

Abstract
Nucleotide-binding oligomerization domain (NOD)-1 and NOD2 are members of the NOD-like receptor family of cytosolic pattern recognition receptors that recognize specific fragments of the bacterial cell wall component peptidoglycan. Neisseria species are unique amongst Gram-negative bacteria in that they turn over large amounts of peptidoglycan during growth. In this study we examined the ability of NOD1 and NOD2 to recognize N. gonorrhoeae, and determined the role of NOD-dependent signaling in regulating the immune response to gonococcal infection. We found that gonococci, as well as conditioned medium from mid-logarithmic phase grown bacteria, were capable of activating both human NOD1 and NOD2, as well as mouse NOD2, leading to the activation of the transcription factor NF-κB and polyubiquitination of the adaptor receptor-interacting serine-threonine kinase 2 (RIPK2). We identified a number of cytokines and chemokines that were differentially expressed in wild type vs. NOD2 deficient macrophages in response to gonococcal infection. Moreover, NOD2 signaling upregulated complement pathway components and cytosolic nucleic acid sensors, suggesting a broad impact of NOD activation on innate immunity. These data demonstrate that NOD1 and NOD2 are important intracellular regulators of the immune response to infection with N. gonorrhoeae. Given the intracellular lifestyle of this pathogen, we believe these cytosolic receptors may provide a key innate immune defense mechanism for the host during gonococcal infection.
Keywords: innate immunity, NOD-like receptors, Neisseria gonorrhoeae
Introduction
Neisseria gonorrhoeae (gonococcus, GC) is the second most commonly reported notifiable disease in the United States 1. Transmission is almost exclusively by close sexual contact, and can occur at any mucosal surface, including the urethra, cervix, rectum and pharynx. In addition to localized disease, certain strains of gonorrhea are capable of dissemination, resulting in bacteremia, septic arthritis, and dermatitis (disseminated gonococcal infection or DGI). Infection with N. gonorrhoeae has also been associated with the development of pelvic inflammatory disease (PID) in women, which can lead to chronic pelvic pain, tubal infertility, and ectopic pregnancy 2. Finally, concomitant gonococcal infection has been shown to increase the risk of HIV transmission both in vivo 3, 4 and in vitro 5-7.
The current model for gonococcal pathogenesis suggests that N. gonorrhoeae colonizes and transmigrates across the columnar epithelium of a mucosal surface, resulting in a localized inflammatory response consisting of polymorphonuclear cells and other professional immune cells. There are a number of virulence factors in the gonococcus that contribute to attachment and invasion of epithelial cells, such as the type IV pili, opa proteins, lipooligosaccharide (LOS), porin, Lip/H.8 lipopeptide, and peptidoglycan [reviewed in 8]. Many of these ligands also activate the innate immune system via pattern recognition receptors. For example, Neisseria LOS is a strong activator of Toll-like receptor (TLR)-4, which has been shown to play a key role in the induction of inflammatory pathways both in vitro 9-11 and in vivo (12), while porin and the H.8/Lip lipopeptide have been shown to be ligands for TLR2 in vitro 13, 14. The role of specific TLRs in response to gonococci may be influenced by the cell type as phagocytic cells are exquisitely sensitive to endotoxin, sensed via TLR4, while cervical epithelial cells appear to respond to gonorrhea via TLR215. In addition to these cell surface receptors, gonococci have also been shown to activate the cytosolic NLRP3/ASC inflammasome pathway 16. Additional innate immune receptors are likely encountered during gonococcal infection.
Nucleotide-binding oligomerization domain (NOD)-1 and NOD2 are cytosolic receptors that belong to the NOD subfamily of the NOD-like receptor family (NLRs)17. They detect peptidoglycan fragments derived from bacteria, triggering a number of cellular signaling events. The receptor-interacting serine-threonine kinase 2 (RIPK2; also known as RICK or RIP2) plays a central role in signaling for both NOD1 and NOD2 18. This signaling cascade eventually results in NF-κB and MAPK activation, and subsequent proinflammatory cytokine, chemokine or antimicrobial peptide production, depending on the cell type 19-21. In cells expressing both TLRs and NOD1/NOD2 receptors, such as macrophages, there is a synergistic effect in cytokine production when both TLRs and NOD1/NOD2 are activated 22, 23. NOD1 and NOD2 have also been shown to activate the autophagic process in cells infected by invasive bacteria, such as S. flexneri and L. monocytogenes or cells exposed to purified NOD ligands24.
NOD1 and NOD2 are activated by a number of different Gram-positive and Gram-negative, intracellular and extracellular bacteria. The smallest peptidoglycan fragment activating NOD1 is the bacterial dipeptide diaminopimelic acid (iEDAP) 25. These dipeptides, as well as other NOD1 activating fragments, are generally derived from Gram-negative bacteria. The smallest fragment activating NOD2 is the muropeptide muramyldipeptide (MDP) 26, a derivative of both Gram-negative and Gram-positive peptidoglycan. Absence of NOD1 or NOD2 has been shown to result in adverse outcomes in a number of infectious disease models, such as infections caused by H. pylori or L. monocytogenes 19. Furthermore, hypomorphic mutations of NOD2 have been implicated in the pathogenesis of Crohn's disease 27.
The turnover of gonococcal peptidoglycan is an active and rapid process, with a large percentage of the peptidoglycan being shed into the surrounding environment during growth. This occurs mainly in the form of peptidoglycan fragments, primarily monomers, released by the activity of lytic transglycosylases 28-30. Additional peptidoglycan fragments can be formed through the action of autolysins released by the gonococci during autolysis or through the action of eukaryotic peptidoglycan lysing enzymes 31. It has been shown that NOD1 and NOD2, as well as the adaptor protein RIPK2, are functionally expressed in the female genital tract 32, and the observation that peptidoglycan fragments derived from gonococci are cytotoxic in an ex vivo human fallopian tube model suggests that they might play a role in gonococcal pathogenesis 33.
In this study we examined the ability of gonococci to activate NOD1 and NOD2 in vitro using cells that are relevant during natural infection, such as epithelial cells and macrophages. We investigated the mechanism of NOD activation and the specific down-stream signaling pathways triggered following activation of the NOD pathways. We observed NOD-dependent activation of a number of inflammatory signaling pathways in response to N. gonorrhoeae and conclude that activation of the NOD1/NOD2 pathway by gonococci modulates the host innate immune response to infection.
