

Abstract
Electroretinograms (ERGs) have been recorded longitudinally in children before and during treatment with the antiepileptic drug vigabatrin for the past 3.5 years. Vigabatrin induced changes in ERG responses occur in children; the most dramatic changes occur in the oscillatory potentials. The purpose of this study was to identify changes in ERG responses associated with discontinuation of vigabatrin treatment. If vigabatrin-induced changes reverse after discontinuation of the drug we infer that the original change is not an indicator of toxicity. ERG data were analyzed from 17 children who discontinued vigabatrin therapy. The duration of treatment ranged from 5 to 52 months, the age for the first ERG ranged from 6 to 38 months (median 10 months). ERGs were tested using the standard protocol established by the International Society for Clinical Electrophysiology of Vision, with Burian-Allen bipolar contact-lens electrodes. In addition to standard responses we recorded photopic oscillatory potentials (OPs). During vigabatrin treatment OPs show a greater change than other ERG responses, with the early occurring wavelets from the photopic OPs showing the greatest change. With discontinuation of vigabatrin the amplitude of the early wavelets of the photopic OPs increased dramatically compared with amplitudes while taking the drug (paired t-test, p = 0.000075). The scotopic oscillatory potentials also show some recovery. Although changes in oscillatory potentials may occur with vigabatrin toxicity, a large change likely occurs with a non-toxic pharmacological effect of vigabatrin on GABAergic amacrine cells in the inner plexiform layer. Reduction of OPs in children on vigabatrin may not be related to toxicity.
Keywords: ERG, pediatric, retinal toxicity, vigabatrin
Introduction
Vigabatrin (γ-vinyl-GABA) is an antiepileptic drug successful in the management of childhood seizures including infantile spasms [1–7]. The anticonvulsant effect of vigabatrin is achieved by irreversible inhibition of the enzyme GABA-transaminase used to break down the inhibitory neurotransmitter GABA. This results in increased levels of GABA in the brain and in the retina.
Vigabatrin has been associated with visual toxicity in the form of irreversible constriction of the visual field in adults [8–15] and in children [16–19]. Electroretinogram (ERG) abnormalities are found in patients taking vigabatrin [8, 13, 14, 20–27]. Longitudinal recording of ERGs in children during vigabatrin treatment has shown significant changes in cone b-wave amplitude [27, 28], flicker amplitude and photopic oscillatory potentials [28] with time on vigabatrin treatment. Vigabatrin attributed visual field loss has been associated with evidence of reduced cone response b-wave [21, 29], decreased amplitude of the 30-Hz flicker response [30] and abnormalities in oscillatory potentials [21, 25, 29].
Some of the vigabatrin attributed ERG changes may result from the changes in GABA levels which might reverse after treatment; i.e., non-toxic change. Alternatively the ERG changes may occur as a result of non-reversible changes arising from retinal toxicity. Identification of those ERG responses, which reverse on cessation of the drug, would provide information on non-toxic changes. In a study of 13 adults on vigabatrin treatment Johnson et al. [31] found there was no significant change in ERG results after cessation of vigabatrin, although there were improvements in some responses in several patients with less marked visual field deficit. Graniewski-Wijnands et al. [32] reported recovery in ERG scotopic b-wave amplitude after cessation of vigabatrin in nine patients previously on the drug. A cross-sectional study compared electrophysiology parameters between patients (over 14 years of age) who were on vigabatrin treatment and seven patients group who had discontinued vigabatrin in the last 6 months (off-vigab). A proportion of patients from both groups showed reduced scotopic and photopic ERG b-wave amplitudes and reduced 30 Hz flicker response. There was no significant difference between the frequency of occurrence of ERG abnormality between the vigabatrin and off-vigabatrin groups [33].
The purpose of the current study was to identify changes in ERG responses associated with discontinuation of vigabatrin treatment in children.
Methods
Subjects
One hundred and five children on vigabatrin treatment, referred for ophthalmological, visual and electrophysiological assessment, have received ERG testing longitudinally between September 1998 and May 2002; the duration between visits was approximately 6 months. Seventeen of these have now discontinued vigabatrin; the data recorded from these children are used for the current analysis. The duration of treatment in these 17 children ranged from 5 to 42 months. Pre-vigabatrin (baseline) ERGs were recorded in five children (age range at first ERG 5–26 months, median 7 months). The remaining 10 children were already on vigabatrin at the time of initial visit (age range at first ERG 7–40 months, median 10.5 months).
