

Abstract
While iron is often a limiting nutrient to Biology, when the element is found in the form of heme cofactors (iron protoporphyrin IX), living systems have exceled at modifying and tailoring the chemistry of the metal. In the context of proteins and enzymes, heme cofactors are increasingly found in stoichiometries greater than one, where a single protein macromolecule contains more than one heme unit. When paired or coupled together, these protein associated heme groups perform a wide variety of tasks, such as redox communication, long range electron transfer and storage of reducing/oxidizing equivalents. Here, we review recent advances in the field of multi-heme proteins, focusing on emergent properties of these complex redox proteins, and strategies found in Nature where such proteins appear to be modular and essential components of larger biochemical pathways.
Keywords: Cytochrome c, Cytochrome c peroxidase, Electron transfer, Dissimilatory metal reduction
1. Introduction: Types of multi-heme proteins used in nature
While biology is efficient at using metals to achieve electron transfer and catalysis, one of the most prevalent metal cofactors is heme iron [1]. The breadth of heme protein structure and function is an immense subject, even when considering only the diversity of proteins and enzymes that contain a single heme cofactor [2]. Here, we will focus on so-called multi-heme proteins that contain multiple units of heme, and in particular heme proteins that can be described as multi-heme cytochromes c, where the vinyl groups of iron-protoporphyrin IX are attached to the two cysteine side chains of a –CXnCH– –residues between the cysteine positions[4], though other c-type heme attachment motifs are known, including –CX3CH- and – CX4CH-[5], as well as a –CX15CH- motif that requires a dedicated maturation enzyme system [6],[7]. However, in the simplest cases, the presence of the –CXXCH– motif itself can be used as sequence-based diagnostic for the c-type covalent attachment of heme units in the context of bacterial organisms. In all cases presented here, multiple heme units appear to be essential for their role in supporting electron transfer chemistry within or between proteins and enzymes. Electron transfer is essential for countless biological processes. Efficient electron transfer occurs when the energy for the electron to be transferred from donor to acceptor is negative meaning that the reduction potential must be fine-tuned to be in between those of the donor and the acceptor. Heme iron reduction potentials of can be affected by ligand type, coordination geometry, and solvent accessibility as well as the pH of the environment [8-11]. In electron transfer through proteins, the distance between cofactors plays a critical role in how quickly electrons are passed. To overcome the challenge of long-range electron transport or multi-electron reactions, Nature has evolved chains of redox cofactors such as iron sulfur (Fe-S) clusters or heme groups, thus enabling electrons to be passed across membranes. With respect to these issues, here we will observe that multi-heme proteins display even more elaborate tricks in their capacity for redox chemistry.
Previous reviews of multi-heme proteins or cytochromes have highlighted their evolutionary relationships and the potential for emerging chemistry [12, 13], while here we will examine recent, emergent properties that appear to be found in multi-heme proteins and enzymes, considering in turn three different abilities where Nature is expert and Man is novice: redox communication and conformational changes of protein structure (bacterial cytochrome c peroxidases), long-range electron transfer through the deployment of many heme cofactors (the multi-heme cytochromes of dissimilatory metal reduction), and storage of multiple reducing and oxidizing equivalents (hydroxylamine oxidoreductase and cytochrome c554). Sadly, many multi-heme proteins and enzymes (e.g. octaheme tetrathionate reductase [14], octaheme nitrite reductase [15, 16], thiosulfate dehydrogenase [17]) are beyond the scope of our current review. Instead, here we will highlight the wealth of biochemical and biophysical information that is now available from increasingly divergent multi-heme proteins, while also underscoring the voids in our understanding of chemistry which need to be filled.
2. Redox communication
2.1. A conversation between cofactors: redox communication
Metalloproteins and metalloenzymes are efficient tools in biology using metals to achieve both electron transfer and catalysis. Electron transfer plays a central role in biological energy conversion, photosynthesis or respiration and also in the regulation of gene expression. Redox communication, in the form of oxidation and reduction reactions, is involved in electron transfer between the redox active cofactors within proteins and enzymes. The charge transfer is either initiated by low molecular redox mediators, including, but not limited to ubiquinone, oxygen/superoxide or the NAD+/NADH couple, or by the direct interaction of the redox centers. Below we discuss examples of conformational changes associated with inter-protein electron transfer reactions from a partner (protein or mediator) into an enzyme and inter-cofactor electron transfer reactions.
2.2. Bacterial Cytochrome c peroxidases
Bacterial diheme cytochrome c peroxidases (bCcPs) differ from the canonical monoheme peroxidases such as horseradish peroxidase (HRP) [18] and yeast cytochrome c peroxidase (yCcP) [19] in their heme cofactor content and catalytic peroxide reduction mechanisms. Found in the periplasmic space of many gram negative microorganisms, the bCcP family includes genuine bCcP enzymes -- the main topic of this section -- but also diheme orthologs such as MauG, a poor peroxidase that is required for the oxidative installation of the tryptophanyl tryptophane quinone (TTQ) cofactor found in methylamine dehydrogenase (recently reviewed in [20]). The two heme cofactors present in all members of the bCcP family are covalently bound within two separate cytochrome c-like domains. In all genuine bCcPs, the high-potential Met-His ligated heme, the H-heme (250-350 mV vs NHE) serves as the electron transfer site, accepting electrons from physiological or artificial electron donors. The other heme, the low-potential bis-His ligated L-heme (∼ -300 mV) serves as the site of peroxide reduction [21]. (In contrast, in the case of MauG the two hemes are known to be very close in potential, displaying redox anti-cooperativity [22]). In the as-isolated state both heme irons are in the ferric oxidation state. The high potential heme, which is not present in monoheme peroxidases, may be responsible for storing a second oxidizing equivalent during the catalytic reaction cycle and is hypothesized to mediate the transfer of electrons from electron donor proteins to the peroxidatic heme [23], potentially abrogating the need for radical-based intermediates in canonical bCcP enzymes.
2.2.1 Redox activation of CcP results in peroxide reduction
The majority of bCcPs are isolated in a catalytically inactive state where both hemes are in the ferric state, exemplified by the diheme peroxidase from Pseudomonas aeruginosa (Pa)[21, 24]. For such bCcP enzymes, the absence of any reducing equivalents provided by an electron donor protein or small molecule redox mediators (such as ascorbate) results in both hemes remaining in the ferric oxidation state where the active site is in a bis-His ligated conformation, preventing binding of the substrate peroxide to the active site heme [24] (Figure 1A, blue). The active form of the enzyme can be achieved by the introduction of one-electron though the H-heme electron transfer site. This initiation of activation by a one electron reduction of the H-heme is termed “reductive activation”: direct reduction of the H-heme causes local conformational changes that reorient the ligand groups on the low-potential heme, allowing for peroxide to access the active site [25] (Figure 1A, gray). The resultant mechanistic implications for canonical bCcP enzymes is schematized in Figure 1B, which suggests that by “banking” an electron prior to catalysis, the first kinetic intermediate after a peroxide substrate binds may be described formally as a ferryl peroxidatic iron, and a ferric high potential iron. There are notable exceptions to the apparent requirement for reductive activation, however, as the enzymes from Nitrosomonas europaea and Methylococcus capsulatus have both been reported to be constitutively active in the fully oxidized form [26, 27]. Thus, the specific role for reductive activation is unclear, though it can be noted that the phenomena may be useful for microbial organisms that undergo fluxuations in the electron acceptors that are used, possibly to only use reducing equivalents from the cytochrome c pool, when the probability of forming hydrogen peroxide from missteps of respiration are high.
Fig. 1.

A. The conformational switch found in the majority of known bCcP enzymes involves the reorganization of the distal face of the peroxidatic heme, as a function of the redox state of a high-potential heme, some 12 Å away. B. The mechanistic impact of reductive activation suggests that by “banking” an electron in the high potential center, the first kinetic intermediate need to involve the build-up of a radical species.
The precise means for redox communication between the heme units is not understood perfectly to date. Physiological electron donors to bCcPs are often small monoheme cytochromes c, or blue copper proteins such as azurin and pseudoazurin [28, 29] that are capable of reducing the high-potential heme, and the heme sites themselves bCcPs are over 12 Å apart (edge-to-edge), such that electron transfer is sill possible between the two heme cofactors via electron tunneling. Four possible through-bond routes for electron transport proposed between the H- an L- hemes of the Pa CcP have been proposed [18, 24] to date: (1) mediated by His201 adjacent to the H-heme via the protein backbone to the L-heme propionate [19]; (2) from the H-heme propionate, through a conserved calcium ion at the interface between the two heme domains, and then using the protein backbone to reach the L-heme [21]; (3) using ligating His261 to connect the H-heme propionate with Asn79 to then use the Ca2+ ion and the protein backbone to reach the L-heme [23]; and, (4) transfer from an H-heme propionate using Trp94 to an L-heme propionate (all residues mentioned use Pa numbering). However, only the last of these four hypotheses has been tested rigorously, where mutations made in Trp94 have resulted in inactive enzyme in steady-state analyses [30, 31].
A further complication in studies of bacterial CcPs stems from their native homodimeric nature, as all known bCcPs exist homodimers in solution, regardless of whether or not the enzyme in question is of the canonical type requiring reductive activation [24, 26, 27, 32-34]. The CcP from P. denitrificans shows a loss of activity upon dilution indicating that an equilibrium may exist between the monomer and the dimer, having the dimer as the active form [34], and dimerization has been further shown to be governed by the presence of a Ca2+ binding site within each protomer, though it is currently unclear whether Ca2+ induces dimerization or if Ca2+ binds after dimerization. It is known, however, that Ca2+ is essential for the reduction and hence activation of the enzyme, as shown with the CcP from P. pantotrophus[35]. In P. nautica, Ca2+ is reported to be needed to induce conformational change around the L-heme [36]. In the Pa, P. nautica, and R. capsulatus enzymes, the positive charge of the calcium ion is not balanced by any negatively charged amino acids, showing that it may be responsible for facilitating electron transport between the H- and L-hemes [24, 30, 32, 36]. Recently, a charge reversal mutant of the enzyme from Shewanella oneidensis(So) has been reported that still maintains considerable activity, though it exists as a monomer in solution [37]. In the E258K mutant of the So enzyme, the same requirement for reductive activation is maintained, but the resulting FeLIIIFeHII state required for catalysis (Figure 1B) is much less kinetically stable than wild-type, emphasizing that bCcP are sensitively tuned masters of redox communication and conformational control.