Materials and Methods
Ethics statement
All human samples used during this study were obtained following written informed consent from healthy donors with approval from the Boston University Institutional Review Board (IRB). The animals described in this study were housed in the AAALAC accredited facilities of Boston University Medical Center, and all animal use protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Boston University, in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Reagents
RPMI1640 was from BioWhittaker® (Lonza Walkersville, MD USA). Fetal bovine serum (FBS; low endotoxin) was from Hyclone (Logan, UT). Ultrapure LPS purified from E. coli serotype O111:B4 was purchased from List Pharmaceuticals (Woburn, MA). MPD (MurNAc-L-Ala-D-isoGln) and iEDAP (D-γ-Glu-mDAP) were purchased from InvivoGen. The synthetic lipopeptide Pam3-Cys-Ser-Lys4 (P3CSK4) was purchased from EMC Microcollections (Tuebingen, Germany). The RIPK2 inhibitor, SB 203580 was from Promega (Madison, WI). The NOD1 inhibitor ML130 (1-(4-methylphenyl)sulfonylbenzimidazol-2-amine) was from Asinex ( Catalog # BAS 07162070). Bafilomycin A1 was from Enzo Life Sciences. Cytochalasin D was from Sigma-Aldrich (St. Louis, MO). Mouse TNF-α ELISA kit was from eBioscience, Inc. (San Diego, CA). All other ELISA kits were obtained from R&D Systems (Minneapolis, MN).
Cell culture and transfection
HeLa cells and HEK 293 cells (ATCC, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (DMEM) (BioWhittaker, Walkersville, MD) containing 10% fetal bovine serum (HyClone, Logan UT) and 20 μg/ml gentamicin. HEK 293 cells were plated at 3×105 cells/well in 6 well plates and were transiently transfected with the use of GeneJuice transfection reagent (Novagen) with plasmids expressing the human NOD1 gene (pUNO-hNOD1), the human NOD2a gene (pUNO-hNOD2a), or a control plasmid (Invivogen, San Diego, CA). For some experiments the plasmids expressing the murine NOD1 gene (pUNO-mNOD1) or the murine NOD2a gene (pUNO-mNOD2a) were used instead (also from Invivogen). Cells were also transfected with the following luciferase plasmids: a 5x-NF-κB luciferase reporter plasmid that reports firefly luciferase in an NF-κB dependent manner; and the pRL Renilla luciferase (Rluc) control reporter vector (Promega), which provides constitutive expression of Renilla luciferase. After resting for 24 hrs, cells were plated in 96 well plates (at 2×104 cells/well) and stimulated for another 24 hrs. Lysates were prepared using the Reporter Lysis Buffer (Promega) and luciferase activity was assessed (Luciferase Assay System, Promega). For each condition, luciferase activity was normalized for Renilla luciferase activity and presented as fold increase over the baseline luciferase activity in HEK 293 cells transfected with an empty vector.
Bacterial preparation
N. gonorrhoeae strain MS11 was kindly provided by Dr. J.P. Dillard (University of Wisconsin, Madison). N. gonorrhoeae was grown with aeration in GC liquid medium supplemented with IsoVitaleX Enrichment (BD) or in Wade-Graver medium 34 to mid-logarithmic phase. Bacterial supernatant from mid-logarithmic phase growth broth culture (O.D.600=0.6 or 2×108 CFU bacteria/ml) was prepared by centrifugation of the bacteria to obtain cell-free supernatant. Supernatant was stored at −80C and used at the indicated dilutions. Bacterial lysates were prepared from the bacterial pellet by repeat freeze-thaw cycles to −80°C of the bacterial suspensions15. For infection of macrophages or HeLa cells with live bacteria, N. gonorrhoeae was grown overnight in chocolate agar plates in a 37°C incubation with 5% CO2.
Human macrophage preparation
Human macrophages were prepared from human peripheral blood mononuclear cells (PBMCs). After obtaining informed consent, PBMCs were obtained from healthy volunteers by differential centrifugation of heparinized whole blood using a Ficoll/Histopaque (Sigma-Aldrich) gradient. Macrophages were separated from blood cells by an initial adherence to plastic culture flasks overnight. After removing nonadherent cells, monocytes were cultured for 5–7 days in RPMI containing 10% fetal bovine serum and 20 μg/ml gentamicin to mature into monocyte-derived macrophages. All studies involving human cells were carried out with the approval of the Institutional Review Board at Boston University Medical Center.
Murine macrophage preparation
Bone marrow derived macrophages (BMDM) were prepared from C57BL/6 mice and NOD2 deficient mice backbred 10 generations onto the same background. Bone marrow cells were cultured in RPMI 1640 supplemented with 20% L929 condition medium and were allowed to differentiate for 7-9 days before use. Immortalized macrophage cell lines from C57BL/6 (wildtype), nod1–/– and nod2–/– mice were kindly provided by Dr. Douglas Golenbock (University of Massachusetts Medical School, Worcester, MA) and have been previously described 35. All mice were backbred onto the C57BL/6 background at least 8-10 generations. Immortalized macrophages were maintained in RPMI containing 10% fetal bovine serum and gentamicin at a concentration of 20 μg/ml. Antibiotics were removed for 24 hours prior to inoculation with live bacteria. All studies involving animals were carried out with the approval of the IACUC at Boston University Medical Center.
Immunoprecipation and Western blot
Differentiated mouse BMDM, immortalized macrophages or human macrophages were plated at a density of 1×107 cells per dish in 10 mm cell culture dishes. HeLa cells were plated at a density of 5×106 cells per dish. Cells were subsequently infected with N. gonorrhoeae at different MOIs or incubated with gonococcal lysates or supernatants for 2 hrs (macrophages) or 3-7 hrs (HeLa cells). Subsequently, lysates were prepared in RIPA buffer, supplemented with a protease mixture inhibitor (Roche Applied Science, Indianapolis, IN) and 5 mM N-ethylmaleimide (Sigma), and immunoprecipitated with anti-RIPK2 antibody (Santa Cruz Biotechnology, Dallas, TX). Polyubiquitinated RIPK2 was detected by immunoblotting with an anti-ubiquitin antibody (Santa Cruz Biotechnology)36.
Microarray
Murine BMDM from C57BL/6 mice or NOD2 deficient mice on C57BL/6 background were infected with N. gonorrhoeae at MOI 10:1 or left uninfected. At 3 hrs after infection, cells lysates were made and RNA was isolated (Rneasy Mini Kit, Qiagen). Whole genome microarray analysis was carried out using the Affymetrix GeneChip Gene 1.0 ST Array System. All microarray studies were carried out with the assistance the BUMC Microarray Core Facility. The raw microarray data are available in GEO (series record GSE37179).
