

Abstract
INTRODUCTION
Although mitochondrial dysfunction is thought to contribute to the development of post-traumatic organ failure, current techniques to assess mitochondrial function in tissues are invasive and clinically impractical. We hypothesized that mitochondrial function in peripheral blood mononuclear cells (PBMCs) would reflect cellular respiration in other organs during hemorrhagic shock and resuscitation (HS&R).
METHODS
Using a fixed pressure HS model, Long Evan’s rats were bled to a mean arterial pressure (MAP) of 40 mmHg. When blood pressure could no longer be sustained without intermittent fluid infusion (Decompensated HS), Lactated Ringer’s (LR) was incrementally infused to maintain the MAP at 40 mmHg until 40% of the shed blood volume was returned (Severe HS). Animals were then resuscitated with 4X total shed volume in LR over 60 minutes (Resuscitation). Control animals underwent the same surgical procedures, but were not hemorrhaged. Animals were randomized to Control (n=6), Decompensated HS (n=6), Severe HS (n=6) or Resuscitation (n=6) groups. Kidney, liver, and heart tissues as well as PBMC’s were harvested from animals in each group to measure mitochondrial oxygen consumption using high resolution respirometry. Flow cytometry was used to assess mitochondrial membrane potential (Ψm) in PBMCs. One-way ANOVA and Pearson correlations were performed.
RESULTS
Mitochondrial oxygen consumption decreased in all tissues, including PBMC’s, following Decompensated HS, Severe HS, and Resuscitation. However, the degree of impairment varied significantly across tissues during HS&R. Of the tissues investigated, PBMC mitochondrial oxygen consumption and Ψm provided the closest correlation to kidney mitochondrial function during HS (complex I: r =0.65; complex II: r=0.65; complex IV: r=0.52; p<0.05). This association, however, disappeared with resuscitation. A weaker association between PBMC and heart mitochondrial function was observed but no association was noted between PBMC and liver mitochondrial function.
CONCLUSION
All tissues including PBMC’s demonstrated significant mitochondrial dysfunction following HS&R. Although PBMC and kidney mitochondrial function correlated well during hemorrhagic shock, the variability in mitochondrial response across tissues over the spectrum of hemorrhagic shock and resuscitation limits the usefulness of using PBMC’s as a proxy for tissue-specific cellular respiration.
Keywords: Hemorrhagic shock, mitochondrial dysfunction, peripheral blood mononuclear cell, resuscitation, vital organ
Introduction
Trauma is the third most common cause of mortality in the industrialized world (1), and the development of multiple organ failure (MOF) following hemorrhagic shock is the leading cause of post-injury death (2). Severe hemorrhage triggers a cascade of intracellular events that can result in MOF independent of aggressive resuscitation and the restoration of tissue oxygen delivery (3). A critical component in this cascade is the development of mitochondrial dysfunction (4). Following ischemia-reperfusion, impaired mitochondrial function contributes to cell injury and organ failure by reducing aerobic ATP production and by increasing the generation of toxic free radical species (5). Mitochondrial dysfunction following hemorrhagic shock has been demonstrated in a variety of animal models and is associated with both impaired organ function and poor outcome (4,6). Persistent mitochondrial dysfunction has also been associated with increased MOF and higher mortality in severely injured patients (3,5,7).
The early detection and serially assessment of mitochondrial dysfunction in organ systems could substantially improve goal-directed resuscitative strategies following hemorrhagic shock. Current strategies to increase oxygen delivery may be beneficial in the earliest phases of resuscitation, but this approach may be less effective, or even harmful, in later stages if mitochondrial oxidative phosphorylation is impaired (8). Currently, there is no practical method available to measure mitochondrial dysfunction within vital organs at the bedside. While blood lactate, base deficit, and venous oxygen saturation can be used to assess impaired aerobic metabolism globally, these biomarkers are neither specific for mitochondrial dysfunction nor informative about the site of cellular injury (9). Although near infrared spectroscopy and electron paramagnetic resonance have been used to assess mitochondrial function following hemorrhagic shock (3), practical and technical challenges have limited their use to research settings (10).
Mitochondrial studies in peripheral blood mononuclear cells (PBMCs) offer pragmatic alternative approach to measure mitochondrial dysfunction in trauma. PBMC’s can be easily isolated following standard phlebotomy and provide a readily accessible tissue that can be repeatedly assayed during the course of resuscitation. In patients with sepsis, reduced oxygen consumption and lower mitochondrial membrane potential in PBMCs have been associated with disease severity, immune dysregulation, and decreased survival (11,12). In a recent study, we have also observed that mitochondrial function is altered in PBMCs following hemorrhagic shock and resuscitation in a rodent model (13). Similar to a report by Belikova et al. (12), we found that PBMCs recovered mitochondrial function when incubated in serum from control animals with control PBMCs developed depressed function when incubated with serum collected following severe shock; suggesting a systemic factor may be responsible for mitochondrial dysfunction in PBMCs. However, the extent to which mitochondrial dysfunction in PBMCs parallels similar changes in other organ systems is not clear. We hypothesized that PBMCs could be used as a proxy to detect mitochondrial dysfunction in vital organs following hemorrhagic shock and early resuscitation.
