was steady and central in both eyes. A binocular preferential looking test score of 0.3 logarithm of the minimum angle of resolution demonstrated a reasonable level of visual acuity (Cardiff Acuity Test, Vistech Consultants Inc, Dayton, OH). On fundus examination at that time, the optic discs showed relative nasal pallor. There was an observable dramatic decrease in the retinal nerve fiber layer in both eyes affecting most of the entire fundus, with the notable relative exception of the macular areas and the temporal aspect of the disc (i.e., no temporal pallor or atrophy). The maculae showed some irregular light reflexes and wrinkling in keeping with some involvement of the nerve fiber layer and deeper layers. While he was under general anesthesia for a repeat ERG, the fundi were examined and photographed to document this unusual form of bilateral retinal and optic nerve atrophy (Figs 5, 6). His ERG at that time showed that the 30-Hz flicker response of 32 μV was markedly reduced (66% lower than age-expected amplitude) compared with age-matched normal data (normal range, 53–176 μV). He was weaned off his 6-year course of vigabatrin as the drug was gradually replaced with topiramate (Topamax, OrthoBiotec, Toronto, Canada). His clinical examination, including fundus appearance, has not changed over 12 months of follow-up examination.
Figure 5.
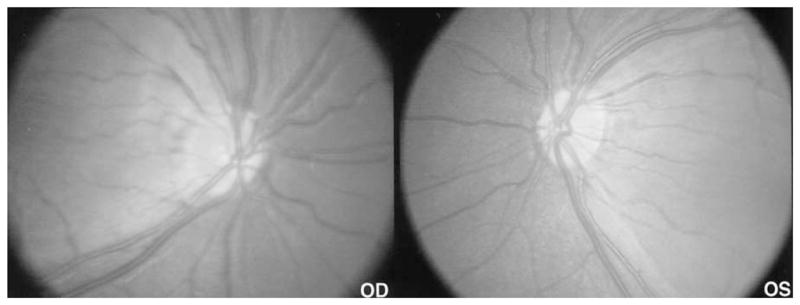
RetCam (Massie Laboratories, Inc., Pleasanton, CA) 30° fundus photos of case 2 showing the areas of retinal atrophy as darker and more granular, well demarcated from the relatively spared central retina. The nasal peripapillary nerve fiber layer is thinned, and nasal disc tissue shows relative loss of small vessels in keeping with early or mild atrophy.
Figure 6.
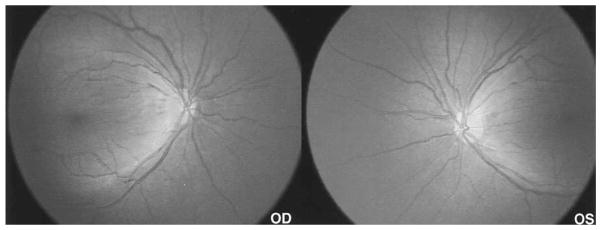
An 80° field RetCam photo demonstrates the large area of retinal atrophy contrasting with the relatively spared macular areas.
Case 3
A 13-month-old boy with trisomy 21, who had infantile spasms develop at 9 months of age in September 1999, was seen for ophthalmic examination. Vigabatrin, 500 mg twice daily, was initiated shortly thereafter, resulting in control of seizures in November 1999. On review in February 2000, 5 months after starting vigabatrin, he had no apparent visual problems. Visual fixation was normal, and his entire ocular examination, including fundus-copy under sedation, was normal. There was no loss of retinal nerve fiber layer in either fundus. An electroretinogram performed under sedation was entirely normal. Vigabatrin was increased to 1250 mg/day. Clinical assessment 6 months later showed normal confrontation visual fields and fixation and no change in his fundus examination. The ERG taken while the patient was under sedation remained within normal limits. Although there was some reduction in the cone-isolated b waves and 30-Hz flicker response, these responses were well within normal limits.