2.2.2 Redox related conformational changes in CcPs
X-ray crystal structures for a bCcP in the canonical Pa-like class of CcPs or the Ne-like class of CcPs has allowed for the comparison of structural changes during different redox phases for the enzymes. Crystal structures for both the oxidized and semireduced states of the CcPs from multiple organisms have allowed for an overall comparison between specific conformational changes in both the oxidized (FeLIIIFeHIII), and semireduced (FeLIIIFeHII) states. The Pa enzyme is commonly used to demonstrate the comparison of the placement of three key loops in the oxidized state (shaded red, blue and green in Figure 2A) against these same loops in the one electron reduced state [25] (Figure 2B). Where Loop 1 (red) bears the universally conserved His residue that may serve as a ligand in the inactivated forms of bCcP, Loop 2 (blue) reorganization is required to form the distal pocket around the peroxidatic heme, while Loop 3 packs around the high-potential heme, and is presumed to present at least part of the surface responsible for interactions with electron donors. The structural changes shown in Figure 2 appear to be uniform amongst the bCcP sub-family that require reductive activation: For example the oxidized P. pantotrophus(Pp) enzyme [38], a canonical family member, closely resembles the oxidized P. aeruginosa(Pa) enzyme [21] while the semi-reduced P. pantotrophus enzyme closely resembles the oxidized N. europaea enzyme [26] and the semi-reduced Pa enzyme. The oxidized Pp enzyme and the oxidized Pa enzyme are closely related in all conformations. There is also very close structural similarity between the mixed valent Pp enzyme and the oxidized Ne enzyme [39]. This structural similarity is consistent with the fully oxidized Ne enzyme being in an active state without the need for a prereduction step [26]. In both cases, there is open access to the active site in either the FeLIIIFeHII form of the Pa-like enzymes, or the FeLIIIFeHIII state of the Ne-like enzymes. It is not known whether any redox state or other condition can compel the Ne enzyme to adopted the closed off, bis-His ligated state that is found in fully oxidized, canonical bCcP (as in Figure 2A). Recent success in manipulating the apparent requirement for reductive activation has been reported recently through the studies of Einsle and co-workers, who have structurally and functionally characterized paralogs of the Pa-type of bCcP from Geobacter sulfurreducens (CcpA and a second diheme peroxidase, MacA) [40, 41]. Indeed the CcpA protomer adopts the same fold as other canonical bCcP enzymes, displays the identical requirement for reductive activation, and has sequence elements in Loops 1, 2 and 3 that suggest a strong similarity to Pa-type of bCcP enzyme. While mutations in Loop 1 what would render the sequence more like the Ne enzyme (and thereby presumably convert the enzyme to be constitutively active) did not perturb the reactivity of CcpA, the S134V/P135K double mutant in Loop 2 did (in part) achieve Ne enzyme-like activity [40, 42]. The resulting structure of the fully oxidized form of S134V/P135K shows a partially converted enzyme, where Loop 1 has shifted to an open conformation such that substrate might bind, yet Loop 2 is only partially reorganized as it should be in semi-reduced bCcPs, and Loop 3 failed to convert to the required, semi-reduced conformation [40]. A further functional result of this partial inter-conversion between enzymatic conformers, was an apparent control of inter-cofactor electron transfer upon electrocatalytic reduction of substrate [42].
Fig. 2.
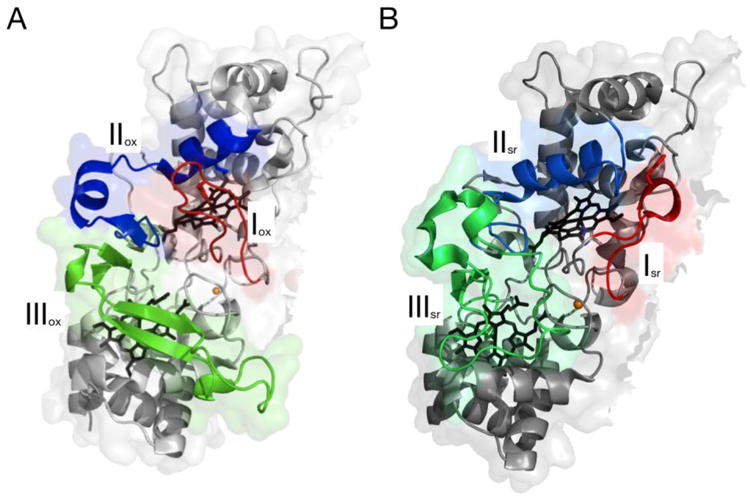
The conformational differences observed in (A) the fully oxidized form of Pa bCcP, and (B) the subsequent reorganization of three loop regions upon chemical preparation of the FeLIIIFeHII redox state. (Figure composed using Pymol, with Protein Data Bank files, 1EB7.pdb and 2VHD.pdb (panel B)).
Thus, it is clear that there is still much to be learned about how a relatively simple switch in loop structures can be manipulated by redox state information contained in two heme cofactors. While bCcPs are one example of such a phenomenon, in the next section we will consider another enzyme, cytochrome cd1, in which conformational dynamics are also controlled by a multi-heme structure.
2.3. Cytochrome cd1: redox activation and nitrite reduction
Cytochrome cd1 nitrite reductase catalyzes the reduction of nitrite to nitric oxide within the denitrification pathway, as well as the four-electron reduction of dioxygen to water [43, 44]. Cytochrome cd1 has been characterized as a heme nitrite reductase, a homodimer with each monomer containing one heme c and one heme d moiety [44]. The two heme groups are located in the hydrophobic pockets of the two domains. The heme c serves as the electron accepting heme and receives electrons in vitro from both cytochrome c551 and azurin. Redox titrations have been used to calculate the redox potential of the heme c on the order of +250 mV [45]. The heme d is the active site heme serving as he binding site for physiological oxidants. The redox potential of the d1 heme has been estimate to be at least +200 mV or greater [46].
Crystal structures have been solved for both the oxidized and reduced structures of the nitrite reductases from Paracoccus denitrificans[47-49] and Pseudomonas aeruginosa[50, 51]. The domains carrying the d1 hemes in the oxidized forms of both the Pd and Pa enzymes are nearly identical, however in the Pa enzyme the N-terminal domain, the c heme domain, of one monomer crosses the interface between the two monomers and is wrapped around the second monomer [51]. The structural differences between the Pd and Pa enzymes are all observed in the N-terminal region. In the oxidized state the Pd c heme is bis-His coordinated, while the Pa enzyme c heme is His/Met coordinated. In the oxidized Pd enzyme the N-terminal arm wraps around the domain of the monomer while the oxidized Pa enzyme has an interchanging of the N-terminal arm and wrapping around the second monomer. The reduced enzymes are very similar in conformation, except for the N-terminal arms. Upon reduction the movement of the N-terminal arm, in both the case of the Pd and Pa enzymes, is required to make the Fe of the d1 heme available for catalysis [48-52].
Kinetics and thermodynamics of the internal electron transfer process in the Pseudomonas stutzeri enzyme have been studied and found to be dominated by pronounced interactions between the c and the d1 hemes [53]. The interactions are expressed both in dramatic changes in the internal electron-transfer rates between these sites and in marked cooperatively in their electron affinity. The results constitute a prime example of intraprotein control of the electron transfer rates by allosteric interactions. The enzyme-reduction state has been analyzed by using a model that involves electron uptake by the c hemes followed by equilibration between hemes c and d1 within the same subunit. The model includes only equilibria in which intrasubunit ET can take place [49].
3. Long-range electron transfer: Molecular “wires” composed of hemes
3.1 Long range electron transfer
Multiheme cytochromes can possess a variety of functions, including the passage of charge over long distances. In such a function, maintaining appropriate reduction potentials that allow for efficient electron transfer is essential. And indeed, many factors can tune the reduction potential[11]: A heme c with bis-histidine ligation has, on average, a midpoint potential range of -400 mV to 0 mV whereas a heme c with histidine/methionine ligation has, on average, a higher midpoint potential range of 0 mV to +400 mV [8, 11]. Interestingly, while multiheme cytochromes are quite prevalent throughout Nature, there is a notably higher incidence of them in gram negative organisms, where they are localized and matured in the periplasmic space, and thereby linked with respiratory processes with either soluble inorganic, or exogenous electron acceptors (like Fe(III) oxides [13]) as described below.
3.2 Dissimilatory metal reduction
In sedimentary environments, such as aquatic sediments and submerged soils, iron-reducing organisms can grow by coupling the oxidation of organic or inorganic compounds to the reduction of iron in a process referred to as dissimilatory metal reduction (DMR) [54, 55]. The DMR process occurs in strictly anaerobic organisms such as Geobacter sulfurreducens to aerobic organisms like Pseudomonas aeruginosa to facultative species such as Shewanella oneidensis[56]. These species are able to use metal as a terminal electron acceptor, which supports the anaerobic growth of the bacteria. Notably, unlike other cases of metal reduction by organisms, metal uptake is not associated with exogenous metal reduction in the DMR chemistry of Shewanella and Geobacter. These organisms can reduce a variety of metals like solid iron and manganese oxides [57-59] as well as soluble metals such as uranium and chromium [60, 61].
Microbes capable of metal reduction have gained a vast following over the last decade because of their possible use in bioremediation and microbial fuel cells. Bioremediation that involves the capabilities of microorganisms in the removal of pollutants is a promising area of study as it could be a relatively inexpensive means to clean up waste. Additionally, the ability of these organisms to grow biofilms and produce current indicates the possibility that microbial fuel cells could be an alternative source of energy. However, the process by which organisms are able to “breathe” metal is still not entirely understood and the current output from biofilms is relatively small. Thus, investigating the mechanisms of metal reduction and pathways of electron transfer in these organisms is necessary before utilizing them in these enticing applications. Two of the most studied DMR organisms, Geobacter sulfurreducens and Shewanella oneidensis, have incredibly high cytochrome content, presumably enabling them to use multiple pathways for electron transfer.
Geobacter species are obligate anaerobes found in aquatic sediments. However, the completed genome of Geobacter sulfurreducens unveiled its ability to tolerate oxygen [62]. The genome also revealed 111 c-type cytochromes, which is higher than what has been reported for any other organism to date [62]. Seventy-three of these contain two or more heme groups, including one that has 27 heme cofactors [62]. The large number of cytochromes suggests that heme-based strategies for electron transport are critical in Geobacter. These species can use insoluble Fe(III) and Mn(IV) oxides as terminal electron acceptors [60]. This process is predominantly believed to occur through the use of outer membrane cytochromes [63]. Additionally, they can precipitate uranium in contaminated environments [60]. As such, Geobacter is considered an excellent organism for use in bioremediation.
Shewanella species are found in a variety of habitats, from deep sea, anaerobic environments to soil to sedimentary locations, and are exemplified by Shewanella oneidensis MR-1. Of its 5,000 genes, 42 encode putative c-type cytochromes, many of which are directly involved in the bacteria's respiratory pathways that lead to a wide variety of terminal electron acceptors. Unusually, 33 of the 42 c-type cytochrome genes in S. oneidensis MR-1 contain more than one heme group [64].