Materials and Methods
Experimental Protocol
Experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and were performed in adherence to the National Institute of Health Guidelines on the Use of Laboratory Animals. Male Long-Evans rats (250–300 grams) were housed in a facility with a 12-hour light/dark cycle, constant temperature and humidity, and access to food and water ad libitum Animals were allowed to acclimate at least 2 days prior to surgery. The relationship between mitochondrial function in PBMC’s and other tissues was investigated using a validated decompensated hemorrhagic shock model (14) in which tissues were sampled at discrete physiologic time points during the development of hemorrhagic shock. Animals were randomized to one of four experimental groups: control (n=6), decompensated hemorrhagic shock (DHS) (n=6), severe hemorrhagic shock (SHS) (n=6) or resuscitation (R) (n=6). All animals were anesthetized using vaporized isofluorane (1–3.5%) and underwent placement of femoral arterial and venous catheters for blood pressure measurement, blood withdrawal, and resuscitation (PE50, Braintree Scientific, Inc.). In order to simulate a clinically relevant scenario in which soft tissue injury frequently accompanies hemorrhage, a laparotomy was performed while the animal was under anesthesia. Wounds were infiltrated with 1% lidocaine and surgically closed in layers. Additional analgesics were not provided. Animals were then placed in a specially designed rodent restraint device and allowed to emerge fully from anesthesia. Mean arterial pressure (MAP) and heart rate (HR) were continuously monitored and recorded throughout the experimental protocol (Digi-Med Signal Analyzers, Louisville, KY, USA). After full reversal of anesthesia and 30 minutes of hemodynamic stability, awake but restrained animals were passively bled via the femoral artery to a MAP of 40 mmHg until this pressure could no longer be maintained without fluid infusion (DHS group). A MAP of 40 mmHg was subsequently maintained by infusing 0.2 ml increments of Lactated Ringer’s (LR) until 40% of the shed blood volume had been returned (SHS group). Animals were subsequently resuscitated with 4 times the total shed blood volume in LR over 60 minutes (R group). Control animals underwent the same anesthetic and surgical procedures, but were not subjected to hemorrhagic shock or resuscitation. Immediately prior to sacrifice, arterial blood samples were taken and assayed for pH, pO2, PCO2, lactate, hemoglobin, glucose, alanine aminotransferase, aspartate aminotransferase, blood urea nitrogen (BUN), creatinine, and electrolytes (i-STAT, Abbot Point of Care Inc., Princeton, NJ). PBMC were also immediately isolated. Animals were euthanized at each of the time points (control, DHS, SHS, or R) and tissues (liver, kidney, and heart) were immediately harvested and processed for mitochondrial isolation.
Isolation of Peripheral Blood Mononuclear Cells
Peripheral blood mononuclear cells were isolated by density gradient centrifugation. Under sterile conditions, blood samples were diluted 1:1 using a balanced salt solution (BSS: 1 volume of anhydrous D-glucose 0.1 %, CaCl2 50 uM, MgCl2 0.98 mM, KCl 5.4 mM, TRIS 145 mM and 9 volumes of NaCl 140 mM). Diluted blood samples (2–4 mL) were layered on top of 3 mL Ficoll-Paque™ PREMIUM 1.084 (GE Healthcare, Pittsburgh, PA) and centrifuged at 400 × g for 40 minutes (18 – 20 °C). The white buffy coat was gently aspirated, mixed with 3 to 4 volumes of BSS, and recentrifuged at 400 × g for 15 minutes. The PBMC pellet was resuspended in 6–8 mL BSS and centrifuged at 400 × g for an additional 10 minutes. The PBMC pellet was resuspended in phosphate buffer solution (PBS) for flow cytometry or in Hanks’ balanced salt solution (pH 7.4) containing 5.5 mM glucose and 1 mM pyruvate for respirometry. Cell count and viability were performed using trypan blue exclusion (Countess, Life Technologies, Grand Island, NY) and ranged between 90 and 97%.
Isolation of Tissue Mitochondria
Liver-Kidney Mitochondrial Isolation
Samples of liver and kidney (4–6 g) were excised immediately after euthanasia at each time point. Samples were weighed and immersed in ice-cold mitochondrial isolation buffer (MIB) composed of 210 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mM EDTA with final pH adjusted to 7.2 using KOH and supplemented with 0.5% BSA (fatty acid-free). Tissue was minced and then homogenized with an additional 10 volumes (wt/vol) of MIB and 5% BSA using Potter Elvehjem homogenizer with a loose-fitting Teflon pestle. Mitochondrial isolation was performed using differential centrifugation (15). The homogenate was centrifuged for 10 minutes at 1000 × g (4°C). The supernatant was collected and recentrifuged for 10 minutes at 9,600 × g. The pellet was then resuspended in 15 ml MIB without BSA and centrifuged for 10 minutes at 9,600 × g for further mitochondrial purification. The final mitochondrial pellet was resuspended in MIB and protein concentration was determined spectrophotometrically using the Biuret method with BSA as standard (16).
Heart Mitochondrial Isolation
Heart tissue (4–6 g) was minced with scissors into small pieces and homogenized with 10 volumes (wt/vol) of Buffer A composed of 100 mM KCl, 50 mM Tris, 5 mM MgCl2, 1 mM EDTA (pH adjusted to 7.5 with HCl on the day of use after supplementing 1 mM ATP and 5% BSA) in a Potter Elvehjem homogenizer with a loose-fitting Teflon pestle. Liquid and tissue were transferred into a beaker and treated with trypsin for 2 minutes at 4°C with constant stirring followed by trypsin inhibitor (4mg/g trypsin and trypsin inhibitor, Sigma Aldrich, St. Louis, MO, USA). The tissue suspension was rehomogenized and then centrifuged for 10 minutes at 1000 × g (4°C) (low spin). The supernatant was again centrifuged for 10 minutes at 14000 × g (4°C). The pellet was resuspended in 10 mL of Buffer B (100 mM KCl, 50 mM Tris, 1 mM MgCl2, 0,2 mM EDTA; pH adjusted to 7.2 with HCl on the day of use) and kept on ice. The low spin pellet was resuspended in10 mL of Buffer A, homogenized and centrifuged for 10 minutes at 1000 × g. The supernatant was recentrifuged again for 10 minutes at 14,000 × g. The two high spin pellets were combined in Buffer B, and centrifuged at 9,600 × g for 10 minutes. The final mitochondrial pellet was suspended in 1 mL MIB. The protein concentration was determined spectrophotometrically with the Biuret method using BSA as standard (15,16).
Mitochondrial Membrane Potential
After isolation of PBMCs using the Ficoll gradient as described above, we evaluated mitochondrial function using flow cytometry. In order to normalize measurement of mitochondrial membrane potential to mitochondrial mass, two-color fluorophore experiments were performed by simultaneously incubating PBMCs (106 – 108 cells/mL) in the dark at 37 °C with MitoTracker Green (MTG) to assess mitochondrial mass (140 nM × 45 min) and tetramethylrhodamine (TMRE) to measure membrane potential (ΔΨm) (75 nM × 30 min) (Life Technologies, New York, NY). PBMCs were then washed twice with PBS. Positive and compensation controls were established for each dye. MTG was excited with a blue laser and TMRE was excited with a green laser using 515/20 and 585/42 filters respectively. PBMCs were identified using Forward (FSC) and Side (SSC) scatter. Only cells that stained positive for MTG, and thus viable, were considered. Data acquisition was performed using DiVa 6.1.2 software (BD Bioscience, San Jose, CA) and 100,000 events were recorded per tube. Data analysis was performed using FlowJo software (Tree Star, Inc, Ashland, Oregon). Data were expressed as a percentage of the cells stained positive for MTG.