Contrast sensitivity (CS), recorded with the sweep VEP method, was reduced at this time (CS = 19.41, which is an 83% reduction from control value), although visual acuity was within normal limits. He was reexamined in January 2001. By that time, he was taking 1500 mg/day of vigabatrin. Again, his clinical fixation, confrontation visual field examination, and fundi all appeared to be within normal limits. The ERG remained within normal limits for most tested stimuli, except for the 30-Hz flicker response, which had decreased dramatically to become 55% lower than the age-expected amplitude. Because seizure control was not complete, lamotrigine (Lamictal, Novartis Pharmaceuticals Canada) and domperidone were added to the vigabatrin, with plans to taper the vigabatrin dosage. On next assessment in September 2001, his mother described his vision as good but noted that he tended to stare straight ahead. She noticed now that she had to attract his attention downward to his food at meal times by tapping on his plate. In general, he was responding more regularly to sound clues rather than to visual ones. Clinical examination showed accurate and central fixation in both eyes. Binocular visual acuity assessed by preferential looking was 9.8 cycles per degree (Teller Acuity Cards, Vistech Consultants Inc, Dayton, OH). This score falls within the lower half of the age-expected normal range. At this stage, the 30-Hz flicker responses were similar to those recorded on the previous visit, but the cone response had decreased to a level 42% lower than that expected for age, thus an abnormal result. He showed no responsiveness to objects in his peripheral fields on confrontation testing in all quadrants, rather tending to stare straight ahead during field testing. Fundus examination showed retinal atrophy involving most of the retinae, with relatively less involvement of the central or macular areas, which showed wrinkling of irregular thickness (Fig 7). His mother and neurologist were advised regarding the evidence for vigabatrin toxicity, and the vigabatrin was further tapered and discontinued. He was retested in December 2001. The cone-isolated responses and 30-Hz flicker showed further reduction in both eyes (Fig 8). In March 2002, a field-specific VEP,25 recorded binocularly, failed to show decreased N135 responses to the peripheral stimuli. The amplitudes of the P100 and N135 responses were within normal limits (10–17 μV).
Figure 7.
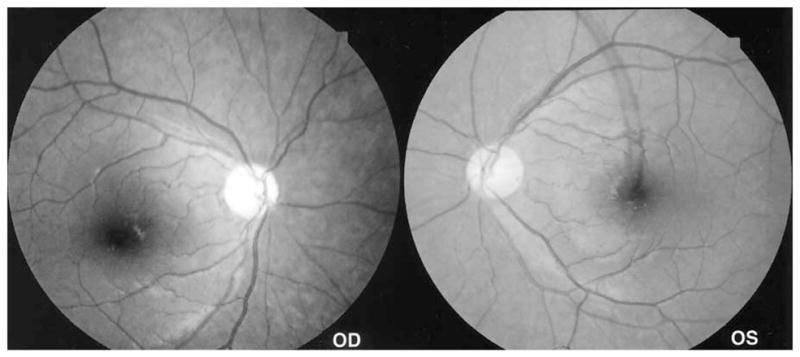
Standard 50° fundus photographs of case 3 showing an “inverse” type of atrophy of the nasal optic nerve, with loss of retinal nerve fibers diffusely throughout the more peripheral retina, with relative sparing of the macular areas and the temporal aspect of the disc in both eyes.
Figure 8.
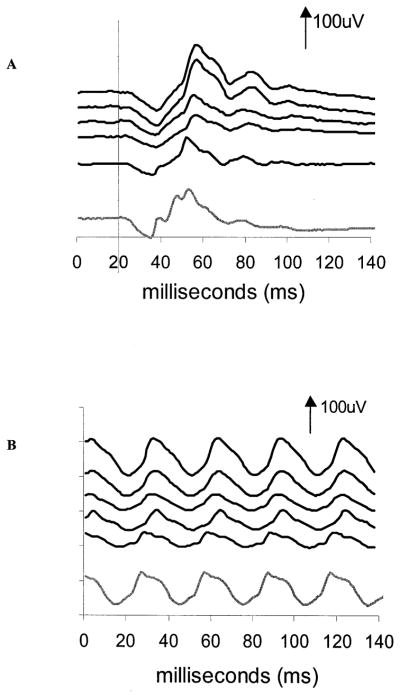
Electroretinogram traces for case 3. A, Sequential recordings of single flash cone response. B, Sequential recordings of the 30-Hz flicker response. The recordings for both occurred from top to bottom at 5 months, 11 months, 16 months, 24 months, and 27 months after vigabatrin was initiated. By the 27-month postvigabatrin visit, vigabatrin had been discontinued. The final trace is from a 34-month-old control. Time is shown in milliseconds. Positive electrical signals are in the upward direction. Vertical arrows and numbers represent microvolts. For the cone response, stimulus onset was at time 20 ms. For the flicker response, the stimulus onset is at time zero.