Patient characteristics, including a description of whether the children were on vigabatrin monotherapy or on a combination of anticonvulsant medications, are shown in Table 1.
Table 1.
Patient characteristics
| Subject | Age start of vigabatrin (months) | Type of seizure | Other health problems | Other meds
|
Total cumulative dosage (g/kg) | Age first ERG (months) | |
|---|---|---|---|---|---|---|---|
| During | After cessation | ||||||
| 19 | 7 | IS | DD | None | None | 23 g | 7 |
| 26 | 6 | IS | None | Nitrazipam | 47 g | 6 | |
| 55 | 26 | IS | None | lamotigrine, clobazam, Valproic acid | 21 g | 26 | |
| 11 | 6 | Seiz | HC, IBD | None | Phenobarb | 21 g | 10 |
| 37 | 7 | IS | TS | None | None | 12 g | 10 |
| 54 | 7 | IS | DD, | None | None | 30 g | 12 |
| 8 | 4 | IS | DD,CP | Phenobarbital, ACTH | None | 28 g | 8 |
| 32 | 30 | Seiz | HC, DW | None | None | 18 g | 38 |
| 40 | 8 | IS | MDF | Fresium, Clobazam, Apival, Depakane | Topamax | 77 g | 19 |
| 10 | 5 | Seiz | TS | None | Lamictal | 60 g | 13 |
| 17 | 5 | IS | DD | None | Carbamazepine, Clobazam | 32 g | 8 |
| 5 | 5 | IS | Trisomy 21 | None | Clobazam, Depakane | 53 g | 5 |
| 15 | 4 | Seiz | DD, ME | None | Phenobarb | 76 g | 18 |
| 44 | 11 | IS | TS,DD | Phenobarbital | Lamictal/mogadon | 28 g | 11 |
| 68 | 7 | IS | None | carbamazepine | 36 g | 8 | |
| 61 | 9 | IS | Trisomy 21 | Lamictal, Domeridone, Losec, Clobazam | Lamictal clobazam | 34 g | 14 |
| 29 | 40 | Seiz | DD | Topamax | None | 35 g | 42 |
IS – infantile spasms; Seiz – seizures; ME – mitochondral encephalopathy; DW – Dandy Walker syndrome; CP – cerebral Palsy; DD –developmental delay; HC – hydrocephalus; IBD – ischemic brain damage; MDF – mild dysmorphic features; TS – tuberous sclerosis; other drugs taken during vigabatrin treatment, and drugs started after discontinuation are shown.
All testing was performed in the Visual Electro-physiology Unit at The Hospital for Sick Children. The research followed the tenets of the Declaration of Helsinki. Parents of subjects gave signed consent for their participation in the study. The consent form acknowledged that research procedures were described, any questions answered and that harms and benefits were fully explained. Approval for this study was obtained from the Research Ethics Board at the Hospital For Sick Children.
Subjects were excluded from the study if they had a retinal dystrophy or a known family history of retinal dystrophy, any eye disease associated with abnormal ERG, previous intraocular surgery or systemic disease known to affect the retina.
Most children were sedated with oral chloral hydrate (80 mg per kg of body weight; maximum single dose of 1 g). ERGs were recorded according to ISCEV standards [34]. Before ERG assessment, both pupils were pharmacologically dilated with 1% cyclopentolate and 2.5% phenylephrine. Each subject was dark-adapted for at least 30 min before recording began. One drop of proparacaine 0.5% was instilled and a bipolar Burian-Allen electrode (Hansen Ophthalmic Development Laboratory, Iowa City, IA) of the appropriate size for the individual subject’s eye-was placed on the corneal eye surface. An electrode attached to the forehead served as the ground.
ERGs were recorded using a Grass PS22 photic stimulator (Grass Instruments, Quincy, MA) and Ganzfeld stimulation (LKC Technologies, Gaithersburg, MD). Neuroscan (Herndon, VA) manufactured the recording equipment and software used. The standard flash intensity was 2 cd·s/m2(Gamma Scientific DR-2000 integrated photometer, San Diego). The rod response was elicited by the standard flash attenuated by 2.6 log units using a neutral density filter mounted on a filter wheel (LKC Technologies).
After dark-adapted responses were recorded, subjects were exposed, for 10 min, to a light-adapting background (Ganzfeld) set at 30 cd/m2. The single-flash cone responses (2.0 cd/m2 and also at 3.6 cd·s/m2), followed by 30-Hz flicker response (2.0 cd·s/m2), were recorded in the presence of the adapting background.