This non-pathogenic, gram-negative microbe can grow both aerobically and anaerobically using an extensive assortment of terminal electron acceptors including iron, manganese, uranium, nitrite, nitrate, sulfate, thiosulfate, fumarate, and DMSO in the absence of oxygen [59, 61, 65-68]. As such, the electron transfer pathway is thought to be highly branched. The anaerobic versatility of S. oneidensis is likely a result of its intricate electron-transfer pathway involving many redox-active proteins, include multi-heme c-type cytochromes. The redox active components of this pathway shuttle electrons to terminal electron acceptors, which are reduced during contact with the proteins located on the outer membrane of the bacteria [69, 70]. When cells are grown under anaerobic conditions, 80% of membrane-bound c-type cytochrome is localized to the outer membrane. This differs from the results of an aerobic growth in which the cytoplasmic membrane has higher cytochrome content than the outer membrane [71]. These findings suggest that c-type cytochromes localized to the outer membrane have a direct role in metal reduction.
Fe(III)-reducing microorganisms are proposed to have developed three different methods by which to transfer electrons from the inside of the cell to extracellular iron: electron shuttling compounds, conductive pili, and finally, direct contact between the ferric iron oxide by c-type cytochromes [72]. Fe(III)-reducing microbes have been shown to form biofilms on the surface of iron(III) oxides [73]. Which mechanism is at work may be influenced by the depth of the biofilm and the availability of nutrients [74]. At very shallow biofilm depths, c-type cytochromes can make direct contact with the iron oxide, whereas at the middle of the biofilm, electron shuttling compounds may be produced where they can be recycled easily. Finally, at a further distance from the iron oxide surface, conductive nanowires could be at work. At this point it is unclear why organisms have evolved to use these three, possibly energetically unfavorable, mechanisms of dissmilatory metal reduction.
When nutrients are scarce, it is believed that these organisms will reduce Fe(III) minerals through direct contact between outer membrane proteins and the mineral surface [58, 75]. In general, cytochromes are not found on the outside of the outer membrane [76]. However, in some DMR organisms, c-type cytochromes are oriented on the cell surface, making a direct interaction with iron minerals and reducing them. For instance, there are cytochromes located on the outside of the outer membrane in Shewanella oneidensis and Geobacter sulfurreducens, which enable direct exchange of electrons with metal oxides [71]. Two Shewanella proteins, MtrC and OmcA, are decaheme cytochromes that have been studied extensively over the last decade. Studies have shown that removal of either of the genes encoding these proteins greatly decreases the Mn(IV) and Fe(III) reductase activity [58, 75]. Additionally, it has been shown that MtrC and OmcA form a high affinity complex with a heightened ability to reduce Fe(III) [64]. A functional investigation of OmcA showed that it was able to bind and reduce hematite through a direct interaction [77]. In order for this mechanism to be viable, there must be a pathway of electron transfer involving periplasmic proteins to form a respiratory chain to the outer membrane cytochromes, which can then directly contact the iron(III) oxide [71].
3.3 Molecular “wires”
The DMR pathway in Shewanella has been studied extensively (see, for example, reference [78]). It is composed of several multiheme cytochromes that form a wire-like pathway from the cell interior to its exterior. Along with cymA, the mtrDEF-omcA-mtrCAB gene cluster has been shown to participate in DMR chemistry directly. The mtrDEF cluster is highly similar to mtrCAB, but has been the focus of few specific studies, aside from the recent biochemical and crystallographic characterization of MtrF [79, 80]. Electrons are generated in the cytoplasmic membrane, entering the quinol pool, at which point they enter the DMR pathway at CymA, a tetraheme protein anchored in the cytoplasmic membrane, which uses menaquinol-7 as an essential cofactor [81]. From CymA, electrons are transferred to periplasmic proteins such as the decaheme cytochrome MtrA or the tetraheme cytochrome STC, and finally, electrons cross the outer membrane through the beta barrel protein, MtrB to MtrC and OmcA, which are thought to directly interact with iron (III) oxides (shown in Figure 3). Importantly, redox chemistry appears to proceed from CymA to the other proposed components of the pathway in a iso-potential fashion, where one impact of the redundancy of a large number of hemes, is that there are no obvious thermodynamic barriers or sinkholes to successful electron transfer [82]. The interaction of quinol anologs with CymA appear to do little to affect the potentials of any of the four CymA heme cofactors (−110, −190, −240 and −265 mV), suggesting equal affinity for the oxidized and reduced states [83]. Further, it has been shown recently that beyond the generation of genetic knock-outs, biochemical studies of the individual components of the Shewanella Mtr-based pathway of electron transfer demonstrate that electrons can be handed off from one protein to another as shown in Figure 3, as monitored by biochemistry [84], direct electrochemistry [85] or NMR studies [86]. Electrochemical analyses further illustrate how nature has tuned the proteins of the Mtr pathway to transfer electrons with a sense of kinetic bias, as electrocatalytic currents generated between proteins occur largely in a single direction [85]. That said, it has also been shown that electron transfer from the outside of Shewanella to the periplasm is also possible, suggesting the possibility of electrosyntheses conducted via multi-heme electron transfer conduits [87].
Fig. 3.
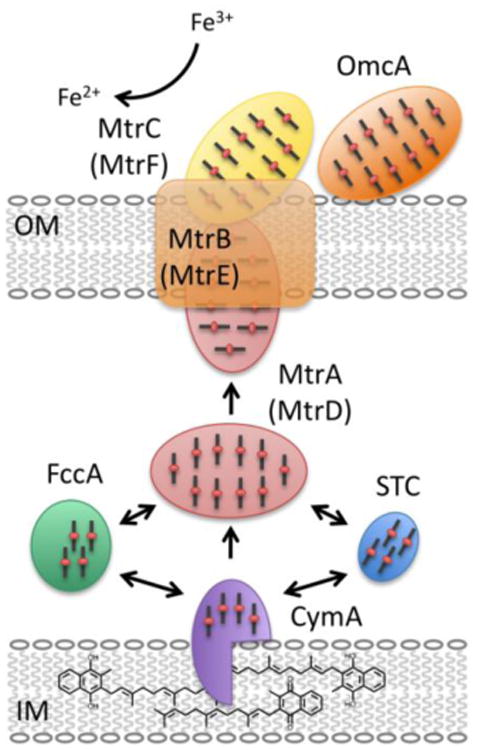
The dissimilatory metal reduction (DMR) pathway in Shewanella oneidensis. A network of multi-heme cytochromes is responsible for the long-range electron transfer from the quinol pool through the tetraheme cytochrome, CymA, across the periplasm via MtrA, and finally to MtrC through the porin protein, MtrB. Arrows indicate substantiated redox reactions through electrochemical and spectroscopic studies (see main text).
In Geobacter, OmcB, OmcE, and OmcS have been implicated in extracellular Fe(III) reduction as removal of the genes encoding these proteins results in decreased Fe(III) reduction [63, 88]. It is proposed that they will transfer electrons directly to the electrode surface similar to the role of MtrC and OmcA [63, 88], but OmcB, OmcE, and OmcS have not been studied to the same extent as the outer membrane cytochromes in Shewanella. In both of these organisms, electrons are transferred via a series of heme cofactors that essentially form a molecular wire from the inside of the cell to the outside.
It is also possible to consider the decaheme cytochromes as individual units. For instance, MtrA itself can be thought of as a molecular wire [79, 89]. MtrA has a remarkably low amino acid to heme ratio, just 34, whereas an average heme containing protein has 60-70 amino acids per heme cofactor [13]. It may perform the unique function of an intermediary periplasmic protein, transporting electrons from CymA to the outer membrane protein MtrC [85]. A structural characterization of MtrA using small-angle X-ray scattering (SAXS) showed that it possesses a rod-like aspect ratio with a maximum dimension of 104 Å (Figure 4) [90]. Crystal structures of other multiheme cytochromes have shown that their hemes are packed as a wire with edge-to-edge distances between 4 and 8 Å [91, 92]. A similar packing of cofactors in MtrA would suggest that the protein is approximately 100 Å in length and thus able to span a substantial portion of the ∼250 Å [93] distance across the periplasm. Overall, these traits are thought to be retained in other paralogs of MtrA, while the other decademe cytochromes involved in long range electron transfer in Shewanella and related organisms (e.g. MtrC, OmcA) are sufficiently divergent to suggest alternative structures [94], as confirmed by the recent structure of MtrF [80] a close paralog of MtrC [95]. Indeed, while MtrA may be thought of best as a wire, the recent x-ray crystal structures of MtrF [80] and an eleven-heme bearing ortholog UndA [96], display a bifurcated cross arrangement, that suggest two intersecting pathways of electron transfer, one that emerges from and is orthogonal to the membrane normal, and another that is parallel to it. While tantalizing, these structures also leave several open questions, including elucidating the nature of interactions with inorganic oxides, redox shuttles, or other proteins. For example it has been established that MtrC can interact tightly with OmcA [58] in Shewanella oneidensis in a 2:1 ratio[64], which presumably facilitates successful direct electron transfer with inorganic materials [77].
Fig. 4.
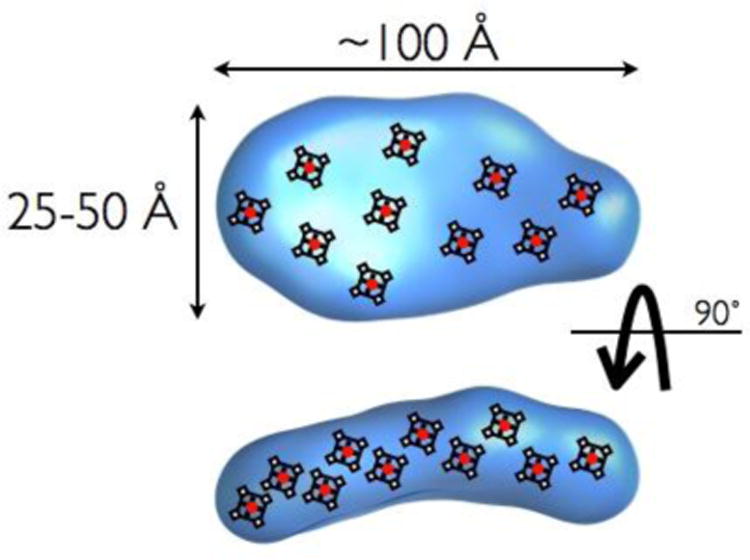
Depiction of MtrA structure, as described by SAXS, by Firer-Sherwood and co-workers [90], with schematized placement of potential porphyrin structures.