PBMC Oxygen Consumption
Given the inability to isolate an adequate amount of mitochondria from PBMCs in this model, PBMC oxygen consumption was measured using high-resolution respirometry (Oxygraph-2k Oroboros Instruments, Innsbruck, Austria) (17). Freshly isolated non-permeabilized PBMC (106 cells/mL) were suspended in the 2 mL glass chamber (37 C°) in Hank’s Balanced Salt Solution (HBSS) containing 5.5 mM glucose, 1 mM pyruvate and 10 mM HEPES (pH 7.4). The oxygen solubility factor was set to 0.92 (relative to pure water) for the HBSS-glucose-pyruvate solution. After PBMC stabilization (3–5 minutes), real-time oxygen concentration and flux data were collected continuously (DatLab software 4.3, Oroboros Instruments, Innsbruck, Austria). After the basal respiration rate was recorded, oligomycin (0.25 μg/ml), a mitochondrial ATP synthase inhibitor, was added to measure respiration independent of ATP production (proton leak). The maximal capacity of the electron transport system (i.e. maximal respiratory rate) was assessed by titrating the protonophore, carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP, 0.75 μM increments) until no further increase in oxygen consumption was detected. The mitochondrial respiratory chain was then inhibited by adding rotenone (complex I inhibitor, 0.5 μM) and antimycin-A (complex III inhibitor, 2.5 μM) in order to assess non-mitochondrial respiration.
Liver-Kidney-Heart Mitochondrial Oxygen Consumption
A standard substrate/inhibitor titration protocol was used for functional analysis of mitochondrial respiratory chain complexes (17). Individual complex activity directly impacts overall mitochondrial function and directly correlates with basal and maximal respiration. Isolated mitochondria (1.5 mg) were resuspended in respiration medium which includes 110mM mannitol, 0.5mM EGTA, 3mM MgCl2, 20mM taurine 10mM KH2PO4, 60mM K lactobionate, 0.3mM DTT, and 0.1% BSA (fatty acid free), adjusted to pH of 7.1 with KOH. Oxygen consumption was measured using high-resolution respirometry (Oxygraph-2k Oroboros Instruments, Innsbruck, Austria).
Following stabilization (3–5 minutes), real-time oxygen concentration and flux data were collected continuously (DatLab software 4.3, Oroboros Instruments, Innsbruck, Austria). After the basal respiration rate was recorded, scomplex I (CI)-dependent mitochondrial respiration was induced by adding 10 mM glutamate, 5 mM malate and 1 mM ADP. In order to determine complex II (CII)-dependent respiration, rotenone (0.5 μM), a selective inhibitor of complex I, was added to the medium followed by 10 mM of succinate. Antimycin A (5 μM) was then added to inhibit complex III (CIII) in order to measure non-mitochondrial respiration; followed by addition of the artificial substrates for complex IV, TMPD (0.5 mM) and ascorbate (2 mM). In order to ensure that the respiratory capacity of complex IV (CIV) was not limited by cytochrome c depletion, respiration was measured after the addition cytochrome c (10 μM) (17). This protocol was completed within 60 to 70 min.
Data analysis
SPSS 15.0 software (Armonk, New York) was used for statistical analysis. Each experiment was performed as an independent assay and results are presented as mean (± standard deviation [SD]) unless otherwise stated. Animal groups were compared across the spectrum of hemorrhagic shock and resuscitation using one-way ANOVA and Pearson’s correlations (significance: p <0.05).
Results
Physiologic and Laboratory Parameters
Physiologic and laboratory parameters for animals in the control, DHS, SHS, and R groups are compared in Table 1. As expected, the mean arterial blood pressure (MAP) was maintained at 40 mmHg in the DHS and SH groups. Although MAP increased following 60 minutes of resuscitation (R groups), it remained lower than the control group (p<0.05) (Table 1). The DHS and SHS groups exhibited severe metabolic acidosis and increased BUN and creatinine levels, but normal liver function tests as measured by alanine aminotransferase and aspartate aminotransferase). With resuscitation (R group), acidosis and kidney dysfunction improved but remained abnormal compared to control animals. Resuscitation was also associated with a marked increase in liver enzymes (p<0.05). Although hyperkalemia and hyponatremia developed in the DHS and SHS groups, electrolytes normalized with resuscitation. As previously described, this model of hemorrhagic shock was associated with a significant decrease in hemoglobin in the DHS and SHS groups that further declined in the R group (18).
Table 1.
Physiologic and Laboratory Parameters
| Groups | ||||
|---|---|---|---|---|
| Control (n=6) | Decompensated Shock (n=6) | Severe Shock (n=6) | Resuscitation (n=6) | |
| MAP (mmHg) | 112 ± 5 | 39 ± 2 § | 41 ± 1 § | 78 ± 11 § |
| HR (bpm) | 447 ± 28 | 506 ± 26 § | 419 ± 19 | 438 ± 56 |
| pH | 7.39 ± 0.03 | 7.10 ± 0.19 | 7.05 ± 0.21 | 7.20 ± 0.11 § |
| Lactate (mmol/L) | 1.6 ± 0.9 | 16.0 ± 1.3 § | 17.2 ± 3.1 § | 10.2 ± 3.0 § |
| pCO2 (mmHg) | 50.9 ± 6.3 | 13.8 ± 5.3 § | 16.6 ± 4.8 § | 28.2 ± 3.4 § |
| pO2 (mmHg) | 84.1 ± 9.3 | 113.5 ± 16.5 § | 127.83 ± 9.4 § | 113.17 ± 13.0 § |
| HCO3− (mmol/L) | 30.8 ± 3.0 | 4.3 ± 1.5 § | 5.4 ± 3.8 § | 11.3 ± 2.8 § |
| Base Excess (mmol/L) | 7.2 ± 1.5 | −25.0 ± 3.7 § | −24.7 ± 6.3 § | −16.7 ± 4.6 § |
| O2Sat (%) | 96 ± 1 | 96 ± 2 | 97 ± 1 | 97 ± 1 |
| BUN (mg/dL) | 22.3 ± 3.1 | 27.2 ± 4.0 | 29.3 ± 3.8 * | 25.3 ± 3.2 |
| Creatinine (mg/dL) | 0.3 ± 0.1 | 0.5 ± 0.1 § | 0.6 ± 0.1 § | 0.5 ± 0.1 § |
| Alanine Aminotransferase (u/L) | 70.3 ± 11.4 | 54.7 ± 11.3 § | 61.2 ± 10.6 | 145.3 ± 47.1 § |
| Aspartate Aminotransferase (u/L) | 90.0 ± 44.0 | 66.7 ± 17.4 | 68.2 ± 21.8 | 241.3 ± 105.5 § |
| Na+ (mmol/L) | 135.2 ± 3.0 | 129.3 ± 2.2 | 128.2 ± 2.7 * | 130.3 ± 6.1 |
| K+ (mmol/L) | 4.4 ± 0.5 | 6.8 ± 0.9 § | 6.4 ± 1.4 | 5.0 ± 0.6 |
| Cl− (mmol/L) | 102.3 ± 1.8 | 101.8 ± 2.6 | 101.2 ± 1.9 | 100.3 ± 4.3 |
| Hb (g/dL) | 12.3 ± 1.0 | 6.5 ± 2.3 § | 5.0 ± 1.2 § | 3.8 ± 0.3 § |
Data was analyzed using ANOVA with * and § representing significance of p<0.05 and p<0.005 respectively compared to baseline values. Data is represented as mean ± SD.