Individual ERG responses were amplified (gain 2833; bandpass 0.3–300 Hz; analogue-to-digital rate, 1000), digitized and saved on a computer disk for subsequent analysis. ERG responses were analyzed off-line. OPs were isolated from the standard flash scotopic ERG response and from the 3.6 cd·s/m2cone response by digital-filtering (bandwidth, 100–300 Hz; roll off 12 dB/octave) and averaging the epochs.
After ERG testing, both direct and indirect ophthalmoscopy was performed through pharmacologically dilated pupils on all patients. Refractive errors were assessed with cycloplegic refraction.
Data analysis
Much of our data were collected from infants while ERG responses were still developing [35]. To avoid confounding effects of age expected developmental changes all data were expressed as relative units to age-expected responses. The age expected responses were derived from the logistic growth curve of normal ERG data (described in Ref. [35]). The upper and lower limits of normal responses had been derived from the root mean-square deviations of amplitude (or implicit times) of ERG components from the logistic growth curve (log amplitude). The limits were set at twice the root mean square deviation above and below the logistic growth curve (i.e., ± 2SD). For amplitude data collected in the current study the log of the age expected amplitude was subtracted from the log of the individual response of the child on vigabatrin; negative values therefore represented reduction in ERG amplitude relative to expected value. For implicit time data, age expected implicit times were subtracted from individual patient values and therefore responses that were delayed relative to expected values were represented by positive values.
Descriptive statistics were used to illustrate changes in ERG responses over time. The visit classification depended on approximate duration of vigabatrin treatment. The baseline visit was defined as the time at initiation of vigabatrin.
Paired t-tests, using the Bonferroni correction to account for multiple testing were used to identify changes between ERG testing during vigabatrin treatment with those following discontinuation of treatment.
Results
Of the 17 subjects who discontinued vigabatrin five had ERGs from the baseline visit; 13 had data from the 6-month visit, 12 had been on vigabatrin for more than 6 months (range 12–30 months). The data from all 17 were included in the vigabatrin-discontinued visit.
Representative data describing ERG responses from two children on vigabatrin treatment are shown in Figure 1. Mixed rod-cone responses, scotopic OPs, cone-isolated responses, photopic oscillatory potentials and the 30-Hz flicker response are shown.
Figure 1.


Sample ERG traces (left column) and percent deviation from control (right column). From top to bottom: mixed rod cone responses, scotopic OPs, cone isolated responses, photopic oscillatory potentials and the 30-Hz flicker. In the right column, 4th graph from the top (photopic OPs) the percent deviation is shown for the late OP (black line) and early OPs (gray line). (a) subject 5, a child who began vigabatrin at 5 months of age and had ERG testing at approximately 6-month intervals during vigabatrin treatment and after vigabatrin treatment (labeled age off). The last waveform (gray ERG trace) is data recorded from a 30-month-old control (with normal vision). (b) subject 61, 14 months of age for his first ERG recorded. By this time he had been on vigabatrin for 5 months. He was tested at approximately 6-month intervals and again after discontinuation of vigabatrin. The last waveform (gray ERG trace) is data recorded from a 36-month-old control (with normal vision) The numbers at the left of the series of graphs shows length of time on vigabatrin. Time is shown in milliseconds. Positive electrical signals are in the upward direction. Vertical arrows and numbers represent microvolts. For the top four graphs stimulus onset was at time 20 ms. For the flicker response the stimulus flash is at time 0. *Abnormally reduced amplitude levels.
Figure 1a shows ERG waveforms (left column) from the right eye of a boy with Trisomy 21 (subject 5) diagnosed with infantile spasms at 5.5 months of age, who previously had been healthy and had a normal birth history. At the time of the initial ERG (5.5 months) he was on no medication. Data recorded from the first four visits have been reported previously [27]. The last waveform is that recorded from a 30-month-old control. Also shown is a profile of relative change of ERG parameters over time (right column). ERG change is expressed as percentage deviation from age expected data. At the initial ERG, responses were within normal limits. After 6 months on vigabatrin treatment most mixed rod-cone and cone b-wave amplitudes, flicker and the photopic late OP had increased in amplitude. By this time the amplitude of the scotopic and the early photopic OPs had decreased and were diminished compared with the lower limits of normal. This pattern continued until the 18-month visit where the mixed rod-cone and cone b-wave amplitudes, flicker and the photopic late OP amplitude had declined, but remained within normal limits. After almost 2 years of vigabatrin treatment, with a cumulative dosage of 53 g of vigabatrin per kilogram body weight, the drug was discontinued. Four months later the ERG was repeated. There was further decline in mixed rod-cone and cone b-wave amplitudes, flicker and the photopic late OP amplitude, such that late OP and flicker amplitude were now lower than the 2.5th percentile of data expected for this 30-month-old child. Scotopic OPs and the early photopic OPs showed recovery, with the scotopic but not photopic OPs being within normal limits. Ophthalmoloical examination was normal.