Discussion
Three children, aged 12 years, 15 years, and 2½ years are described, in whom a severe and atypical form of retinal and optic atrophy has been recognized in association with extended use of the anticonvulsant vigabatrin, peripheral field loss (presumed or clinically detectable), and ERG abnormalities. Because clinical assessment of peripheral visual loss, especially in its milder forms, is so difficult and unreliable in young children, clinicians have turned to investigational support of electrophysiologic tests (i.e., the VEP and ERG) for early detection of optic toxicity. Vigabatrin has been demonstrated in animals to accumulate differentially in many cell types of the retina, particularly in the inner retinal GABAergic amacrine cells and displaced amacrine cells in retinae of rabbits, cats, chickens, fish, and monkeys.26,27 In addition, vigabatrin has been shown to accumulate preferentially in the peripheral portion of the retina and in different proportions in the retinal center compared with the periphery (Pow DV. Differences in cellular sites of accumulation of vigabatrin in central versus peripheral primate retina. International Society for Clinical Electrophysiology of Vision XXXVIII Symposium. Sydney, Australia, 2000). The relative toxic effects on various cell types have yet to be differentiated and demonstrated fully. The HSC longitudinal study of ERGs in children taking vigabatrin has shown that the ERG responses change with duration of vigabatrin treatment. Some of these electrophysiologic indicators are likely to reflect the changing retinal nontoxic GABA levels.20,21 For example, oscillatory potentials decrease in amplitude during drug treatment but recover after discontinuation of the drug, unlike the decrease in flicker amplitude, which is less likely to recover.22 Most children in the HSC longitudinal study do not show the form of retinal nerve fiber layer atrophy described in these 3 more advanced cases of retinal toxicity. Of interest is the tendency, reported by the parents of these children, to stare straight ahead rather than look normally about their visual environment. This may be a useful sign or symptom suggesting field loss when accurate field assessment is clinically not possible or reliable.
Whatever the underlying cellular pathophysiology, vigabatrin toxicity has resulted in a permanent and differential loss of retinal fibers in all 3 children, producing a recognizable peripheral retinal and nasal pattern of optic atrophy. The relatively greater loss of nerve fiber layer nasal to the optic nerve head results in a subtle pallor of the nasal aspect of the disc and relative preservation of the macular fibers, the macular appearance, and the temporal aspect of the disc. The area of peripheral nerve fiber layer atrophy is well demarcated from the central nonatrophic portion along the course of the temporal branching of the central retinal vessels. There may be, however, definite, although subtle, involvement of macular retina at this stage of toxicity, which can be identified clinically as wrinkling. This macular change probably represents spotty or irregular atrophy of a more evolved stage of toxicity. Diagnostically, it is important to recognize that funduscopy of the nonmacular portion of the retina shows the vascular net located relatively superficially on the residual retinal surface, as evidenced by the more obvious and continuous linear paravascular light reflexes and clarity of the uncovered vessels themselves. This change is most readily identifiable in the peripapillary area. The retinal light reflexes, which are strikingly more obvious in children than in adults, help identify the loss of nerve fiber layer by their changed relationship to the arteriolar tree in the posterior pole of the fundus. As the nerve fiber layer decreases, the youthful sheen of the smooth-flowing retinal light reflexes becomes segmentalized by the vascular net. The relative decrease in the nerve fiber layer thickness and its opalescence caused by atrophy renders a darker, granular appearance to the remaining retina and choroid. The authors term the observable and identifiable pattern of tissue loss at the level of the optic nerve head as an “inverse” optic atrophy to distinguish it from the more common temporal and macular atrophy seen with other toxic and nutritional optic neuropathies (Fig 9). This pattern of retinal injury corresponds to the characteristic diffuse 360° peripheral visual field loss documented in older patients with perimetric evidence of vigabatrin toxicity and emphasizes that the retina, not the optic nerve, is the main site of the drug’s toxic effect.
Figure 9.