Unfortunately, the ability to manipulate the chemistry of the multi-heme cytochromes involved in extra-cellular metal reduction has been limited due to the lack of structural data. While the majority of the information that is known about these proteins, or in the identification of their orthologs, is derived from bioinformatics studies that rely upon the identification of c-type cytochromes via their –CXXCH- heme binding motif. As such, it is hard to identify other types of “nanowire” elements from Nature that may also engage in long-range electron transfer, because they lack a defining amino acid sequence. So while is it possible, and likely, that there are other molecular wires comprised of non-c-type hemes, this approach makes it difficult to identify them. Further, the individual structures that do exist for components of long-range electron transfer pathways often leave more questions than answers. For example, in the case of recent structure of MtrF and its ortholog UndA [96], the potential enzymatic activity, assemblage into larger structures, interaction/recognition of inorganic materials, and ability to participate in redox cycling of flavin compounds (known redox shuttles in Shewanella[97, 98]), and overall relevance to the similar pathways found in Geobacter are all open questions.
4. Storage of reducing equivalents
4.1. Multi-heme enzymes: multi-electron catalysis
Multi-heme containing proteins have the ability to store multiple reducing equivalents [84, 99, 100], which is useful in Nature and in the biomimetic world. Section 3.3 highlights the ability of multiple neighboring c-type hemes to act as a conductive “molecular wire”, and here, we will discuss how a string of neighboring hemes can be used to store electron equivalents for catalysis, using well illustrated examples of catalysis found in biological nitrification and nitrogen metabolism: cytochrome c nitrite reductase (ccNir) and hydroxylamine oxidoreductase (HAO). Intriguingly, the evolutionary relationship between ccNir, HAO and cytochrome c554 has been described recently, suggesting a model for an ancient event of truncation that separated the tetraheme proteins from octaheme ancestors, and that pentaheme ccNir proteins resulted from a distinct event of branching from the HAO family [7, 12]. The substrate specificities of these enzymes, along with other multi-heme cytochromes c has also been recently reviewed [101].
Cytochrome c nitrite reductase (ccNir, or NrfA) is a homo-dimeric protein containing five heme groups per monomer [102]. ccNir is able to catalyze the six electron reduction of nitrite to ammonia, the five electron reduction of nitric oxide to ammonia, as well as the two electron reduction of hydroxylamine to ammonia [103]. Hydroxylamine oxidoreductase is a 24 heme containing homo-trimer that catalyzes the four electron oxidation of hydroxylamine to nitrite [92, 104-106]. There is a design feature that is shared between these two enzymes: the presence of two pathways for electrons to flow to/from the active site [92, 106]. This feature aids in the ability of these enzymes to hold charge generated during catalysis. And, as in the case of bCcP enzymes described above, the role of protein quaternary structure may play a role of some significance: ccNir is an obligate dimer, where it has been recently suggested that negative cooperativity between the active sites of the dimer may be due to redox communication between the two protomers [107]; as discussed below, the organization of hydroxylamine oxidoreductase is cearly significant for the organization of the heme units within the protein trimer. The specific example of hydroxylamine oxidoreductase and its partner protein, cytochrome c554, is discussed in further detail below.
4.2 Ammonia oxidation: hydroxylamine oxidoreductase and cytochrome c554
4.2.1 Ammonia oxidation
The enzymatic system responsible for extracting energy from ammonia in Nitrosomonas europeaea has two catalytic enzymes (ammonia monooxygenase (AMO) and hydroxylamine oxidoreductase (HAO)) and two electron transfer proteins (c554 and c552) [108]. Ammonia is oxidized to hydroxylamine by AMO, which is subsequently oxidized to nitrite by HAO [109]. Reducing equivalents are shuttled from HAO to c554, c552 and ultimately to cytochrome c oxidase (Figure 5). It is interesting to note that HAO, c554 and c552 have different isoelectric points, which may facilitate the protein-protein interactions necessary for electron transfer. HAO, c554 and c552 have pI values of 5.3, 10.7 and 3.7 respectively [108, 110]. Cytochrome c554 has been shown to have a basic patch that is thought to interact directly with HAO [111].
Fig. 5.
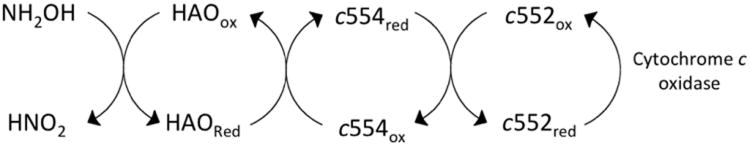
Ammonia oxidation pathway in Nitrosomonas europaea. Hydroxylamine (which is produced from the oxidation of ammonia from ammonia monooxygenase) is oxidized by hydroxylamine oxidoreductase (HAO). Electrons are then transferred through cytochromes c554, c552, and finally to cytochrome c oxidase.
HAO is one of the most complex multi-heme enzymes found in Nature, as the homo-trimer contains 24 heme groups [92, 106]. The crystal structure has been solved (Figure 6A) and shows a complex heme arrangement (Figure 6B) [92]. The trimer holds 18 of the bis-His, c-type hemes in a ring, with the catalytic P460 hemes (one per monomer) slightly above the ring. The additional three c-type heme groups (one per monomer, Heme 1) are outside of the ring structure. P460 is a unique heme that is covalently attached to the protein backbone by two cysteine residues (in a typical -CXXCH- motif) plus an unusual tyrosine residue. P460 is pentacoodinate and is the site of hydroxylamine binding.
Fig. 6.
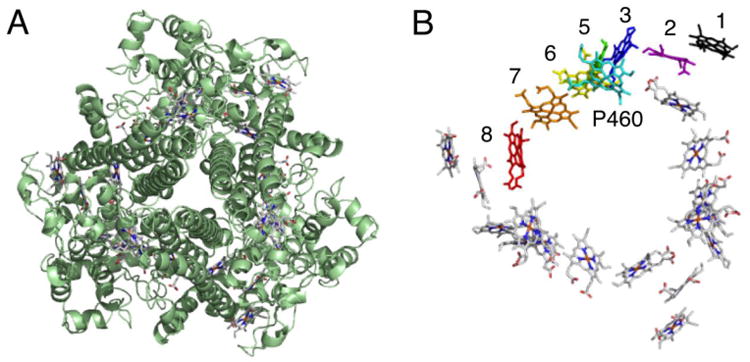
Hydroxylamine oxidoreductase (HAO) is a homotrimer containing 24 heme groups (A). Each monomer contains 8 heme groups with a unique P460 heme (shown in cyan, B). Figure constructed from Protein Data Bank file 1FGJ.pdb.
4.2.2 Hydroxylamine oxidoreductase: electron storage
As mentioned in Section 4.2.1, the heme configuration in HAO is complex and it is this complexity that allows HAO to be an efficient catalyst. Hydroxylamine binds to P460 and the electrons released during the oxidation are quickly transferred from heme to heme in the trimer ring [112]. Recently, the non-equilibrium redox potentials of the hemes were modeled to predict what happens biologically [113]. Previous redox titrations were performed [114, 115], yet have not lead to significant insight into the mechanism of electron transfer. However, the results of the spectropotentiometric titrations, do show that all 8 hemes can be reduced [114], further advancing the concept of using HAO as a reducing equivalent storage unit.
The non-equilibrium model predicts that the redox potentials of any given heme group change depending on the oxidation state of the holo-protein and the protein environment (i.e. binding of c554) [113]. This current model suggests that Heme 2 has the highest potential (between +203 mV and +264 mV vs NHE) and is always reduced. The additional electrons will then be able to flow freely through the other hemes, whose redox potentials are modulated to be isopotential, and settle on Hemes 3 and 8. The ring structure allows electron flow between the subunits and supports the “multiple pathway” idea of electron transfer. The solvent exposed Heme 1 has a negative redox potential (-225 mV) and it is the cooperativity of Heme 1 (lowest potential) and Heme 2 (highest potential) that “locks” the electrons within the homo-trimer. HAO's partner protein, c554, is then able to retrieve these electrons by docking near Heme 1, changing the electrostatic environment and increasing the potential of Heme 1, thus releasing its electrons to the hemes in c554[113, 116]. This electron transfer has been postulated to be a two-electron transfer. Two electrons are thought to enter c554 through heme II and the subsequent intramolecular electron transfer to heme I is fast, where the two hemes operate as a single redox unit. Stopped-flow experiments have shown that HAO and c554 do exchange electrons in vitro: Two electrons have been found to be transferred, each at a different rate: the first at rate of 250-300 sec-1 and the second at a rate of 25-30 sec-1[116].
Spectroelectrochemical analysis of cytochrome c554 revealed the presence of three unique midpoint potentials: +47, -147, and -276 mV (vs NHE) [117, 118], which are depicted in Figure 7A. Further analysis using a coulometric titration showed that the high potential couple was associated with two electrons stoichiometrically, thus accounting for all four redox active heme groups [117]. Protein film voltammetry (PFV) experiments have further resolved the midpoint potentials and the number of electrons in each couple [119]. Hemes I to IV are found to have potentials of +32 , +50, -183 and -283 mV, respectively. Additionally, the cooperativity between heme I/II was observed from direct electrochemistry (Figure 8), as an apparent “narrowing” of the voltammograms associated with the hemes I/II couples. Such behavior verifies the observation of a desirable trait in many bioenergetics systems: the ability to selectively deliver or accept multiple redox equivalents. Figure 8 further illustrates the challenge associated with observing such cooperativity: here the crossing of the redox potentials results in narrowing of the electrochemical signatures, but the effect is subtle. Clearly such phenomena may be lost in cases where there are more than two redox couples in close proximity, such as the cases of the Shewanella cytochromes discussed above [82], [79], or ccNiR [120, 121]: in all of these cases, heme redox potentials appear to be closely spaced in terms of overall thermodynamics, and no cooperativity has been reported to date. The molecular basis for redox cooperativity is poorly understood, and may arise in part from fast intramolecular electron transfer (the hypothesized ET route is from heme II to IV to III to I [118, 122]). This intramolecular electron transfer appears to be much faster than intermolecular or interfacial ET [119]. The coupling of heme I and II along with observed gating may partially support the strict two-electron ET observed in nature between HAO and c554[116, 119].
Fig. 7.
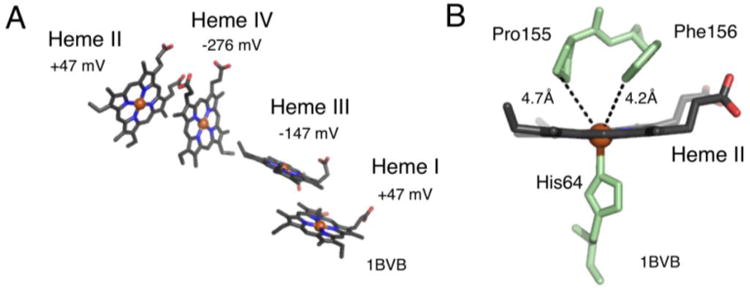
Heme configuration of c554 shows two sets of parallel heme groups (A). The high potential heme groups (Hemes I and II) are the furthest apart, spatially (28.1 Å, Fe-Fe). The heme groups are bis-His ligated, except for Heme II. Heme II is penta-coordinate with one ligated histidine (B). (Protein Data Bank entry 1BVB.pdb).
Fig. 8.