Figure 1b shows data from the right eye of a boy who eventually developed vigabatrin attributed visual toxicity (Subject 61). He has trisomy 21 and was diagnosed with infantile spasms at 9 months of age and was started on vigabatrin therapy (500 mg bid at that time). On his first two visits (vigab 5 and 10 months responses) the ERG was within normal limits except for scotopic and early photopic OPs. The late OP declined in amplitude between visits 1 and 2 (gray line in percent reduction graph). At his second visit his ophthalmological examination was normal. The ERG was normal apart from OPs, which were reduced. By visit 3 (16 months vigabatrin treatment) the flicker amplitude had reduced and was now lower than expected age matched normal limits. The fourth visit (21 months on vigabatrin) was in September 2001, the cone-isolated b-wave response had decreased and was now diminished in both eyes and the 30-Hz flicker response was diminished and delayed. Binocular visual acuity by preferential looking (Teller Acuity Cards) was 9.8 cpd, placing his score in the lower half of the normal range. The ophthalmological examination conducted by one of the authors (RB) revealed accurate and central fixation in both eyes. He failed to respond to objects in his peripheral fields on confrontation testing in all quadrants and tended to stare straight ahead. Fundus exam showed retinal atrophy involving most of the retina with relative sparing of the macular areas in both eyes. He had taken 34 g/kg vigabatrin by this time. His mother and neurologist were advised regarding the evidence for vigabatrin toxicity and the vigabatrin was tapered and discontinued. The main difference between the ERGs for subject 5 and subject 61 was reduction of the photopic ERG and flicker response after 16 months of vigabatrin in the latter.
Descriptive results of ERG responses from ISCEV recommended stimulus conditions are shown in Figure 2. The p-value for the paired t-test (>6 months on vigabatrin versus off vigabatrin) are shown when the p-value nears significance. To correct for multiple testing using the Bonferoni adjustment, a significance value of p < 0.0045 needs to be reached. The error bars describe the standard deviation for each visit. Grey shading describes the upper and lower limits of lab normal control data as described previously [35]. Scotopic ERG amplitudes are described in Figure 2a–c. The mean scotopic a-wave amplitude was below the lower limit of the normal range before vigabatrin treatment, but showed slight improvement after 6 months of vigabatrin treatment. The scotopic rod-cone b-wave amplitude data showed a small increase from baseline with subsequent reduction (not significant; p = 0.018) after 6 months of vigabatrin treatment. During vigabatrin treatment most children had rod-cone b-wave amplitudes within normal limits. These decreased after treatment was discontinued. The mean of the sum of scotopic oscillatory potentials was below the lower limits of normal for baseline and subsequent visits, but increased to lie within normal limits after vigabatrin was discontinued. The paired t-test comparison between the last on-vigabatrin visit and the off-vigabatrin visit was close to significance (p = 0.0078).
Figure 2.

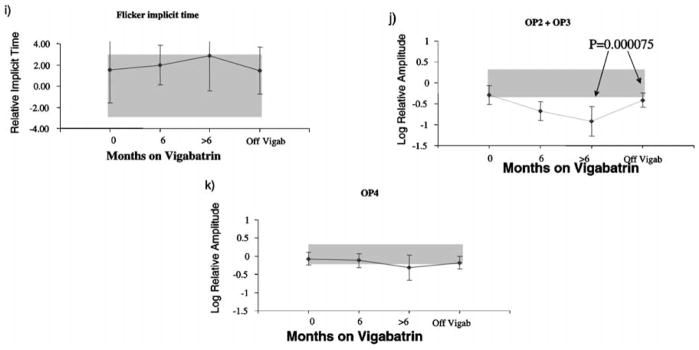
Mean ERG responses from the 17 children. Log relative amplitudes are plotted against length of time on vigabatrin (months). Black lines represent mean ERG amplitude. The error bars represent the standard deviation. Shaded areas represent lab normal ranges containing 95% of control data. (a) rod-cone a-wave amplitude, (b) mixed rod-cone b-wave amplitude, (c) scotopic sum of OPs (d) cone a-wave amplitude, (e) cone b-wave amplitude responses, (f) flicker amplitude data, (g) photopic sum of OPs, (h) photopic OP2 implicit time, (i) flicker implicit time, (j) photopic sum of early OPs, (k) photopic OP4 amplitude. Upward direction reflects increase in both amplitude and implicit time data. (i) flicker implicit time, (j) photopic sum of early OPs, (k) photopic OP4 amplitude. Upward direction reflects increase in both amplitude and implicit time data.