PBMC Mitochondrial Dysfunction
PBMC mitochondrial membrane potential (ΔΨm) decreased sequentially from the control to R groups (Figure 1). The ΔΨm in the SHS group also trended lower than controls but did not reach statistical significance. The R group exhibited the lowest ΔΨm. As shown in Table 2, basal, maximal, and ATP-linked PBMC oxygen consumption were significantly decreased in the DHS group compared to controls (p<0.05 for all). All measures of PBMC oxygen consumption improved in the SHS group and were not significantly different from that of control animals. A substantial decline in PBMC oxygen consumption was noted in the R group; with lower basal, maximal, and ATP-linked PBMC oxygen consumption rates compared to both control and DHS animals. The pattern of change in PBMC ΔΨm paralleled the changes in PBMC oxygen consumption across the spectrum of hemorrhage severity from control to DHS, SHS, and R animals
Figure 1. PBMC Mitochondrial Oxygen Consumption and Membrane Potential During Hemorrhagic Shock and Resuscitation.
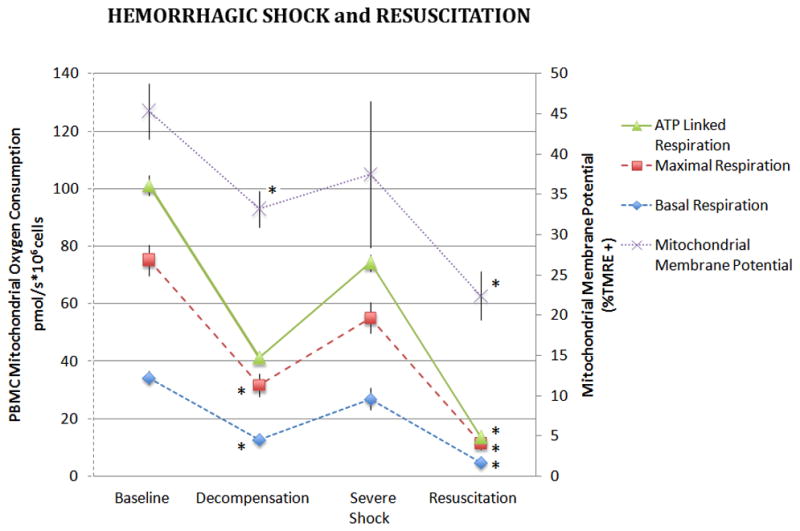
Basal, maximal and ATP-linked respiration rates were measured in freshly isolated PBMCs during hemorrhagic shock and following 60 minutes of resuscitation. Basal oxygen consumption of PBMC’s are also depicted. Data was analyzed using ANOVA with * representing significance of p<0.005 compared to baseline values. Data is represented as means ± SEM.
Table 2.
PBMC Oxygen Consumption.
| Oxygen consumption parameters (pmol O2/s × 106 PBMC) | Groups | |||
|---|---|---|---|---|
| Control (n = 6) | Decompensated Shock (n = 6) | Severe Shock (n = 6) | Resuscitation (n = 6) | |
| Basal Respiration | 34.3 ± 10.1 | 12.9 ± 3.9* | 26.9 ± 7.5 | 4.8 ± 2.2* |
| Maximal Respiration | 40.9 ± 10.9 | 18.9 ± 7.8* | 28.3 ± 10.9 | 6.7 ± 4.3* |
| ATP-linked Respiration | 25.9 ± 7.2 | 9.4 ± 4.4 | 16.7 ± 7.8 | 1.9 ± 3.0* |
Data was analyzed using ANOVA with * representing significance of p<0.005 compared to baseline values. Data is represented as means ± SEM.
Kidney, Liver and Heart Mitochondrial Dysfunction
Kidney mitochondrial CI, CII, and CIV-dependent oxygen consumption decreased significantly in the DHS group compared to controls and oxygen consumption remained significantly depressed in the SHS and R groups (p<0.005, Figure 2). The largest decrease in mitochondrial complex-dependent respiration was noted in CIV.
Figure 2. Kidney and PBMC Mitochondrial Oxygen Consumption During Hemorrhagic Shock and Resuscitation.
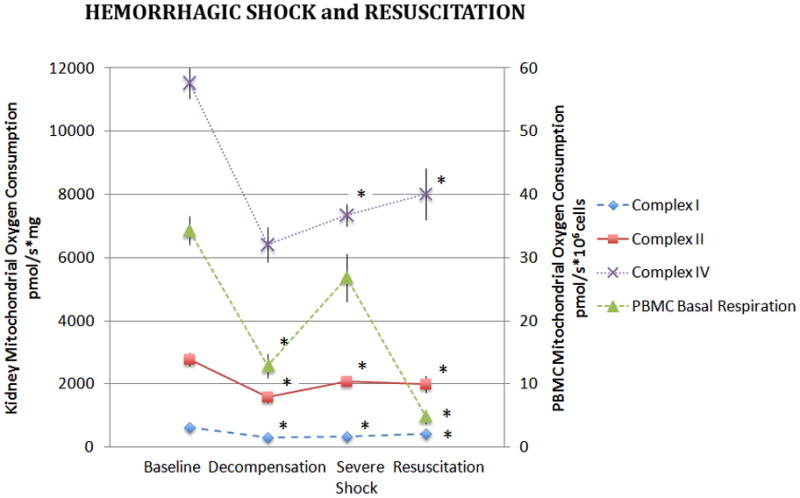
Isolated kidney mitochondria were assayed for complex I, II, and IV dependent oxygen consumption during hemorrhagic shock and following 60 minutes of resuscitation. Basal oxygen consumption of PBMC’s are also depicted. Data was analyzed using ANOVA with * representing significance of p<0.005 compared to baseline values. Data is represented as means ± SEM.