PCR and Real Time PCR
Total RNA was extracted from infected primary BMDM or HeLa cells using the Rneasy Mini Kit (Qiagen). Complementary cDNA was synthesized using the RT2 First Strand Kit (SABiosciences). The following oligonucleotide primers were purchased from SABiosciences: Mouse C3 (PPM04471E), Mouse Cfb (PPM24558A), Mouse Csf1 (PPM031116B), Mouse IL23a (PPM03763E), Mouse Actb (PPM02945A). The RT2 SYBR® Green/Rox™ qPCR Master Mix was purchased from SABiosciences. The following primers were used for PCR experiments with HeLa cells: huNOD1 F: TCA AGT TGG GGA TGA AGG AG; Nod1 R: GCC AAA CTC TCT GCC ACT TC; huNOD2 F: CTC CAT GGC TAA GCT CCT TG; Nod2 R: CAC ACT GCC AAT GTT GTT CC; huGAPDH F: GAG TCA ACG GAT TTG; GAPDH R: GAC AAG CTT CCC GTT CTC AG.
Statistical analysis
Each data point was assayed in triplicate, and graphed as the mean +/− standard deviation (SD), and compared using an unpaired t-test. GraphPad software was used for calculation of all unpaired t-tests. For the microarray data, a 2-way ANOVA was applied to analyze the effects of genotype and infection by N. gonorrhoeae as well as the interaction between genotype and infection by N. gonorrhoeae. Raw affinity data was normalized using a Robust Multichip Analysis (RMA) method at probe level. To assess the results, a false discovery rate (fdr) < 0.01 was used as a cutoff for infection effect and genotype effect. For interaction effect, an fdr < 0.05 was used.
Results
Neisseria gonorrhoeae activates human NOD1 and NOD2
In order to determine if N. gonorrhoeae is capable of activating human NOD1 and NOD2, we utilized a luciferase reporter system driven by the transcription factor NF-κB. HEK293 cells were transiently transfected with plasmids containing human NOD1 or NOD2, together with the NF-κB reporter as described in the Methods section, and luciferase activity was determined as a marker of NF-κB activation. As HEK293 cells are not naturally phagocytic, we used crude gonococcal lysates as a source of membrane fragments containing peptidoglycan. For comparison, we also stimulated cells with conditioned medium from logarithmic-phase broth culture of gonorrhea, since this has been shown to be a source of secreted peptidoglycan fragments 37. As shown in Fig. 1, we observed that preparations of crude gonococcal lysates were able to activate human NOD1 and NOD2, as measured by fold increase in luciferase activity over baseline, demonstrating that peptidoglycan fragments released from N. gonorrhoeae can drive both human NOD1 and NOD2 activation. Interestingly, gonococcal supernatant from mid-logarithmic phase grown broth cultures activated NOD1 but not NOD2, suggesting the supernatant contains peptidoglycan fragments of different size and composition than the crude whole cell lysates. In contrast, neither the TLR4 ligand LPS, nor the TLR2 ligand Pam3-Cys-SK4 resulted in NOD-dependent activation of the luciferase reporter.
Figure 1. N. gonorrhoeae activates human NOD1 and NOD2.
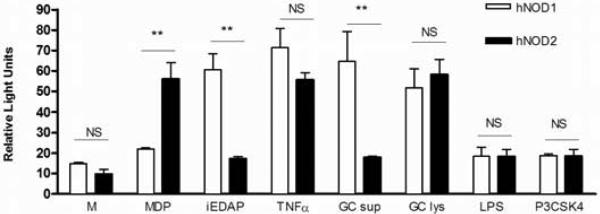
HEK293 cells, transiently transfected with human NOD1 (hNOD1) or NOD2 (hNOD2) and an NF-κB driven luciferase reporter, were incubated for 24 hrs in the presence of N. gonorrhoeae supernatant (GC sup) or N. gonorrhoeae crude lysate (GC lys), prepared as described in the Methods; or in the presence of the NOD1 ligand iEDAP (10 μg/ml), the NOD2 ligand MDP (10 μg/ml), human TNF-α (50ng/ml), LPS (100 ng/ml), P3CSK4 (100 ng/ml), or medium only (M). All conditions were tested in triplicate. Cell lysates were prepared and assessed for luciferase activity as described in the Methods. Data is presented as fold increase over the baseline luciferase activity in HEK 293 cells transfected with an empty vector. *=p<0.05; **=p<0.005; NS=not statistically significant. Each experiment was repeated at least 3 times with similar findings.
Neisseria gonorrhoeae activates the NOD adaptor RIPK2 in human cervical epithelial cells and human macrophages
Next, we examined whether endogenous NOD1 and NOD2 could be activated by gonococci and gonococcal membrane fragments during infection. We first examined HeLa cells as an in vitro model for cervical epithelial cells. HeLa cells have been previously shown to express endogenous NOD1 and NOD2, although not to the same levels 38. To assess for a specific NOD1 or NOD2 signature, we examined activation of RIPK2, an adaptor that is specific for NOD1 and NOD2 signaling 39. Upon activation of NOD1 or NOD2, RIPK2 undergoes stable Lys-63 polyubiquitination, which acts as scaffolding for recruitment of additional adaptor proteins such as the kinase TAK1 19, 36, 40. Thus, this Lys-63 linked polyubiquitination is crucial for further downstream signaling events and has been used as a marker for NOD-RIPK2 activation 36. As shown in Fig. 2A, we were able to detect RIPK2 polyubiquitination occurring upon treatment of cells with gonococcal supernatants, gonococcal lysates, and live N. gonorrhoeae. Of note, only the NOD1 ligand C12-iEDAP but not the NOD2 ligand MDP induced RIPK2 polyubiquitination in HeLa cells, suggesting that only the NOD1 signaling pathway was intact, consistent with the previous report that HeLa cells primarily express NOD1 38. Taken together, these data suggest that gonococcal activation of RIPK2 polyubiquitination in HeLa cells occurs through NOD1.
Figure 2. N. gonorrhoeae activates the NOD1/2 adapter RIPK2 in human cervical epithelial cells and human macrophages.

(A) HeLa cells or (B) human macrophages were incubated with the indicated Neisseria gonorrhoeae preparations, or NOD1 ligand C12-iEDAP (10 μg/ml; iEDAP), the NOD2 ligand MDP (10 μg/ml), or medium alone (M) for 24 hr, unless otherwise indicated. Protein lysates were made and subjected to immunoprecipitation with an anti-RIPK2 antibody, followed by immunoblot with an anti-ubiquitin antibody, as described in the Methods. Activation of NOD1 or NOD2 results in polyubiquitination of the adaptor protein RIPK2, which appears as a smear in this assay. Immunoblot for RIPK2 is shown below as an indicator of protein loading. Each experiment was repeated at least 2 times with similar findings.