Photopic responses are described in Figure 2d–g. The cone b-wave and flicker amplitudes show a pattern decrease over time with no change after discontinuation of vigabatrin. The sum of the photopic OPs amplitude was towards the lower limit of normal before treatment and declined over time, recovering to the pre-vigabatrin level after discontinuation of the drug.
There was a slight trend for the photopic OP2 implicit time and flicker implicit time to increase with length of vigabatrin treatment and decrease to pre-vigabatrin levels after discontinuation of vigabatrin treatment (Figures 2h, i).
The sum of early OPs (OP2 +OP3) showed marked reduction with treatment, with recovery after the discontinuation of vigabatrin; paired t-test between on-vigabatrin and off-vigabatrin results was highly significant (p = 0.000075). At the last on-vigabatrin visit the early OPs recorded from all 17 children were below the lower limits of normal; after discontinuation of the drug 12 had shown improvement although OPs from 13 children were still below normal. OP4 showed a mild reduction with very small recovery after vigabatrin was discontinued.
Regression analysis determined that recovery of the OPs was not dependent on drug dosage. Recovery was defined as OP amplitude at the OFF vigabatrin visit minus that for the last ON vigabatrin visit; recovery = −0.0009 (drug dosage) + 0.4264. The coefficient of determiniation (R2) was = 0.005 and indicated lack of association.
Discussion
Descriptive statistics indicate that after cessation of the drug vigabatrin, children demonstrate no change in the cone b-wave amplitude while photopic and scotopic OP amplitudes increase.
The most dramatic change occurs with the early photopic OPs, which show significant recovery after cessation of vigabatrin. Synaptic activity from amacrine cells of the inner retina are involved in the inhibitory feedback loops that form the OPs [36]. Vigabatrin results in an increase in GABA and there is a high density of GABA in amacrine cells of the inner plexiform layer of the retina [37]. Connections between amacrine cell processes, bipolar cells and dendrites to ganglion cells occur within the inner plexiform layer. The retinal feedback circuitry responsible for OPs is likely to be disrupted by the GABAergic amacrine cells in the inner plexiform layer.
However, in response to vigabatrin, the later OP did not respond in the same way as the early OPs. Is it possible that the later OP is less responsive to GABA? In the retina different neurotransmitters are associated with ON and OFF pathways. ON bioplar cells (cone and rod) are depolarized to the onset of light and are dominated by GABAergic input [38]; while OFF cone bipolar cells depolarize to light offset and are dominated by glycinergic input [38]. Kojima and Zrenner [39] have shown that the early photopic OPs result from the onset of the light stimulus while the later OP is evoked by the offset of the light. In the human retina many amacrine cells respond to GABA as an inhibitory neurotransmitter; those that do not, use the inhibitory neurotransmitter glycine [40].
The differences in response to vigabatrin of the early versus the late OPs found in the current study may be explained by differences in neurotransmission between the two categories of OPs. Watchmeister shows that in mudpuppy retina the earlier OPs arise closer to the ganglion cell layer than the later OPs [36]. Adding to the evidence that different neurotransmitters may be responsible for early versus late OPs there seems be some physical separation between the laminae responsible for ON and OFF responses in the inner plexiform layer [37]. In cat, glycine is concentrated in the outer two-thirds of the inner plexiform layer while GABA is localized closer to the ganglion cell layer [41].
The early OPs are reduced dramatically in children taking vigabatrin. As there is significant recovery after cessation of the drug it is likely that some of the change was due to a temporary increase in retinal GABA levels during drug treatment. Subject 61 developed retinal toxicity. His OPs were greatly reduced during vigabatrin treatment, with some improvement after treatment. We cannot rule out the possibility that the process of retinal toxicity caused a further reduction to the OPs. Therefore our results do not contradict studies showing correlation of OPs with visual field defect [21, 25, 29].
The reason that photopic OP amplitudes remained lower than normal limits after cessation of vigabatrin may be related to the finding that the OP summation was abnormal before initiation of vigabatrin treatment. This suggests that the retina may be compromised by abnormal neurotransmission associated with the underlying pathology responsible for the seizure disorder. Although other medications may affect the OPs, subjects 26 and 55 had abnormal OPs at their pre-vigabatrin visit and were on no other medication at that time.