Unlike kidney mitochondria, complex-dependent oxygen consumption in liver mitochondria decreased less acutely during hemorrhagic shock. Although liver mitochondrial CI oxygen consumption decreased significantly in the DHS group, CII, and CIV-dependent oxygen consumption became statistically different from controls at the SHS time point (p<0.05, Figure 3). A subsequent increase in mitochondrial oxygen consumption was observed with resuscitation, although CI-dependent respiration remained significantly lower than the control group.
Figure 3. Liver and PBMC Mitochondrial Oxygen Consumption During Hemorrhagic Shock and Resuscitation.
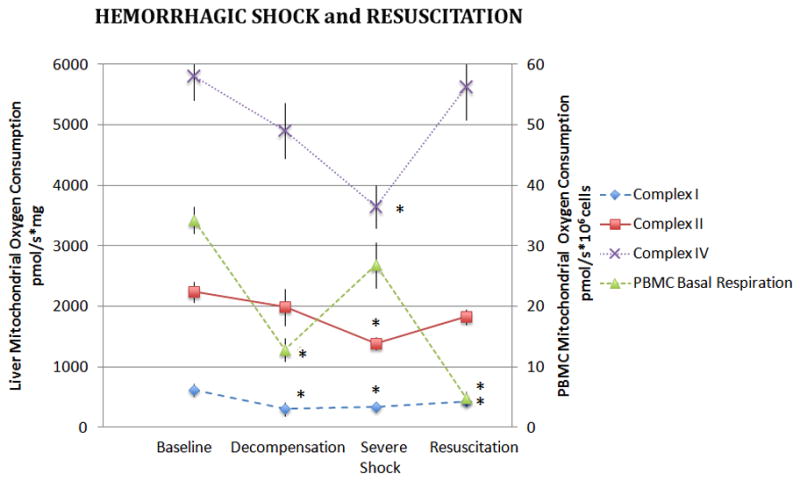
Isolated liver mitochondria were assayed for complex I, II, and IV dependent oxygen consumption during hemorrhagic shock and following 60 minutes of resuscitation. Basal oxygen consumption of PBMC’s are also depicted. Data was analyzed using ANOVA with * representing significance of p<0.05 compared to baseline values. Data is represented as means ± SEM.
In the heart, only CIV-dependent oxygen consumption decreased significantly during hemorrhagic shock (p<0.05, Figure 4). However, unlike both kidney and liver mitochondria, in the heart oxygen consumption through all three complexes was significantly depressed following resuscitation.
Figure 4. Heart and PBMC mitochondrial oxygen consumption During Hemorrhagic Shock and Resuscitation.
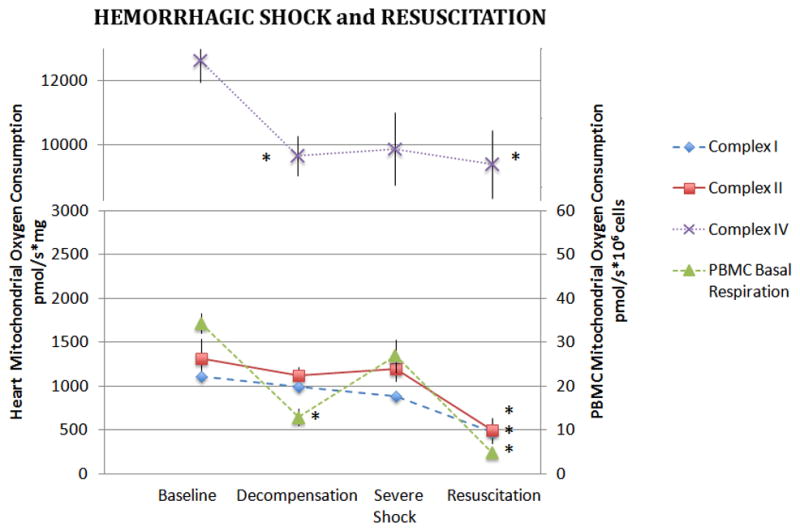
Isolated heart mitochondria were assayed for complex I, II, and IV dependent oxygen consumption during hemorrhagic shock and following 60 minutes of resuscitation. Basal oxygen consumption of PBMC’s are also depicted. Data was analyzed using ANOVA with * representing significance of p<0.05 compared to baseline values. Data is represented as means ± SEM.
Relationship Between PBMC and Tissue Mitochondrial Function
Of the tissues investigated, kidney mitochondrial complex dependent respiration (I, II and IV) significantly correlated with both PBMC basal and maximal respirations during hemorrhagic shock (Figure 5 and 6). At the point of decompensation, kidney mitochondrial complex I, II, and IV significantly correlated with both PBMC basal respiration (r=0.795, 0.841, and 0.669, p<0.05 respectively) and PBMC maximal respiration (r=0.726, r=0.688, and 0.658, p<0.05 respectively). As the severity of shock increased, the correlation between kidney and PBMC mitochondrial function persisted. With severe shock, kidney mitochondrial complex I, II, and IV significantly correlated with both PBMC basal respiration (r=0.653, 0.655, and 0.523 p<0.05 respectively) and PBMC maximal respiration (r=0.621, r=0.456, and 0.512, p<0.05 respectively). Additionally, kidney complex II and IV dependent respiration were noted to correlate with PBMC ΔΨm during acute blood loss (r=0.631; 0.549 respectively, p<0.05). However, with resuscitation, the correlation between kidney and PBMC mitochondrial function disappeared.
Figure 5. Correlation Between Kidney Mitochondrial Complex Dependent Respiration and PBMC Mitochondrial Basal Oxygen Consumption.

Kidney complex I, II and IV dependent respiration rates in freshly isolated PBMCs. Basal oxygen consumptions was measured during hemorrhagic shock and following 60 minutes of resuscitation. Correlations between were analyzed using Pearson’s correlations test (significance: p <0.05).