Next, we examined primary human macrophages and observed that infection with live N. gonorrhoeae (Fig. 2B) resulted in RIPK2 polyubiquitination; similar results were found using gonococcal lysates (data not shown). Unlike the HeLa cells, the macrophages responded to both the NOD1 and NOD2 ligands, demonstrating functional pathways for both receptors.
N. gonorrhoeae induce IL-8 secretion in HeLa cells via NOD1
In order to examine signaling events downstream of NOD1/2 and the adaptor RIPK2, we returned to HeLa cells. As shown in Fig. 3A, we found that infection of HeLa cells with live gonococci, but not heat killed gonococci or crude gonococcal lysates, resulted in the upregulation of IL-8 secretion. The production of IL-8 with live N. gonorrhoeae infection was enhanced even further if the HeLa cells were primed with IFN-γ, which has been shown to upregulate NOD1 expression 41. It should be noted that a higher MOI of gonococci was required in the HeLa cells as compared with macrophages, perhaps reflecting low expression of the receptor as well as inefficient uptake of ligand compared to phagocytic cells. Priming with IFN-γ also resulted in IL-8 upregulation in response to the NOD1 ligand C12-iEDAP, but had no effect on the response to TLR2 and TLR4 ligands, Pam3-C-SK4 and LPS. In fact, our HeLa cells under the conditions of this assay were unresponsive to both LPS and Pam3-C-SK4, thus eliminating confounding activation of TLR4 and TLR2 pathways in response to the gonococcal preparations. Likewise, HeLa cells failed to respond to the NOD2 ligand, MDP, in the presence or absence of IFN-γ priming (data not shown). To test whether the IL-8 production by N. gonorrhoeae was specifically a result of NOD1 activation, we treated HeLa cells with the RIPK2 inhibitor SB 203580. This is a potent RIPK2 inhibitor, although at higher concentrations it has been shown to inhibit p38α MAPK 42. Treatment with the inhibitor resulted in a dose dependent reduction in both N. gonorrhoeae and C12-iEDAP-induced IL-8 production, without affecting IL-8 induction by TNF-α, except at the higher concentration (Fig. 3B). Finally, treatment of cells with the NOD1 inhibitor ML130 43 resulted in similar reduction in N. gonorrhoeae and C12-iEDAP induced IL-8 production (Fig. 3C). Thus, gonococci appear to activate HeLa cells via NOD1 to upregulate secretion of IL-8.
Figure 3. NOD activation by N. gonorrhoeae alters chemokine induction in cervical epithelial cells.

HeLa cells were primed overnight with IFN-γ (100 U/ml), except as indicated in (A), followed by incubation with live Neisseria gonorrhoeae (GC) or heat killed Neisseria gonorrhoeae (HK GC) (both MOI 50:1), the NOD1 ligand C12iEDAP (10 μg/ml), human TNF-α (100 ng/ml), LPS (100 ng/ml), P3CSK4 (100 ng/ml), or medium only (M). Where indicated, the RIPK2 inhibitor SB 203580 (B) or the Nod1 inhibitor ML130 (Fig. 3C) was added 1 hour prior to treatment at the indicated concentrations. Cell supernatants were collected after 22 hrs and analyzed for the presence of IL-8 by ELISA. *=p<0.05; **=p<0.005; ***=p<0.0005; NS=not statistically significant; ND=not detected. All conditions were tested in triplicates. Each experiment was repeated 2 times with similar findings.
Neisseria gonorrhoaeae activates murine NOD2 but not murine NOD1
To further examine the mechanism and the effect of NOD1 and NOD2 activation by N. gonorrhoeae, we turned to murine macrophages, where we could take advantage of the availability of defined knockout mice. First, we tested if RIPK2 polyubiquitination in response to gonococci was dependent on expression of NOD1 or NOD2 in murine macrophages. We observed RIPK2 polyubiquitination in both primary BMDM, as well as immortalized BMDM, upon treatment with gonococcal supernatants, gonococcal lysates or live N. gonorrhoeae (Fig. 4A), demonstrating that the NOD1/NOD2-RIPK2 pathway was activated upon exposure to gonococci. In contrast, LPS failed to activate RIPK2 polyubiquitination, confirming that ultrapure LPS preparations do not activate NOD signaling pathways. We next determined if RIPK2 polyubiquitination in response to N. gonorrhoeae was dependent on the expression of either NOD1 or NOD2. As shown in Fig. 4B, N. gonorrhoeae failed to stimulate RIPK2 polyubiquitination in the absence of NOD2 expression, while NOD1 deficient macrophages remained responsive to N. gonorrhoeae stimulation. This suggested that activation of the NOD-specific RIPK2 pathway by gonococci in murine cells was dependent on murine NOD2 but not NOD1. To confirm our findings, we transiently transfected HEK293 cells with plasmids containing either mouse NOD1 or NOD2, together with the NF-κB luciferase reporter. As shown in Fig.4C, unlike human NOD1, there was no activation in response to expression of mouse NOD1 by gonococcal supernatants or gonococcal lysates. In contrast, mouse NOD2 was activated by gonococcal lysates, similarly to human NOD2. Thus, N. gonorrhoeae is capable of activating murine NOD2 but not mouse NOD1, demonstrating a species-specific response to gonococcal peptidoglycan fragments.
Figure 4. N. gonorrhoeae activates NOD2 but not NOD1 in murine macrophages.

(A) Primary murine BMDM or (B) immortalized murine BMDM derived from wild type C57BL/6 mice (WT), NOD1 knockout mice (NOD1 KO) or NOD2 knockout mice (NOD2 KO) were incubated for 2 hrs with either live Neisseria gonorrhoeae (Live GC) at the indicated MOIs, gonococcal lysates (GC Lys), supernatant from mid-log phase grown Neisseria gonorrhoeae (GC sup), the NOD2 ligand MDP (10 μg/ml), the TLR4 ligand LPS (100 ng/ml) or medium only (M). Protein lysates were made and subjected to immunoprecipitation with an anti-RIPK2 antibody, followed by immunoblot with an anti-ubiquitin antibody. Polyubiquitination of the adaptor protein RIPK2 appears as a smear in this assay. Each experiment was repeated at least 2 times with similar findings. (C) HEK293 cells transiently transfected with murine NOD1 (mNOD1) or NOD2 (mNOD2) and a NF-κB driven luciferase reporter, were incubated for 24 hrs in the presence of Neisseria gonorrhoeae supernatant (GC sup) or Neisseria gonorrhoeae crude lysate (GC lys), both diluted 1:10 or in the presence of the NOD1 ligand iEDAP (10 μg/ml), the NOD2 ligand MDP (10 μg/ml), human TNF-α (50 ng/ml), or medium only (M). All conditions were tested in triplicate. Cell lysates were prepared and assessed for luciferase activity as described in the Methods. Data is presented as fold increase over the baseline luciferase activity in HEK 293 cells transfected with an empty vector. *=p<0.05; **=p<0.005; NS=not statistically significant. Each experiment was repeated at least 3 times with similar findings.