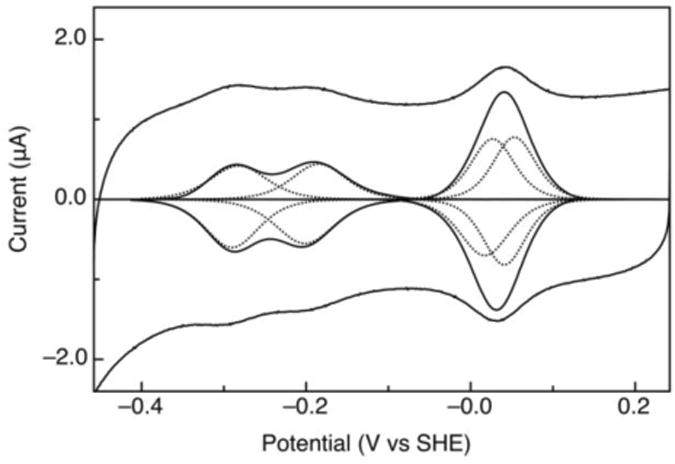
A cyclic voltammogram of c554 on 4-mercaptobenzoic acid modified gold (scan rate of 1V, 4°C, pH 7); both raw and baseline subtracted data. The four heme groups are modeled in with dashed lines.
N. europaea has optimized this ammonia oxidation system in vivo. When taken out of context, it has been found that HAO is not quite an efficient machine, as side reactions have been observed, including it's ability to run in reverse and be a cytochrome c nitrite reductase [123]. HAO functions differently in the presence or absence of oxygen. The oxidation of hydroxylamine by HAO occurs aerobically. Anaerobically, HAO has the ability to reduce nitric oxide or hydroxylamine [124], at least in vitro. This leaky system poses challenges to both in vitro studies and its potential off-label utilization as an electron storage unit.
4.2.3 Cytochrome c554: alternative function
Some researchers have speculated that c554 may have another function within the cell; that it may moonlight as an NO reductase. Joint work from the Hendrich Laboratory (Carnegie Mellon University) and the Hooper Laboratory (University of Minnesota) have shown that heme II from c554 has the ability to bind and reduce nitric oxide [125].
The crystal structure of both oxidized and fully reduced c554 have been solved. The overall fold is predominately alpha helical, and the four heme groups are orientated in two sets of parallel molecules (Figure 7A). The numbering of the hemes is from the order in which they appear in the sequence. Heme groups I, III and IV are bis-His ligated, while heme II is pentacoordinated (one His ligand) (Figure 7B). From the structure alone it is difficult to conjecture why heme II has an open coordination site (Figure 7). It is open in the sense that there is not a sixth ligand, yet Pro155 and Phe156 are in close proximity [111]; either blocking access completely or adding specificity to which small molecules can bind. One challenge is to determine if this penta-coordinate heme is an advantageous product of evolution, or if it is an accidental artifact of the evolutionary process. However it came to be, this penta-coordinate site could be exploited for new reactivity. It is not unrealistic to think the coordination sphere can be tuned in such a way to make c554 a designer catalyst.
5. Concluding remarks: multi-heme proteins as molecular tools
The traits of multi-heme proteins highlighted in this review (redox linked conformational changes, the capacity to yield vectorial electron transfer over the length scale of 102 Å, and the ability to store and manipulate n > 1 redox equivalents) all point the potential utility multi-heme proteins may provide to the emerging field of synthetic biology, where either multi-electron redox catalysis, long-range electron transfer or redox-based conformational dynamics may be required as an a component within a novel biological process. For example, the possibility of using reverse electron flow to achieve electrosynthesis in Shewanella oneidensis via electron transfer into the cellular interior has already been realized [73], and seems an attractive approach to achieve electrosyntheses in other host organisms. At the same time, several challenges remain in the ability to rationalize the electronic and chemical properties of multi-heme cytochromes c, whether as enzymes or as electron transfer elements. For example, while the ability to incorporate the MtrCAB complex from Shewanella oneidensis into Escherichia coli to provide for an exogenous redox conduit has been demonstrated [126], a way forward to improve upon the multi-heme electron transfer complex, by design, is still lacking in our attempts to improve upon Nature's apparent mastery.
Highlights for “Multi-heme proteins: Nature's multi-purpose tool”.
Bacterial cytochrome c peroxidases and cytochrome cd1 reveal redox-linked conformation changes
Multi-heme redox proteins of Shewanella show a bias for long range, unidirectional electron transfer reactions
Hydroxylamine oxidoreductase and cytochrome c554 reveal specificity in multi-electron redox processes
Acknowledgments
The authors wish to acknowledge the support of a Scialog® Award from the Research Corporation for the Advancement of Science, the National Science Foundation (MCB 0546323 and 1122977) and the NIH (GM07266).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Banci L, Bertini I, Luchinat C, Turano P. Special Cofactors and Metal Clusters. In: Bertini I, Gray H, Stiefel E, Valentine J, editors. Biological Inorganic Chemistry: structure and reactivity. University Science Books; Sausalito, California: 2007. pp. 43–56. [Google Scholar]
- 2.Messerschmidt A, Huber R, Poulos T, Wieghardt K. Handbook of Metalloproteins. John Wiley & Sons, Ltd; New York, NY: 2001. [Google Scholar]
- 3.Sanders C, Turkarslan S, Lee DW, Daldal F. Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 2010;18:266–274. doi: 10.1016/j.tim.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartshorne S, Richardson DJ, Simon J. Multiple haem lyase genes indicate substrate specificity in cytochrome c biogenesis. Biochem Soc Trans. 2006;34:146–149. doi: 10.1042/BST0340146. [DOI] [PubMed] [Google Scholar]
- 5.Herbaud ML, Aubert C, Durand MC, Guerlesquin F, Thony-Meyer L, Dolla A. Escherichia coli is able to produce heterologous tetraheme cytochrome c(3) when the ccm genes are co-expressed. Biochim Biophys Acta. 2000;1481:18–24. doi: 10.1016/s0167-4838(00)00117-5. [DOI] [PubMed] [Google Scholar]
- 6.Hartshorne RS, Kern M, Meyer B, Clarke TA, Karas M, Richardson DJ, Simon J. A dedicated haem lyase is required for the maturation of a novel bacterial cytochrome c with unconventional covalent haem binding. Mol Microbiol. 2007;64:1049–1060. doi: 10.1111/j.1365-2958.2007.05712.x. [DOI] [PubMed] [Google Scholar]
- 7.Kern M, Klotz MG, Simon J. The Wollinella succinogenes mcc gene cluster encodes an unconventional respiratory sulphite reduction system. Mol Microbiol. 2011;82:1515–1530. doi: 10.1111/j.1365-2958.2011.07906.x. [DOI] [PubMed] [Google Scholar]
- 8.Bertini I, Gray HB, Stiefel EI, Valentine JS. Biological Inorganic Chemistry: Structure and Reactivity. University Science Books; Sausalito, CA: 2007. [Google Scholar]
- 9.Michel LV, Ye T, Bowman SE, Levin BD, Hahn MA, Russell BS, Elliott SJ, Bren KL. Heme attachment motif mobility tunes cytochrome c redox potential. Biochemistry. 2007;46:11753–11760. doi: 10.1021/bi701177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye T, Kaur R, Wen X, Bren KL, Elliott SJ. Redox properties of wild-type and heme-binding loop mutants of bacterial cytochromes C measured by direct electrochemistry. Inorg Chem. 2005;44:8999–9006. doi: 10.1021/ic051003l. [DOI] [PubMed] [Google Scholar]
- 11.Reedy CJ. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 12.Klotz MG, Schmid MC, Strous M, Op Den Camp HJM, Jetten MSM, Hooper AB. Evolution of an octahaem cytochrome c protein family that is key to aerobic and anaerobic ammonia oxidation by bacteria. Environ Microbiol. 2008;10:3150–3163. doi: 10.1111/j.1462-2920.2008.01733.x. [DOI] [PubMed] [Google Scholar]
- 13.Sharma S, Cavallaro G, Rosato A. A systematic investigation of multiheme c-type cytochromes in prokaryotes. J Biol Inorg Chem. 2010;15:559–571. doi: 10.1007/s00775-010-0623-4. [DOI] [PubMed] [Google Scholar]
- 14.Mowat CG, Rothery E, Miles CS, McIver L, Doherty MK, Drewette K, Taylor P, Walkinshaw MD, Chapman SK, Reid GA. Octaheme tetrathionate reductase is a respiratory enzyme with novel heme ligation. Nat Struct Mol Biol. 2004;11:1023–1024. doi: 10.1038/nsmb827. [DOI] [PubMed] [Google Scholar]
- 15.Boyko KM, Polyakov KM, Tikhonova TV, Slutsky A, Antipov AN, Zvyagilskaya RA, Bourenkov GP, Popov AN, Lamzin VS, Popov VO. Crystallization and preliminary X-ray analysis of cytochrome c nitrite reductase from Thioalkalivibrio nitratireducens. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:215–217. doi: 10.1107/S174430910600296X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tikhonova TV, Slutsky A, Antipov AN, Boyko KM, Polyakov KM, Sorokin DY, Zvyagilskaya RA, Popov VO. Molecular and catalytic properties of a novel cytochrome c nitrite reductase from nitrate-reducing haloalkaliphilic sulfur-oxidizing bacterium Thioalkalivibrio nitratireducens. Biochim Biophys Acta. 2006;1764:715–723. doi: 10.1016/j.bbapap.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 17.Hensen D, Sperling D, Truper HG, Brune DC, Dahl C. Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Mol Microbiol. 2006;62:794–810. doi: 10.1111/j.1365-2958.2006.05408.x. [DOI] [PubMed] [Google Scholar]
- 18.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Crystal structure of horseradish peroxidase C at 2.15 A resolution. Nat Struct Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 19.Finzel BC, Poulos TL, Kraut J. Crystal structure of yeast cytochrome c peroxidase refined at 1.7-A resolution. J Biol Chem. 1984;259:13027–13036. [PubMed] [Google Scholar]