Figure 6. Correlation Between Kidney Mitochondrial Complex Dependent Respiration and PBMC Mitochondrial Maximal Oxygen Consumption.

Kidney complex I, II and IV dependent respiration rates in freshly isolated PBMCs. Maximal oxygen consumption was measured during hemorrhagic shock and following 60 minutes of resuscitation. Correlations between were analyzed using Pearson’s correlations test (significance: p <0.05).
We also observed a weaker relationship between heart mitochondrial complex II O2 consumption and PBMC Basal and Maximal Respirations, but only following resuscitation (r=0.451 and r=0.436, p<0.05 respectively, Figure 7). Interestingly, heart mitochondrial complex IV dependent O2 consumption was correlated with PBMC ΔΨm following decompensation (r=0.858, p<0.005), severe hemorrhagic shock (r=0.667, p<0.01) and following resuscitation (r=0.509, p<0.05).
Figure 7. Correlation Between Heart Mitochondrial Complex Dependent Respiration, and PBMC Mitochondrial Oxygen Consumption.

Heart Complex II and IV dependent respiration rates were correlated with freshly isolated PBMCs basal and maximal oxygen consumption rates. Correlations between were analyzed using Pearson’s correlations test (significance: p <0.05).
No significant correlation between liver mitochondrial function (I, II and IV) and PBMC mitochondrial respiration was observed during either hemorrhagic shock or with resuscitation.
Discussion
Multiple organ failure remains a significant cause of in-hospital morbidity and mortality following traumatic injury (2,19). Although the underlying cellular events leading to MOF are incompletely understood, impaired mitochondrial function and the depletion of cellular energy stores appear to play a central role (3–5). In this study, we investigated the potential utility of using PBMCs as a proxy for measuring mitochondrial function in more vital organs such as liver, kidney and heart during hemorrhagic shock and resuscitation. While significant mitochondrial dysfunction was noted during acute blood loss and resuscitation, the degree of impairment varied significantly between tissues. Moreover, while PBMC mitochondrial membrane potential and function appeared to correlate well with kidney mitochondrial function during hemorrhagic shock, this association dissipated with resuscitation. Using PBMC’s as a proxy for tissue-specific mitochondrial dysfunction, therefore, is limited because the mitochondrial response between PBMCs and different vital organs is not linear following acute blood loss and resuscitation.
Although mitochondrial dysfunction following hemorrhagic shock has been demonstrated in a number of experimental and clinical studies, current resuscitation strategies do not target mitochondrial performance (20). Under normal physiologic conditions, mitochondria provide almost 95% of the body’s energy needs via oxidative phosphorylation; and oxygen utilization by the mitochondrial electron transport system is tightly coupled to oxygen availability (21). With severe hemorrhagic shock and reperfusion, however, alterations in the mitochondrial electron transport complexes can lead to decreased electron flow, decreased ATP production and increased generation of damaging free radical oxygen species (20,22). With prolonged hemorrhagic shock, there is a progression from decreased oxidative phosphorylation to frank and irreversible, structural damage to the mitochondria (23,24). Moreover, organ failure from hemorrhagic shock appears to coincide with the development of bioenergetic failure and mitochondrial dysfunction (4). Recognizing, and treating, mitochondrial dysfunction before the damage becomes irreversible could potentially mitigate the development of cellular dysfunction and decrease the incidence of MOF.
Previous investigators have used near-infrared spectroscopy (NIRS) as a non-invasive means of assessing tissue mitochondrial function during resuscitation. Not only can NIRS measure the adequacy of tissue oxygen delivery, it can also be used to assess mitochondrial oxygen consumption and redox status (25). In an animal model of severe hemorrhagic shock, Rhee et al noted that despite adequate resuscitation, as measured by the normalization of blood pressure, cardiac output and tissue oxygen delivery, mitochondrial cytochrome a,a3 oxidation remained significantly decreased in splanchnic and skeletal muscle beds (26). In a similar experiment, decoupling of the hepatic cytochrome a,a3 redox state from oxygen delivery was associated with increased early mortality following hemorrhagic shock (27). In a prospective study of 24 severely injured patients monitored continuously with NIRS, Cairns et al noted that patients who developed MOF demonstrated a significantly higher incidence of mitochondrial decoupling early in the course of their resuscitation when compared to those who did not develop MOF (89% vs 13%, p<0.05) (3). Moreover, severely injured trauma patients with impaired hepatic mitochondrial redox status were more likely to develop liver dysfunction despite adequate resuscitation (28). Although promising, NIRS is technically challenging and not clinically practical. In addition to the potential difficulty in interpreting spectral overlap between hemoglobin, myoglobin, and mitochondrial cytochromes, significant artifact from the variability in depth and composition of subcutaneous tissues among patients as well as increasing tissue edema following trauma may make accurate measurements challenging (3,10).
Recently, blood samples have been used to assess mitochondrial function in variety of pathologic states (29–31). In particular, PBMC respiratory function may reflect disease severity in sepsis (12) and we have noted that PBMC mitochondrial function is dramatically altered during hemorrhagic shock and resuscitation (13). As such, monitoring PBMC respiratory capacity may provide a minimally invasive surrogate for end-organ mitochondrial function. Although we noted a clear correlation between PBMC respiratory capacity and kidney mitochondrial function (complexes I, II, and IV) during hemorrhagic shock in our current experiments, this association was not universally observed in other tissues and unfortunately dissipated with resuscitation. Given the lack of linear response throughout the spectrum of shock and resuscitation, as well as the variability in mitochondrial response between different tissues, we must conclude that PBMCs cannot be used clinically as a proxy for tissue mitochondrial function during the acute resuscitation of hemorrhagic shock.
The differences in mitochondrial function observed in our study, however, likely reflects tissue-specific variability in oxidative capacity and the ability to respond to ischemia-reperfusion. Although the majority of mammalian cells contain mitochondria, there is striking diversity across tissue types in terms of mitochondrial protein expression, function, and capacity for oxidative phosphorylation (32). As such, it is not surprising that the different organs in our study demonstrated a variable response to hemorrhagic shock and resuscitation. What is somewhat intriguing, however, is the significant decrease in PBMC and cardiac mitochondrial function observed following resuscitation. In contrast to both liver and kidney, PBMC and cardiac mitochondrial function declined dramatically with reperfusion. While direct reperfusion injury with the generation of toxic reactive oxygen species and peroxynitrite may account for the acute changes seen in cardiac mitochondrial function (32,33), PBMC’s in our model were not subjected to hypoxia and the serum pO2 did not decline during hemorrhagic shock or resuscitation. Although it is unclear how resuscitation directly impairs PBMC mitochondrial function, the release of nitric oxide and IL-6 into the circulation upon reperfusion could potentially inhibit mitochondrial function directly (34,35). Further research is needed to determine if the observed decrease in PBMC respiratory capacity persists or predicts the later development of MOF.