NOD2 activation by N. gonorrhoeae alters levels of cytokine and chemokine induction and affects multiple signaling pathways in macrophages
Because gonococci, like all bacteria, express a number of ligands for TLRs and other innate immune signaling pathways, the question remained as to the significance of NOD-dependent signaling in the global host inflammatory response to infection. In other words, could we identify a NOD-dependent gene expression profile in response to gonococcal infection? As our data suggested that murine macrophage responses were primarily driven by NOD2 and not NOD1, we compared the response of wild-type (WT) C57BL/6 macrophages with macrophages derived from NOD2 KO mice on the same background. We first examined the upregulation of TNF-α using immortalized macrophages in response to a number of ligands as well as bacterial preparations. As shown in Fig. 5A, we predictably observed no significant change in LPS-dependent TNF-α secretion between the WT and NOD2 deficient cells, while MDP was a surprisingly weak agonist when used alone. However, when the combination of LPS plus MDP was used, there was a clear and statistically significant difference in the TNF-α inducing capability in the WT and KO macrophages. Likewise, the induction of TNF-α by both live and heat-killed gonococci was modulated by NOD2 expression, with NOD2 sufficient macrophages secreting significantly more TNF-α compared to NOD2 deficient macrophages. We next confirmed this data using primary murine BMDM, and this time surveyed for a variety of pro-inflammatory cytokines, including IL-6 and IL-10, as well as the chemokine RANTES, in addition to TNF-α. As shown in Fig. 5B-D, while both WT and NOD2 KO macrophages secreted high amounts of TNFα, IL-6, IL-10, and RANTES, there was again a reproducible and statistically significant decrease in the production of these cytokines in response to N. gonorrhoeae in the absence of NOD2 expression. These data suggest that NOD2 activation by N. gonorrhoeae has an additive effect on the predominantly TLR driven production of cytokines in macrophages.
Figure 5. NOD2 activation by N. gonorrhoeae alters levels of TNF-α, IL-6, IL-10, and RANTES in macrophages.

(A) Immortalized murine BMDM derived from wild type C57BL/6 mice (WT) or NOD2 knockout mice (NOD2 KO) incubated with the following: live N. gonorrhoeae (MOI = 1 or 10); heat killed N. gonorrhoeae (MOI = 1 or 10), MDP (10 μg/ml), LPS (100 ng/ml), or a combination of the two; or medium alone (M). Cell supernatants were collected at 24 hours and assayed for TNF-α by ELISA. (B-E) Primary murine BMDM derived from wild type C57BL/6 mice (WT) or NOD2 knockout mice (NOD2 KO) were incubated with the indicated treatments. Cell supernatants were collected at 6 hours post infection, and assessed for presence of (B) TNF-α, (C) IL-6, (D) IL-10, and (E) RANTES by ELISA. *= p<0.05; **=p<0.005; ***=p<0.0005; NS=non statistically significant; ND=not detected. All conditions were tested in triplicate. Each experiment was repeated at least 3 times with similar findings.
In order to identify a NOD2-specific gene expression signature, we conducted whole genome microarray analysis in N. gonorrhoeae-infected BMDM from WT C57BL/6 mice and NOD2 deficient mice on the same background. Data was analyzed as described in the Methods section. There were over 10,000 genes that were differentially expressed as a result of infection with gonorrhea alone and 1600 genes differentially expressed between WT and NOD2 KO BMDM independently of infection. After taking into consideration the interaction of both infection and genotype, we were left with 127 genes that were differentially expressed in the absence of NOD2 (Fig. 6). Among those identified, there were many genes for cytokines and chemokines, such as CCL17, CCL22, and CCL5/RANTES, as well as growth factors such as CSF1. While most genes demonstrated lower expression in the infected NOD2 KO macrophages compared to infected WT macrophages, others, such as IL-23A, had a higher expression in the absence of NOD2. Neither TNF-α nor IL-6 came up as differentially expressed at the level of RNA. Two other pathways that seemed to be affected in the absence of NOD2 were the alternative complement pathway, where both complement component 3 (C3) as well as complement factor B (CFB) gene expression was decreased, and the cytosolic DNA sensing pathway, where RIG-I and ZBP1 (DAI) gene expression were both decreased as well.
Figure 6. Differential gene expression in response to N. gonorrhoeae infection in vitro.

Representative genes out of the 127 genes found to be differentially expressed between wild type (WT) and NOD2 knockout (NOD2 KO) primary murine Bone BMDM infected with N. gonorrhoeae (GC) or medium only (M). Each condition was tested in triplicate. Highlighted in yellow are noted genes of particular interest, such as cytokines, chemokines, cytokine, chemokine receptors and growth factors (CSF1, Lif, IL23a, Ccl5, Tnfsf9, Cxcr4, Ccl24, Ccl22, Ccl17, Tnfsf4, Cx3cl1, Cxcl1), complement cascade related genes (Cfb, C3) and cytosolic DNA sensing pathway related genes (Zbp1=DAI, Dhx58=RIG-I).
To validate the microarray findings we performed real time PCR using primers specific for a subset of representative genes among those that were identified in the microarray. As shown in Fig. 7, we were able to confirm differential expression of Cfb, C3, CSF1 and IL23a in infected cells in an independent experiment.
Figure 7. NOD activation by N. gonorrhoeae affects multiple signaling pathways in macrophages.

Primary murine BMDM from C57BL/6 mice (WT) or NOD2 knock out (NOD2−/−) mice were prepared as described in the Methods and incubated with live N. gonorrhoeae at an MOI 10:1 or medium only for 3 hrs. Each condition was tested in triplicate. Total RNA was extracted from the cell lysates and subjected to real time PCR as described in the Methods. Shown is the relative expression for Cfb, C3, CSF1 and IL23a normalized for GAPDH expression. *= p<0.05; **=p<0.005; ***=p<0.0005.