It is not clear why the OPs failed to recover in five children. Child 15 had a higher cumulative dosage (76 g/kg) than other children whereas child 37 only had 12 g/kg vigabatrin. Ten children were on other medications after cessation of vigabatrin (Table 1). There may have been some effect on OPs due to these drugs. Although all children underwent ophthalmological exams at all visits, it may be that some are in a clinically undetected stage of early vigabatrin toxicity. It is not possible to record visual fields due to the age of the children in this study. Those with abnormal results will continued to be followed in our eye clinic. When it becomes possible to perform a field specific VEP [42] then those data may help provide evidence of toxicity.
In our sample of 17 children the improvement in amplitude of the early photopic OPs was not associated with cumulative dosage of vigabatrin. It is possible that in some cases recovery is related to non-toxic pharmacological effects of change in vigabatrin level. The pharmaco-dynamics of vigabatrin are fast as is evidenced by a rapid vigabatrin-induced increase in photopic b-wave implicit time, thought to be caused by alterations in retinal GABA levels [43]. The lack of effect of total vigabatrin dosage on recovery of OP amplitude may be related to the relative importance of the daily dosage.
Acknowledgments
We are grateful to Rita Buffa for help with data collection, to Ajmal Kahn for help with deriving ERG normal ranges according to age and Dena Hamoudi for helping determine drug dosage and helping with figures.
References
- 1.Fois A, Buoni S, Bartolo R, Marco V, Mostardini R. Vigabatrin treatment in children. Child’s Nerv Syst. 1994;10:244–8. doi: 10.1007/BF00301162. [DOI] [PubMed] [Google Scholar]
- 2.Kwong L. Vigabatrin as first line therapy in infantile spasms: review of seven patients. J Paediatr Child Health. 1997;33(2):121–4. doi: 10.1111/j.1440-1754.1997.tb01013.x. [DOI] [PubMed] [Google Scholar]
- 3.Uldall P, Alving J, Gram L, Beck S. Vigabatrin in pediatric epilepsy – An open study. J Child Neurol. 1991;6:2S38–44. [PubMed] [Google Scholar]
- 4.Uldall P, Alving J, Gram L, Hogenhaven H. Vigabatrin in Childhood Epilepsy: A 5-Year Follow-Up Study. Neuropediatrics. 1995;26:253–6. doi: 10.1055/s-2007-979766. [DOI] [PubMed] [Google Scholar]
- 5.Nabbout R, Melki I, Gerbaka B, Dulac O, Akatcherian C. Infantile spasms in Down syndrome: good response to a short course of vigabatrin. Epilepsia. 2001;42(12):1580–3. doi: 10.1046/j.1528-1157.2001.13501.x. [DOI] [PubMed] [Google Scholar]
- 6.Mackay M, Weiss S, Snead OC., 3rd Treatment of infantile spasms: an evidence-based approach. Int Rev Neurobiol. 2002;49:157–84. doi: 10.1016/s0074-7742(02)49012-5. [DOI] [PubMed] [Google Scholar]
- 7.Elterman RD, Shields WD, Mansfield KA, Nakagawa J. Randomized trial of vigabatrin in patients with infantile spasms. Neurology. 2001;57(8):1416–21. doi: 10.1212/wnl.57.8.1416. [DOI] [PubMed] [Google Scholar]
- 8.Eke T, Talbot JF, Lawden MC. Severe persistent visual field constriction associated with vigabatrin. Br Med J. 1997;314:180–1. doi: 10.1136/bmj.314.7075.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawden MC, Eke T, Degg C, Harding GF, Wild JM. Visual field defects associated with vigabatrin therapy [see comments] J Neurol Neurosurg Psychiatry. 1999;67(6):716–22. doi: 10.1136/jnnp.67.6.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wild JM, Martinez C, Reinshagen G, Harding GF. Characteristics of a unique visual field defect attributed to vigabatrin. Epilepsia. 1999;40(12):1784–94. doi: 10.1111/j.1528-1157.1999.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 11.Kalviainen R, Nousiainen I, Mantyjarvi M, Nikoskelainen E, Partanen J, Partanen K, Riekkinen P., Sr Vigabatrin, a gabaergic antiepileptic drug, causes concentric visual field defects. Neurology. 1999;53(5):922–6. doi: 10.1212/wnl.53.5.922. [DOI] [PubMed] [Google Scholar]