- 20.Davidson VL, Liu A. Tryptophan tryptophylquinone biosynthesis: A radical approach to posttranslational modification. Biochim Biophys Acta. 2012;1824:1299–1305. doi: 10.1016/j.bbapap.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellfolk N, Ronnberg M, Aasa R, Andreasson LE, Vanngard T. Properties and function of the two hemes in Pseudomonas cytochrome c peroxidase. Biochim Biophys Acta. 1983;743:23–30. doi: 10.1016/0167-4838(83)90413-2. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Evidence for redox cooperativity between c-type hemes of MauG which is likely coupled to oxygen activation during tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:821–828. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettigrew GW, Echalier A, Pauleta SR. Structure and mechanism in the bacterial dihaem cytochrome c peroxidases. J Inorg Biochem. 2006;100:551–567. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Fulop V, Ridout CJ, Greenwood C, Hajdu J. Crystal structure of the di-haem cytochrome c peroxidase from Pseudomonas aeruginosa. Structure. 1995;3:1225–1233. doi: 10.1016/s0969-2126(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 25.Echalier A, Brittain T, Wright J, Boycheva S, Mortuza GB, Fulop V, Watmough NJ. Redox-linked structural changes associated with the formation of a catalytically competent form of the diheme cytochrome c peroxidase from Pseudomonas aeruginosa. Biochemistry. 2008;47:1947–1956. doi: 10.1021/bi702064f. [DOI] [PubMed] [Google Scholar]
- 26.Arciero DM, Hooper AB. A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J Biol Chem. 1994;269:11878–11886. [PubMed] [Google Scholar]
- 27.Zahn JA, Arciero DM, Hooper AB, Coats JR, DiSpirito AA. Cytochrome c peroxidase from Methylococcus capsulatus Bath. Arch Microbiol. 1997;168:362–372. doi: 10.1007/s002030050510. [DOI] [PubMed] [Google Scholar]
- 28.Pauleta SR, Cooper A, Nutley M, Errington N, Harding S, Guerlesquin F, Goodhew CF, Moura I, Moura JJ, Pettigrew GW. A copper protein and a cytochrome bind at the same site on bacterial cytochrome c peroxidase. Biochemistry. 2004;43:14566–14576. doi: 10.1021/bi0485833. [DOI] [PubMed] [Google Scholar]
- 29.Ronnberg M, Araiso T, Ellfolk N, Dunford HB. The catalytic mechanism of Pseudomonas cytochrome c peroxidase. Arch Biochem Biophys. 1981;207:197–204. doi: 10.1016/0003-9861(81)90025-4. [DOI] [PubMed] [Google Scholar]
- 30.De Smet L, Savvides SN, Van Horen E, Pettigrew G, Van Beeumen JJ. Structural and mutagenesis studies on the cytochrome c peroxidase from Rhodobacter capsulatus provide new insights into structure-function relationships of bacterial di-heme peroxidases. J Biol Chem. 2006;281:4371–4379. doi: 10.1074/jbc.M509582200. [DOI] [PubMed] [Google Scholar]
- 31.Lee Y, Boycheva S, Brittain T, Boyd PD. Intramolecular electron transfer in the dihaem cytochrome c peroxidase of Pseudomonas aeruginosa. ChemBioChem. 2007;13:1440–1446. doi: 10.1002/cbic.200700159. [DOI] [PubMed] [Google Scholar]
- 32.De Smet L, Pettigrew GW, Van Beeumen JJ. Cloning, overproduction and characterization of cytochrome c peroxidase from the purple phototrophic bacterium Rhodobacter capsulatus. Eur J Biochem. 2001;268:6559–6568. doi: 10.1046/j.0014-2956.2001.02610.x. [DOI] [PubMed] [Google Scholar]
- 33.Alves T, Besson S, Duarte LC, Pettigrew GW, Girio FM, Devreese B, Vandenberghe I, Van Beeumen J, Fauque G, Moura I. A cytochrome c peroxidase from Pseudomonas nautica 617 active at high ionic strength: expression, purification and characterization. Biochim Biophys Acta. 1999;1434:248–259. doi: 10.1016/s0167-4838(99)00188-0. [DOI] [PubMed] [Google Scholar]
- 34.Gilmour R, Goodhew CF, Pettigrew GW, Prazeres S, Moura JJ, Moura I. The kinetics of the oxidation of cytochrome c by Paracoccus cytochrome c peroxidase. Biochem J. 1994;300:907–914. doi: 10.1042/bj3000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prazeres S, Moura JJ, Moura I, Gilmour R, Goodhew CF, Pettigrew GW, Ravi N, Huynh BH. Mossbauer characterization of Paracoccus denitrificans cytochrome c peroxidase. Further evidence for redox and calcium binding-induced heme-heme interaction. J Biol Chem. 1995;270:24264–24269. doi: 10.1074/jbc.270.41.24264. [DOI] [PubMed] [Google Scholar]
- 36.Dias JM, Alves T, Bonifacio C, Pereira AS, Trincao J, Bourgeois D, Moura I, Romao MJ. Structural basis for the mechanism of Ca(2+) activation of the di-heme cytochrome c peroxidase from Pseudomonas nautical 617. Structure. 2004;12:961–973. doi: 10.1016/j.str.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 37.Ellis KE, Frato KE, Elliott SJ. Impact of Quaternary Structure upon Bacterial Cytochrome c Peroxidases: Does Homodimerization Matter? Biochemistry. 2012;51:10008–10016. doi: 10.1021/bi301150n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Echalier A, Goodhew CF, Pettigrew GW, Fulop V. Activation and catalysis of the di-heme cytochrome c peroxidase from Paracoccus pantotrophus. Structure. 2006;14:107–117. doi: 10.1016/j.str.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 39.Shimizu H, Schuller DJ, Lanzilotta WN, Sundaramoorthy M, Arciero DM, Hooper AB, Poulos T. Crystal structure of Nitrosomonas europaea cytochrome c peroxidase and the structural basis for ligand switching in bacterial di-heme peroxidases. Biochemistry. 2001;40:13483–13490. doi: 10.1021/bi011481h. [DOI] [PubMed] [Google Scholar]
- 40.Hoffmann M, Seidel J, Einsle O. CcpA from Geobacter sulfurreducens is a basic di-heme cytochrome c peroxidase. J Mol Biol. 2009;393:951–965. doi: 10.1016/j.jmb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Seidel J, Hoffmann M, Ellis KE, Seidel A, Spatzal T, Gerhardt S, Elliott SJ, Einsle O. MacA is a second cytochrome c peroxidase of Geobacter sulfurreducens. Biochemistry. 2012;51:2747–2756. doi: 10.1021/bi300249u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis KE, Seidel J, Einsle O, Elliott SJ. Geobacter sulfurreducens cytochrome c peroxidases: electrochemical classification of catalytic mechanisms. Biochemistry. 2011;50:4513–4520. doi: 10.1021/bi200399h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Averill BA. Dissimilatory nitrite and nitric oxide reductases. Chem Rev (Washington, DC) 1996;96:2951–2964. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 44.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koppenhofer A, Turner KL, Allen JW, Chapman SK, Ferguson SJ. Cytochrome cd(1) from Paracoccus pantotrophus exhibits kinetically gated, conformationally dependent, highly cooperative two-electron redox behavior. Biochemistry. 2000;39:4243–4249. doi: 10.1021/bi000192a. [DOI] [PubMed] [Google Scholar]
- 46.Besson S, Carneiro C, Moura JJ, Moura I, Fauque G. A cytochrome cd1-type nitrite reductase isolated from the marine denitrifier Pseudomonas nautical 617: purification and characterization. Anaerobe. 1995;1:219–226. doi: 10.1006/anae.1995.1021. [DOI] [PubMed] [Google Scholar]
- 47.Williams PA, Fulop V, Garman EF, Saunders NF, Ferguson SJ, Hajdu J. Haem-ligand switching during catalysis in crystals of a nitrogen-cycle enzyme. Nature. 1997;389:406–412. doi: 10.1038/38775. [DOI] [PubMed] [Google Scholar]
- 48.Baker SC, Saunders NFW, Willis AC, Ferguson SJ, Hajdu J, Fülöp V. Cytochrome cd1 Structure: unusual haem environments in a nitrite reductase and analysis of factors contributing to [beta]-propeller folds1. J Mol Biol. 1997;269:440–455. doi: 10.1006/jmbi.1997.1070. [DOI] [PubMed] [Google Scholar]
- 49.Fulop V, Moir JW, Ferguson SJ, Hajdu J. The anatomy of a bifunctional enzyme: structural basis for reduction of oxygen to water and synthesis of nitric oxide by cytochrome cd1. Cell. 1995;81:369–377. doi: 10.1016/0092-8674(95)90390-9. [DOI] [PubMed] [Google Scholar]
- 50.Nurizzo D, Cutruzzola F, Arese M, Bourgeois D, Brunori M, Cambillau C, Tegoni M. Conformational changes occurring upon reduction and NO binding in nitrite reductase from Pseudomonas aeruginosa. Biochemistry. 1998;37:13987–13996. doi: 10.1021/bi981348y. [DOI] [PubMed] [Google Scholar]
- 51.Nurizzo D, Silvestrini MC, Mathieu M, Cutruzzola F, Bourgeois D, Fulop V, Hajdu J, Brunori M, Tegoni M, Cambillau C. N-terminal arm exchange is observed in the 2.15 A crystal structure of oxidized nitrite reductase from Pseudomonas aeruginosa. Structure. 1997;5:1157–1171. doi: 10.1016/s0969-2126(97)00267-0. [DOI] [PubMed] [Google Scholar]
- 52.Silvestrini M, Falcinelli S, Ciabatti I, Cutruzzolą F, Brunori M. Pseudomonas aeruginosa nitrite reductase (or cytochrome oxidase): an overview. Biochimie. 1994;76:641–654. doi: 10.1016/0300-9084(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 53.Farver O, Kroneck PM, Zumft WG, Pecht I. Allosteric control of internal electron transfer in cytochrome cd1 nitrite reductase. Proc Natl Acad Sci U S A. 2003;100:7622–7625. doi: 10.1073/pnas.0932693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kappler A, Straub KL. Geomicrobiological cycling of iron. Rev Mineral Geochem. 2005;59:85–108. [Google Scholar]
- 55.Lovley DR. Dissimilatory metal reduction. Annu Rev Microbiol. 1993;47:263–290. doi: 10.1146/annurev.mi.47.100193.001403. [DOI] [PubMed] [Google Scholar]
- 56.Nealson KH, Saffarini D. Iron and manganese in anaerobic respiration: Environmental significance, physiology, and regulation. Annu Rev Microbiol. 1994;48:311–343. doi: 10.1146/annurev.mi.48.100194.001523. [DOI] [PubMed] [Google Scholar]