Discussion
Activation of the innate immune system is clearly essential for the survival of a host following an infectious challenge. However, inflammatory signaling pathways must be carefully regulated to avoid excessive bystander injury, as well as to instruct the development of a polarized adaptive immune response. In contrast to activation with purified ligands, the response to whole bacteria must also reflect the integration of numerous innate immune signaling pathways that are activated simultaneously or sequentially. While a number of studies have investigated the role of the transmembrane Toll-like receptors (TLR), TLR4 and TLR2, in response to N. gonorrhoeae, little has been written on the role of cytosolic innate immune receptors, despite the fact that gonococci can reside and replicate within the cytosolic compartment.
In this manuscript, we demonstrate that N. gonorrhoeae activates the cytosolic receptors NOD1 and NOD2, and that this contributes to the immune response, regulating not only the induction of inflammatory cytokines and chemokines, but also growth factors, complement proteins and other cytosolic pattern recognition receptors. N. gonorrhoeae was capable of activating both human NOD1 and NOD2, demonstrating that polymeric peptidoglycan derived from the Gram-negative cell wall can result in formation of both NOD1 and NOD2 activating peptides and muropeptides. Likely, these peptidoglycan fragments are formed under the action of different peptidoglycan degrading enzymes, which originate either from the bacteria, such as autolysins, or the host cells, such as lysozyme. Similar to our findings, NOD1 and NOD2 activation has been shown to occur with other Gram-negative bacteria, including heat killed E. coli and A. actinomycetemcomitans 44 or with undigested or digested E.coli peptidoglycan 45. We did not see major differences in terms of the ability of live or killed N. gonorrhoeae to activate NOD receptors in macrophages, suggesting that intracellular growth of N. gonorrhoeae is not required for NOD activation in professional phagocytes. On the other hand, we only observed NOD dependent IL-8 induction in HeLa cells upon exposure to live bacteria, suggesting that gonococci must actively invade and replicate within epithelial cells in order for ligands to efficiently access these cytosolic receptors. While the ability of a Gram-negative cell wall to activate these peptidoglycan-recognition receptors may not be surprising, we were struck by the ability of conditioned supernatant from logarithmic-phase grown bacteria to activate NOD1 and NOD2. This confirms previous reports that peptidoglycan fragments are secreted during gonococcal growth 28-30 and suggests that these cytosolic receptors could be activated not only upon uptake and processing of bacterial membranes, but by replicating bacteria residing in the cytosol.
The smallest peptidoglycan fragment which can activate NOD1 is the dipeptide iEDAP 25, while there are also other NOD1 activating fragments, notably different diaminopimelic acid (DAP) containing tripeptides 45. The smallest fragments capable of activating NOD2 are the muropeptide MDP 26 and other associated muropeptides 46. We found that N. gonorrhoeae conditioned supernatant during growth in broth cultures activated primarily NOD1, similar to what has been shown for other Gram-negative bacteria such Shigella flexneri or Bortetella pertussis 47. Although we did not identify the major NOD1 activating fragment from gonococci, we hypothesize that it is a 1,6-anhydro disaccharide tripeptide, one of the major fragments released by N. gonorrhoeae into the environment during periods of exponential growth 30. In agreement with this is the fact that purified peptidoglycan from the closely related species N. meningitidis was reported to activate human NOD1 through an identical diaminopimelate-containing N-acetylglucosamine–N-acetylmuramic acid tripeptide motif 48.
While we found that both human NOD1 and NOD2 were capable of detecting gonococci, our data from knockout mice suggests that mouse NOD1 is not activated by gonococcal preparations. Previous reports suggest that murine NOD1 is activated primarily by tetrapeptide structures while human NOD1 is activated by tripeptide fragments 49. Thus, one explanation is that gonococci release primarily tripeptide fragments. In addition to this species specificity, the ability of N. gonorrhoeae to activate NOD1 or NOD2 may also depend on the cell type. For example, while Nod1 is ubiquitously expressed, Nod2 expression is believed to be more restricted to bone marrow-derived cells such as macrophages and dendritic cells, although it has also been identified in epithelial cells from the intestine, lung, and oral cavity [reviewed in 23]. Thus, while transfected HEK293 cells over expressing NOD1 and NOD2 could be activated by ligands specific to both receptors, in our hands HeLa cells only responded to purified NOD1 ligands while human macrophages responded to both NOD1 and NOD2 ligands. Thus in the reproductive tract, the role of NOD1 vs. NOD2 in detecting gonococci likely varies with the cellular composition of the tissue. It should be noted that, because of the species-specific ability of human but not mouse NOD1 to detect gonococci, infection of mice may not mimic infection in humans given that NOD1-dependent signaling pathways may be absent in epithelial cells. The implications of this remain to be tested.
The mechanism by which bacterial peptidoglycan fragments can access NOD1/NOD2 and other cytosolic receptors is not always fully understood and likely varies with the bacterial species. For example, the intracellular bacterium H. pylori appears to use a type IV secretion system to inject peptidoglycan fragments in the cytosol. Another mechanism of entry into cells is via outer membrane vesicles (OMVs). It has been shown that OMVs from different Gram-negative bacteria such as Helicobacter pylori, Pseudomonas aeruginosa and notably N. gonorrhoeae enter epithelial cells through lipid rafts and are able to induce NOD1 activation 50. There is also evidence that peptidoglycan fragments enter the cytosol through endocytosis or through epithelial transporters, such as the transporter pepT1 for MDP 51 and the transporter pepT2 for iEDAP 52. Finally, there is also some evidence that a fraction of NOD1 might also localize to the plasma membrane 53.
Finally, we observed NOD-dependent regulation of cytokine induction in response to N. gonorrhoeae, as well as alterations in other cellular immune processes. Specifically, NOD2 activation by N. gonorrhoeae in murine macrophages contributed to the upregulation of TNF-α and IL-6 secretion, suggesting that cells integrate NOD and TLR-dependent signaling pathways. In addition, we identified a number of genes that were regulated by NOD2-dependent signaling in response to N. gonorrhoeae. Some, such as the Th2-associated chemokines CCL17 and CCL22 were not surprising given previous reports of NOD2-dependent Th2 polarization 54; NOD1 and NOD2 have also been previously shown to upregulate the chemokine CCL5/RANTES 55. The significance of enhanced IL-23 induction in the absence of NOD2 is difficult to explain given that others have reported that NOD2 deficient dendritic cells have reduced capacity to activate the IL-23-IL-17 axis in response to NOD2/TLR2 co-stimulation 56. In our case, the enhanced IL-23 response in the absence of NOD2 might reflect exaggerated LPS/TLR4 signaling by gonococci, and thus might lead to more Th17 polarization 57. What is most interesting is that we identified several unexpected genes whose expression was influenced by NOD2 expression, novel findings that may be relevant to understanding NOD2 function as well as gonococcal pathogenesis. This includes gene products involved in the complement pathway, C3 and CFB, and given the important role of complement in gonococcal pathogenesis, these findings warrant further investigation. The specific role of NOD1 signaling in the global response to gonococcal infection remains undefined.