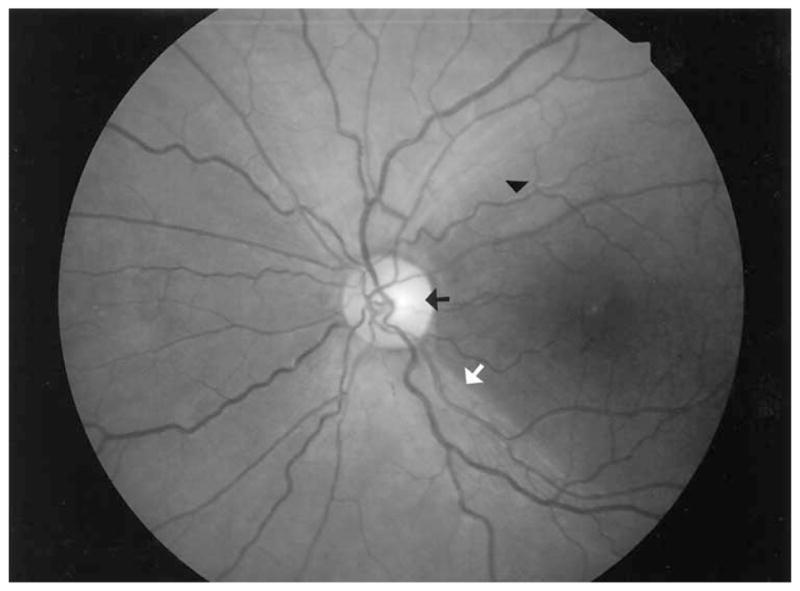
In contrast to the pattern of retinal nerve fiber layer loss in the 3 cases, this fundus photo of a 15-year-old boy in the chronic phase of Leber hereditary optic atrophy shows the typical pattern of retinal nerve fiber loss affecting the macula, with the typical temporal disc atrophy, accompanying a central scotoma. This is the pattern of atrophy seen more commonly in optic neuropathies. In this photo, there is obvious normality or sparing of the nasal peripapillary nerve fiber layer as evidenced by the present retinal opalescence and normal superficial light reflexes. The black arrow indicates temporal disc pallor, the white arrow the demarcation between atrophic and nonaffected nerve fiber layer, and the black triangle the telltale change in superficial light reflex aligning along the course of the exposed small vessel of the atrophic macula.
This retinal and inverse optic atrophy pattern is an important, although rather late, clinical diagnostic sign in the assessment of progressive drug toxicity, especially in young or unmanageable children on vigabatrin. Although there may be some potential for improvement in functional vision (i.e., improvement in the visual field test performance), the atrophic change that is evident clinically is a permanent one and limits the degree of potential peripheral vision restoration. That there have been reported more complete forms of diffuse optic atrophy in the literature18 suggests that this inverse form is not an end-stage picture but rather represents an approximate midstage or phase in the progression of toxic deterioration with continued drug use. The inverse optic atrophy seems to be the clinical correlate of the histologic atrophy reported by Ravindran et al16 in their pathologic study of the eyes and optic nerves in an adult male with vigabatrin toxicity. In this case, the retina showed general atrophy, but peripheral axons of the optic nerve were significantly affected, whereas the central ones were relatively spared. The VEP results were normal in case 1 (sweep VEP), and the levels of visual acuity for cases 2 and 3 were reasonable. These results were recorded after the loss in peripheral vision had become apparent and thus support the notion that the central pathways usually involved in other toxic optic neuropathies have been spared and that the main vigabatrin toxic site is at the level of the retina. The field-specific VEP has a sensitivity of 75% in detecting visual field loss.25 The normal result in case 3 might result from less advanced toxicity combined with reduced sensitivity, because monocular recording was not possible in this child. The authors interpret the optic nerve atrophy as being secondary to the retinal ganglion cell damage and nerve fiber layer atrophy. It would seem logical that as drug toxicity continues, the effect in the macular area would increase to the point that the optic atrophy or pallor becomes more diffuse and appears as predominantly temporal, with the more typical form of diffuse optic atrophy that is seen in advanced cases.
Unfortunately, the early stage of this form of retinal and optic change is difficult to recognize in children because of its relative subtlety. This challenge is compounded by the problem of the examiner getting a sufficiently long critical look at the retina of a moving infant or toddler. Moreover, these authors’ experience within the longitudinal study is that many children with seizure disorders, not surprisingly, have diffuse retinal nerve fiber loss involving the macula as well, apart from drug toxicity, with some visible, although mild, optic atrophy present even at initial assessment. In addition, it is clinically more challenging to recognize retinal nerve fiber loss in “blond” or lightly pigmented fundi compared with more darkly pigmented ones. For these reasons, superimposition of a toxic effect may be even more difficult to recognize, and, hence, clinical judgments need to be made on the basis of changes in sequential ERGs and follow-up funduscopy, as well as photographic comparison with pretreatment fundus appearance when possible.