- 57.Arnold RG, Hoffman MR, DiChristina TJ, Picardal FW. Regulation of dissimilatory Fe(III) reduction activity in Shewanella putrefaciens. Appl Environ Microbiol. 1990;56:2811–2817. doi: 10.1128/aem.56.9.2811-2817.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myers JM, Myers CR. Role for outer membrane cytochromes OmcA and OmcB of Shewanella putrefaciens MR-1 in reduction of manganese dioxide. Appl Environ Microbiol. 2001;67:260–269. doi: 10.1128/AEM.67.1.260-269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Myers CR, Nealson KH. Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor. Science. 1988;240:319–1321. doi: 10.1126/science.240.4857.1319. [DOI] [PubMed] [Google Scholar]
- 60.Lovley DR. Dissimilatory Fe(III) and Mn(IV) reduction. Microbiol Mol Biol Rev. 1991;55:259–287. doi: 10.1128/mr.55.2.259-287.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Myers CR, Carstens BP, Antholine WE, Myers JM. Chromium(VI) reductase activity is associated with the cytoplasmic membrane of anaerobically grown Shewanella putrefaciens MR-1. J Appl Microbiol. 2000;88:98–106. doi: 10.1046/j.1365-2672.2000.00910.x. [DOI] [PubMed] [Google Scholar]
- 62.Methe BA, Nelson KE, Eisen JA, Paulsen IT, Nelson W, Heidelberg JF, Wu D, Wu M, Ward N, Beanan MJ, Dodson RJ, Madupu R, Brinkac LM, Daugherty SC, DeBoy RT, Durkin AS, Gwinn M, Kolonay JF, Sullivan SA, Haft DH, Selengut J, Davidsen TM, Zafar N, White O, Tran B, Romero C, Forberger HA, Weidman J, Khouri H, Feldblyum TV, Utterback TR, Van Aken SE, Lovley DR, Fraser CM. Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science. 2003;302:1967–1969. doi: 10.1126/science.1088727. [DOI] [PubMed] [Google Scholar]
- 63.Mehta T, Coppi MV, Childers SE, Lovley DR. Outer membrane c-type cytochromes required for Fe(III) and Mn(IV) oxide reduction in Geobacter sulfurreducens. Appl Environ Microbiol. 2005;71:8634–8641. doi: 10.1128/AEM.71.12.8634-8641.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi L, Chem B, Wang Z, Elias DA, Mayer MU, Gorby YA, Ni S, Lower BH, Kennedy DW, Wunschel DS, Mottaz HM, Marshall MJ, Hill EA, Beliaev AS, Zachara JM, Fredrickson JK, Squier TC. Isolation of a high-affinity functional protein complex between OmcA and MtrC: two outer membrane decaheme c-type cytochromes of Shewanella oneidensis MR-1. J Bacteriol. 2006;188:4705–4714. doi: 10.1128/JB.01966-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Myers CR, Nealson KH. Respiration-linked proton translocation coupled to anaerobic reduction of manganese(IV) and iron(III) in Shewanella putrefaciens MR-1. J Bacteriol. 1990;172:6232–6238. doi: 10.1128/jb.172.11.6232-6238.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Myers CR, Myers JM. Cloning and sequence of cymA, a gene encoding a tetraheme cytochrome c required for reduction of iron(III), fumarate, and nitrate by Shewanella putrefaciens MR-1. J Bacteriol. 1997;179 doi: 10.1128/jb.179.4.1143-1152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tiedje JM. Shewanella-the environmentally versatile genome. Nat Biotechnol. 2002;20:1093–1094. doi: 10.1038/nbt1102-1093. [DOI] [PubMed] [Google Scholar]
- 68.Schwalb C, Chapman SK, Reid GA. The tetraheme cytochrome CymA is required for anaerobic respiration with dimethyl sulfoxide and nitrite in Shewanella oneidensis. Biochemistry. 2003;42:9491–9497. doi: 10.1021/bi034456f. [DOI] [PubMed] [Google Scholar]
- 69.Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, Eisen JA, Seshadri R, Ward N, Methe B, Clayton RA, Meyer T, Tsapin A, Scott J, Beanan M, Brinkac L, Daugherty S, DeBoy RT, Dodson RJ, Durkin AS, Haft DH, Kolonay JF, Madupu R, Peterson JD, Umayam LA, White O, Wolf AM, Vamathevan J, Weidman J, Impraim M, Lee K, Berry K, Lee C, Mueller J, Khouri H, Gill J, Utterback TR, McDonald LA, Feldblyum TV, Smith HO, Venter JC, Nealson KH, Fraser CM. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat Biotechnol. 2002;20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- 70.Lower SK, Hochella MF, Beveridge TJ. Bacterial recognition of mineral surfaces: nanoscale interactions between Shewanella and α-FeOOH. Science. 2001;292:1360–1363. doi: 10.1126/science.1059567. [DOI] [PubMed] [Google Scholar]
- 71.Myers CR, Myers JM. Localization of cytochromes to the outer membrane of anaerobically grown Shewanella putrefaciens MR-1. J Bacteriol. 1992;174:3429–3438. doi: 10.1128/jb.174.11.3429-3438.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weber KA, Achenbach LA, Coates JD. Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction. Nat Rev Microbiol. 2006;4:752–764. doi: 10.1038/nrmicro1490. [DOI] [PubMed] [Google Scholar]
- 73.Reguera G, Nevin KP, Nicoll JS, Covalla SF, Woodard TL, Lovley DR. Biofilm and nanowire production leads to increased current in Geobacter sulfurreducens fuel cells. Appl Environ Microbiol. 2006;72:7345–7348. doi: 10.1128/AEM.01444-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hernandez ME, Newman DK. Extracellular electron transfer. Cell Mol Life Sci. 2001;58:1562–1571. doi: 10.1007/PL00000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beliaev AS, Saffarini DA, McLaughlin JL, Hunnicutt D. MtrC, an outer membrane decahaem c cytochrome required for metal reduction in Shewanella putrefaciens MR-1. Mol Microbiol. 2001;39:722–730. doi: 10.1046/j.1365-2958.2001.02257.x. [DOI] [PubMed] [Google Scholar]
- 76.Thony-Meyer L. Biogenesis of respiratory cytochromes in bacteria. Microbiol Mol Biol Rev. 1997;61:337–376. doi: 10.1128/mmbr.61.3.337-376.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiong Y, Shi L, Baowei C, Mayer MU, Lower BH, Londer Y, Bose S, Hochella MF, Fredrickson JK, Squier TC. High-affinity binding and direct electron transfer to solid metals by the Shewanella oneidensis MR-1 outer membrane c-type cytochrome OmcA. J Am Chem Soc. 2006;128:13978–13979. doi: 10.1021/ja063526d. [DOI] [PubMed] [Google Scholar]
- 78.Richardson DJ, Butt JN, Fredrickson JK, Zachara JM, Shi L, Edwards MJ, White G, Baiden N, Gates AJ, Marritt SJ, Clarke TA. The porin-cytochrome' model for microbe-to-mineral electron transfer. Mol Microbiol. 2012;85:201–212. doi: 10.1111/j.1365-2958.2012.08088.x. [DOI] [PubMed] [Google Scholar]
- 79.Hartshorne RS, L RC, Ross D, Nuester J, Clarke TA, Gates AJ, Mills PC, Fredrickson JK, Zarchara JM, Shi L, Beliaev AS, Marshall MJ, Tien M, Brantley S, Butt JN, Richardson DJ. Characterization of an electron conduit between bacteria and the extracellular environment. Proc Natl Acad Sci U S A. 2009;106:22169–22174. doi: 10.1073/pnas.0900086106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clarke TA, Edwards MJ, Gates AJ, Hall A, White GF, Bradley J, Reardon CL, Shi L, Beliaev AS, Marshall MJ, Wang Z, Watmough NJ, Frederickson JK, Zachara JM, Butt JN, Richardson DJ. Structure of a bacterial cell surface decaheme electron conduit. Proc Natl Acad Sci U S A. 2011;108:9384–9389. doi: 10.1073/pnas.1017200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McMillan DG, Marritt SJ, Butt JN, Jeuken LJ. Menaquinone-7 is specific cofactor in tetraheme quinol dehydrogenase CymA. J Biol Chem. 2012;287:14215–14225. doi: 10.1074/jbc.M112.348813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Firer-Sherwood MA, Pulcu GS, Elliott SJ. Electrochemical interrogations of the Mtr cytochromes from Shewanella: opening a potential window. J Biol Inorg Chem. 2008;13:849–854. doi: 10.1007/s00775-008-0398-z. [DOI] [PubMed] [Google Scholar]
- 83.Marritt SJ, Lowe TG, Bye J, McMillan DG, Shi L, Fredrickson J, Zachara J, Richardson DJ, Cheesman MR, Jeuken LJ, Butt JN. A functional description of CymA, an electron-transfer hub supporting anaerobic respiratory flexibility in Shewanella. Biochem J. 2012;444:465–474. doi: 10.1042/BJ20120197. [DOI] [PubMed] [Google Scholar]
- 84.Schuetz B, Schicklberger M, Kuermann J, Spormann AM, Gescher J. Periplasmic electron transfer via the c-type cytochromes MtrA and FccA of Shewanella oneidensis MR-1. Appl Environ Microbiol. 2009;75:7789–7796. doi: 10.1128/AEM.01834-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Firer-Sherwood MA, Bewley KD, Mock JY, Elliott SJ. Tools for resolving complexity in the electron transfer networks of multiheme cytochromes c. Metallomics. 2011;3:344–348. doi: 10.1039/c0mt00097c. [DOI] [PubMed] [Google Scholar]
- 86.Fonseca BM, Paquete CM, Neto SE, Pacheco I, Soares CM, Louro RO. Mind the gap: cytochrome interactions reveal electron pathways across the periplasm of Shewanella oneidensis MR-1. Biochem J. 2013;449:101–108. doi: 10.1042/BJ20121467. [DOI] [PubMed] [Google Scholar]
- 87.Ross DE, Flynn JM, Baron DB, Gralnick JA, Bond DR. Towards electrosynthesis in Shewanella: energetics of reversing the mtr pathway for reductive metabolism. PLoS One. 2011;6:e16649. doi: 10.1371/journal.pone.0016649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holmes DE, Chaudhuri SK, Nevin KP, Mehta T, Methe BA, Liu A, Ward JE, Woodard TL, Webster J, Lovley DR. Microarray and genetic analysis of electron transfer to electrodes in Geobacter sulfurreducents. Environ Microbiol. 2006;8:1805–1815. doi: 10.1111/j.1462-2920.2006.01065.x. [DOI] [PubMed] [Google Scholar]
- 89.Clarke TA, Holley T, Hartshorne RS, Fredrickson JK, Zachara JM, Shi L, Richardson DJ. The role of multihaem cytochromes in the respiration of nitrite in Escherichia coli and Fe(III) in Shewanella oneidensis. Biochem Soc Trans. 2008;36:1005–1010. doi: 10.1042/BST0361005. [DOI] [PubMed] [Google Scholar]