In conclusion, we have demonstrated that the human pathogen N. gonorrhoeae activates the peptidoglycan sensing cytosolic receptors NOD1 and NOD2, and that this impacts the inflammatory response of epithelial cells and macrophages to infection with gonococci by altering the immune response, including changes in the expression of cytokines, complement related proteins and innate immune receptors. Moreover, the inability of murine NOD1 to detect gonococcal peptidoglycan fragments may limit the use of existing mouse models to examine the role of in vivo NOD signaling in gonococcal pathogenesis. While cell surface TLR2 and TLR4 are key innate immune receptors during the host response to gonococcal infection, we believe these intracellular receptors also play an important role in the cytosolic recognition of this pathogen given its intracellular lifestyle. Thus, we propose a model where the global host response to N. gonorrhoeae during infection integrates both TLR and NLR signaling pathways.
Acknowledgements
This work was supported by the following grants from the National Institutes of Health: U19 AI084048 (RRI); KL2RR025770 (NM); and AI075118 (MAK).
References
- 1.CDC . Sexually Transmitted Disease Surveillance, 2009. U.S. Department of Health and Human Services; Atlanta: 2010. [Google Scholar]
- 2.Hook EW, Handsfield HH. Gonococcal infections in the adult. In: Holmes KK, Sparling PF, Mardh PA, et al., editors. Sex Transm Dis. McGraw-Hill Companies, Inc; New York: 1999. pp. 451–66. [Google Scholar]
- 3.Cohen MS, Hoffman IF, Royce RA, et al. Reduction of concentration of HIV-1 in semen after treatment of urethritis: implications for prevention of sexual transmission of HIV-1. AIDSCAP Malawi Research Group. Lancet. 1997;349:1868–73. doi: 10.1016/s0140-6736(97)02190-9. [DOI] [PubMed] [Google Scholar]
- 4.Levine WC, Pope V, Bhoomkar A, et al. Increase in endocervical CD4 lymphocytes among women with nonulcerative sexually transmitted diseases. The Journal of infectious diseases. 1998;177:167–74. doi: 10.1086/513820. [DOI] [PubMed] [Google Scholar]
- 5.Chen A, Boulton IC, Pongoski J, Cochrane A, Gray-Owen SD. Induction of HIV-1 long terminal repeat-mediated transcription by Neisseria gonorrhoeae. Aids. 2003;17:625–8. doi: 10.1097/00002030-200303070-00019. [DOI] [PubMed] [Google Scholar]
- 6.Klotman ME, Rapista A, Teleshova N, et al. Neisseria gonorrhoeae-induced human defensins 5 and 6 increase HIV infectivity: role in enhanced transmission. Journal of immunology. 2008;180:6176–85. doi: 10.4049/jimmunol.180.9.6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Li G, Bafica A, et al. Neisseria gonorrhoeae enhances infection of dendritic cells by HIV type 1. Journal of immunology. 2005;174:7995–8002. doi: 10.4049/jimmunol.174.12.7995. [DOI] [PubMed] [Google Scholar]
- 8.Merz AJ, So M. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu Rev Cell Dev Biol. 2000;16:423–57. doi: 10.1146/annurev.cellbio.16.1.423. [DOI] [PubMed] [Google Scholar]
- 9.Ingalls RR, Lien E, Golenbock DT. Membrane-Associated Proteins of a Lipopolysaccharide-Deficient Mutant of Neisseria meningitidis Activate the Inflammatory Response through Toll-Like Receptor 2. Infect Immun. 2001;69:2230–6. doi: 10.1128/IAI.69.4.2230-2236.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu M, John CM, Jarvis GA. Phosphoryl moieties of lipid A from Neisseria meningitidis and N. gonorrhoeae lipooligosaccharides play an important role in activation of both MyD88- and TRIF-dependent TLR4-MD-2 signaling pathways. Journal of immunology. 2010;185:6974–84. doi: 10.4049/jimmunol.1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pridmore AC, Wyllie DH, Abdillahi F, et al. A Lipopolysaccharide-Deficient Mutant of Neisseria meningitidis Elicits Attenuated Cytokine Release by Human Macrophages and Signals via Toll-like Receptor (TLR) 2 but Not via TLR4/MD2. J Infect Dis. 2001;183:89–96. doi: 10.1086/317647. [DOI] [PubMed] [Google Scholar]
- 12.Packiam M, Wu H, Veit SJ, Mavrogiorgos N, Jerse AE, Ingalls RR. Protective role of Toll-like receptor 4 in experimental gonococcal infection of female mice. Mucosal Immunol. 2011 doi: 10.1038/mi.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisette PL, Ram S, Andersen JM, Guo W, Ingalls RR. The Lip lipoprotein from Neisseria gonorrhoeae stimulates cytokine release and NF-kB activation in epithelial cells in a TLR2-dependent manner. J Biol Chem. 2003;278:46252–60. doi: 10.1074/jbc.M306587200. [DOI] [PubMed] [Google Scholar]
- 14.Massari P, Henneke P, Ho Y, Latz E, Golenbock DT, Wetzler LM. Cutting edge: immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J Immunol. 2002;168:1533–7. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 15.Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of Toll-like receptor 4-mediated signaling. J Immunol. 2002;168:2424–32. doi: 10.4049/jimmunol.168.5.2424. [DOI] [PubMed] [Google Scholar]
- 16.Duncan JA, Gao X, Huang MT, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. Journal of immunology. 2009;182:6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magalhaes JG, Sorbara MT, Girardin SE, Philpott DJ. What is new with Nods? Curr Opin Immunol. doi: 10.1016/j.coi.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 18.McDonald C, Inohara N, Nunez G. Peptidoglycan signaling in innate immunity and inflammatory disease. The Journal of biological chemistry. 2005;280:20177–80. doi: 10.1074/jbc.R500001200. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Shaw MH, Kim YG, Nunez G. Nod-like Receptors: Role in Innate Immunity and Inflammatory Disease. Annu Rev Pathol. 2008 doi: 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
- 20.Voss E, Wehkamp J, Wehkamp K, Stange EF, Schroder JM, Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. The Journal of biological chemistry. 2006;281:2005–11. doi: 10.1074/jbc.M511044200. [DOI] [PubMed] [Google Scholar]
- 21.Boughan PK, Argent RH, Body-Malapel M, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. The Journal of biological chemistry. 2006;281:11637–48. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- 22.Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infection and immunity. 2005;73:7967–76. doi: 10.1128/IAI.73.12.7967-7976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 25.Chamaillard M, Hashimoto M, Horie Y, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–7. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 26.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–12. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 27.Bonen DK, Ogura Y, Nicolae DL, et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003;124:140–6. doi: 10.1053/gast.2003.50019. [DOI] [PubMed] [Google Scholar]