Close examination with the young child sedated or anesthetized, combined with the routine use of photography concentrating on the papillary and peripapillary areas, provides useful baseline data in follow-up assessments and in the early diagnosis of vigabatrin toxicity. Early diagnosis will depend on identifying parameters that become measurably abnormal, before the fundus atrophy becomes evident, to preserve peripheral vision by early discontinuation of vigabatrin. The most likely parameters will be the electroretinographic ones. The results from these 3 cases are consistent with a toxic drug effect evident clinically as peripheral retinal atrophy initially, with progression to involve the central retina with time. These dramatic changes may be the result of the relatively large doses of vigabatrin used over an extended period of 2.5 to 5 years. Recognition of this characteristic pattern seen on funduscopy enables the clinician to identify the presence of a moderate degree of significant clinical peripheral visual field loss. This fact taken with maintained reduction in ERG responses should serve as the criterion for considering the discontinuation of vigabatrin.
Acknowledgments
Technical support for electroretinogram and visual evoked potential measures was partially funded by The Hospital for Sick Children Research Institute, Toronto, Canada (seed grant), and the University of Toronto Vision Science Research Program, Toronto, Canada.
Footnotes
Presented at: Neuro-ophthalmology Society of Australia meeting, April 21, 2002; Sydney, Australia, and Canadian Ophthalmological Society meeting, June 26, 2003; Ottawa, Canada.
References
- 1.Uldall P, Alving J, Gram L, Hogenhaven H. Vigabatrin in childhood epilepsy: a 5-year follow-up study. Neuropediatrics. 1995;26:253–6. doi: 10.1055/s-2007-979766. [DOI] [PubMed] [Google Scholar]
- 2.Mackay M, Weiss S, Snead OC., III Treatment of infantile spasms: an evidence-based approach. Int Rev Neurobiol. 2002;49:157–84. doi: 10.1016/s0074-7742(02)49012-5. [DOI] [PubMed] [Google Scholar]
- 3.Curatolo P. Vigabatrin for refractory partial seizures in children with tuberous sclerosis [letter] Neuropediatrics. 1994;25:55. doi: 10.1055/s-2008-1071586. [DOI] [PubMed] [Google Scholar]
- 4.Guberman A, Bruni J The Canadian Vigabatrin Study Group. Long-term open multicentre, add-on trial of vigabatrin in adult resistant partial epilepsy. Seizure. 2000;9:112–8. doi: 10.1053/seiz.2000.0382. [DOI] [PubMed] [Google Scholar]
- 5.Eke T, Talbot JF, Lawden MC. Severe persistent visual field constriction associated with vigabatrin. BMJ. 1997;314:180–1. doi: 10.1136/bmj.314.7075.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawden MC, Eke T, Degg C, et al. Visual field defects associated with vigabatrin therapy. J Neurol Neurosurg Psychiatry. 1999;67:716–22. doi: 10.1136/jnnp.67.6.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wild JM, Martinez C, Reinshagen G, Harding GF. Characteristics of a unique visual field defect attributed to vigabatrin. Epilepsia. 1999;40:1784–94. doi: 10.1111/j.1528-1157.1999.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 8.Kalviainen R, Nousiainen I. Visual field defects with vigabatrin: epidemiology and therapeutic implications. CNS Drugs. 2001;15:217–30. doi: 10.2165/00023210-200115030-00005. [DOI] [PubMed] [Google Scholar]
- 9.Krauss GL, Johnson MA, Miller NR. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings. Neurology. 1998;50:614–8. doi: 10.1212/wnl.50.3.614. [DOI] [PubMed] [Google Scholar]
- 10.Daneshvar H, Racette L, Coupland SG, et al. Symptomatic and asymptomatic visual loss in patients taking vigabatrin. Ophthalmology. 1999;106:1792–8. doi: 10.1016/S0161-6420(99)90345-7. [DOI] [PubMed] [Google Scholar]