- 12.Manuchehri K. Visual field defect associated with vigabatrin. Method of estimating prevalence was inappropriate [letter] Br Med J. 2000;320(7246):1403–4. discussion 1404. [PubMed] [Google Scholar]
- 13.van der Torren K, Graniewski-Wijnands HS, Polak BC. Visual field and electrophysiological abnormalities due to vigabatrin. Doc Ophthalmol. 2002;104(2):181–8. doi: 10.1023/a:1014615517996. [DOI] [PubMed] [Google Scholar]
- 14.Hardus P, Verduin WM, Berendschot TT, Kamermans M, Postma G, Stilma JS, van Veelen CW. The value of electrophysiology results in patients with epilepsy and vigabatrin associated visual field loss. Acta Ophthalmol Scand. 2001;79(2):169–74. doi: 10.1034/j.1600-0420.2001.079002169.x. [DOI] [PubMed] [Google Scholar]
- 15.Kalviainen R, Nousiainen I. Visual field defects with vigabatrin: epidemiology and therapeutic implications. CNS Drugs. 2001;15(3):217–30. doi: 10.2165/00023210-200115030-00005. [DOI] [PubMed] [Google Scholar]
- 16.Russell-Eggitt IM, Mackey DA, Taylor DS, Timms C, Walker JW. Vigabatrin-associated visual field defects in children. Eye. 2000;14(Pt 3a):334–9. doi: 10.1038/eye.2000.83. [DOI] [PubMed] [Google Scholar]
- 17.Iannetti P, Spalice A, Perla FM, Conicella E, Raucci U, Bizzarri B. Visual field constriction in children with epilepsy on vigabatrin treatment. Pediatrics. 2000;106(4):838–42. doi: 10.1542/peds.106.4.838. [DOI] [PubMed] [Google Scholar]
- 18.Gross-Tsur V, Banin E, Shahar E, Shalev RS, Lahat E. Visual impairment in children with epilepsy treated with vigabatrin. Ann Neurol. 2000;48(1):60–4. [PubMed] [Google Scholar]
- 19.Luchetti A, Amadi A, Gobbi G. Visual field defects associated with vigabratin monotherapy in children. J Neurol Neurosurg Psychiatry. 2000;69(4):566. doi: 10.1136/jnnp.69.4.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daneshvar H, Racette L, Coupland SG, Kertes PJ, Guberman A, Zackon D. Symptomatic and asymptomatic visual loss in patients taking vigabatrin. Ophthalmology. 1999;106(9):1792–8. doi: 10.1016/S0161-6420(99)90345-7. [DOI] [PubMed] [Google Scholar]
- 21.Krauss GL, Johnson MA, Miller NR. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings [see comments] Neurology. 1998;50(3):614–8. doi: 10.1212/wnl.50.3.614. [DOI] [PubMed] [Google Scholar]
- 22.Miller NR, Johnson MA, Paul SR, Girkin CA, Perry JD, Endres M, Krauss GL. Visual dysfunction in patients receiving vigabatrin: clinical and electrophysiologic findings. Neurology. 1999;53(9):2082–7. doi: 10.1212/wnl.53.9.2082. [DOI] [PubMed] [Google Scholar]
- 23.Arndt CF, Derambure P, Defoort-Dhellemmes S, Hache JC. Outer retinal dysfunction in patients treated with vigabatrin. Neurology. 1999;52(6):1201–5. doi: 10.1212/wnl.52.6.1201. [DOI] [PubMed] [Google Scholar]
- 24.Harding GF, Wild JM, Robertson KA, Rietbrock S, Martinez C. Separating the retinal electrophysiologic effects of vigabatrin: treatment versus field loss [In Process Citation] Neurology. 2000;55(3):347–52. doi: 10.1212/wnl.55.3.347. [DOI] [PubMed] [Google Scholar]
- 25.Besch D, Kurtenbach A, Apfelstedt-Sylla E, Sadowski B, Dennig D, Asenbauer C, Zrenner E, Schiefer U. Visual field constriction and electrophysiological changes associated with vigabatrin. Doc Ophthalmol. 2002;104(2):151–70. doi: 10.1023/a:1014644307518. [DOI] [PubMed] [Google Scholar]
- 26.Jensen H, Sjo O, Uldall P, Gram L. Vigabatrin and retinal changes. Doc Ophthalmol. 2002;104(2):171–80. doi: 10.1023/a:1014679804792. [DOI] [PubMed] [Google Scholar]