- 28.Cloud KA, Dillard JP. A lytic transglycosylase of Neisseria gonorrhoeae is involved in peptidoglycan-derived cytotoxin production. Infect Immun. 2002;70:2752–7. doi: 10.1128/IAI.70.6.2752-2757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenthal RS. Release of soluble peptidoglycan from growing gonococci: hexaminidase and amidase activities. Infect Immun. 1979;24:869–78. doi: 10.1128/iai.24.3.869-878.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha RK, Rosenthal RS. Release of soluble peptidoglycan from growing conococci: demonstration of anhydro-muramyl-containing fragments. Infect Immun. 1980;29:914–25. doi: 10.1128/iai.29.3.914-925.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Striker R, Kline ME, Haak RA, Rest RF, Rosenthal RS. Degradation of gonococcal peptidoglycan by granule extract from human neutrophils: demonstration of N-acetylglucosaminidase activity that utilizes peptidoglycan substrates. Infection and immunity. 1987;55:2579–84. doi: 10.1128/iai.55.11.2579-2584.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart KM, Murphy AJ, Barrett KT, Wira CR, Guyre PM, Pioli PA. Functional expression of pattern recognition receptors in tissues of the human female reproductive tract. Journal of reproductive immunology. 2009;80:33–40. doi: 10.1016/j.jri.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melly MA, McGee ZA, Rosenthal RS. Ability of monomeric peptidoglycan fragments from Neisseria gonorrhoeae to damage human fallopian-tube mucosa. J Infect Dis. 1984;149:378–86. doi: 10.1093/infdis/149.3.378. [DOI] [PubMed] [Google Scholar]
- 34.Wade JJ, Graver MA. A fully defined, clear and protein-free liquid medium permitting dense growth of Neisseria gonorrhoeae from very low inocula. FEMS Microbiol Lett. 2007;273:35–7. doi: 10.1111/j.1574-6968.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- 35.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA. NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. The Journal of biological chemistry. 2007;282:36223–9. doi: 10.1074/jbc.M703079200. [DOI] [PubMed] [Google Scholar]
- 37.Garcia DL, Dillard JP. Mutations in ampG or ampD affect peptidoglycan fragment release from Neisseria gonorrhoeae. J Bacteriol. 2008;190:3799–807. doi: 10.1128/JB.01194-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44:3100–11. doi: 10.1016/j.molimm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 39.Park JH, Kim YG, McDonald C, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. Journal of immunology. 2007;178:2380–6. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- 40.Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. Embo J. 2008;27:373–83. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe T, Asano N, Fichtner-Feigl S, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120:1645–62. doi: 10.1172/JCI39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Windheim M, Lang C, Peggie M, Plater LA, Cohen P. Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem J. 2007;404:179–90. doi: 10.1042/BJ20061704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magnuson G, Khan P, Yuan H, et al. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. High Throughput Screening Assays for NOD1 Inhibitors. [Google Scholar]
- 44.Okugawa T, Kaneko T, Yoshimura A, Silverman N, Hara Y. NOD1 and NOD2 mediate sensing of periodontal pathogens. J Dent Res. 2010;89:186–91. doi: 10.1177/0022034509354843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girardin SE, Travassos LH, Herve M, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. The Journal of biological chemistry. 2003;278:41702–8. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- 46.Coulombe F, Divangahi M, Veyrier F, et al. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med. 2009;206:1709–16. doi: 10.1084/jem.20081779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nigro G, Fazio LL, Martino MC, et al. Muramylpeptide shedding modulates cell sensing of Shigella flexneri. Cell Microbiol. 2008;10:682–95. doi: 10.1111/j.1462-5822.2007.01075.x. [DOI] [PubMed] [Google Scholar]
- 48.Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584–7. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 49.Magalhaes JG, Philpott DJ, Nahori MA, et al. Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep. 2005;6:1201–7. doi: 10.1038/sj.embor.7400552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaparakis M, Turnbull L, Carneiro L, et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010;12:372–85. doi: 10.1111/j.1462-5822.2009.01404.x. [DOI] [PubMed] [Google Scholar]
- 51.Ismair MG, Vavricka SR, Kullak-Ublick GA, Fried M, Mengin-Lecreulx D, Girardin SE. hPepT1 selectively transports muramyl dipeptide but not Nod1-activating muramyl peptides. Can J Physiol Pharmacol. 2006;84:1313–9. doi: 10.1139/y06-076. [DOI] [PubMed] [Google Scholar]
- 52.Swaan PW, Bensman T, Bahadduri PM, et al. Bacterial peptide recognition and immune activation facilitated by human peptide transporter PEPT2. Am J Respir Cell Mol Biol. 2008;39:536–42. doi: 10.1165/rcmb.2008-0059OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. 2008;10:477–86. doi: 10.1111/j.1462-5822.2007.01062.x. [DOI] [PubMed] [Google Scholar]
- 54.Magalhaes JG, Fritz JH, Le Bourhis L, et al. Nod2-dependent Th2 polarization of antigen-specific immunity. Journal of immunology. 2008;181:7925–35. doi: 10.4049/jimmunol.181.11.7925. [DOI] [PubMed] [Google Scholar]
- 55.Werts C, le Bourhis L, Liu J, et al. Nod1 and Nod2 induce CCL5/RANTES through the NF-kappaB pathway. Eur J Immunol. 2007;37:2499–508. doi: 10.1002/eji.200737069. [DOI] [PubMed] [Google Scholar]
- 56.van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity. 2007;27:660–9. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 57.Feinen B, Jerse AE, Gaffen SL, Russell MW. Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal Immunol. 2010;3:312–21. doi: 10.1038/mi.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]