- 11.Besch D, Kurtenbach A, Apfelstedt-Sylla E, et al. Visual field constriction and electrophysiological changes associated with vigabatrin. Doc Ophthalmol. 2002;104:151–70. doi: 10.1023/a:1014644307518. [DOI] [PubMed] [Google Scholar]
- 12.Harding GF, Wild JM, Robertson KA, et al. Separating the retinal electrophysiologic effects of vigabatrin: treatment versus field loss. Neurology. 2000;55:347–52. doi: 10.1212/wnl.55.3.347. [DOI] [PubMed] [Google Scholar]
- 13.Comaish IF, Gorman C, Brimlow GM, et al. The effects of vigabatrin on electrophysiology and visual fields in epileptics: a controlled study with a discussion of possible mechanisms. Doc Ophthalmol. 2002;104:195–212. doi: 10.1023/a:1014603229383. [DOI] [PubMed] [Google Scholar]
- 14.Koul R, Chacko A, Ganesh A, et al. Vigabatrin associated retinal dysfunction in children with epilepsy. Arch Dis Child. 2001;85:469–73. doi: 10.1136/adc.85.6.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson EA, Brodie MJ. Severe persistent visual field constriction associated with vigabatrin [letter] BMJ. 1997;314:1693. [PMC free article] [PubMed] [Google Scholar]
- 16.Ravindran J, Blumbergs P, Crompton J, et al. Visual field loss associated with vigabatrin: pathological correlations. J Neurol Neurosurg Psychiatry. 2001;70:787–9. doi: 10.1136/jnnp.70.6.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crofts K, Brennan R, Kearney P, O’Connor G. Vigabatrin-induced optic neuropathy. J Neurol. 1997;244:666–7. doi: 10.1007/s004150050165. [DOI] [PubMed] [Google Scholar]
- 18.Russell-Eggitt IM, Mackey DA, Taylor DS, et al. Vigabatrin-associated visual field defects in children. Eye. 2000;14:334–9. doi: 10.1038/eye.2000.83. [DOI] [PubMed] [Google Scholar]
- 19.Marmor MF, Zrenner E International Society for Clinical Electrophysiology of Vision. Standard for clinical electroretinography (1999 update) Doc Ophthalmol. 1999;97:143–56. doi: 10.1023/a:1002016531591. [DOI] [PubMed] [Google Scholar]
- 20.Westall CA, Logan WJ, Smith K, et al. The Hospital for Sick Children, Toronto, longitudinal ERG study of children on vigabatrin. Doc Ophthalmol. 2002;104:133–49. doi: 10.1023/a:1014656626174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morong S, Westall CA, Nobile R, et al. Longitudinal changes in photopic OPs occurring with vigabatrin treatment. Doc Ophthalmol. 2003;107:289–97. doi: 10.1023/b:doop.0000005338.51554.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westall CA, Nobile R, Morong S, et al. Changes in the electroretinogram resulting from discontinuation of vigabatrin in children. Doc Ophthalmol. 2003;107:299–309. doi: 10.1023/b:doop.0000005339.23258.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norcia AM, Tyler CW, Allen D. Electrophysiological assessment of contrast sensitivity in human infants. Am J Optom Physiol Opt. 1986;63:12–5. doi: 10.1097/00006324-198601000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Power Diva [computer program]. Version 1.0. San Francisco: Smith Kettlewell Eye Research Institute; 2004. [Google Scholar]
- 25.Harding GF, Spencer EL, Wild JM, et al. Field-specific visual-evoked potentials. identifying field defects in vigabatrin-treated children. Neurology. 2002;58:1261–5. doi: 10.1212/wnl.58.8.1261. [DOI] [PubMed] [Google Scholar]
- 26.Pow DV, Baldridge W, Crook DK. Activity-dependent transport of GABA analogues into specific cell types demonstrated at high resolution using a novel immunocytochemical strategy. Neuroscience. 1996;73:1129–43. doi: 10.1016/0306-4522(96)00097-8. [DOI] [PubMed] [Google Scholar]
- 27.Neal MJ, Shah MA. Development of tolerance to the effects of vigabatrin (gamma-vinyl-GABA) on GABA release from rat cerebral cortex, spinal cord and retina. Br J Pharmacol. 1990;100:324–8. doi: 10.1111/j.1476-5381.1990.tb15803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]