While this study is the first to explore using PBMC’s as a proxy for mitochondrial function in more vital tissues, there are several limitations. Most importantly, our controlled model of decompensated shock does not recapitulate the typical clinical scenario where patients are frequently bleeding and resuscitated simultaneously. That being said, if we were not able to appreciate a linear relationship between PBMC respiratory function and tissue mitochondrial function during controlled blood loss and resuscitation, it is unlikely a strong association would emerge under less structured circumstances. Secondly, we investigated the effects of hemorrhagic shock and resuscitation on mitochondrial function over a short duration. While isolated measurements of PBMC respiratory capacity may not reflect acute changes in tissue mitochondrial function following hemorrhagic shock, impaired PBMC mitochondrial function does appear to correlate with disease severity in other disease states such as sepsis (12). Longer term studies are need to determine if PBMC mitochondrial function following resuscitation can also be used to predict the development of MOF. Finally, we investigated mitochondrial function in PBMCs as a whole. It is likely that the composition of PBMCs (i.e. monocytes, T cells, NK cells, etc.) may shift during hemorrhagic shock and resuscitation. As such, because different cell types may have different mitochondrial function, the shift in subgroup populations may impact the results.
Conclusion
Hemorrhagic shock and resuscitation are associated with significant mitochondrial dysfunction in a variety of tissues including PBMCs. Although PBMCs represent an easily assessable tissue that could be readily and repeatedly measured during the course of resuscitation, several issues limit the usefulness of PBMCs as a surrogate for organ mitochondrial function. In particular, the variability in organ-specific mitochondrial response to acute blood loss and resuscitation limit the generalizability. In addition, the lack of linear correlation over the spectrum of shock and resuscitation limits the usefulness of using PBMC’s as a proxy for tissue-specific mitochondrial function in the acute setting.
Acknowledgments
The present study was supported, in part, by grant 1K08GM097614-01 from the National Institute of Health.
Footnotes
This paper will be presented in the 36th Annual Conference on Shock as one of the finalist of the New Investigator Awards Competition
The presented authors declare no conflicts of interest.
Contributor Information
Mehmet Akif Karamercan, Email: makaramercan@gazi.edu.tr.
Scott L. Weiss, Email: weisss@email.chop.edu.
Jose Paul Perales Villarroel, Email: jose.peralesvillarroel@uphs.upenn.edu.
Yuxia Guan, Email: Yuxia.Guan@uphs.upenn.edu.
Evan Werlin, Email: evan.werlin@gmail.com.
Ronald Figueredo, Email: Ronald.Figueredo@uphs.upenn.edu.
Lance B. Becker, Email: Lance.Becker@uphs.upenn.edu.
Carrie Sims, Email: Carrie.Sims@uphs.upenn.edu.
References
- 1.Pfeifer R, Tarkin IS, Rocos B, Pape HC. Patterns of mortality and causes of death in polytrauma patients--has anything changed? Injury. 2009;40(9):907–911. doi: 10.1016/j.injury.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Minei JP, Cuschieri J, Sperry J, Moore EE, West MA, Harbrecht BG, O’Keefe GE, Cohen MJ, Moldawer LL, Tompkins RG, Maier RV. The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Crit Care Med. 2012;40(4):1129–1135. doi: 10.1097/CCM.0b013e3182376e9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cairns CB, Moore FA, Haenel JB, Gallea BL, Ortner JP, Rose SJ, Moore EE. Evidence for early supply independent mitochondrial dysfunction in patients developing multiple organ failure after trauma. J Trauma. 1997;42:532–536. doi: 10.1097/00005373-199703000-00023. [DOI] [PubMed] [Google Scholar]
- 4.Cairns CB. Rude unhinging of the machinery of life: metabolic approaches to hemorrhagic shock. Curr Opin Crit Care. 2001;7(6):437–443. doi: 10.1097/00075198-200112000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Kozlov AV, Bahrami S, Calzia E, Dungel P, Gille L, Kuznetsov AV, Troppmair J. Mitochondrial dysfunction and biogenesis: do ICU patients die from mitochondrial failure? Ann Intensive Care. 2011;41:1–13. doi: 10.1186/2110-5820-1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tobin MJ. Critical care medicine in AJRCCM 2001. Am J Respir Crit Care Med. 2002;165(5):565–583. doi: 10.1164/ajrccm.165.5.2201060. [DOI] [PubMed] [Google Scholar]
- 7.Moore FA, Haenel JB, Moore EE, Whitehill TA. Incommensurate oxygen consumption in response to maximal oxygen availability predicts postinjury multiple organ failure. J Trauma. 1992;33(1):58–65. doi: 10.1097/00005373-199207000-00012. [DOI] [PubMed] [Google Scholar]
- 8.Hayes MA, Timmins AC, Yau EH, Palazzo M, Hinds CJ, Watson D. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330(24):1717–1722. doi: 10.1056/NEJM199406163302404. [DOI] [PubMed] [Google Scholar]
- 9.Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg. 2003;185(5):485–491. doi: 10.1016/s0002-9610(03)00044-8. [DOI] [PubMed] [Google Scholar]
- 10.Cooper CE, Springett R. Philos. Measurement of cytochrome oxidase and mitochondrial energetics by near-infrared spectroscopy. Trans R Soc Lond B Biol Sci. 1997;352(1354):669–676. doi: 10.1098/rstb.1997.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Japiassú AM, Santiago AP, d’Avila JC, Garcia-Souza LF, Galina A, Castro Faria-Neto HC, Bozza FA, Oliveira MF. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5′-triphosphate synthase activity. Crit Care Med. 2011;39(5):1056–1063. doi: 10.1097/CCM.0b013e31820eda5c. [DOI] [PubMed] [Google Scholar]