- 27.Westall CA, Logan WJ, Smith K, Buncic JR, Panton CM, Abdolell M. The Hospital for Sick Children, Toronto, Longitudinal ERG study of children on vigabatrin. Doc Ophthalmol. 2002;104(2):133–49. doi: 10.1023/a:1014656626174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morong S, Nobile R, Westall CA, Buncic JR, Logan WJ, Panton CM, Abdolell M. Longitudinal changes in photopic OPs occurring with vigabatrin treatment. Doc Ophthalmol. 2003;107:289–297. doi: 10.1023/b:doop.0000005338.51554.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comaish IF, Gorman C, Brimlow GM, Barber C, Orr GM, Galloway NR. The effects of vigabatrin on electrophysiology and visual fields in epileptics: a controlled study with a discussion of possible mechanisms. Doc Ophthalmol. 2002;104(2):195–212. doi: 10.1023/a:1014603229383. [DOI] [PubMed] [Google Scholar]
- 30.Harding GFA. Electrophysiological methods for assessing field losses associated with Vigabatrin. International Societry for Clinical Electrophysiology of Vision XXXVIII Symposium; 2000; Sydney, Australia. [Google Scholar]
- 31.Johnson MA, Krauss GL, Miller NR, Medura M, Paul SR. Visual function loss from vigabatrin: effect of stopping the drug. Neurology. 2000;55(1):40–5. doi: 10.1212/wnl.55.1.40. [DOI] [PubMed] [Google Scholar]
- 32.Graniewski-Wijnands HS, van der Torren K. Electro-ophthalmological recovery after withdrawal from vigabatrin. Doc Ophthalmol. 2002;104(2):189–94. doi: 10.1023/a:1014607331200. [DOI] [PubMed] [Google Scholar]
- 33.Coupland SG, Zackon DH, Leonard BC, Ross TM. Vigabatrin effect on inner retinal function. Ophthalmology. 2001;108(8):1493–6. doi: 10.1016/s0161-6420(01)00638-8. discussion 1497–8. [DOI] [PubMed] [Google Scholar]
- 34.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update) Doc Ophthalmol. 1999;97:143–56. doi: 10.1023/a:1002016531591. [DOI] [PubMed] [Google Scholar]
- 35.Westall CA, Panton CM, Levin AV. Time course for maturation of electroretinogram response, from infancy to adulthood. Doc Ophthalmol. 1999;96:355–79. doi: 10.1023/a:1001856911730. [DOI] [PubMed] [Google Scholar]
- 36.Watchtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res. 1998;17(4):485–521. doi: 10.1016/s1350-9462(98)00006-8. [DOI] [PubMed] [Google Scholar]
- 37.Nelson R, Famiglietti EV, Jr, Kolb H. Intracellular staining reveals different levels of stratification for on- and off-center ganglion cells in cat retina. J Neurophysiol. 1978;41(2):472–83. doi: 10.1152/jn.1978.41.2.472. [DOI] [PubMed] [Google Scholar]
- 38.Grunert U. Distribution of GABA and glycine receptors on bipolar and ganglion cells in the mammalian retina. Microsc Res Tech. 2000;50(2):130–40. doi: 10.1002/1097-0029(20000715)50:2<130::AID-JEMT5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 39.Kojima M, Zrenner E. Off-components in response to brief light flashes in the oscillatory potential of the human electroretinogram. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1978;206(2):107–20. doi: 10.1007/BF00414619. [DOI] [PubMed] [Google Scholar]
- 40.Marc RE, Liu WL. (3H) glycine-accumulating neurons of the human retina. J Comp Neurol. 1985;232(2):241–60. doi: 10.1002/cne.902320209. [DOI] [PubMed] [Google Scholar]
- 41.Pourcho RG. Uptake of [3H]glycine and [3H]GABA by amacrine cells in the cat retina. Brain Res. 1980;198(2):33–46. doi: 10.1016/0006-8993(80)90748-9. [DOI] [PubMed] [Google Scholar]
- 42.Harding GF, Spencer EL, Wild JM, Conway M, Bohn RL. Field-specific visual-evoked potentials: identifying field defects in vigabatrin-treated children. Neurology. 2002;58(8):1261–5. doi: 10.1212/wnl.58.8.1261. [DOI] [PubMed] [Google Scholar]
- 43.Harding GF, Robertson KA, Edson AS, Barnes P, Wild J. Visual electrophysiological effect of a GABA transaminase blocker. Doc Ophthalmol. 1999;97(2):179–88. doi: 10.1023/a:1002045223358. [DOI] [PubMed] [Google Scholar]