- 90.Firer-Sherwood MA, Ando N, Drennan CL, Elliott SJ. Solution-based structural analysis of the decaheme cytochrome, MtrA, by small-angle X-ray scattering and analytical ultracentrifugation. J Phys Chem B. 2011;115:11208–11214. doi: 10.1021/jp203603r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Taylor P, Pealing SL, Reid GA, Chapman SK, Walkinshaw MD. Structural and mechanistic mapping of a unique fumarate reductase. Nat Struct Biol. 1999;6:1108–1112. doi: 10.1038/70045. [DOI] [PubMed] [Google Scholar]
- 92.Igarashi N, Moriyama H, Fujiwara T, Fukumori Y, Tanaka N. The 2.8 A structure of hydroxylamine oxidoreductase from a nitrifying chemoautotrophic bacterium, Nitrosomonas europaea. Nat Struct Biol. 1997;4:276–284. doi: 10.1038/nsb0497-276. [DOI] [PubMed] [Google Scholar]
- 93.Seltmann G, Holst O. The Bacterial Cell Wall. Springer; 2002. Periplasmic Space and Rigid Layer; p. 103. [Google Scholar]
- 94.Bewley KD, Firer-Sherwood MA, Mock JY, Ando N, Drennan CL, Elliott SJ. Mind the gap: diversity and reactivty relationships among multihaem cytochromes of the MtrA/DmsE family. Biochem Soc Trans. 2012;40:1268–1273. doi: 10.1042/BST20120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Richardson DJ, Edwards MJ, White GF, Baiden N, Hartshorne RS, Fredrickson J, Shi L, Zachara J, Gates AJ, Butt JN, Clarke TA. Exploring the biochemistry at the extracellular redox frontier of bacterial mineral Fe(III) respiration. Biochem Soc Trans. 2012:1–15. doi: 10.1042/BST20120018. [DOI] [PubMed] [Google Scholar]
- 96.Edwards MJ, Hall A, Shi L, Frederickson JK, Zachara JM, Butt JN, Richardson DJ, Clarke TA. The crystal structure of the extracellular 11-heme cytochrome UndA reveals a conserved 10-heme motif and defined binding site for soluble iron chelates. Structure. 2012;20:1275–1284. doi: 10.1016/j.str.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 97.Brutinel ED, Gralnick JA. Shuttling happens: soluble flavin mediators of extracellular electron transfer in Shewanella. Appl Microbiol Biotechnol. 2012;93:41–48. doi: 10.1007/s00253-011-3653-0. [DOI] [PubMed] [Google Scholar]
- 98.Marsili E, Baron DB, Shikhare ID, Courselle D, Gralnick JA, Bond DR. Shewanella secretes flavins that mediate extracellular electron transfer. Proc Natl Acad Sci U S A. 2008;105:3968–3973. doi: 10.1073/pnas.0710525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodrigues ML, Oliveira TF, Pereira IAC, Archer M. X-ray structure of the membrane-bound cytochrome c quinol dehydrogenase NrfH reveals novel haem coordination. EMBO J. 2006;25:5951–5960. doi: 10.1038/sj.emboj.7601439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Esteve-Núñez A, Sosnik J, Visconti P, Lovley DR. Fluorescent properties of c-type cytochromes reveal their potential role as an extracytoplasmic electron sink in Geobacter sulfurreducens. Environ Microbiol. 2008;10:497–505. doi: 10.1111/j.1462-2920.2007.01470.x. [DOI] [PubMed] [Google Scholar]
- 101.Simon J, Kern M, Hermann B, Einsle O, Butt JN. Physiological function and catalytic versatility of bacterial multihaem cytochromes c involved in nitrogen and sulfur cycling. Biochem Soc Trans. 2011;39:1864–1870. doi: 10.1042/BST20110713. [DOI] [PubMed] [Google Scholar]
- 102.Einsle O, Messerschmidt A, Stach P, Bourenkov GP, Bartunik HD, Huber R, Kroneck PMH. Structure of cytochrome c nitrite reductase. Nature. 1999;400:476–480. doi: 10.1038/22802. [DOI] [PubMed] [Google Scholar]
- 103.Einsle O, Messerschmidt A, Huber R, Kroneck PMH, Neese F. Mechanism of the six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase. J Am Chem Soc. 2002;124:11737–11745. doi: 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- 104.Sayavedra-Soto LA, Hommes NG, Arp DJ. Characterization of the gene encoding hydroxylamine oxidoreductase in Nitrosomonas europaea. J Bacteriol. 1994;176:504–510. doi: 10.1128/jb.176.2.504-510.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Andersson KK, Kent TA, Lipscomb JD, Hooper AB, Münck E. Mössbauer, EPR, and optical studies of the P-460 center of hydroxylamine oxidoreductase from Nitrosomonas. A ferrous heme with an unusually large quadrupole splitting. J Biol Chem. 1984;259:6833–6840. [PubMed] [Google Scholar]
- 106.Arciero DM, Hooper AB. Hydroxylamine oxidoreductase from Nitrosomonas europaea is a multimer of an octa-heme subunit. J Biol Chem. 1993;268:14645–14654. [PubMed] [Google Scholar]
- 107.Judd ET, Youngblut M, Pacheco AA, Elliott SJ. Direct Electrochemistry of Shewanella oneidensis Cytochrome c Nitrite Reductase: Evidence of Interactions across the Dimeric Interface. Biochemistry. 2012;51:10175–10185. doi: 10.1021/bi3011708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yamanaka T, Shinra M. Cytochrome c-552 and cytochrome c-554 derived from Nitrosomonas europaea. J Biochem. 1974;75:1265–1273. doi: 10.1093/oxfordjournals.jbchem.a130510. [DOI] [PubMed] [Google Scholar]
- 109.Arp D, Sayavedra-Soto L, Hommes N. Molecular biology and biochemistry of ammonia oxidation by Nitrosomonas europaea. Arch Microbiol. 2002;178:250–255. doi: 10.1007/s00203-002-0452-0. [DOI] [PubMed] [Google Scholar]
- 110.Hooper AB, Maxwell PC, Terry KR. Hydroxylamine oxidoreductase from Nitrosomonas: absorption spectra and content of heme and metal. Biochemistry. 1978;17:2984–2989. doi: 10.1021/bi00608a007. [DOI] [PubMed] [Google Scholar]
- 111.Iverson TM, Arciero DM, Hsu BT, Logan MSP, Hooper AB, Rees DC. Heme packing motifs revealed by the crystal structure of the tetra-heme cytochrome c554 from Nitrosomonas europaea. Nat Struct Mol Biol. 1998;5:1005–1012. doi: 10.1038/2975. [DOI] [PubMed] [Google Scholar]
- 112.Hooper AB, Debey P, Andersson KK, Balny C. Heme P460 of hydroxylamine oxidoreductase of Nitrosomonas. Eur J Biochem. 1983;134:83–87. doi: 10.1111/j.1432-1033.1983.tb07534.x. [DOI] [PubMed] [Google Scholar]
- 113.Kurnikov IV, Ratner MA, Pacheco AA. Redox equilibria in hydroxylamine oxidoreductase. Electrostatic control of electron redistribution in multielectron oxidative processes. Biochemistry. 2005;44:1856–1863. doi: 10.1021/bi048060v. [DOI] [PubMed] [Google Scholar]
- 114.Collins MJ, Arciero DM, Hooper AB. Optical spectropotentiometric resolution of the hemes of hydroxylamine oxidoreductase. Heme quantitation and pH dependence of Em. J Biol Chem. 1993;268:14655–14662. [PubMed] [Google Scholar]
- 115.Prince RC, Hooper AB. Resolution of the hemes of hydroxylamine oxidoreductase by redox potentiometry and electron spin resonance spectroscopy. Biochemistry. 1987;26:970–974. doi: 10.1016/0014-5793(83)81154-5. [DOI] [PubMed] [Google Scholar]
- 116.Arciero DM, Balny C, Hooper AB. Spectroscopic and rapid kinetic studies of reduction of cytochrome c554 by hydroxylamine oxidoreductase from Nitrosomonas europaea. Biochemistry. 1991;30:11466–11472. doi: 10.1021/bi00112a014. [DOI] [PubMed] [Google Scholar]
- 117.Arciero DM, Collins MJ, Haladjian J, Bianco P, Hooper AB. Resolution of the four hemes of cytochrome c554 from Nitrosomonas europaea by redox potentiometry and optical spectroscopy. Biochemistry. 1991;30:11459–11465. doi: 10.1021/bi00112a013. [DOI] [PubMed] [Google Scholar]
- 118.Upadhyay AK, Petasis DT, Arciero DM, Hooper AB, Hendrich MP. Spectroscopic characterization and assignment of reduction potentials in the tetraheme cytochrome c554 from Nitrosomonas Europaea. J Am Chem Soc. 2003;125:1738–1747. doi: 10.1021/ja020922x. [DOI] [PubMed] [Google Scholar]
- 119.Pulcu GS, Elmore BL, Arciero DM, Hooper AB, Elliott SJ. Direct electrochemistry of tetraheme cytochrome c554 from Nitrosomonas europaea: Redox cooperativity and gating. J Am Chem Soc. 2007;129:1838–1839. doi: 10.1021/ja065657k. [DOI] [PubMed] [Google Scholar]
- 120.Youngblut M, Judd ET, Srajer V, Sayyed B, Goelzer T, Elliott SJ, Schmidt M, Pacheco AA. Laue crystal structure of Shewanella oneidensis cytochrome c nitrite reductase from a high-yield expression system. J Biol Inorg Chem. 2012;17:647–662. doi: 10.1007/s00775-012-0885-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gates AJ, Kemp GL, To CY, Mann J, Marritt SJ, Mayes AG, Richardson DJ, Butt JN. The relationship between redox enzyme activity and electrochemical potential-cellular and mechanistic implications from protein film electrochemistry. Phys Chem Chem Phys. 2011;13:7720–7731. doi: 10.1039/c0cp02887h. [DOI] [PubMed] [Google Scholar]
- 122.Iverson TM, Arciero DM, Hooper AB, Rees DC. High-resolution structures of the oxidized and reduced states of cytochrome c554 from Nitrosomonas europaea. J Biol Inorg Chem. 2001;6:390–397. doi: 10.1007/s007750100213. [DOI] [PubMed] [Google Scholar]
- 123.Kostera J, McGarry J, Pacheco AA. Enzymatic interconversion of ammonia and nitrite: The right tool for the job. Biochemistry. 2010;49:8546–8553. doi: 10.1021/bi1006783. [DOI] [PubMed] [Google Scholar]
- 124.Kostera J, Youngblut MD, Slosarczyk JM, Pacheco AA. Kinetic and product distribution analysis of NO* reductase activity in Nitrosomonas europaea hydroxylamine oxidoreductase. J Biol Inorg Chem. 2008;13:1073–1083. doi: 10.1007/s00775-008-0393-4. [DOI] [PubMed] [Google Scholar]
- 125.Upadhyay AK, Hooper AB, Hendrich MP. NO reductase activity of the tetraheme cytochrome c554 of Nitrosomonas europaea. J Am Chem Soc. 2006;128:4330–4337. doi: 10.1021/ja055183+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jensen HM, Albers AE, Malley KR, Londer YY, Cohen BE, Helms BA, Weigele P, Groves JT, Ajo-Franklin CM. Engineering of a synthetic electron conduit in living cells. Proc Natl Acad Sci U S A. 2010;107:19213–19218. doi: 10.1073/pnas.1009645107. [DOI] [PMC free article] [PubMed] [Google Scholar]