- 12.Belikova I, Lukaszewicz AC, Faivre V, Damoisel C, Singer M, Payen D. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit Care Med. 2007;35(12):2702–2708. doi: 10.1097/01.ccm.0000295593.25106.c4. [DOI] [PubMed] [Google Scholar]
- 13.Villarroeal JPP, Guan Y, Werlin E, et al. Hemorrhagic shock and resuscitation are associated with peripheral mononuclear cell mitochondrial dysfunction and immunosuppression. Journal of Trauma and Acute Care Surgery. doi: 10.1097/TA.0b013e3182988b1f. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayala A, Wang P, Chaudry IH. Shock Models: Hemorrhage. In: Souba W, Wilmore D, editors. Surgical Research. San Diego, CA: Academic Press; 2001. pp. 325–327. [Google Scholar]
- 15.Pesta D, Gnaiger E. High-resolution respirometry. OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. In: Palmeria C, Moreno A, editors. Mitochondrial bioenergetics: methods and protocols. New York, NY: Humana Press; 2012. pp. 25–58. [DOI] [PubMed] [Google Scholar]
- 16.Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- 17.Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3(6):965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 18.Ba ZF, Wang P, Koo DJ, Cioffi WG, Bland KI, Chaudry IH. Alterations in tissue oxygen consumption and extraction after trauma and hemorrhagic shock. Crit Care Med. 2000;28:2837–2842. doi: 10.1097/00003246-200008000-00026. [DOI] [PubMed] [Google Scholar]
- 19.Ciesla DJ, Moore EE, Johnson JL, Burch JM, Cothren CC, Sauaia A. A 12-year prospective study of postinjury multiple organ failure: has anything changed? Arch Surg. 2005;140:432–438. doi: 10.1001/archsurg.140.5.432. [DOI] [PubMed] [Google Scholar]
- 20.Hubbard WJ, Bland KI, Chaudry IH. The role of the mitochondrion in trauma and shock. Shock. 2004;22:395–402. doi: 10.1097/01.shk.0000143407.90473.cc. [DOI] [PubMed] [Google Scholar]
- 21.Williamson JR. Mitochondrial metabolism and cell regulation. In: Packer L, Gomez-Poyon A, editors. Mitochondria: Bioenergetics, Biogenesis and Membrane Structure. New York: Academic; 1976. pp. 79–107. [Google Scholar]
- 22.Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radical Biol Med. 1999;26:463–471. doi: 10.1016/s0891-5849(98)00216-0. [DOI] [PubMed] [Google Scholar]
- 23.Taylor JH, Beilman GJ, Conroy MJ, Mulier KE, Myers D, Gruessner A, Hammer BE. Tissue energetics as measured by nuclear magnetic resonance spectroscopy during hemorrhagic shock. Shock. 2004;21:58–64. doi: 10.1097/01.shk.0000101674.49265.93. [DOI] [PubMed] [Google Scholar]
- 24.Shimahara Y, Ozawa K, Ida T, Ukikusa M, Tobe T. Four stages of mitochondrial deterioration in hemorrhagic shock. Res Exp Med. 1981;179:23–33. doi: 10.1007/BF01852122. [DOI] [PubMed] [Google Scholar]
- 25.Ryan TE, Erikson Ml, Brizendine JT, Young HJ, McCully KK. Noninvasive evaluation of skeletal muscle mitochondrial capacity with near-infrared spectroscopy: correcting for blood volume changes. J Appl Phys. 2012;113:175–183. doi: 10.1152/japplphysiol.00319.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhee P, Langdale L, Charles M, Gentilello LM. Near-infrared spectroscopy: continuous measurement of cytochrome oxidation during hemorrhagic shock. Crit Care Med. 1997;25:166–170. doi: 10.1097/00003246-199701000-00030. [DOI] [PubMed] [Google Scholar]
- 27.Ketcham EM, Bell G, Petterson J, Williams BT, Veeramacheni N, Walther JM, Hamiel C, Carins CB. Decoupling of hepatic oxidative metabolism after hemorrhagic shock presages short-term mortality. Acad Emerg Med. 1998;5:454. [Google Scholar]
- 28.Nakatani T, Endoh Y, Kobayashi K. Significance of the hepatic mitochondrial redox state in the development of posttraumatic jaundice. Surgery Today. 1995;25:490–497. doi: 10.1007/BF00311304. [DOI] [PubMed] [Google Scholar]
- 29.Ijsselmuiden AJ, Musters RJ, de Ruiter G, van Heerebeek L, Alderse-Baas F, van Schilfgaarde M, Leyte A, Tangelder GJ, Laarman GJ, Paulus WJ. Circulating white blood cells and platelets amplify oxidative stress in heart failure. Nature Clin Practice Cardiovasc Med. 2008;5:811–820. doi: 10.1038/ncpcardio1364. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Ramirez M, Francisco G, Garcia-Arumi E, Hernandez C, Martinez R, Andreu AL, Simo R. Mitochondrial DNA oxidation and manganese superoxide dismutase activity in peripheral blood mononuclear cells from type 2 diabetic patients. Diabetes Metab. 2008;34:117–124. doi: 10.1016/j.diabet.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Kong CH, Huang CH, Hsu TG, Tsai KKC, Huse CF, Huang MC, Chen LC. Leukocyte mitochondrial alterations after cardiac surgery involving cardiopulmonary bypass: clinical correlations. Shock. 2004;21:315–319. doi: 10.1097/00024382-200404000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics. 2006;5:608–619. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Kaminski KA, Bonda TA, Korecki J, Musial WJ. Oxidative stress and neutrophil activation--the two keystones of ischemia/reperfusion injury. Int J Cardiol. 2002;86(1):41–59. doi: 10.1016/s0167-5273(02)00189-4. [DOI] [PubMed] [Google Scholar]
- 34.Torres A, Bentley T, Bartels J, Sarkar J, Barclay D, Namas R, Constantine G, Zamora R, Puyana JC, Vodovotz Y. Mathematical modeling of posthemorrhage inflammation in mice: studies using a novel, computer-controlled, closed-loop hemorrhage apparatus. Shock. 2009;32(2):172–178. doi: 10.1097/SHK.0b013e318193cc2b. [DOI] [PubMed] [Google Scholar]
- 35.Raffaella T, Fiore F, Fabrizia M, Francesco P, Arcangela I, Salvatore S, Luigi S, Nicola B. Induction of mitochondrial dysfunction and oxidative stress in human fibroblast cultures exposed to serum from septic patients. Life Sci. 2012;91(7–8):237–243. doi: 10.1016/j.lfs.2012.06.041. [DOI] [PubMed] [Google Scholar]